The Influence of Gene Aberrations on Survival in Resected IDH Wildtype Glioblastoma Patients: A Single-Institution Study

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
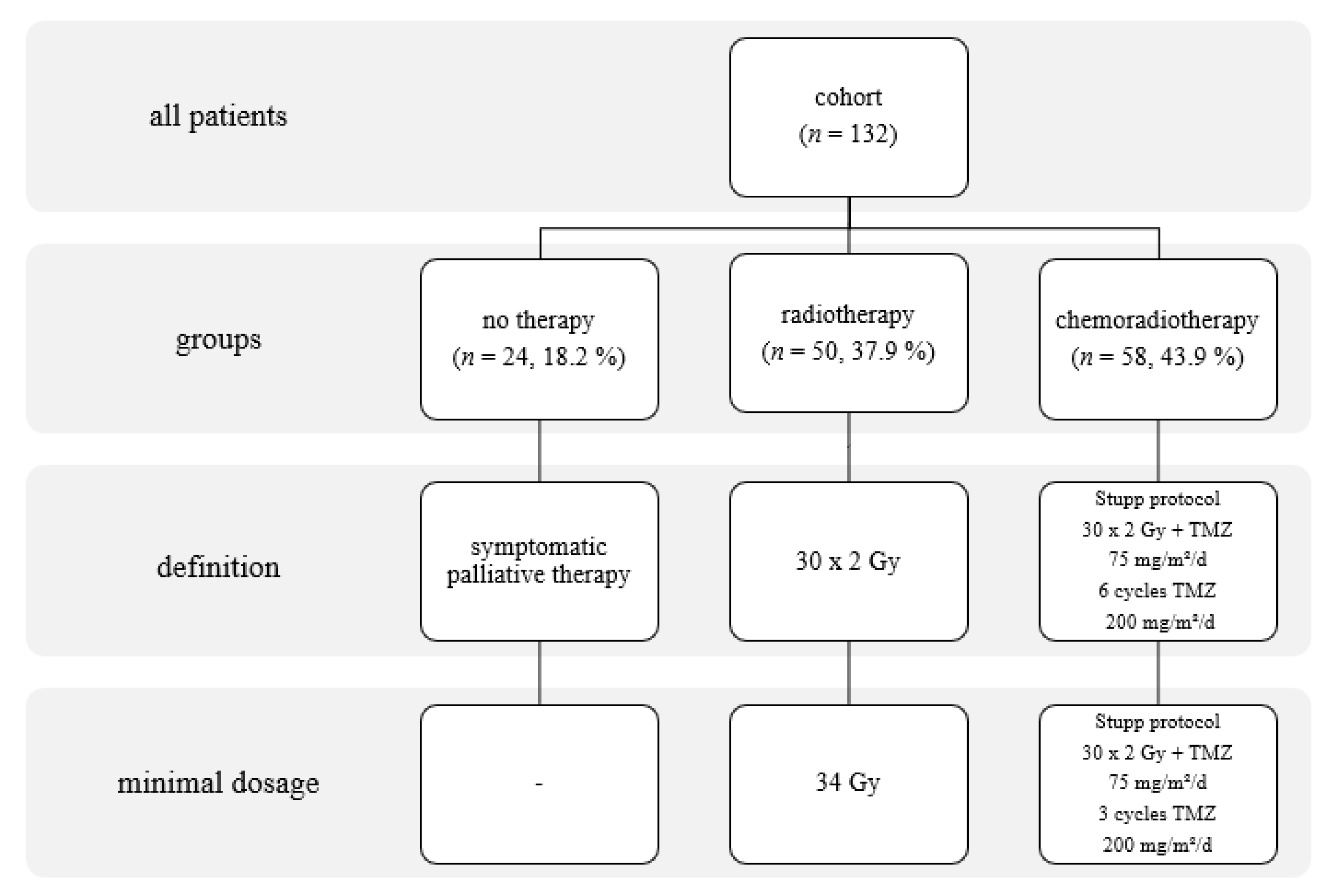
2.1. Patients
2.2. Immunohistochemical Determination of IDH1 R132H Status
2.3. Immunohistochemical Determination of Ki67 Status
2.4. IDH1 R132 and IDH2 R172 Genotyping
2.5. FISH Analysis
2.6. Setting Copy Number Cutoff Values
2.7. MGMT Methylation Status
2.8. Statistical Analysis
3. Results
3.1. Baseline Patient Characteristics
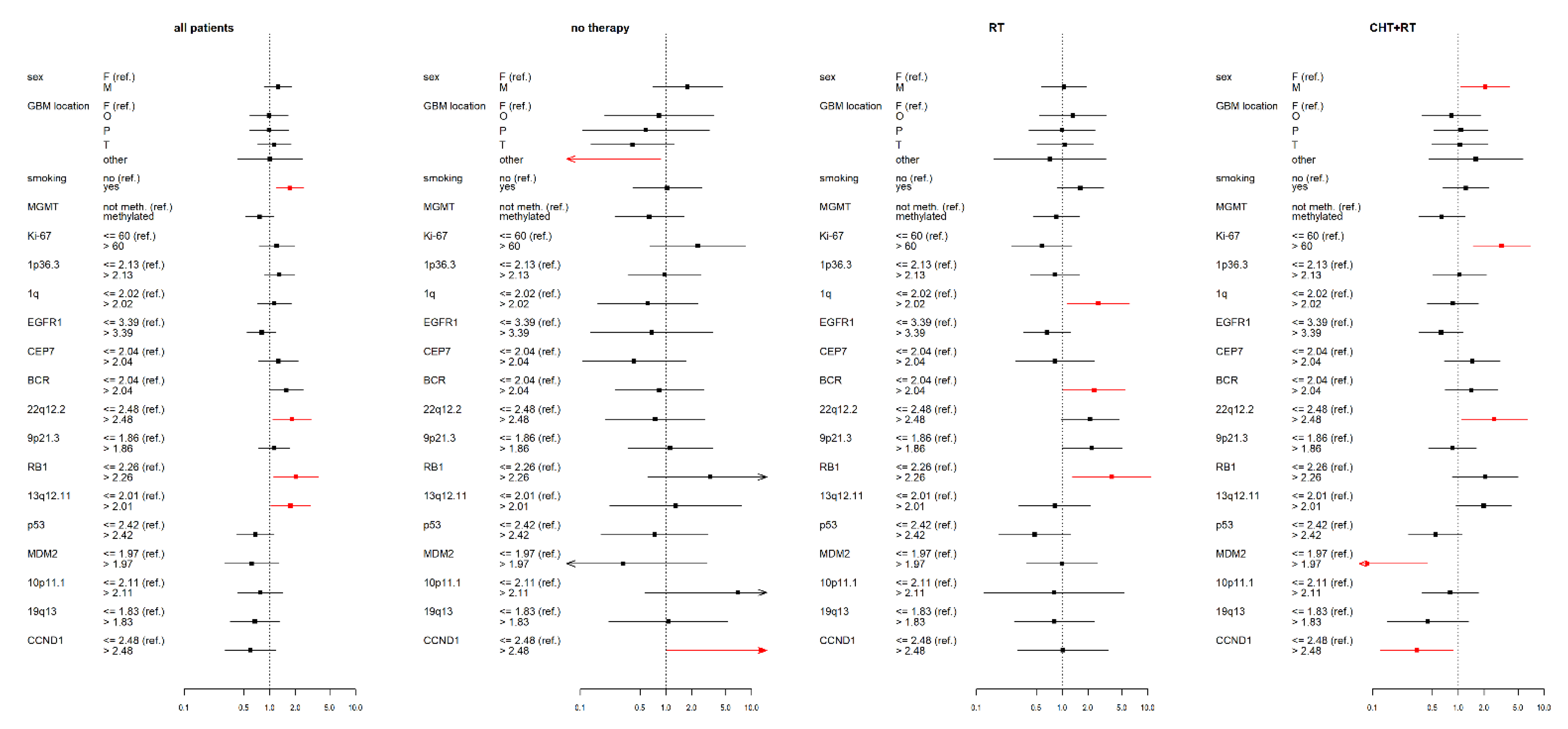
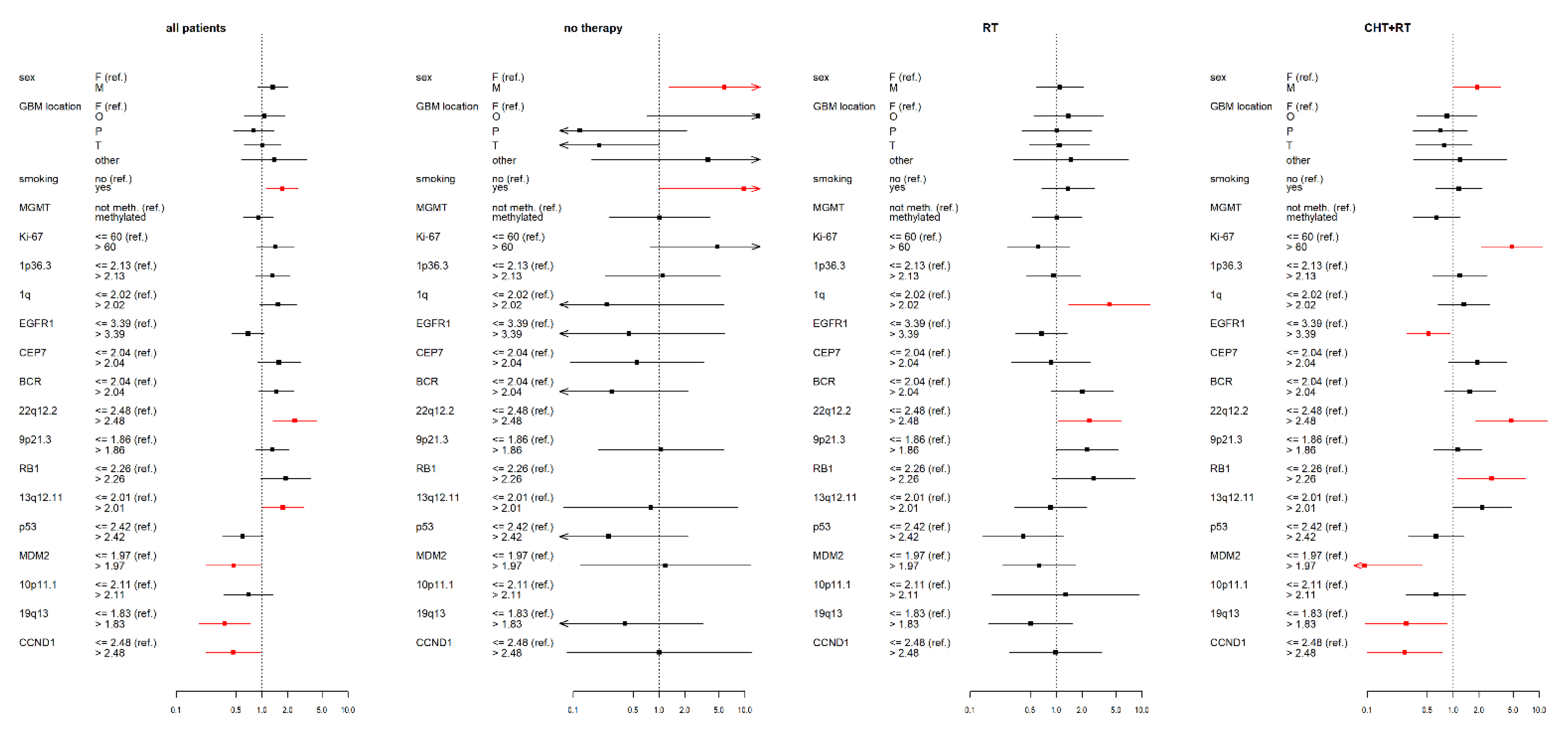
3.2. IDH wt GBM Patients Receiving Chemoradiotherapy
3.3. IDH wt GBM Patients Receiving Radiotherapy
3.4. IDH wt GBM Patients with No Therapy
3.5. Statistical Data for the Complete IDH wt GBM Cohort
3.6. Summary of Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. Available online: http://pmc/articles/PMC1929165/?report=abstract (accessed on 23 October 2020). [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.V. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. Available online: http://pmc/articles/PMC4185005/?report=abstract (accessed on 14 November 2020). [CrossRef] [PubMed]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. Available online: http://pmc/articles/PMC3337451/?report=abstract (accessed on 14 November 2020). [CrossRef]
- Braganza, M.Z.; Rajaraman, P.; Park, Y.; Inskip, P.D.; Freedman, N.D.; Hollenbeck, A.R.; De González, A.B.; Kitahara, C.M. Cigarette smoking, alcohol intake, and risk of glioma in the NIH-AARP Diet and Health Study. Br. J. Cancer 2013, 110, 242–248. Available online: https://pubmed.ncbi.nlm.nih.gov/24335921/ (accessed on 14 November 2020). [CrossRef]
- Hou, L.; Jiang, J.; Liu, B.; Han, W.; Wu, Y.; Zou, X.; Nasca, P.C.; Xue, F.; Chen, Y.; Zhang, B.; et al. Smoking and adult glioma: A population-based case-control study in China. Neuro Oncol. 2016, 18, 105–113. Available online: https://academic.oup.com/neuro-oncology/article-lookup/doi/10.1093/neuonc/nov146 (accessed on 12 December 2020). [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Bent, M.J.V.D.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. Available online: https://pubmed.ncbi.nlm.nih.gov/19269895/ (accessed on 24 October 2020). [CrossRef]
- Liang, J.; Lv, X.; Lu, C.; Ye, X.; Chen, X.; Fu, J.; Luo, C.; Zhao, Y. Prognostic factors of patients with Gliomas—An analysis on 335 patients with Glioblastoma and other forms of Gliomas. BMC Cancer 2020, 20, 35. Available online: https://bmccancer.biomedcentral.com/articles/10.1186/s12885-019-6511-6 (accessed on 15 November 2020). [CrossRef]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019, 9, 963. Available online: http://www.frontiersin.org (accessed on 15 November 2020). [CrossRef] [PubMed]
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863. Available online: https://pubmed.ncbi.nlm.nih.gov/31405017/ (accessed on 15 November 2020). [CrossRef] [PubMed]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. Available online: http://pmc/articles/PMC7286557/?report=abstract (accessed on 13 December 2020). [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. Available online: https://pubmed.ncbi.nlm.nih.gov/27157931/ (accessed on 23 October 2020). [CrossRef]
- DeWitt, J.C.; Jordan, J.T.; Frosch, M.P.; Samore, W.R.; Iafrate, A.J.; Louis, D.N.; Lennerz, J.K. Cost-effectiveness of IDH testing in diffuse gliomas according to the 2016 WHO classification of tumors of the central nervous system recommendations. Neuro Oncol. 2017, 19, 1640–1650. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29016871 (accessed on 28 January 2019). [CrossRef]
- Sanai, N.; Berger, M.S. Glioma extent of resection and its impact on patient outcome. Neurosurgery 2008, 62, 753–764. [Google Scholar] [CrossRef]
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef]
- Ziu, M.; Kim, B.Y.S.; Jiang, W.; Ryken, T.; Olson, J.J. The role of radiation therapy in treatment of adults with newly diagnosed glioblastoma multiforme: A systematic review and evidence-based clinical practice guideline update. J. Neuro Oncol. 2020, 150, 215–267. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Geng, R.; Yuan, F.; Sun, Q.; Liu, B.; Chen, Q. Identification of differentially expressed key genes between glioblastoma and low-grade glioma by bioinformatics analysis. PeerJ 2019, 7, e6560. Available online: http://pmc/articles/PMC6409090/?report=abstract (accessed on 10 December 2020).
- Li, L.; Liu, X.; Ma, X.; Deng, X.; Ji, T.; Hu, P.; Wan, R.; Qiu, H.; Cui, D.; Gao, L. Identification of key candidate genes and pathways in glioblastoma by integrated bioinformatical analysis. Exp. Ther. Med. 2019, 18, 3439–3449. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn (accessed on 10 December 2020). [CrossRef]
- Galbraith, K.; Kumar, A.; Abdullah, K.G.; Walker, J.M.; Adams, S.H.; Prior, T.; Dimentberg, R.; Henderson, F.C.; Mirchia, K.; Sathe, A.A.; et al. Molecular Correlates of Long Survival in IDH-Wildtype Glioblastoma Cohorts. J. Neuropathol. Exp. Neurol. 2020, 79, 843–854. Available online: https://pubmed.ncbi.nlm.nih.gov/32647886/ (accessed on 10 December 2020). [CrossRef] [PubMed]
- Dmitrenko, V.; Iershov, A.V.; Stetsyuk, P.I.; Lykhovid, A.P.; Laptin, Y.P.; Schwartz, D.R.; Mekler, A.A.; Kavsan, V.M. Determination of molecular glioblastoma subclasses on the basis of analysis of gene expression. Cytol. Genet. 2014, 48, 383–391. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma Subclasses Can Be Defined by Activity among Signal Transduction Pathways and Associated Genomic Alterations. PLoS ONE 2009, 4, e7752. Available online: http://pmc/articles/PMC2771920/?report=abstract (accessed on 22 November 2020). [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Q.; Wu, F.; Li, J.-J.; Li, Y.-F.; Liu, X.; Wang, Z.; Chai, R.-C. Gene Expression Profiling Stratifies IDH-Wildtype Glioblastoma with Distinct Prognoses. Front. Oncol. 2019, 9, 1433. Available online: http://pmc/articles/PMC6929203/?report=abstract (accessed on 10 December 2020). [CrossRef] [PubMed]
- Ma, S.; Rudra, S.; Campian, J.L.; Dahiya, S.; Dunn, G.P.; Johanns, T.; Goldstein, M.; Kim, A.H.; Huang, J. Prognostic impact of CDKN2A/B deletion, TERT mutation, and EGFR amplification on histological and molecular IDH-wildtype glioblastoma. Neuro Oncol. Adv. 2020, 2. Available online: https://academic.oup.com/noa/article/doi/10.1093/noajnl/vdaa126/5908751 (accessed on 10 December 2020). [CrossRef]
- Christians, A.; Adel-Horowski, A.; Banan, R.; Lehmann, U.; Bartels, S.; Behling, F.; Barrantes-Freer, A.; Stadelmann, C.; Rohde, V.; Stockhammer, F.; et al. The prognostic role of IDH mutations in homogeneously treated patients with anaplastic astrocytomas and glioblastomas. Acta Neuropathol. Commun. 2019, 7, 156. Available online: http://pmc/articles/PMC6798425/?report=abstract (accessed on 19 January 2021). [CrossRef] [PubMed]
- Biau, J.; Chautard, E.; de Schlichting, E.; Dupic, G.; Pereira, B.; Fogli, A.; Müller-Barthélémy, M.; Dalloz, P.; Khalil, T.; Dillies, A.F.; et al. Radiotherapy plus temozolomide in elderly patients with glioblastoma: A “real-life” report. Radiat. Oncol. 2017, 12, 197. Available online: http://pmc/articles/PMC5719937/?report=abstract (accessed on 19 January 2021). [CrossRef] [PubMed]
- Incekara, F.; van der Voort, S.R.; Dubbink, H.J.; Atmodimedjo, P.N.; Nandoe Tewarie, R.; Lycklama, G.; Vincent, A.J.P.E.; Kros, J.M.; Klein, S.; van den Bent, M.; et al. Topographical Mapping of 436 Newly Diagnosed IDH Wildtype Glioblastoma With vs. Without MGMT Promoter Methylation. Front. Oncol. 2020, 10, 596. Available online: https://www.frontiersin.org/article/10.3389/fonc.2020.00596/full (accessed on 19 January 2021). [CrossRef] [PubMed]
- Amelot, A.; de Cremoux, P.; Quillien, V.; Polivka, M.; Adle-Biassette, H.; Lehmann-Che, J.; Françoise, L.; Carpentier, A.F.; George, B.; Mandonnet, E.; et al. IDH-Mutation Is a Weak Predictor of Long-Term Survival in Glioblastoma Patients. PLoS ONE 2015, 10, e0130596. Available online: http://pmc/articles/PMC4497660/?report=abstract (accessed on 19 January 2021). [CrossRef] [PubMed]
- Sheikh, S.; Radivoyevitch, T.; Barnholtz-Sloan, J.S.; Vogelbaum, M. Long-term trends in glioblastoma survival: Implications for historical control groups in clinical trials. Neuro Oncol. Pract. 2020, 7, 158–163. Available online: https://pubmed.ncbi.nlm.nih.gov/32626584/ (accessed on 1 January 2021). [CrossRef] [PubMed]
- Stensjøen, A.L.; Solheim, O.; Kvistad, K.A.; Håberg, A.K.; Salvesen, Ø.; Berntsen, E.M. Growth dynamics of untreated glioblastomas in vivo. Neuro Oncol. 2015, 17, 1402–1411. Available online: https://pubmed.ncbi.nlm.nih.gov/25758748/ (accessed on 10 December 2020). [CrossRef]
- Pardee, A.B. G1 events and regulation of cell proliferation. Science 1989, 246, 603–608. Available online: https://pubmed.ncbi.nlm.nih.gov/2683075/ (accessed on 24 November 2020). [CrossRef]
- Zhang, D.; Dai, D.; Zhou, M.; Li, Z.; Wang, C.; Lu, Y.; Li, Y.; Wang, J. Inhibition of Cyclin D1 Expression in Human Glioblastoma Cells is Associated with Increased Temozolomide Chemosensitivity. Cell. Physiol. Biochem. 2018, 51, 2496–2508. Available online: https://www.karger.com/Article/FullText/495920 (accessed on 24 November 2020). [CrossRef] [PubMed]
- Laigle-Donadey, F.; Crinière, E.; Benouaich, A.; Lesueur, E.; Mokhtari, K.; Hoang-Xuan, K.; Sanson, M. Loss of 22q Chromosome is Related to Glioma Progression and Loss of 10q. J. Neuro Oncol. 2005, 76, 265–268. Available online: https://link.springer.com/article/10.1007/s11060-005-7019-2 (accessed on 24 November 2020). [CrossRef] [PubMed]
- Nakamura, M.; Ishida, E.; Shimada, K.; Kishi, M.; Nakase, H.; Sakaki, T.; Konishi, N. Frequent LOH on 22q12.3 and TIMP-3 inactivation occur in the progression to secondary glioblastomas. Lab. Investig. 2004, 85, 165–175. Available online: https://pubmed.ncbi.nlm.nih.gov/15592495/ (accessed on 24 November 2020). [CrossRef] [PubMed][Green Version]
- Oskam, N.T.; Bijleveld, E.H.; Hulsebos, T.J.M. A region of common deletion in 22q13.3 in human glioma associated with astrocytoma progression. Int. J. Cancer 2000, 85, 336–339. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/%28SICI%291097-0215%2820000201%2985%3A3%3C336%3A%3AAID-IJC7%3E3.0.CO%3B2-9 (accessed on 24 November 2020). [CrossRef]
- Fisher, C. The diversity of soft tissue tumours with EWSR1 gene rearrangements: A review. Histopathology 2014, 64, 134–150. Available online: https://pubmed.ncbi.nlm.nih.gov/24320889/ (accessed on 24 November 2020). [CrossRef]
- Pandita, A.; Balasubramaniam, A.; Perrin, R.; Shannon, P.; Guha, A. Malignant and benign ganglioglioma: A pathological and molecular study1. Neuro Oncol. 2007, 9, 124–134. Available online: http://academic.oup.com/neuro-oncology/article/9/2/124/1116583/Malignant-and-benign-ganglioglioma-A-pathological (accessed on 24 November 2020). [CrossRef] [PubMed]
- Cimino, P.J.; McFerrin, L.; Wirsching, H.-G.; Arora, S.; Bolouri, H.; Rabadan, R.; Weller, M.; Holland, E.C. Copy number profiling across glioblastoma populations has implications for clinical trial design. Neuro Oncol. 2018, 20, 1368–1373. Available online: https://academic.oup.com/neuro-oncology/article/20/10/1368/5047422 (accessed on 10 December 2020). [CrossRef]
- Geisenberger, C.; Mock, A.; Warta, R.; Rapp, C.; Schwager, C.; Korshunov, A.; Nied, A.-K.; Capper, D.; Brors, B.; Jungk, C.; et al. Molecular profiling of long-term survivors identifies a subgroup of glioblastoma characterized by chromosome 19/20 co-gain. Acta Neuropathol. 2015, 130, 419–434. Available online: https://pubmed.ncbi.nlm.nih.gov/25931051/ (accessed on 10 December 2020). [CrossRef]
- Mirchia, K.; Richardson, T.E. Beyond IDH-mutation: Emerging molecular diagnostic and prognostic features in adult diffuse gliomas. Cancers 2020, 12, 1817. Available online: https://www.mdpi.com/journal/cancers (accessed on 10 December 2020). [CrossRef]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; I Lindeman, N.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro Oncol. 2019, 21, 167–178. Available online: https://pubmed.ncbi.nlm.nih.gov/30189035/ (accessed on 27 December 2020). [CrossRef]
- Radke, J.; Koch, A.; Pritsch, F.; Schumann, E.; Misch, M.; Hempt, C.; Lenz, K.; Löbel, F.; Paschereit, F.; Heppner, F.L.; et al. Predictive MGMT status in a homogeneous cohort of IDH wildtype glioblastoma patients. Acta Neuropathol. Commun. 2019, 7, 89. Available online: https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-019-0745-z (accessed on 27 December 2020). [CrossRef] [PubMed]
- Marchi, F.; Sahnane, N.; Cerutti, R.; Cipriani, D.; Barizzi, J.; Stefanini, F.M.; Epistolio, S.; Cerati, M.; Balbi, S.; Mazzucchelli, L.; et al. The Impact of Surgery in IDH 1 Wild Type Glioblastoma in Relation with the MGMT Deregulation. Front Oncol. 2020, 9, 1569. Available online: https://www.frontiersin.org/article/10.3389/fonc.2019.01569/full (accessed on 27 December 2020). [CrossRef] [PubMed]
- Li, H.-X.; Peng, X.-X.; Zong, Q.; Zhang, K.; Wang, M.-X.; Liu, Y.; Han, G.-I. Cigarette smoking and risk of adult glioma: A meta-analysis of 24 observational studies involving more than 2.3 million individuals. Onco Targets Ther. 2016, 9, 3511–3523. Available online: http://pmc/articles/PMC4913539/?report=abstract (accessed on 22 November 2020). [PubMed]
- Gittleman, H.; Cioffi, G.; Chunduru, P.; Molinaro, A.M.; Berger, M.S.; E Sloan, A.; Barnholtz-Sloan, J.S. An independently validated nomogram for isocitrate dehydrogenase-wild-type glioblastoma patient survival. Neuro Oncol. Adv. 2019, 1. Available online: https://pubmed.ncbi.nlm.nih.gov/31608326/ (accessed on 10 December 2020). [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | DNA Sequence | Final Concentration (µM) |
|---|---|---|---|
| MGMT promoter methylation | Forward | 5′-CGAATATACTAAAACAACCCGCG-3′ | 1.0 |
| Reverse | 5′-GTATTTTTTCGGGAGCGAGGC-3′ | 1.0 | |
| Probe | FAM-BHQ-CAAATCCTCGCGATACGCACCGTTTACG | 0.2 | |
| E-cadherin promoter methylation | Forward | 5′-AATTTTAGGTTAGAGGGTTATCGCGT-3′ | 1.0 |
| Reverse | 5′-TCCCCAAAACGAAACTAACGAC-3′ | 1.0 | |
| Probe | FAM-BHQ-CGCCCACCCGACCTCGCAT | 0.2 | |
| Alu-M5 promoter methylation | Forward | 5′-GGTATGATGGCGTATGTTTGT-3′ | 0.17 |
| Reverse | 5′-GACTCACCACAACTTCCAC-3′ | 0.17 | |
| Probe | FAM-BHQ-AAACGATTCTCCTACCTCAACCTCCCGAA | 0.03 |
| Characteristics | Level | No Therapy | RT | CHT+RT | All Patients |
|---|---|---|---|---|---|
| Gender | F | 10/24 (41.7%) | 25/50 (50%) | 17/58 (29.3%) | 52/132 (39.4%) |
| M | 14/24 (58.3%) | 25/50 (50%) | 41/58 (70.7%) | 80/132 (60.6%) | |
| GBM location | F | 8/24 (33.3%) | 14/50 (28%) | 20/58 (34.5%) | 42/132 (31.8%) |
| O | 3/24 (12.5%) | 10/50 (20%) | 10/58 (17.2%) | 23/132 (17.4%) | |
| P | 2/24 (8.3%) | 9/50 (18%) | 13/58 (22.4%) | 24/132 (18.2%) | |
| T | 10/24 (41.7%) | 15/50 (30%) | 12/58 (20.7%) | 37/132 (28%) | |
| other | 1/24 (4.2%) | 2/50 (4%) | 3/58 (5.2%) | 6/132 (4.5%) | |
| Smoker * | No | 9/24 (37.5%) | 33/50 (66%) | 41/58 (70.7%) | 83/132 (62.9%) |
| Yes | 15/24 (62.5%) | 17/50 (34%) | 17/58 (29.3%) | 49/132 (37.1%) | |
| MGMT | Not methylated | 16/23 (69.6%) | 31/48 (64.6%) | 33/52 (63.5%) | 80/123 (65%) |
| Methylated | 7/23 (30.4%) | 17/48 (35.4%) | 19/52 (36.5%) | 43/123 (35%) | |
| Age category * | ≤55 | 2/24 (8.3%) | 2/50 (4%) | 19/58 (32.8%) | 23/132 (17.4%) |
| >55 | 22/24 (91.7%) | 48/50 (96%) | 39/58 (67.2%) | 109/132 (82.6%) | |
| Karnofsky score * | 0–79 | 16/24 (66.7%) | 18/50 (36%) | 12/58 (20.7%) | 46/132 (34.8%) |
| 80–100 | 8/24 (33.3%) | 32/50 (64%) | 46/58 (79.3%) | 86/132 (65.2%) | |
| Ki67 | ≤60 | 20/24 (83.3%) | 41/49 (83.7%) | 49/58 (84.5%) | 110/131 (84%) |
| >60 | 4/24 (16.7%) | 8/49 (16.3%) | 9/58 (15.5%) | 21/131 (16%) | |
| 1q CN | ≤2.02 | 5/15 (33.3%) | 12/33 (36.4%) | 13/39 (33.3%) | 30/87 (34.5%) |
| >2.02 | 10/15 (66.7%) | 21/33 (63.6%) | 26/39 (66.7%) | 57/87 (65.5%) | |
| 22q CN | ≤2.48 | 13/16 (81.2%) | 30/39 (76.9%) | 33/41 (80.5%) | 76/96 (79.2%) |
| >2.48 | 3/16 (18.8%) | 9/39 (23.1%) | 8/41 (19.5%) | 20/96 (20.8%) | |
| CEP7 CN | ≤2.04 | 4/16 (25%) | 4/41 (9.8%) | 10/50 (20%) | 18/107 (16.8%) |
| >2.04 | 12/16 (75%) | 37/41 (90.2%) | 40/50 (80%) | 89/107 (83.2%) | |
| BCR CN | ≤2.04 | 6/15 (40%) | 11/31 (35.5%) | 14/39 (35.9%) | 31/85 (36.5%) |
| >2.04 | 9/15 (60%) | 20/31 (64.5%) | 25/39 (64.1%) | 54/85 (63.5%) | |
| EGFR1 CN | ≤3.39 | 6/16 (37.5%) | 25/44 (56.8%) | 26/53 (49.1%) | 57/113 (50.4%) |
| >3.39 | 10/16 (62.5%) | 19/44 (43.2%) | 27/53 (50.9%) | 56/113 (49.6%) | |
| 9p21.3 CN | ≤1.86 | 6/17 (35.3%) | 10/41 (24.4%) | 16/49 (32.7%) | 32/107 (29.9%) |
| >1.86 | 11/17 (64.7%) | 31/41 (75.6%) | 33/49 (67.3%) | 75/107 (70.1%) | |
| 1p36.3 CN | ≤2.13 | 12/20 (60%) | 29/45 (64.4%) | 43/52 (82.7%) | 84/117 (71.8%) |
| >2.13 | 8/20 (40%) | 16/45 (35.6%) | 9/52 (17.3%) | 33/117 (28.2%) | |
| 13q12.11 CN | ≤2.01 | 3/10 (30%) | 6/31 (19.4%) | 14/41 (34.1%) | 23/82 (28%) |
| >2.01 | 7/10 (70%) | 25/31 (80.6%) | 27/41 (65.9%) | 59/82 (72%) | |
| RB1 CN | ≤2.26 | 14/16 (87.5%) | 37/42 (88.1%) | 42/49 (85.7%) | 93/107 (86.9%) |
| >2.26 | 2/16 (12.5%) | 5/42 (11.9%) | 7/49 (14.3%) | 14/107 (13.1%) | |
| P53 CN | ≤2.42 | 14/18 (77.8%) | 38/44 (86.4%) | 41/52 (78.8%) | 93/114 (81.6%) |
| >2.42 | 4/18 (22.2%) | 6/44 (13.6%) | 11/52 (21.2%) | 21/114 (18.4%) | |
| 10p11.1 | ≤2.11 | 13/14 (92.9%) | 33/36 (91.7%) | 35/47 (74.5%) | 81/97 (83.5%) |
| >2.11 | 1/14 (7.1%) | 3/36 (8.3%) | 12/47 (25.5%) | 16/97 (16.5%) | |
| 19q13 CN | ≤1.83 | 2/18 (11.1%) | 4/43 (9.3%) | 4/53 (7.5%) | 10/114 (8.8%) |
| >1.83 | 16/18 (88.9%) | 39/43 (90.7%) | 49/53 (92.5%) | 104/114 (91.2%) | |
| MDM2 CN | ≤1.97 | 1/17 (5.9%) | 5/42 (11.9%) | 3/51 (5.9%) | 9/110 (8.2%) |
| >1.97 | 16/17 (94.1%) | 37/42 (88.1%) | 48/51 (94.1%) | 101/110 (91.8%) | |
| CCND1 CN | ≤2.48 | 12/14 (85.7%) | 29/32 (90.6%) | 40/47 (85.1%) | 81/93 (87.1%) |
| >2.48 | 2/14 (14.3%) | 3/32 (9.4%) | 7/47 (14.9%) | 12/93 (12.9%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalita, O.; Sporikova, Z.; Hajduch, M.; Megova Houdova, M.; Slavkovsky, R.; Hrabalek, L.; Halaj, M.; Klementova, Y.; Dolezel, M.; Drabek, J.; et al. The Influence of Gene Aberrations on Survival in Resected IDH Wildtype Glioblastoma Patients: A Single-Institution Study. Curr. Oncol. 2021, 28, 1280-1293. https://doi.org/10.3390/curroncol28020122
Kalita O, Sporikova Z, Hajduch M, Megova Houdova M, Slavkovsky R, Hrabalek L, Halaj M, Klementova Y, Dolezel M, Drabek J, et al. The Influence of Gene Aberrations on Survival in Resected IDH Wildtype Glioblastoma Patients: A Single-Institution Study. Current Oncology. 2021; 28(2):1280-1293. https://doi.org/10.3390/curroncol28020122
Chicago/Turabian StyleKalita, Ondrej, Zuzana Sporikova, Marian Hajduch, Magdalena Megova Houdova, Rastislav Slavkovsky, Lumir Hrabalek, Matej Halaj, Yvona Klementova, Martin Dolezel, Jiri Drabek, and et al. 2021. "The Influence of Gene Aberrations on Survival in Resected IDH Wildtype Glioblastoma Patients: A Single-Institution Study" Current Oncology 28, no. 2: 1280-1293. https://doi.org/10.3390/curroncol28020122
APA StyleKalita, O., Sporikova, Z., Hajduch, M., Megova Houdova, M., Slavkovsky, R., Hrabalek, L., Halaj, M., Klementova, Y., Dolezel, M., Drabek, J., Tuckova, L., Ehrmann, J., Jr., Vrbkova, J., Trojanec, R., & Vaverka, M. (2021). The Influence of Gene Aberrations on Survival in Resected IDH Wildtype Glioblastoma Patients: A Single-Institution Study. Current Oncology, 28(2), 1280-1293. https://doi.org/10.3390/curroncol28020122