
C-phycoerythrin from Phormidium persicinum Prevents Acute Kidney Injury by Attenuating Oxidative and Endoplasmic Reticulum Stress


 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
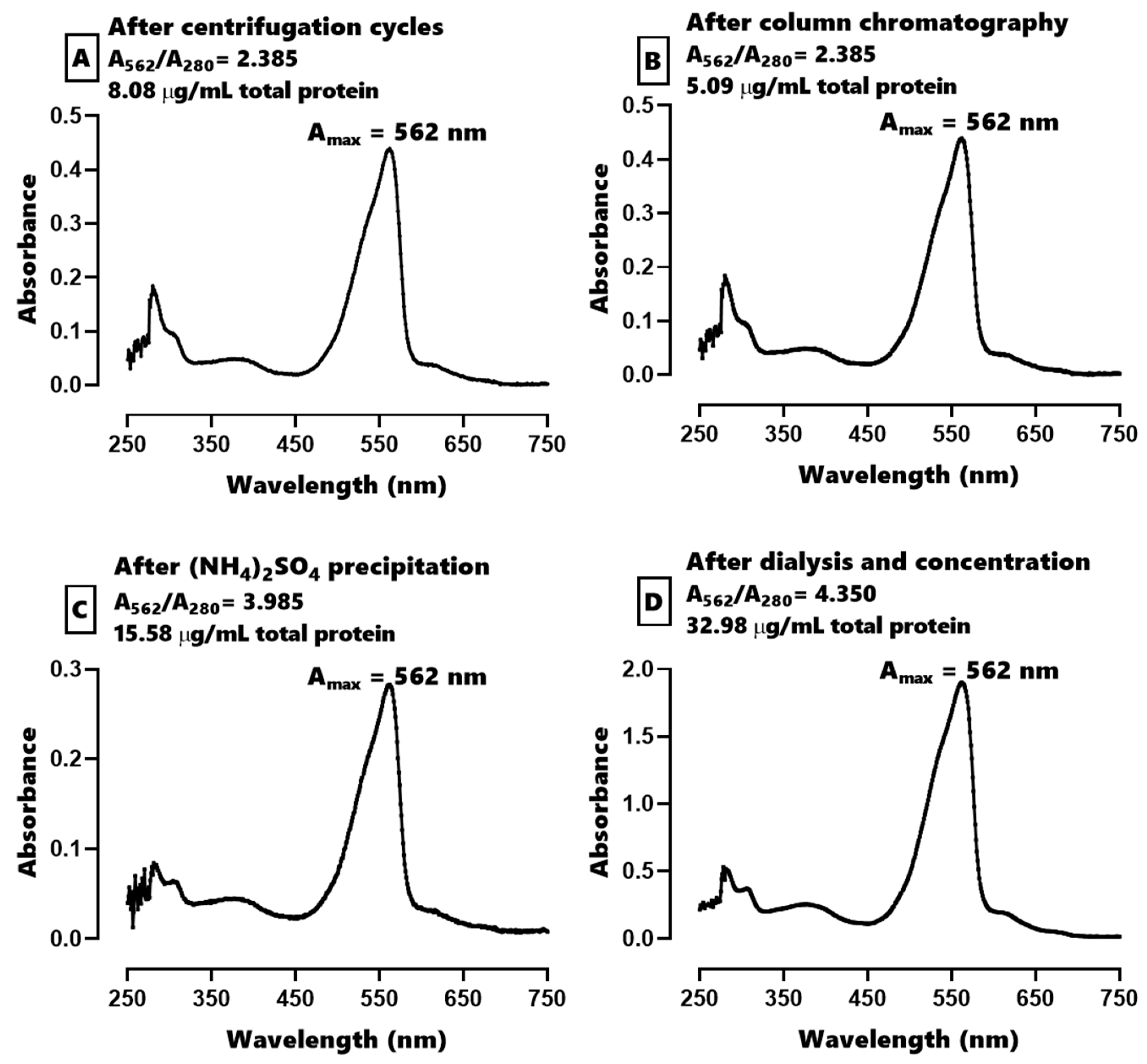
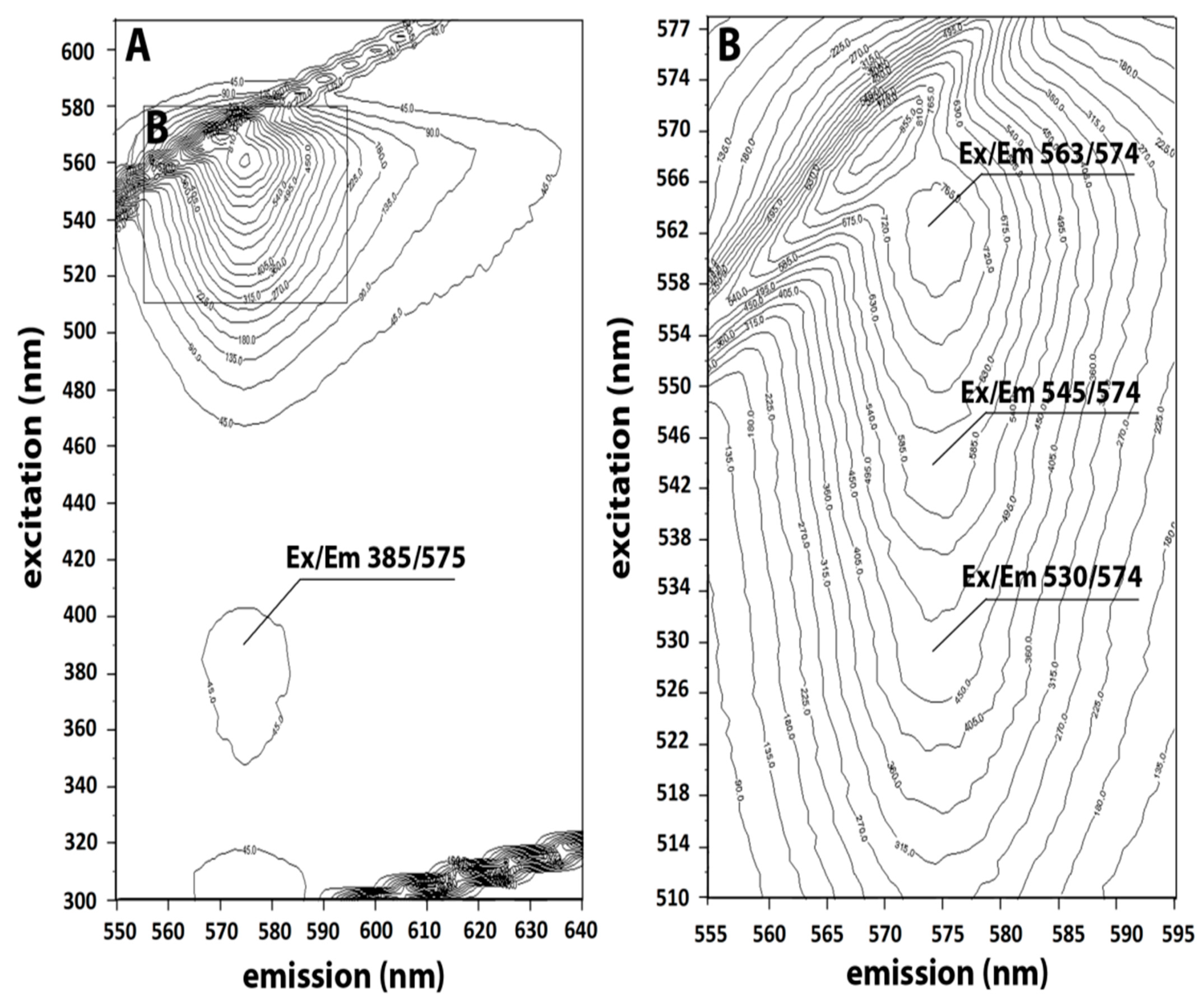
2.1. Characterization of C-PE from Phormidium persicinum
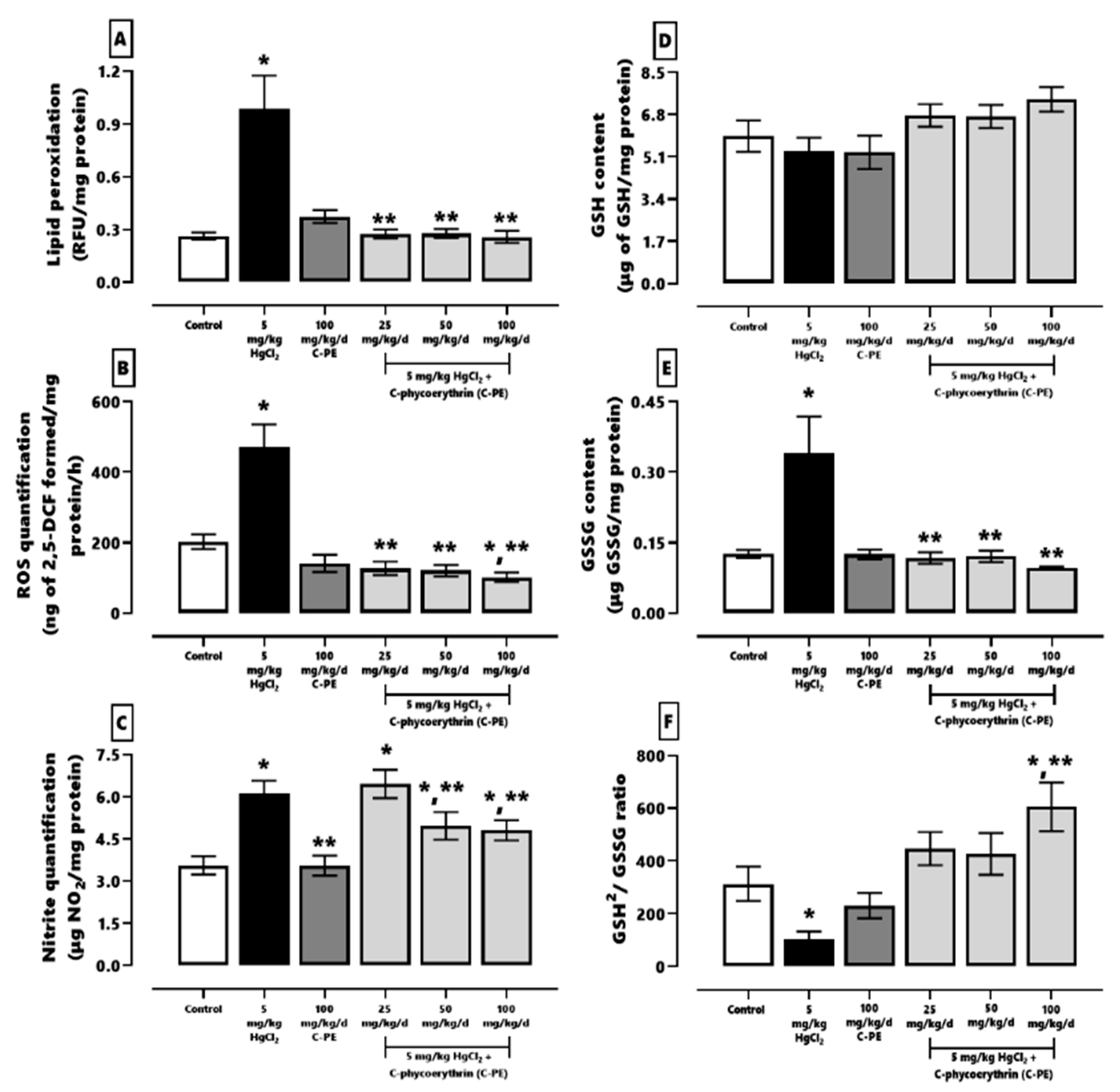
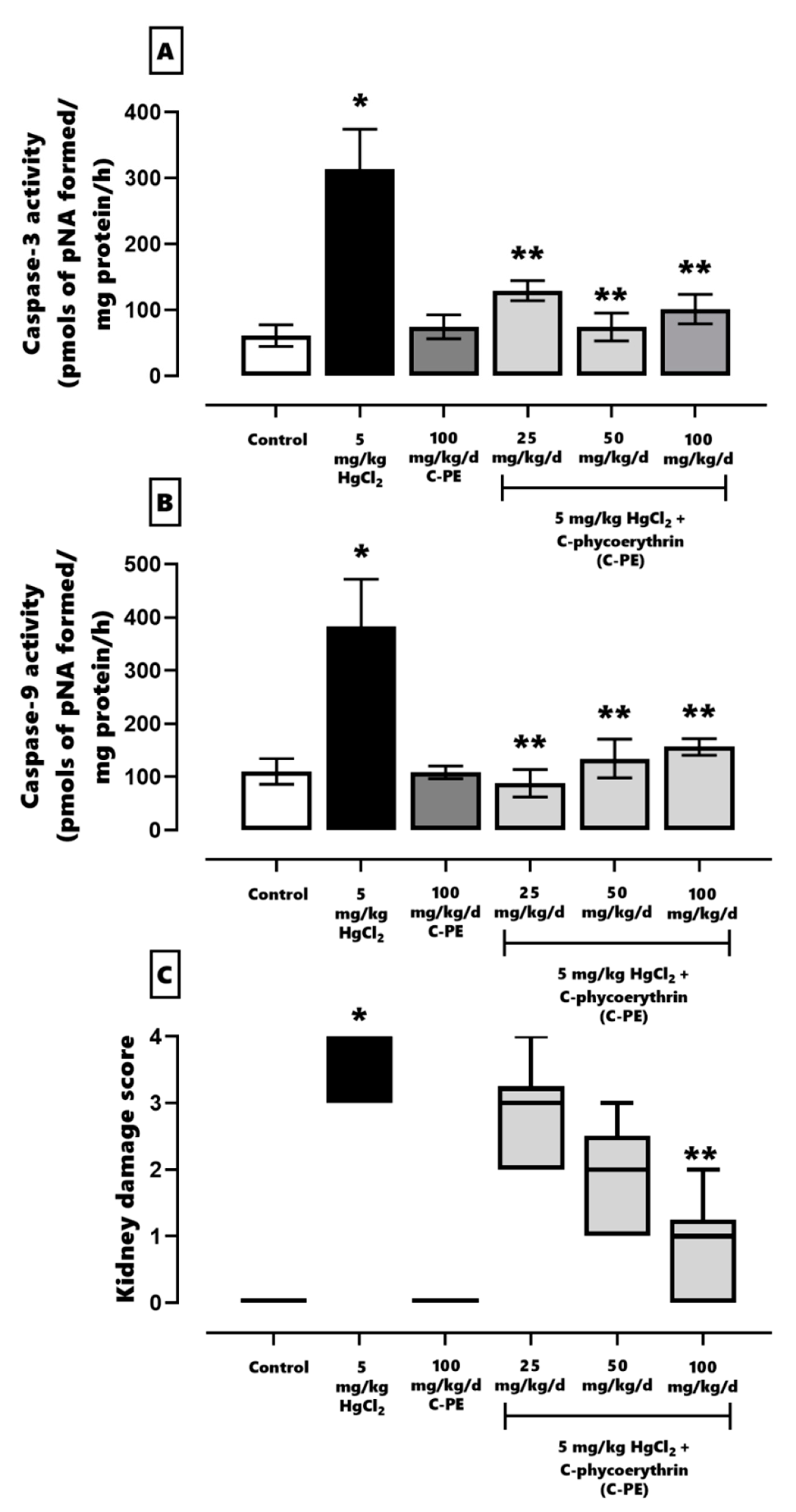
2.2. Evaluation of Oxidative Stress, the Redox Environment, the Activity of Effector Caspases 3 and 9, the Expression of Nephrin and Podocin, and Renal Damage
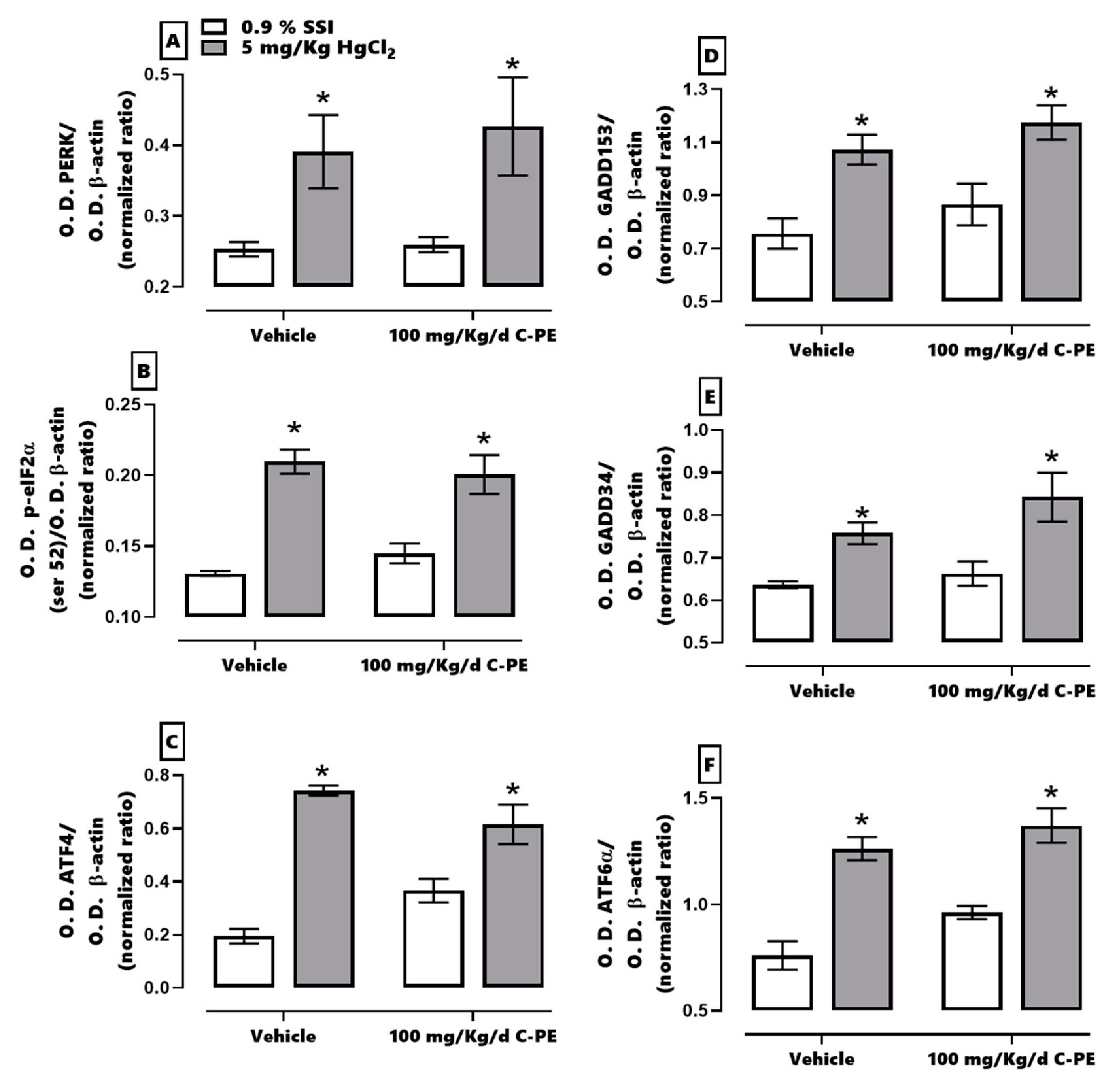
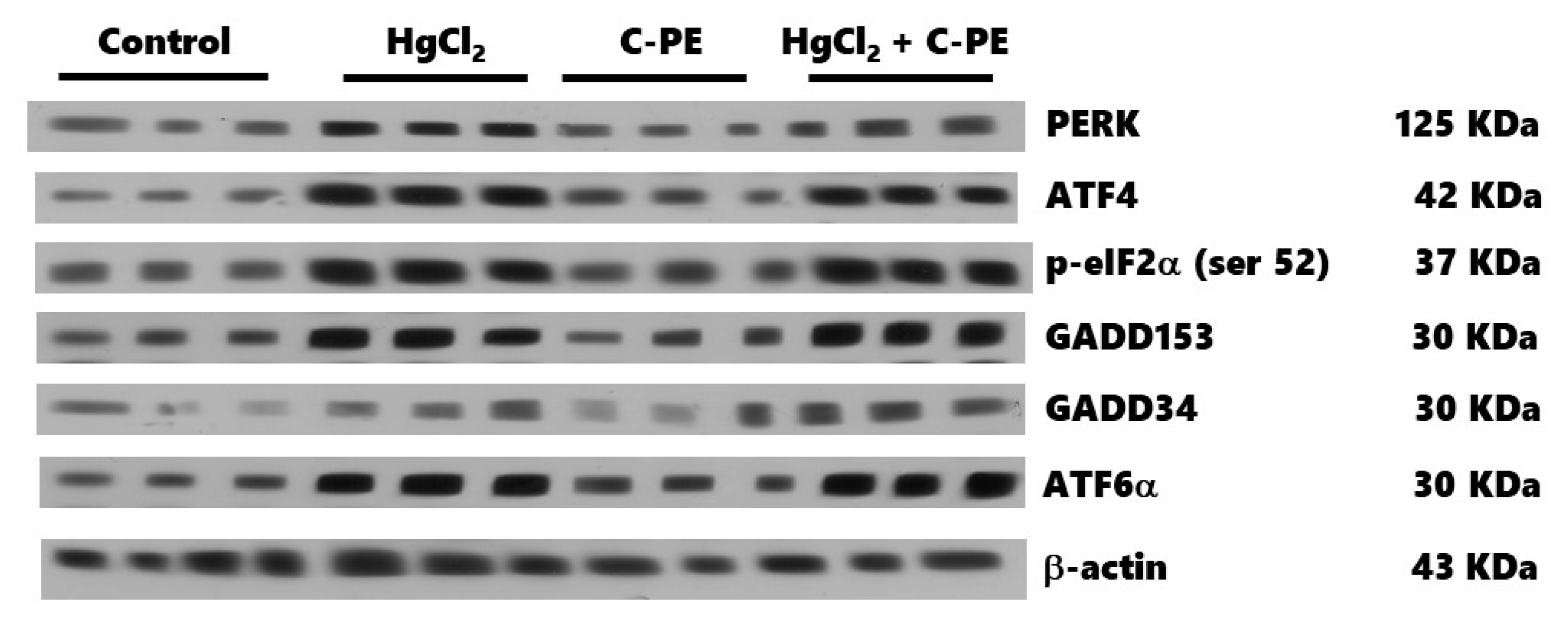
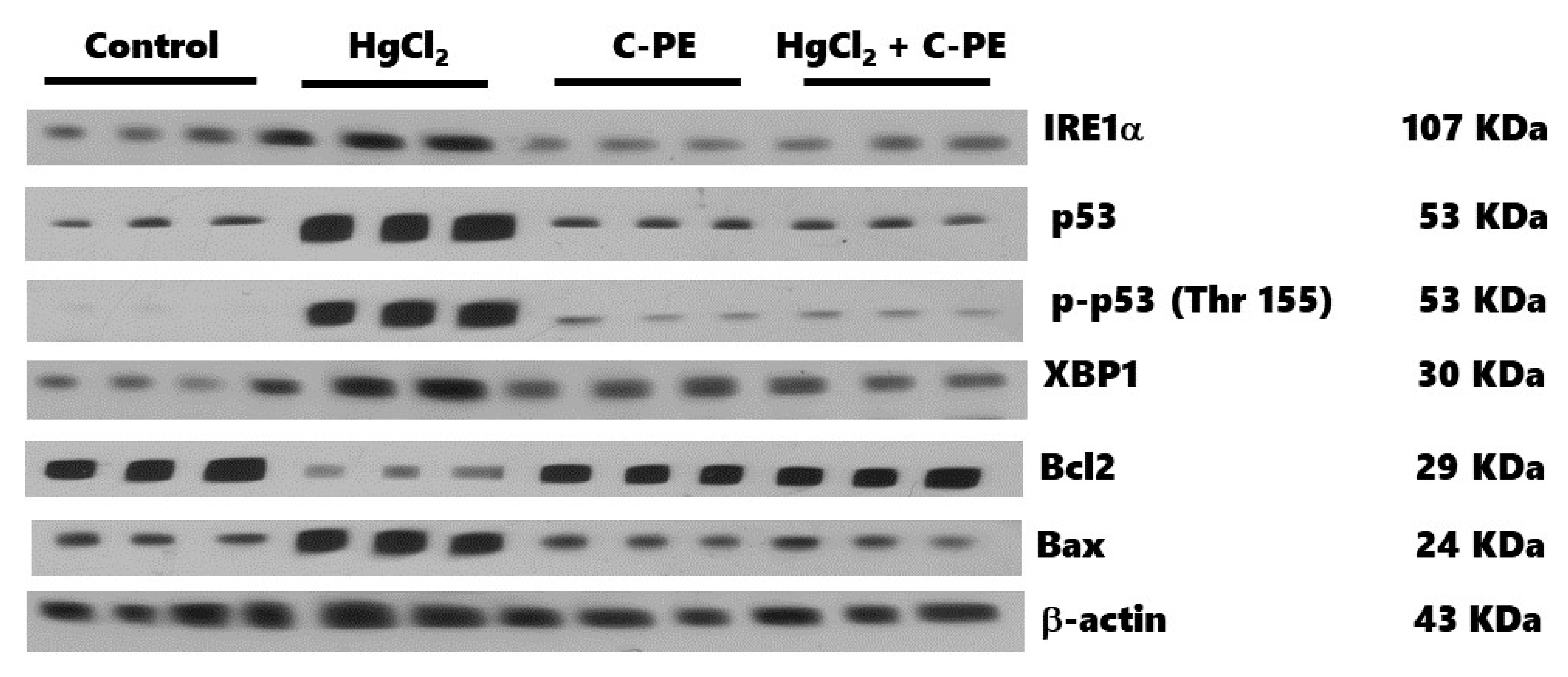
2.3. Evaluation of ER Stress
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cultivation, Purification, and Characterization of C-PE from Phormidium persicinum
4.3. Evaluation of Oxidative Stress, the Redox Environment, and the Activity of Effector Caspases 3 and 9
4.4. Examination of Kidney Damage
4.5. Western Blot Analysis for Nephrin, Podocin, and ER Stress Markers
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Peyrou, M.; Hanna, P.E.; Cribb, A.E. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol. Sci. 2007, 99, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Franco, P.; Franco-Colín, M.; Torres-Manzo, A.P.; Blas-Valdivia, V.; Thompson-Bonilla, M.R.; Kandir, S.; Cano-Europa, E. Endoplasmic reticulum stress participates in the pathophysiology of mercury-caused acute kidney injury. Ren. Fail. 2019, 41, 1001–1010. [Google Scholar] [CrossRef]
- Bridges, C.C.; Zalups, R.K. Transport of inorganic mercury and methylmercury in target tissues and organs. J. Toxicol. Environ. Health-Part B Crit. Rev. 2010, 13, 385–410. [Google Scholar] [CrossRef]
- Zalups, R.K. Molecular interactions with mercury in the kidney. Pharmacol. Rev. 2000, 52, 113–144. [Google Scholar]
- Rodriguez-Sánchez, R.; Ortiz-Butrón, R.; Blas-Valdivia, V.; Hernández-García, A.; Cano-Europa, E.; Rodríguez-Sánchez, R.; Ortiz-Butrón, R.; Blas-Valdivia, V.; Hernández-García, A.; Cano-Europa, E.; et al. Phycobiliproteins or C-phycocyanin of Arthrospira (Spirulina) maxima protect against HgCl2-caused oxidative stress and renal damage. Food Chem. 2012, 135, 2359–2365. [Google Scholar] [CrossRef]
- Smedley, G.D.; Walker, K.E.; Yuan, S.H. The role of PERK in understanding development of neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 8146. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Franco, P.; Franco-Colín, M.; Camargo, M.E.; Estévez Carmona, M.M.; Ortíz-Butrón, M.R.; Blas-Valdivia, V.; Cano-Europa, E. Phycobiliproteins and phycocyanin of Arthrospira maxima (Spirulina) reduce apoptosis promoters and glomerular dysfunction in mercury-related acute kidney injury. Toxicol. Res. Appl. 2018, 2, 2397847318805070. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Franco, P.; Franco-Colín, M.; Blas-Valdivia, V.; Melendez-Camargo, M.E.; Cano-Europa, E. Arthrospira maxima (Spirulina) prevents endoplasmic reticulum stress in the kidney through its C-phycocyanin. J. Zhejiang Univ. Sci. B 2021, 22, 603–608. [Google Scholar] [CrossRef]
- Soni, B.; Visavadiya, N.P.; Madamwar, D. Attenuation of diabetic complications by C-phycoerythrin in rats: Antioxidant activity of C-phycoerythrin including copper-induced lipoprotein and serum oxidation. Br. J. Nutr. 2009, 102, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, B.; Visavadiya, N.P.; Madamwar, D. Ameliorative action of cyanobacterial phycoerythrin on CCl4-induced toxicity in rats. Toxicology 2008, 248, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Sonani, R.R.; Sharma, M.; Gupta, G.D.; Kumar, V.; Madamwar, D. Phormidium phycoerythrin forms hexamers in crystals: A crystallographic study. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Sonani, R.R.; Singh, N.K.; Kumar, J.; Thakar, D.; Madamwar, D. Concurrent purification and antioxidant activity of phycobiliproteins from Lyngbya sp. A09DM: An antioxidant and anti-aging potential of phycoerythrin in Caenorhabditis elegans. Process Biochem. 2014, 49, 1757–1766. [Google Scholar] [CrossRef]
- Cano-Europa, E.; Ortiz-Butrón, R.; Gallardo-Casas, C.A.; Blas-Valdivia, V.; Pineda-Reynoso, M.; Olvera-Ramírez, R.; Franco-Colin, M. Phycobiliproteins from Pseudanabaena tenuis rich in c-phycoerythrin protect against HgCl2-caused oxidative stress and cellular damage in the kidney. J. Appl. Phycol. 2010, 22, 495–501. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Patrick, L. Mercury toxicity and antioxidants: Part I: Role of glutathione and alpha-lipoic acid in the treatment of mercury toxicity. Altern. Med. Rev. 2002, 7, 456–471. [Google Scholar]
- Nechemia-arbely, Y.; Barkan, D.; Pizov, G.; Shriki, A.; Rose-john, S.; Galun, E.; Axelrod, J.H. IL-6/IL-6R axis plays a critical role in acute kidney injury. J. Am. Soc. Nephrol. 2008, 19, 1106–1115. [Google Scholar] [CrossRef]
- Sharma, M.K.; Sharma, A.; Kumar, A.; Kumar, M. Evaluation of protective efficacy of Spirulina fusiformis against mercury induced nephrotoxicity in Swiss albino mice. Food Chem. Toxicol. 2007, 45, 879–887. [Google Scholar] [CrossRef]
- Memije-Lazaro, I.N.I.N.; Blas-Valdivia, V.; Franco-Colín, M.; Cano-Europa, E. Arthrospira maxima (Spirulina) and C-phycocyanin prevent the progression of chronic kidney disease and its cardiovascular complications. J. Funct. Foods 2018, 43, 37–43. [Google Scholar] [CrossRef]
- Garcia-Pliego, E.; Franco-Colin, M.; Rojas-Franco, P.; Blas-Valdivia, V.; Serrano-Contreras, J.I.; Pentón-Rol, G.; Cano-Europa, E. Phycocyanobilin is the molecule responsible for the nephroprotective action of phycocyanin in acute kidney injury caused by mercury. Food Funct. 2021, 12, 2985–2994. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.N.; Sonani, R.R.; Jakharia, K.; Bhastana, B.; Patel, H.M.; Chaubey, M.G.; Singh, N.K.; Madamwar, D. Antioxidant activity and associated structural attributes of Halomicronema phycoerythrin. Int. J. Biol. Macromol. 2018, 111, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Yabuta, Y.; Fujimura, H.; Kwak, C.S.; Enomoto, T.; Watanabe, F. Antioxidant activity of the phycoerythrobilin compound formed from a dried Korean purple laver (Porphyra sp.) during in vitro digestion. Food Sci. Technol. Res. 2010, 16, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Minic, S.L.; Stanic-Vucinic, D.; Mihailovic, J.; Krstic, M.; Nikolic, M.R.; Cirkovic Velickovic, T. Digestion by pepsin releases biologically active chromopeptides from C-phycocyanin, a blue-colored biliprotein of microalga Spirulina. J. Proteom. 2016, 147, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Radibratovic, M.; Minic, S.; Stanic-Vucinic, D.; Nikolic, M.; Milcic, M.; Cirkovic Velickovic, T. Stabilization of human serum albumin by the binding of phycocyanobilin, a bioactive chromophore of blue-green alga Spirulina: Molecular dynamics and experimental study. PLoS ONE 2016, 11, e0167973. [Google Scholar] [CrossRef]
- Sakai, S.; Komura, Y.; Nishimura, Y.; Sugawara, T.; Hirata, T. Inhibition of mast cell degranulation by phycoerythrin and its pigment moiety phycoerythrobilin, prepared from Porphyra yezoensis. Food Sci. Technol. Res. 2011, 17, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, T.; Chatterjee, S.; Bhayani, K.; Mishra, S. A natural cyanobacterial protein C-phycoerythrin as an Hg2+ selective fluorescent probe in aqueous systems. New J. Chem. 2020, 44, 6601–6609. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2018, 70, 24–31. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.H.; Kim, E.-Y.; Nam, T.-J. Phycoerythrin peptide from Pyropia yezoensis Alleviates endoplasmic reticulum stress caused by perfluorooctane sulfonate-induced calcium dysregulation. Mar. Drugs 2018, 16, 44. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.-W.; Oh, W.; Song, H.P.; Kim, W.K. Phosphorylation of p53 at threonine 155 is required for Jab1-mediated nuclear export of p53. BMB Rep. 2017, 50, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Norma Oficial Mexicana NOM-062-ZOO-1999: Especificaciones Técnicas para la Producción, Cuidado y Uso de los Animales de Laboratorio. 1999. Available online: https://www.fmvz.unam.mx/fmvz/principal/archivos/062ZOO.PDF (accessed on 12 October 2021).
- Kannaujiya, V.K.; Sinha, R.P. Thermokinetic stability of phycocyanin and phycoerythrin in food-grade preservatives. J. Appl. Phycol. 2016, 28, 1063–1070. [Google Scholar] [CrossRef]
- Sfriso, A.A.; Gallo, M.; Baldi, F. Phycoerythrin productivity and diversity from five red macroalgae. J. Appl. Phycol. 2018, 30, 2523–2531. [Google Scholar] [CrossRef]
- Madamwar, D.; Patel, D.K.; Desai, S.N.; Upadhyay, K.K.; Devkar, R. V Apoptotic potential of C-phycoerythrin from Phormidium sp. A27DM and Halomicronema sp. A32DM on human lung carcinoma cells. EXCLI J. 2015, 14, 527–539. [Google Scholar] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blas-Valdivia, V.; Rojas-Franco, P.; Serrano-Contreras, J.I.; Sfriso, A.A.; Garcia-Hernandez, C.; Franco-Colín, M.; Cano-Europa, E. C-phycoerythrin from Phormidium persicinum Prevents Acute Kidney Injury by Attenuating Oxidative and Endoplasmic Reticulum Stress. Mar. Drugs 2021, 19, 589. https://doi.org/10.3390/md19110589
Blas-Valdivia V, Rojas-Franco P, Serrano-Contreras JI, Sfriso AA, Garcia-Hernandez C, Franco-Colín M, Cano-Europa E. C-phycoerythrin from Phormidium persicinum Prevents Acute Kidney Injury by Attenuating Oxidative and Endoplasmic Reticulum Stress. Marine Drugs. 2021; 19(11):589. https://doi.org/10.3390/md19110589
Chicago/Turabian StyleBlas-Valdivia, Vanessa, Plácido Rojas-Franco, Jose Ivan Serrano-Contreras, Andrea Augusto Sfriso, Cristian Garcia-Hernandez, Margarita Franco-Colín, and Edgar Cano-Europa. 2021. "C-phycoerythrin from Phormidium persicinum Prevents Acute Kidney Injury by Attenuating Oxidative and Endoplasmic Reticulum Stress" Marine Drugs 19, no. 11: 589. https://doi.org/10.3390/md19110589
APA StyleBlas-Valdivia, V., Rojas-Franco, P., Serrano-Contreras, J. I., Sfriso, A. A., Garcia-Hernandez, C., Franco-Colín, M., & Cano-Europa, E. (2021). C-phycoerythrin from Phormidium persicinum Prevents Acute Kidney Injury by Attenuating Oxidative and Endoplasmic Reticulum Stress. Marine Drugs, 19(11), 589. https://doi.org/10.3390/md19110589