Enzymatic Modification of Native Chitin and Conversion to Specialty Chemical Products



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
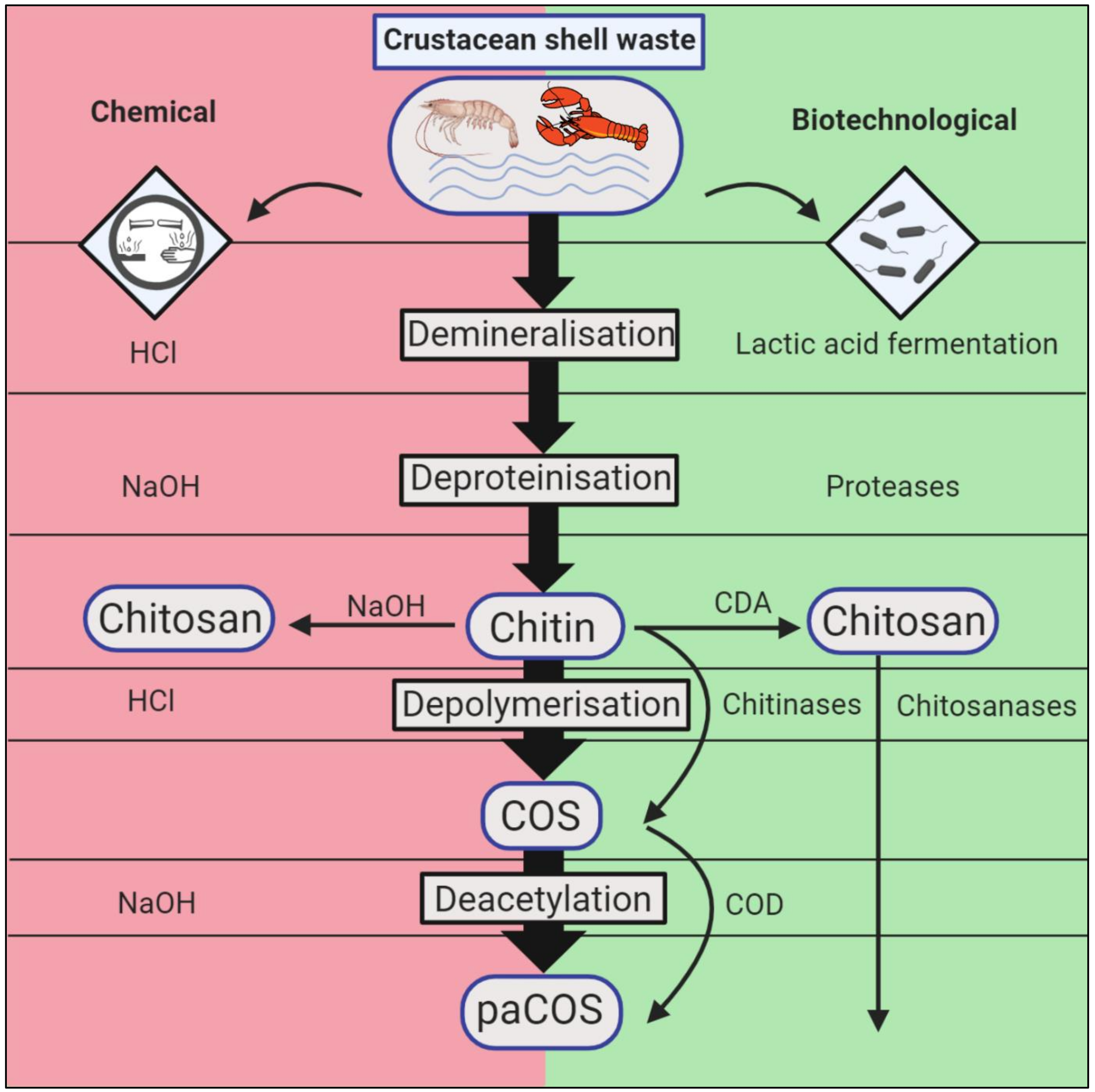
2. Conventional and Biotechnological Methods of Chitin Extraction and Conversion
2.1. Chemical Chitin Extraction
2.2. Chemical Processing of Chitin into Chitosan
2.3. Biotechnological Chitin Extraction
2.4. Biotechnological Chitin Conversion
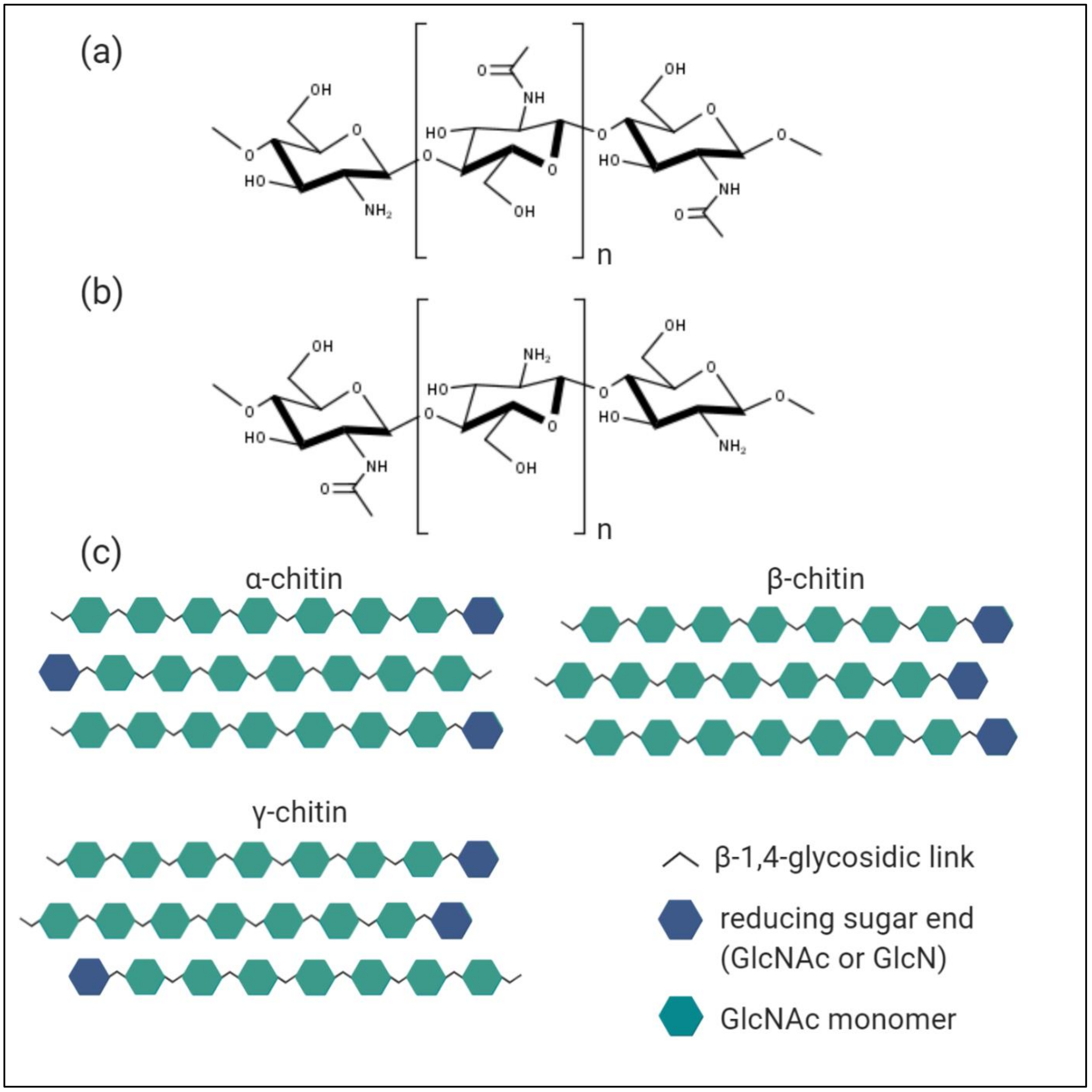
3. Characterisation of Chitin, Chitosan, and Chitooligosaccharides
3.1. Chitin
3.2. Chitosan
3.3. Chitooligosaccharides
4. Characterization of Enzymes Acting on Chitin and its Derivatives
4.1. Chitinases
4.1.1. Processive and Non-Processive Chitinases
4.1.2. Endo- and Exo-Chitinases
4.1.3. Chitinases with Transglycosylation Capabilities
4.2. Chitosanases
4.3. Chitin Deacetylases
- Enzymes with the multiple attack mechanism bind to their respective recognition site in the polysaccharide chain and process a number of sequential deacetylations, after which they detach and bind to a different region. In the case of high Mw substrate so-called block–copolymer structures arise, in which several adjacent, consecutive GlcN subunits alternate with regions of ambiguous DA or GlcNAc, where the CDA is not active. Shorter polymer chains or COS become deacetylated entirely [92].
- Enzymes following the multiple chain mechanism tightly bind to their recognition site of the substrate, resulting in an enzyme–polymer complex. In contrast to the multiple attack mechanism, the complex already dissociates after a single deacetylation reaction, with the enzyme binding to another recognition site afterwards [93]. For polymeric substrates this results in random distribution of deacetylated subunits and therefore PA. For COS no obvious proposition can be made, depending on the specific substrate length and involved enzyme; either a full deacetylation or a specific pattern can be generated. It is of peculiar interest to discover CDAs with new and unique patterns of deacetylation, since the influence of PA on the physio-chemical properties of paCOS remains to be further elucidated in detail. Furthermore, the discovery of more CDAs might also benefit our understanding of their modes of action and in doing so, our ability to tailor chitin- and chitosan oligomers with defined DA and PA.
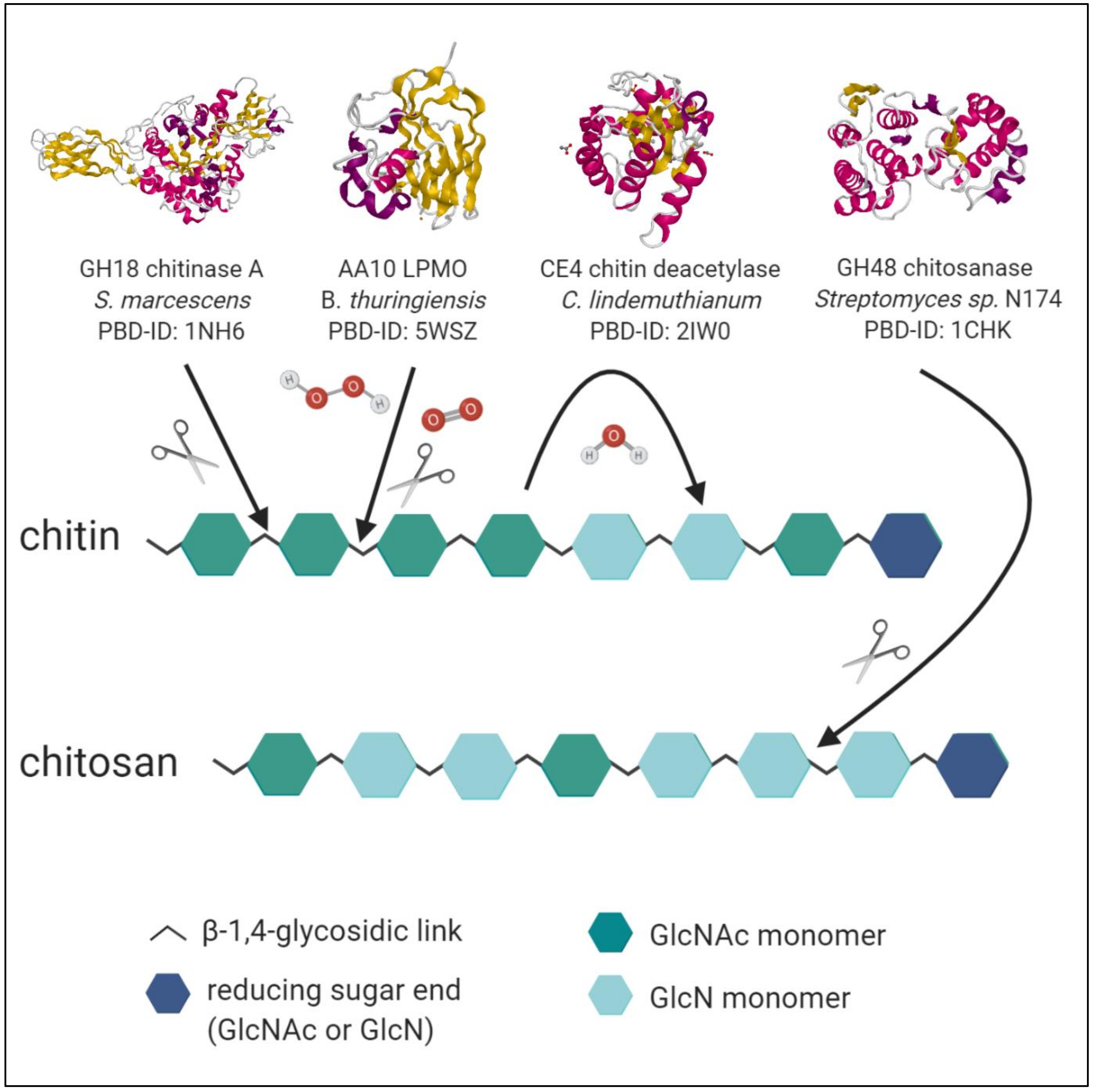
- Ultimately, single-chain-acting CDAs are processive enzymes that deacetylate a single substrate molecule sequentially by means of several catalytic events. Some bacterial CODs, which are specific for a single position, resulting in mono-deacetylated products (e.g., Rhizobium sp. NodB or Vibrio sp. CDA and COD) also belong to this group [94]. See Figure 3 for an overview of enzymatic activities of chitinases, LPMO, CDA and chitosanases towards chitin and chitosan polymer chains.
4.4. Lytic Polysaccharide Monooxygenases
4.4.1. LPMO Mechanisms with O2 as Co-substrate
4.4.2. LPMO Mechanisms with H2O2 as Co-substrate
5. Pretreatment of Native Chitin and COS Synthesis
5.1. Physical Pretreatment Methods and COS Synthesis
5.2. Chemical COS Synthesis
5.3. Biotechnological Chitin Pretreatment
5.4. Biotechnological COS Synthesis
6. Quantitative and Qualitative Separation of COS Mixtures
7. Perspectives and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tharanathan, R.N.; Kittur, F.S. Chitin--the undisputed biomolecule of great potential. Crit. Rev. Food Sci. Nutr. 2003, 43, 61–87. [Google Scholar] [CrossRef]
- Shirai, K.; Guerrero, I.; Huerta, S.; Saucedo, G.; Castillo, A.; Gonzalez, R.O.; Hall, G.M. Effect of initial glucose concentration and inoculation level of lactic acid bacteria in shrimp waste ensilation. Enzym. Microb. Technol. 2001, 28, 446–452. [Google Scholar] [CrossRef]
- Dhillon, G.S.; Kaur, S.; Brar, S.K.; Verma, M. Green synthesis approach: Extraction of chitosan from fungus mycelia. Crit. Rev. Biotechnol. 2013, 33, 379–403. [Google Scholar] [CrossRef] [PubMed]
- Younes, I.; Rinaudo, M. Chitin and Chitosan Preparation from Marine Sources. Structure, Properties and Applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Beaney, P.; Lizardi-Mendoza, J.; Healy, M. Comparison of chitins produced by chemical and bioprocessing methods. J. Chem. Technol. Biotechnol. 2005, 80, 145–150. [Google Scholar] [CrossRef]
- Blair, H.S.; Ho, T.-C. Studies in the adsorption and diffusion of ions in chitosan. J. Chem. Technol. Biotechnol. Biotechnol. 2007, 31, 6–10. [Google Scholar] [CrossRef]
- Weinhold, M.X.; Sauvageau, J.C.; Kumirska, J.; Thöming, J. Studies on acetylation patterns of different chitosan preparations. Carbohydr. Polym. 2009, 78, 678–684. [Google Scholar] [CrossRef]
- Omara, N.; Elsebaie, E.; Kassab, H.; Salama, A. Production of chitosan from shrimp shells by microwave technique and its use in minced beef preservation. Slov. Veter. Res. 2019, 56, 773–780. [Google Scholar] [CrossRef]
- El Knidri, H.; El Khalfaouy, R.; Laajeb, A.; Addaou, A.; Lahsini, A. Eco-friendly extraction and characterization of chitin and chitosan from the shrimp shell waste via microwave irradiation. Process. Saf. Environ. Prot. 2016, 104, 395–405. [Google Scholar] [CrossRef]
- Healy, M.; Green, A.; Healy, A. Bioprocessing of Marine Crustacean Shell Waste. Acta Biotechnol. 2003, 23, 151–160. [Google Scholar] [CrossRef]
- Hajji, S.; Ghorbel-Bellaaj, O.; Younes, I.; Jellouli, K.; Nasri, M. Chitin extraction from crab shells by Bacillus bacteria. Biological activities of fermented crab supernatants. Int. J. Boil. Macromol. 2015, 79, 167–173. [Google Scholar] [CrossRef] [PubMed]
- A Cira, L.; Huerta, S.; Hall, G.M.; Shirai, K. Pilot scale lactic acid fermentation of shrimp wastes for chitin recovery. Process. Biochem. 2002, 37, 1359–1366. [Google Scholar] [CrossRef]
- Harkin, C.; Lynch, C.; Brück, W. Isolation & identification of bacteria for the treatment of brown crab (Cancer pagurus) waste to produce chitinous material. J. Appl. Microbiol. 2015, 118, 954–965. [Google Scholar] [PubMed]
- Xu, Y.; Gallert, C.; Winter, J. Chitin purification from shrimp wastes by microbial deproteination and decalcification. Appl. Microbiol. Biotechnol. 2008, 79, 687–697. [Google Scholar] [CrossRef]
- Castro, R.; Guerrero-Legarreta, I.; Bórquez, R. Chitin extraction from Allopetrolisthes punctatus crab using lactic fermentation. Biotechnol. Rep. 2018, 20, e00287. [Google Scholar] [CrossRef]
- Jung, W.J.; Jo, G.H.; Kuk, J.H.; Kim, K.Y.; Park, R.D. Extraction of chitin from red crab shell waste by cofermentation with Lactobacillus paracasei subsp. tolerans KCTC-3074 and Serratia marcescens FS-3. Appl. Microbiol. Biotechnol. 2006, 71, 234–237. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, Y.; Deng, Y.; Wang, D.; Zhao, Y. Production of chitin from shrimp shell powders using Serratia marcescens B742 and Lactobacillus plantarum ATCC 8014 successive two-step fermentation. Carbohydr. Res. 2012, 362, 13–20. [Google Scholar] [CrossRef]
- Liu, P.; Liu, S.; Guo, N.; Mao, X.; Lin, H.; Xue, C.; Wei, D. Cofermentation of Bacillus licheniformis and Gluconobacter oxydans for chitin extraction from shrimp waste. Biochem. Eng. J. 2014, 91, 10–15. [Google Scholar] [CrossRef]
- Kaur, S.; Dhillon, G.S. Recent trends in biological extraction of chitin from marine shell wastes: A review. Crit. Rev. Biotechnol. 2013, 35, 44–61. [Google Scholar] [CrossRef]
- Bajaj, M.; Freiberg, A.; Winter, J.; Xu, Y.; Gallert, C. Pilot-scale chitin extraction from shrimp shell waste by deproteination and decalcification with bacterial enrichment cultures. Appl. Microbiol. Biotechnol. 2015, 99, 9835–9846. [Google Scholar] [CrossRef]
- Sarai, S.; Ram, A.; Peña-chora, G.; Hern, M. Enhanced Tolerance against a Fungal Pathogen and Insect Resistance in Transgenic Tobacco Plants Overexpressing an Endochitinase Gene from Serratia marcescens. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Park, B.K.; Kim, M.-M. Applications of Chitin and Its Derivatives in Biological Medicine. Int. J. Mol. Sci. 2010, 11, 5152–5164. [Google Scholar] [CrossRef]
- Dutta, P.K.; Duta, J.; Tripathi, V.S. Chitin and Chitosan: Chemistry, properties and applications. J. Sci. Ind. Res. 2004, 63, 20–31. [Google Scholar]
- Harikrishnan, R.; Kim, J.-S.; Balasundaram, C.; Heo, M.-S. Dietary supplementation with chitin and chitosan on haematology and innate immune response in Epinephelus bruneus against Philasterides dicentrarchi. Exp. Parasitol. 2012, 131, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.-S.; Jung, M.-W.; Paeng, K.-J. Evaluation of Chitin and Chitosan as a Sorbent for the Preconcentration of Phenol and Chlorophenols in Water. Anal. Sci. 1998, 14, 1089–1092. [Google Scholar] [CrossRef]
- Bloch, R.; Burger, M.M. Purification of wheat germ agglutinin using affinity chromatography on chitin. Biochem. Biophys. Res. Commun. 1974, 58, 13–19. [Google Scholar] [CrossRef]
- Bartnicki-Garcia, S.; Nickerson, W.J. Isolation, composition, and structure of cell walls of filamentous and yeast-like forms of Mucor rouxii. Biochim. Biophys. Acta 1962, 58, 102–119. [Google Scholar] [CrossRef]
- Younes, I.; Hajji, S.; Frachet, V.; Rinaudo, M.; Jellouli, K.; Nasri, M. Chitin extraction from shrimp shell using enzymatic treatment. Antitumor, antioxidant and antimicrobial activities of chitosan. Int. J. Boil. Macromol. 2014, 69, 489–498. [Google Scholar] [CrossRef]
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Goy, R.C.; De Britto, D.; Assis, O.B.G. A review of the antimicrobial activity of chitosan. Polímeros 2009, 19, 241–247. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Sorlier, P.; Denuzière, A.; Viton, C.; Domard, A. Relation between the degree of acetylation and the electrostatic properties of chitin and chitosan. Biomacromolecules 2001, 2, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-C.; Su, Y.-P.; Chen, C.-C.; Jia, G.; Wang, H.-L.; Wu, J.C.G.; Lin, J.-G. Relationship between antibacterial activity of chitosan and surface characteristics of cell wall. Acta Pharmacol. Sin. 2004, 25, 932–936. [Google Scholar] [PubMed]
- Aam, B.B.; Heggset, E.B.; Norberg, A.L.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G.H. Production of Chitooligosaccharides and Their Potential Applications in Medicine. Mar. Drugs 2010, 8, 1482–1517. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, G.; Kim, Y.-S.; Hwang, J.-W.; Kim, S.-K.; Jeon, Y.-J.; Je, J.-Y.; Ahn, C.-B.; Moon, S.-H.; Jeon, B.-T.; Park, P.-J. Chitooligosaccharide and Its Derivatives: Preparation and Biological Applications. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liaqat, F.; Eltem, R. Chitooligosaccharides and their biological activities: A comprehensive review. Carbohydr. Polym. 2018, 184, 243–259. [Google Scholar] [CrossRef]
- Kim, S.-K.; Rajapakse, N. Enzymatic production and biological activities of chitosan oligosaccharides (COS): A review. Carbohydr. Polym. 2005, 62, 357–368. [Google Scholar] [CrossRef]
- Bahrke, S. Mass Spectrometric Analysis of Chitooligosachharides and their Interaction with proteins. Ph.D. Thesis, Universität Potsdam, Potsdam, Germany, 2008; pp. 1–232. [Google Scholar]
- Elieh-Ali-Komi, D.; Hamblin, M.R. Chitin and Chitosan: Production and Application of Versatile Biomedical Nanomaterials. Int. J. Adv. Res. 2016, 4, 411–427. [Google Scholar]
- Hamed, I.; Özogul, F.; Regenstein, J.M. Industrial applications of crustacean by-products (chitin, chitosan, and chitooligosaccharides): A review. Trends Food Sci. Technol. 2016, 48, 40–50. [Google Scholar] [CrossRef]
- Casadidio, C.; Peregrina, D.V.; Gigliobianco, M.R.; Deng, S.; Censi, R.; Di Martino, P. Chitin and Chitosans: Characteristics, Eco-Friendly Processes, and Applications in Cosmetic Science. Mar. Drugs 2019, 17, 369. [Google Scholar] [CrossRef]
- Malerba, M.; Cerana, R. Recent Advances of Chitosan Applications in Plants. Polym. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Velappan, S.P. Recent developments in chitosanase research and its biotechnological applications: A review. Food Chem. 2014, 150, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, M.B.; Struszczyk-Swita, K.; Li, X.; Szczęsna-Antczak, M.; Daroch, M. Enzymatic Modifications of Chitin, Chitosan, and Chitooligosaccharides. Front. Bioeng. Biotechnol. 2019, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ríos, D.; Barrera-Zapata, R.; Ríos-Estepa, R. Comparison of process technologies for chitosan production from shrimp shell waste: A techno-economic approach using Aspen Plus ®. Food Bioprod. Process. 2017, 103, 49–57. [Google Scholar] [CrossRef]
- Yan, N.; Chen, X. Sustainability: Don’t waste seafood waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Van Aalten, D.M.F.; Komander, D.; Synstad, B.; Gåseidnes, S.; Peter, M.G.; Eijsink, V.G.H. Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc. Natl. Acad. Sci. USA 2001, 98, 8979–8984. [Google Scholar] [CrossRef]
- Hamre, A.G.; Frøberg, E.E.; Eijsink, V.G.; Sørlie, M. Thermodynamics of tunnel formation upon substrate binding in a processive glycoside hydrolase. Arch. Biochem. Biophys. 2017, 620, 35–42. [Google Scholar] [CrossRef]
- Davies, G.; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef]
- Zakariassen, H.; Aam, B.B.; Horn, S.J.; Vårum, K.M.; Sørlie, M.; Eijsink, V.G.H. Aromatic Residues in the Catalytic Center of Chitinase A from Serratia marcescens Affect Processivity, Enzyme Activity, and Biomass Converting Efficiency. J. Boil. Chem. 2009, 284, 10610–10617. [Google Scholar] [CrossRef]
- Oyeleye, A.; Normi, Y.M. Chitinase: Diversity, limitations, and trends in engineering for suitable applications. Biosci. Rep. 2018, 38, 1–21. [Google Scholar] [CrossRef]
- Slámová, K.; Bojarová, P.; Petrásková, L.; Křen, V. β-N-Acetylhexosaminidase: What’s in a name...? Biotechnol. Adv. 2010, 28, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.; Sørbotten, A.; Horn, S.J.; Eijsink, V.G.H.; Vårum, K.M. Serratia marcescens chitinases with tunnel-shaped substrate-binding grooves show endo activity and different degrees of processivity during enzymatic hydrolysis of chitosan. Biochemistry 2006, 45, 9566–9574. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.B.; Chen, S.H.; Peng, K.C. Preparation of antibacterial chito-oligosaccharide by altering the degree of deacetylation of β-chitosan in a Trichoderma harzianum chitinase-hydrolysing process. J. Sci. Food Agric. 2009, 89, 238–244. [Google Scholar] [CrossRef]
- Eweis, M.; Elkholy, S.; Elsabee, M. Antifungal efficacy of chitosan and its thiourea derivatives upon the growth of some sugar-beet pathogens. Int. J. Boil. Macromol. 2006, 38, 1–8. [Google Scholar] [CrossRef]
- Tikhonov, V.E.; Stepnova, E.A.; Babak, V.G.; Yamskov, I.A.; Palma-Guerrero, J.; Jansson, H.-B.; Lopez-Llorca, L.V.; Salinas, J.; Gerasimenko, D.V.; Avdienko, I.D.; et al. Bactericidal and antifungal activities of a low molecular weight chitosan and its N-/2(3)-(dodec-2-enyl)succinoyl/-derivatives. Carbohydr. Polym. 2006, 64, 66–72. [Google Scholar] [CrossRef]
- Ribeiro, M.P.; Espiga, A.; Silva, D.; Henriques, J.; Ferreira, C.; Silva, J.C.; Pires, E.; Chaves, P. Development of a new chitosan hydrogel for wound dressing. Wound Repair Regen. 2009, 17, 817–824. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Park, W.H.; Ko, B.M.; Min, B.-M. Effects of PVA sponge containing chitooligosaccharide in the early stage of wound healing. J. Mater. Sci. Mater. Electron. 2004, 15, 297–301. [Google Scholar] [CrossRef]
- Dai, T.; Tanaka, M.; Huang, Y.-Y.; Hamblin, M.R. Chitosan preparations for wounds and burns: antimicrobial and wound-healing effects. Expert Rev. Anti-Infect. Ther. 2011, 9, 857–879. [Google Scholar] [CrossRef]
- Köping-Höggård, M.; Mel’nikova, Y.S.; Vårum, K.M.; Lindman, B.; Artursson, P. Relationship between the physical shape and the efficiency of oligomeric chitosan as a gene delivery system in vitro and in vivo. J. Gene Med. 2003, 5, 130–141. [Google Scholar] [CrossRef]
- Köping-Höggård, M.; Tubulekas, I.; Guan, H.; Edwards, K.; Nilsson, M.; Vårum, K.M.; Artursson, P. Chitosan as a nonviral gene delivery system. Structure–property relationships and characteristics compared with polyethylenimine in vitro and after lung administration in vivo. Gene Ther. 2001, 8, 1108–1121. [Google Scholar] [CrossRef]
- Strand, S.P.; Lélu, S.; Reitan, N.K.; Davies, C.D.L.; Artursson, P.; Vårum, K.M. Molecular design of chitosan gene delivery systems with an optimized balance between polyplex stability and polyplex unpacking. Biomaterials 2010, 31, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zheng, L.; Yang, S.; Niu, R.; Chu, E.; Lin, X. N-Acetylchitooligosaccharide is a potent angiogenic inhibitor both in vivo and in vitro. Biochem. Biophys. Res. Commun. 2007, 357, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Wu, H.; Wei, P.; Pan, M.; Tuo, Y.; Kusakabe, I.; Du, Y. Potent angiogenic inhibition effects of deacetylated chitohexaose separated from chitooligosaccharides and its mechanism of action in vitro. Carbohydr. Res. 2009, 344, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Min, M.; Du, N.; Gu, Y.; Hode, T.; Naylor, M.; Chen, D.; Nordquist, R.E.; Chen, W.R. Chitin, Chitosan, and Glycated Chitosan Regulate Immune Responses: The Novel Adjuvants for Cancer Vaccine. Clin. Dev. Immunol. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Purushotham, P.; Podile, A.R. Synthesis of Long-Chain Chitooligosaccharides by a Hypertransglycosylating Processive Endochitinase of Serratia proteamaculans 568. J. Bacteriol. 2012, 194, 4260–4271. [Google Scholar] [CrossRef]
- Madhuprakash, J.; Singh, A.; Kumar, S.; Sinha, M.; Kaur, P.; Sharma, S.; Podile, A.R.; Singh, T.P. Structure of chitinase D from Serratia proteamaculans reveals the structural basis of its dual action of hydrolysis and transglycosylation. Int. J. Biochem. Mol. Boil. 2013, 4, 166–178. [Google Scholar]
- Mallakuntla, M.K.; Vaikuntapu, P.R.; Bhuvanachandra, B.; Das, S.N.; Podile, A.R. Transglycosylation by a chitinase from Enterobacter cloacae subsp. cloacae generates longer chitin oligosaccharides. Sci. Rep. 2017, 7, 5113. [Google Scholar] [CrossRef]
- Bojarová, P.; Kren, V. Glycosidases: A key to tailored carbohydrates. Trends Biotechnol. 2009, 27, 199–209. [Google Scholar] [CrossRef]
- Mangas-Sanchez, J.; Adlercreutz, P. Enzymatic preparation of oligosaccharides by transglycosylation: A comparative study of glucosidases. J. Mol. Catal. B Enzym. 2015, 122, 51–55. [Google Scholar] [CrossRef]
- Tanaka, T.; Fukui, T.; Atomi, H.; Imanaka, T. Characterization of an Exo-ß-D-Glucosaminidase Involved in a Novel Chitnolytic Pathway from the Hyperthermophilic Archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 2003, 185, 5175–5181. [Google Scholar] [CrossRef]
- Fukamizo, T.; Fleury, A.; Côté, N.; Mitsutomi, M.; Brzezinski, R. Exo-β-d-glucosaminidase from Amycolatopsis orientalis: catalytic residues, sugar recognition specificity, kinetics, and synergism. Glycobiology 2006, 16, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Viens, P.; Lacombe-Harvey, M.-È.; Brzezinski, R. Chitosanases from Family 46 of Glycoside Hydrolases: From Proteins to Phenotypes. Mar. Drugs 2015, 13, 6566–6587. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, H.; Yamaguchi, T.; Fukamizo, T.; Brzezinski, R. Mechanism of chitosanase-oligosaccharide interaction: Subsite structure of Streptomyces sp. N174 chitosanase and the role of Asp57 carboxylate. J. Biochem. 2001, 130, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Fukamizo, T.; Amano, S.; Yamaguchi, K.; Yoshikawa, T.; Katsumi, T.; Saito, J.-I.; Suzuki, M.; Miki, K.; Nagata, Y.; Ando, A. Bacillus circulans MH-K1 Chitosanase: Amino Acid Residues Responsible for Substrate Binding. J. Biochem. 2005, 138, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Saito, J.-I.; Kita, A.; Higuchi, Y.; Nagata, Y.; Ando, A.; Miki, K. Crystal structure of chitosanase from Bacillus circulans MH-K1 at 1.6-A resolution and its substrate recognition mechanism. J. Boil. Chem. 1999, 274, 30818–30825. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, E.M.; Monzingo, A.F.; Ernst, S.R.; Brzezinski, R.; Robertas, J.D. X-ray structure of an anti-fungal chitosanase from streptomyces N174. Nat. Genet. 1996, 3, 155–162. [Google Scholar] [CrossRef]
- Monzingo, A.F.; Marcotte, E.M.; Hart, P.J.; Robertas, J.D. Chitinases, chitosanases, and lysozymes can be divided into procaryotic and eucaryotic families sharing a conserved core. Nat. Genet. 1996, 3, 133–140. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Chang, C.H.; Wu, Y.J.; Li, Y.K. Exploration of glycosyl hydrolase family 75, a chitosanase from Aspergillus fumigatus. J. Biol. Chem. 2006, 281, 3137–3144. [Google Scholar] [CrossRef]
- Hoell, I.A.; Vaaje-Kolstad, G.; Eijsink, V.G. Structure and function of enzymes acting on chitin and chitosan. Biotechnol. Genet. Eng. Rev. 2010, 27, 331–366. [Google Scholar] [CrossRef]
- McCarter, J.D.; Withers, G.S. Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Boil. 1994, 4, 885–892. [Google Scholar] [CrossRef]
- McIntosh, L.P.; Hand, G.; Johnson, P.E.; Joshi, M.D.; Körner, M.; Plesniak, L.A.; Ziser, L.; Wakarchuk, W.W.; Withers, S.G. The pK(a) of the general acid/base carboxyl group of a glycosidase cycles during catalysis: A 13C-NMR study of Bacillus circulans xylanase. Biochemistry 1996, 35, 9958–9966. [Google Scholar] [CrossRef] [PubMed]
- Fukamizo, T.; Ohkawa, T.; Ikeda, Y.; Goto, S. Specificity of chitosanase from Bacillus pumilus. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1994, 1205, 183–188. [Google Scholar] [CrossRef]
- Mitsutomi, M.; Isono, M.; Uchiyama, A.; Nikaidou, N.; Ikegami, T.; Watanabe, T. Chitosanase activity of the enzyme previously reported as β-1,3-1,4-glucanase from Bacillus circulans WL-12. Biosci. Biotechnol. Biochem. 1998, 62, 2107–2114. [Google Scholar] [CrossRef] [PubMed]
- Fenton, D.M.; Eveleigh, D.E. Purification and Mode of Action of a Chitosanase from Penicillium islandicum. Microbiology 1981, 126, 151–165. [Google Scholar] [CrossRef]
- Hirano, K.; Watanabe, M.; Seki, K.; Ando, A.; Saito, A.; Mitsutomi, M. Classification of Chitosanases by Hydrolytic Specificity towardN1,N4-Diacetylchitohexaose. Biosci. Biotechnol. Biochem. 2012, 76, 1932–1937. [Google Scholar] [CrossRef]
- Heggset, E.B.; Dybvik, A.I.; Hoell, I.A.; Norberg, A.L.; Sørlie, M.; Eijsink, V.G.H.; Vårum, K.M. Degradation of Chitosans with a Family 46 Chitosanase fromStreptomyces coelicolorA3(2). Biomacromolecules 2010, 11, 2487–2497. [Google Scholar] [CrossRef]
- Zhao, Y.; Park, R.-D.; Muzzarelli, R.A.A. Chitin Deacetylases: Properties and Applications. Mar. Drugs 2010, 8, 24–46. [Google Scholar] [CrossRef]
- Blair, D.E.; Schüttelkopf, A.W.; Macrae, J.I.; Van Aalten, D.M.F. Structure and metal-dependent mechanism of peptidoglycan deacetylase, a streptococcal virulence factor. Proc. Natl. Acad. Sci. USA 2005, 102, 15429–15434. [Google Scholar] [CrossRef]
- Araki, Y.; Ito, E. A Pathway of Chitosan Formation in Mucor rouxii. Enzymatic Deacetylation of Chitin. JBIC J. Boil. Inorg. Chem. 1975, 55, 71–78. [Google Scholar] [CrossRef]
- Tsigos, I.; Martinou, A.; Kafetzopoulos, D.; Bouriotis, V. Chitin deacetylases: New, versatile tools in biotechnology. Trends Biotechnol. 2000, 18, 305–312. [Google Scholar] [CrossRef]
- Martinou, A.; Bouriotis, V.; Stokke, B.T.; Vårum, K.M. Mode of action of chitin deacetylase from Mucor rouxii on partially N-acetylated chitosans. Carbohydr. Res. 1998, 311, 71–78. [Google Scholar] [CrossRef]
- Hekmat, O.; Tokuyasu, K.; Withers, S.G. Subsite structure of the endo-type chitin deacetylase from a Deuteromycete, Colletotrichum lindemuthianum: An investigation using steady-state kinetic analysis and MS. Biochem. J. 2003, 374, 369–380. [Google Scholar] [CrossRef]
- Grifoll-Romero, L.; Pascual, S.; Aragunde, H.; Biarnés, X.; Planas, A. Chitin Deacetylases: Structures, Specificities, and Biotech Applications. Polymers 2018, 10, 352. [Google Scholar] [CrossRef]
- Aronson, N.N.; Halloran, B.A.; Alexyev, M.F.; Amable, L.; Madura, J.D.; Pasupulati, L.; Worth, C.; Van Roey, P. Family 18 chitinase–oligosaccharide substrate interaction: Subsite preference and anomer selectivity of Serratia marcescens chitinase A. Biochem. J. 2003, 376, 87–95. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, Y.; Cao, H.; Mou, G.; Yin, H. Expression and characterization of a lytic polysaccharide monooxygenase from Bacillus thuringiensis. Int. J. Boil. Macromol. 2015, 79, 72–75. [Google Scholar] [CrossRef]
- Blair, D.E.; Hekmat, O.; Schüttelkopf, A.W.; Shrestha, B.; Tokuyasu, K.; Withers, S.G.; Van Aalten, D.M.F. Structure and Mechanism of Chitin Deacetylase from the Fungal PathogenColletotrichum lindemuthianum. Biochemistry 2006, 45, 9416–9426. [Google Scholar] [CrossRef]
- Liu, Z.; Gay, L.M.; Tuveng, T.R.; Agger, J.W.; Westereng, B.; Mathiesen, G.; Horn, S.J.; Vaaje-Kolstad, G.; Van Aalten, D.M.F.; Eijsink, V.G.H. Structure and function of a broad-specificity chitin deacetylase from Aspergillus nidulans FGSC A4. Sci. Rep. 2017, 7, 1746. [Google Scholar] [CrossRef] [PubMed]
- Hoßbach, J.; Bußwinkel, F.; Kranz, A.; Wattjes, J.; Cord-Landwehr, S.; Moerschbacher, B.M. A chitin deacetylase of Podospora anserina has two functional chitin binding domains and a unique mode of action. Carbohydr. Polym. 2018, 183, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Forsberg, Z.; Loose, J.S.; Bissaro, B.; Eijsink, V.G. Structural diversity of lytic polysaccharide monooxygenases. Curr. Opin. Struct. Boil. 2017, 44, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Tandrup, T.; Frandsen, K.E.H.; Johansen, K.S.; Berrin, J.-G.; Leggio, L.L. Recent insights into lytic polysaccharide monooxygenases (LPMOs). Biochem. Soc. Trans. 2018, 46, 1431–1447. [Google Scholar] [CrossRef] [PubMed]
- Crouch, L.I.; Labourel, A.; Walton, P.H.; Davies, G.J.; Gilbert, H.J. The Contribution of Non-catalytic Carbohydrate Binding Modules to the Activity of Lytic Polysaccharide Monooxygenases. J. Boil. Chem. 2016, 291, 7439–7449. [Google Scholar] [CrossRef]
- Hansson, H.; Karkehabadi, S.; Mikkelsen, N.; Douglas, N.R.; Kim, S.; Lam, A.; Kaper, T.; Kelemen, B.; Meier, K.K.; Jones, S.M.; et al. High-resolution structure of a lytic polysaccharide monooxygenase from Hypocrea jecorina reveals a predicted linker as an integral part of the catalytic domain. J. Boil. Chem. 2017, 292, 19099–19109. [Google Scholar] [CrossRef] [PubMed]
- Hangasky, J.A.; Marletta, M.A. A Random-Sequential Kinetic Mechanism for Polysaccharide Monooxygenases. Biochemistry 2018, 57, 3191–3199. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, T.; Westereng, B.; Aachmann, F.L.; Agger, J.W.; Kracher, D.; Kittl, R.; Ludwig, R.; Haltrich, D.; Eijsink, V.G.; Horn, S.J. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 2014, 289, 2632–2642. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef]
- Beeson, W.T.; Phillips, C.M.; Cate, J.H.D.; Marletta, M.A. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 2012, 134, 890–892. [Google Scholar] [CrossRef]
- Hangasky, J.A.; Iavarone, A.T.; Marletta, M.A. Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc. Natl. Acad. Sci. USA 2018, 115, 4915–4920. [Google Scholar] [CrossRef]
- Bissaro, B.; Várnai, A.; Røhr, A.K.; Eijsink, V.G.H. Oxidoreductases and Reactive Oxygen Species in Conversion of Lignocellulosic Biomass. Microbiol. Mol. Boil. Rev. 2018, 82, e00029-18. [Google Scholar]
- Hedegård, E.D.; Ryde, U. Molecular mechanism of lytic polysaccharide monooxygenases†. †Electronic supplementary information (ESI) available: Detailed description of the employed computational methods and protein setup. QM and MM energy components and B3LYP energies for most reaction paths. Chem. Sci. 2018, 9, 3866–3880. [Google Scholar] [CrossRef]
- Bertini, L.; Breglia, R.; Lambrughi, M.; Fantucci, P.; De Gioia, L.; Borsari, M.; Sola, M.; Bortolotti, C.A.; Bruschi, M. Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases Investigated by First-Principles Calculations. Inorg. Chem. 2018, 57, 86–97. [Google Scholar] [CrossRef]
- Hemsworth, G.R.; Davies, G.J.; Walton, P.H. Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr. Opin. Struct. Boil. 2013, 23, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, B.; Røhr, Å.K.; Müller, G.; Chylenski, P.; Skaugen, M.; Forsberg, Z.; Horn, S.J.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Methods 2017, 13, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Kuusk, S.; Bissaro, B.; Kuusk, P.; Forsberg, Z.; Eijsink, V.G.H.; Sørlie, M.; Väljamäe, P. Kinetics of H2O2-driven degradation of chitin by a bacterial lytic polysaccharide monooxygenase. J. Boil. Chem. 2018, 293, 12284. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.G.; Crouch, L.; Labourel, A.; Forsberg, Z.; Bukhman, Y.V.; Gilbert, H.J.; Keating, D.H.; Vaaje-Kolstad, G. Systems biology defines the biological significance of redox-active proteins during cellulose degradation in an aerobic bacterium. Mol. Microbiol. 2014, 94, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Johnston, E.M.; Li, P.; Shaik, S.; Davies, G.J.; Walton, P.H.H.; Rovira, C. QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate. ACS Catal. 2018, 8, 1346–1351. [Google Scholar] [CrossRef]
- Cao, L.; Caladararu, O.; Rosenzweig, A.C.; Ryde, U. Quantum Refinement Does Not Support Dinuclear Copper Sites in Crystal Structures of Particulate Methane Monooxygenase. Angew. Chem. Int. Ed. Engl. 2018, 57, 162–166. [Google Scholar] [CrossRef]
- Nakagawa, Y.S.; Oyama, Y.; Kon, N.; Nikaido, M.; Tanno, K.; Kogawa, J.; Inomata, S.; Masui, A.; Yamamura, A.; Kawaguchi, M.; et al. Development of innovative technologies to decrease the environmental burdens associated with using chitin as a biomass resource: Mechanochemical grinding and enzymatic degradation. Carbohydr. Polym. 2011, 83, 1843–1849. [Google Scholar] [CrossRef]
- Jung, W.-J.; Park, R.-D. Bioproduction of Chitooligosaccharides: Present and Perspectives. Mar. Drugs 2014, 12, 5328–5356. [Google Scholar] [CrossRef]
- Li, K.; Xing, R.; Liu, S.; Qin, Y.; Meng, X.; Li, P. Microwave-assisted degradation of chitosan for a possible use in inhibiting crop pathogenic fungi. Int. J. Boil. Macromol. 2012, 51, 767–773. [Google Scholar] [CrossRef]
- Sahu, A.; Goswami, P.; Bora, U. Microwave mediated rapid synthesis of chitosan. J. Mater. Sci. Mater. Med. 2009, 20, 171–175. [Google Scholar] [CrossRef]
- Hai, L.; Diep, T.B.; Nagasawa, N.; Yoshii, F.; Kume, T. Radiation depolymerization of chitosan to prepare oligomers. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interactions Mater. Atoms 2003, 208, 466–470. [Google Scholar] [CrossRef]
- Yoksan, R.; Akashi, M.; Miyata, M.; Chirachanchai, S. Optimal γ-Ray Dose and Irradiation Conditions for Producing Low-Molecular-Weight Chitosan that Retains its Chemical Structure. Radiat. Res. 2004, 161, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Liu, S.; Yu, H.; Guo, Z.; Wang, P.; Li, C.; Li, Z.; Li, P. Salt-assisted acid hydrolysis of chitosan to oligomers under microwave irradiation. Carbohydr. Res. 2005, 340, 2150–2153. [Google Scholar] [CrossRef] [PubMed]
- Villa-Lerma, G.; González-Márquez, H.; Gimeno, M.; López-Luna, A.; Bárzana, E.; Shirai, K. Ultrasonication and steam-explosion as chitin pretreatments for chitin oligosaccharide production by chitinases of Lecanicillium lecanii. Bioresour. Technol. 2013, 146, 794–798. [Google Scholar] [CrossRef]
- Husson, E.; Hadad, C.; Huet, G.; Laclef, S.; Lesur, D.; Lambertyn, V.; Jamali, A.; Gottis, S.; Sarazin, C.; Van Nhien, A.N. The effect of room temperature ionic liquids on the selective biocatalytic hydrolysis of chitin via sequential or simultaneous strategies. Green Chem. 2017, 19, 4122–4131. [Google Scholar] [CrossRef]
- Berton, P.; Shamshina, J.L.; Ostadjoo, S.; King, C.A.; Rogers, R.D. Enzymatic hydrolysis of ionic liquid-extracted chitin. Carbohydr. Polym. 2018, 199, 228–235. [Google Scholar] [CrossRef]
- Zdanowicz, M.; Wilpiszewska, K.; Spychaj, T. Deep eutectic solvents for polysaccharides processing. A review. Carbohydr. Polym. 2018, 200, 361–380. [Google Scholar] [CrossRef]
- Roda, A.; Matias, A.A.; Paiva, A.; Duarte, A.R.C. Polymer Science and Engineering Using Deep Eutectic Solvents. Polymers 2019, 11, 912. [Google Scholar] [CrossRef]
- Dadi, A.P.; Schall, C.A.; Varanasi, S. Mitigation of cellulose recalcitrance to enzymatic hydrolysis by ionic liquid pretreatment. Appl. Biochem. Biotechnol. 2007, 137, 407–421. [Google Scholar]
- Zhu, P.; Gu, Z.; Hong, S.; Lian, H. One-pot production of chitin with high purity from lobster shells using choline chloride–malonic acid deep eutectic solvent. Carbohydr. Polym. 2017, 177, 217–223. [Google Scholar] [CrossRef]
- Hong, S.; Yuan, Y.; Yang, Q.; Zhu, P.; Lian, H. Versatile acid base sustainable solvent for fast extraction of various molecular weight chitin from lobster shell. Carbohydr. Polym. 2018, 201, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Einbu, A.; Vårum, K.M. Depolymerization and De-N-acetylation of Chitin Oligomers in Hydrochloric Acid. Biomacromolecules 2007, 8, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Einbu, A.; Vårum, K.M. Characterization of Chitin and Its Hydrolysis to GlcNAc and GlcN. Biomacromolecules 2008, 9, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Arai, Y.; Itoh, T. A microfibril formation from depolymerized chitosan by N-acetylation. Agric. Boil. Chem. 1982, 46, 2379–2381. [Google Scholar]
- Il’Ina, A.V.; Varlamov, V.P. Hydrolysis of chitosan in lactic acid. Прикладная биoхимия и микрoбиoлoгия 2004, 40, 300–303. [Google Scholar] [CrossRef]
- Tian, F.; Liu, Y.; Hu, K.; Zhao, B. Study of the depolymerization behavior of chitosan by hydrogen peroxide. Carbohydr. Polym. 2004, 57, 31–37. [Google Scholar] [CrossRef]
- Prashanth, K.V.H.; Dharmesh, S.M.; Rao, K.S.J.; Tharanathan, R.N. Free radical-induced chitosan depolymerized products protect calf thymus DNA from oxidative damage. Carbohydr. Res. 2007, 342, 190–195. [Google Scholar] [CrossRef]
- Trombotto, S.; Ladaviere, C.; Delolme, F.; Domard, A. Chemical Preparation and Structural Characterization of a Homogeneous Series of Chitin/Chitosan Oligomers. Biomacromolecules 2008, 9, 1731–1738. [Google Scholar] [CrossRef]
- Mourya, V.K.; Inamdar, N.N.; Choudhari, Y.M. Chitooligosaccharides: Synthesis, characterization and applications. Polym. Sci. Ser. A 2011, 53, 583–612. [Google Scholar] [CrossRef]
- Chylenski, P.; Petrović, D.M.; Müller, G.; Dahlström, M.; Bengtsson, O.; Lersch, M.; Siika-Aho, M.; Horn, S.J.; Eijsink, V.G.H. Enzymatic degradation of sulfite-pulped softwoods and the role of LPMOs. Biotechnol. Biofuels 2017, 10, 177. [Google Scholar] [CrossRef]
- Müller, G.; Várnai, A.; Johansen, K.S.; Eijsink, V.G.H.; Horn, S.J. Harnessing the potential of LPMO-containing cellulase cocktails poses new demands on processing conditions. Biotechnol. Biofuels 2015, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, A.; Han, H.; Liu, T.; Yang, Q. A potent chitinase from Bacillus subtilis for the efficient bioconversion of chitin-containing wastes. Int. J. Boil. Macromol. 2018, 116, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wei, G.; Mo, X.; Zhou, N.; Chen, K.; Ouyang, P. Enzymatic hydrolysis of chitin pretreated by bacterial fermentation to obtain pure N-acetyl-d-glucosamine. Green Chem. 2018, 20, 2320–2327. [Google Scholar] [CrossRef]
- Naqvi, S.; Moerschbacher, B.M. The cell factory approach toward biotechnological production of high-value chitosan oligomers and their derivatives: An update. Crit. Rev. Biotechnol. 2017, 37, 11–25. [Google Scholar] [CrossRef]
- Kadokawa, J.-I. Precision Polysaccharide Synthesis Catalyzed by Enzymes. Chem. Rev. 2011, 111, 4308–4345. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A. Enzymatic Polymer Synthesis: An Opportunity for Green Polymer Chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Samain, E.; Drouillard, S.; Heyraud, A.; Driguez, H.; Geremia, R.A. Gram-scale synthesis of recombinant chitooligosaccharides in Escherichia coli. Carbohydr. Res. 1997, 302, 35–42. [Google Scholar] [CrossRef]
- Biely, P.; Kratky, Z.; Vrsanska, M. Substrate-Binding Site of Endo-1,4-P-Xylanase of the Yeast. Eur. J. Biochem. 1981, 119, 559–564. [Google Scholar] [CrossRef]
- Pantaleone, D.; Yalpani, M.; Scollar, M. Unusual susceptibility of chitosan to enzymic hydrolysis. Carbohydr. Res. 1992, 237, 325–332. [Google Scholar] [CrossRef]
- Zhang, H.; Neau, S.H. In vitro degradation of chitosan by a commercial enzyme preparation: Effect of molecular weight and degree of deacetylation. Biomaterials 2001, 22, 1653–1658. [Google Scholar] [CrossRef]
- Kittur, F.S.; Kumar, A.B.V.; Varadaraj, M.C.; Tharanathan, R.N. Chitooligosaccharides—preparation with the aid of pectinase isozyme from Aspergillus niger and their antibacterial activity. Carbohydr. Res. 2005, 340, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-X.; Xia, W.-S.; Zhang, J.-L. Enzymatic preparation of chitooligosaccharides by commercial lipase. Food Chem. 2008, 111, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V. Low molecular weight chitosans: preparation with the aid of papain and characterization. Biochim. Biophys. Acta (BBA) Gen. Subj. 2004, 1670, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-S.; Ahn, K.-J.; Lee, D.-W.; Byun, M.-W.; Park, H.-J. Preparation of chitosan oligomers by irradiation. Polym. Degrad. Stab. 2002, 78, 533–538. [Google Scholar] [CrossRef]
- Lopatin, S.A.; Derbeneva, M.S.; Kulikov, S.N.; Varlamov, V.P.; Shpigun, O.A. Fractionation of chitosan by ultrafiltration. J. Anal. Chem. 2009, 64, 648–651. [Google Scholar] [CrossRef]
- Hattori, T.; Anraku, N.; Kato, R. Capillary electrophoresis of chitooligosaccharides in acidic solution: Simple determination using a quaternary-ammonium-modified column and indirect photometric detection with Crystal Violet. J. Chromatogr. B 2010, 878, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, Y.; Zhao, L.; Zhou, J.; Xia, Q.; Jiang, L.; Fan, L. Purification of DP 6 to 8 chitooligosaccharides by nanofiltration from the prepared chitooligosaccharides syrup. Bioresour. Bioprocess. 2014, 1, 170. [Google Scholar] [CrossRef][Green Version]
- Le Dévédec, F.; Bazinet, L.; Furtos, A.; Venne, K.; Brunet, S.; Mateescu, M.A. Separation of chitosan oligomers by immobilized metal affinity chromatography. J. Chromatogr. A 2008, 1194, 165–171. [Google Scholar] [CrossRef]
- Lv, M.; Hu, Y.; Gänzle, M.G.; Lin, J.; Wang, C.; Cai, J. Preparation of chitooligosaccharides from fungal waste mycelium by recombinant chitinase. Carbohydr. Res. 2016, 430, 1–7. [Google Scholar] [CrossRef]
- Gao, X.A.; Jung, W.J.; Kuk, J.H.; Park, R.D. Reaction pattern of Bacillus cereus D-11 Chitosanase on chitooligosaccharide alchols. J. Microbiol. Biotechnol. 2009, 19, 358–361. [Google Scholar] [CrossRef]
- Yi, L.; Sun, X.; Du, K.; Ouyang, Y.; Wu, C.; Xu, N.; Linhardt, R.J.; Zhang, Z. UP-HILIC-MS/MS to Determine the Action Pattern of Penicillium sp. Dextranase. J. Am. Soc. Mass Spectrom. 2015, 26, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Sørbotten, A.; Horn, S.J.; Eijsink, V.G.H.; Varum, K.M. Degradation of chitosans with chitinase B from Serratia marcescens: Production of chito-oligosaccharides and insight into enzyme processivity. FEBS J. 2005, 272, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Haebel, S.; Bahrke, S.; Peter, M.G. Quantitative Sequencing of Complex Mixtures of Heterochitooligosaccharides by vMALDI-Linear Ion Trap Mass Spectrometry. Anal. Chem. 2007, 79, 5557–5566. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Shen, A.; Gou, Z.; Chen, D.; Liang, X. Hydrophilic interaction/weak cation-exchange mixed-mode chromatography for chitooligosaccharides separation. Carbohydr. Res. 2012, 361, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Santos-Moriano, P.; Woodley, J.M.; Plou, F.J. Continuous production of chitooligosaccharides by an immobilized enzyme in a dual-reactor system. J. Mol. Catal. B Enzym. 2016, 133, 211–217. [Google Scholar] [CrossRef]
- Sasaki, C.; Kristiansen, A.; Fukamizo, T.; Vårum, K.M. Biospecific Fractionation of Chitosan. Biomacromolecules 2003, 4, 1686–1690. [Google Scholar] [CrossRef]
- Yoon, J.H. Enzymatic synthesis of chitooligosaccharides in organic cosolvents. Enzym. Microb. Technol. 2005, 37, 663–668. [Google Scholar] [CrossRef]
- Lopatin, S.; Ilyin, M.; Pustobaev, V.; Bezchetnikova, Z.; Varlamov, V.; Davankov, V. Mass-Spectrometric Analysis of N-Acetylchitooligosaccharides Prepared through Enzymatic Hydrolysis of Chitosan. Anal. Biochem. 1995, 227, 285–288. [Google Scholar] [CrossRef]
- Jung, W.-J.; Souleimanov, A.; Park, R.-D.; Smith, D.L. Enzymatic production of N-acetyl chitooligosaccharides by crude enzyme derived from Paenibacillus illioisensis KJA-424. Carbohydr. Polym. 2007, 67, 256–259. [Google Scholar] [CrossRef]
- Cao, L.; Wu, J.; Li, X.; Zheng, L.; Wu, M.; Liu, P.; Huang, Q. Validated HPAEC-PAD Method for the Determination of Fully Deacetylated Chitooligosaccharides. Int. J. Mol. Sci. 2016, 17, 1699. [Google Scholar] [CrossRef]
- Wu, H.; Yao, Z.; Bai, X.; Du, Y.; Lin, B. Anti-angiogenic activities of chitooligosaccharides. Carbohydr. Polym. 2008, 73, 105–110. [Google Scholar] [CrossRef]
- Jiang, M.; Guo, Z.; Wang, C.; Yang, Y.; Liang, X.; Ding, F. Neural activity analysis of pure chito-oligomer components separated from a mixture of chitooligosaccharides. Neurosci. Lett. 2014, 581, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Hamer, S.N.; Cord-Landwehr, S.; Biarnés, X.; Planas, A.; Waegeman, H.; Moerschbacher, B.M.; Kolkenbrock, S. Enzymatic production of defined chitosan oligomers with a specific pattern of acetylation using a combination of chitin oligosaccharide deacetylases. Sci. Rep. 2015, 5, 8716. [Google Scholar] [CrossRef] [PubMed]
- Paul, T.; Halder, S.K.; Das, A.; Ghosh, K.; Mandal, A.; Payra, P.; Barman, P.; Das Mohapatra, P.K.; Pati, B.R.; Mondal, K.C. Production of chitin and bioactive materials from Black tiger shrimp (Penaeus monodon) shell waste by the treatment of bacterial protease cocktail. 3 Biotech 2015, 5, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Doan, C.T.; Tran, T.N.; Nguyen, V.B.; Nguyen, A.D.; Wang, S.-L. Reclamation of Marine Chitinous Materials for Chitosanase Production via Microbial Conversion by Paenibacillus macerans. Mar. Drugs 2018, 16, 429. [Google Scholar] [CrossRef]
- Bosquez-Molina, E.; Zavaleta-Avejar, L. New Bioactive Biomaterials Based on Chitosan. In Chitosan in the Preservation of Agricultural Commodities; Elsevier: Amsterdam, The Netherlands, 2016; pp. 33–64. [Google Scholar]
- Jiang, Y.; Fu, C.; Wu, S.; Liu, G.; Guo, J.; Su, Z. Determination of the Deacetylation Degree of Chitooligosaccharides. Mar. Drugs 2017, 15, 332. [Google Scholar] [CrossRef]
- Limón, M.C.; Margolles-Clark, E.; Benítez, T.; Penttilä, M. Addition of substrate-binding domains increases substrate-binding capacity and specific activity of a chitinase from Trichoderma harzianum. FEMS Microbiol. Lett. 2001, 198, 57–63. [Google Scholar] [CrossRef]
- Delgado-Jarana, J.; Limon, M.C.; Chacon, M.R.; Mejias, R.; Rincón, A.M.; Codón, A.C.; Benítez, T. Increased antifungal and chitinase specific activities of Trichoderma harzianum CECT 2413 by addition of a cellulose binding domain. Appl. Microbiol. Biotechnol. 2004, 64, 675–685. [Google Scholar]
- Kowsari, M.; Motallebi, M.; Zamani, M. Protein engineering of chit42 towards improvement of chitinase and antifungal activities. Curr. Microbiol. 2014, 68, 495–502. [Google Scholar] [CrossRef]
- Matroodi, S.; Motallebi, M.; Zamani, M.; Moradyar, M. Designing a new chitinase with more chitin binding and antifungal activity. World J. Microbiol. Biotechnol. 2013, 29, 1517–1523. [Google Scholar] [CrossRef]
- Kurašin, M.; Kuusk, S.; Kuusk, P.; Sørlie, M.; Väljamäe, P. Slow Off-rates and Strong Product Binding Are Required for Processivity and Efficient Degradation of Recalcitrant Chitin by Family 18 Chitinases. J. Boil. Chem. 2015, 290, 29074–29085. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Fang, W.; Xiao, Y.; Yang, X.; Zhang, Y.; Bidochka, M.J.; Pei, Y. Directed evolution for increased chitinase activity. App. Microbiol. Biotechnol. 2007, 76, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Songsiriritthigul, C.; Pesatcha, P.; Eijsink, V.G.H.; Yamabhai, M. Directed evolution of a Bacillus chitinase. Biotechnol. J. 2009, 4, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Mekasha, S.; Byman, I.R.; Lynch, C.; Toupalová, H.; Anděra, L.; Næs, T.; Vaaje-Kolstad, G.; Eijsink, V.G. Development of enzyme cocktails for complete saccharification of chitin using mono-component enzymes from Serratia marcescens. Process. Biochem. 2017, 56, 132–138. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, J.; Xu, N.; Zhang, A.; Chen, K.; Xin, F.; Zhang, W.; Ma, J.; Fang, Y.; Jiang, M.; et al. The broad-specificity chitinases: their origin, characterization, and potential application. Appl. Microbiol. Biotechnol. 2019, 103, 3289–3295. [Google Scholar] [CrossRef]
- Pérez-Martínez, A.S.; De León-Rodríguez, A.; Harris, L.J.; Herrera-Estrella, A.; De La Rosa, A.P.B. Overexpression, purification and characterization of the Trichoderma atroviride endochitinase, Ech42, in Pichia pastoris. Protein Expr. Purif. 2007, 55, 183–188. [Google Scholar] [CrossRef]
- Dahiya, N.; Tewari, R.; Tiwari, R.P.; Hoondal, G.S. Chitinase Production in Solid-State Fermentation by Enterobacter sp. NRG4 Using Statistical Experimental Design. Curr. Microbiol. 2005, 51, 222–228. [Google Scholar] [CrossRef]
- Nidheesh, T.; Pal, G.K.; Suresh, P. Chitooligomers preparation by chitosanase produced under solid state fermentation using shrimp by-products as substrate. Carbohydr. Polym. 2015, 121, 1–9. [Google Scholar] [CrossRef]
- Orikoshi, H.; Nakayama, S.; Miyamoto, K.; Hanato, C.; Yasuda, M.; Inamori, Y.; Tsujibo, H. Roles of Four Chitinases (ChiA, ChiB, ChiC, and ChiD) in the Chitin Degradation System of Marine Bacterium Alteromonas sp. Strain O-7. Appl. Environ. Microbiol. 2005, 71, 1811–1815. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, N.D.; Brück, W.M.; Garbe, D.; Brück, T.B. Enzymatic Modification of Native Chitin and Conversion to Specialty Chemical Products. Mar. Drugs 2020, 18, 93. https://doi.org/10.3390/md18020093
Arnold ND, Brück WM, Garbe D, Brück TB. Enzymatic Modification of Native Chitin and Conversion to Specialty Chemical Products. Marine Drugs. 2020; 18(2):93. https://doi.org/10.3390/md18020093
Chicago/Turabian StyleArnold, Nathanael D., Wolfram M. Brück, Daniel Garbe, and Thomas B. Brück. 2020. "Enzymatic Modification of Native Chitin and Conversion to Specialty Chemical Products" Marine Drugs 18, no. 2: 93. https://doi.org/10.3390/md18020093
APA StyleArnold, N. D., Brück, W. M., Garbe, D., & Brück, T. B. (2020). Enzymatic Modification of Native Chitin and Conversion to Specialty Chemical Products. Marine Drugs, 18(2), 93. https://doi.org/10.3390/md18020093