Abstract
Side-chain derivatives of eurotiumide A, a dihydroisochroman-type natural product, have been synthesized and their antimicrobial activities described. Sixteen derivatives were synthesized from a key intermediate of the total synthesis of eurotiumide A, and their antimicrobial activities against two Gram-positive bacteria, methicillin-susceptible and methicillin-resistant Staphylococcus aureus (MSSA and MRSA), and a Gram-negative bacterium, Porphyromonas gingivalis, were evaluated. The results showed that derivatives having an iodine atom on their aromatic ring instead of the prenyl moiety displayed better antimicrobial activity than eurotiumide A against MSSA and P. gingivalis. Moreover, we discovered that a derivative with an isopentyl side chain, which is a hydrogenated product of eurotiumide A, is the strongest antimicrobial agent against all three strains, including MRSA.
1. Introduction
Humans have always struggled against infectious diseases [1,2,3,4,5] and in relatively recent times have developed various antimicrobial therapies [6,7,8]. Since the discovery of penicillin [9], various natural products having antimicrobial activity have been discovered [10,11,12,13,14,15,16], and the majority of clinically used antibiotics are either natural products, semisynthetic derivatives, or compounds derived from them [17,18,19]. Despite the presence of many excellent antibiotics, multidrug-resistant bacterial pathogens have emerged all over the world [20,21,22], and the development of novel and effective antimicrobial agents against many kinds of pathogenic bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), should remain a continuous mission for medicinal chemists. In 2014, Wang and co-workers discovered eurotiumides, which are novel dihydroisocoumarin-type natural products, from a gorgonian-derived fungus, Eurotium sp. XS-200900E6 [23]. Among the series of eurotiumides, eurotiumide A (1), having cis configurations at H3/H4, exhibited potent antimicrobial activities against Staphylococcus epidermidis, Bacillus cereus, Vibrio anguillarum, and Escherichia coli. Based on that report, although 1 seems to be an attractive seed compound for antibiotics, further antimicrobial investigation and a structure–activity relationship study of 1 are needed. In particular, because there is a chance that modification of the side chain of the aromatic ring could improve antimicrobial activity and the spectrum, a structure–activity relationship study of the substituent effect of the aromatic ring is essential for discovering promising candidates for antimicrobial agents. Recently, we reported the first asymmetric total syntheses of (−)-eurotiumide A (1) and (+)-eurotiumide B and revised their reported structures [24]. In our synthetic route, the prenyl side chain of the aromatic ring was introduced in the late stage by the Stille coupling reaction with the key intermediate 2. Based on our previous results, we considered that a number of derivatives of 1, which have a variety of kinds of side-chain moiety, could be obtained from the common intermediate 2 and non-substituted compound 3 in the late stage of synthesis (Figure 1).
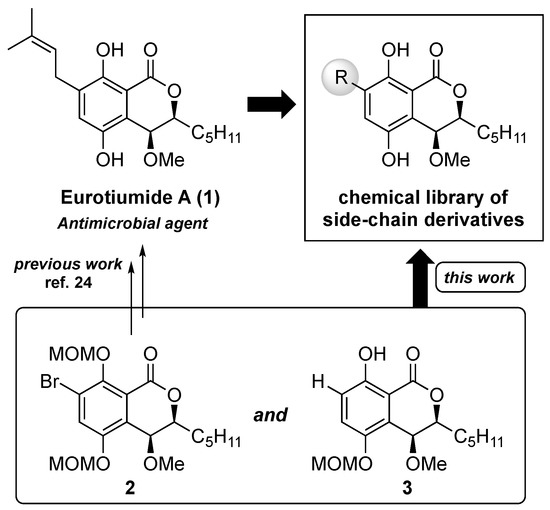
Figure 1.
Concept of construction of the chemical library of the side chain-derivatives of eurotiumide A (1).
In this work, as part of our continuing research [24,25], we constructed a chemical library of the side-chain derivatives of eurotiumide A (1) to elucidate the effects of the side chains of the aromatic rings and to develop antimicrobial agents against methicillin-susceptible S. aureus (MSSA) and methicillin-resistant S. aureus (both Gram-positive bacteria), as well as Porphyromonas gingivalis (a Gram-negative bacterium).
2. Results and Discussion
2.1. Synthesis of the Side-Chain Derivatives of Eurotiumide A
Our synthetic plan is shown in Figure 2. We planned to introduce three types of functional groups: a hydrocarbon group, including hydrogen, alkyl, and aromatic rings (Type A); a heteroatom and heteroatom-containing alkyl group (Type B); and halogen atoms group (Type C). The derivatives of groups A and B could be derived from 2 by the cross-coupling reaction and functional group transformation. The halogenated derivatives (Type C) would be obtained from 3 by direct introduction of the halogen atoms. Although Wang et al. isolated the natural eurotiumide A (1) as a racemic form, they evaluated the antimicrobial activities of its enantiomers after separation by chiral HPLC and revealed that there was no significant difference between the enantiomers [23]. From the viewpoint of the efficiency of compound supply, we decided to make racemic compounds.
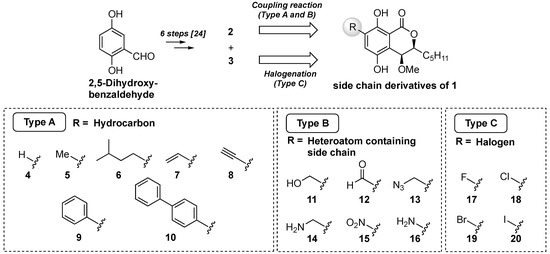
Figure 2.
Synthetic plan of the side-chain derivatives of eurotiumide A (1).
First, we initiated the syntheses of the derivatives of group A (Scheme 1). The non-substituted derivative 4 was obtained from 3 by deprotection of the diMOM group with aqueous 6 M HCl in methanol at 40 °C in 79% yield. Catalytic hydrogenation of eurotiumide A (1) gave the isopentyl derivative 6 in quantitative yield. Methyl and vinyl groups were introduced by the Stille coupling reaction with 2 to afford methyl derivative 5a and styrene derivative 7a in 83% and quantitative yields, respectively. Phenyl derivative 9a and biphenyl derivative 10a were obtained from 2 by the Suzuki–Miyaura cross coupling reaction with the corresponding boronic acids in 75% and 77% yields, respectively. Deprotection of the diMOM group of derivatives 5a, 7a, 9a, and 10a then gave the corresponding desired products (5, 7, 9, and 10). We tried to introduce the alkyne group by the Sonogashira coupling reaction; however, the desired alkyne product was obtained in only 12% yield. To improve the reaction yield, the Seyferth–Gilbert homologation using the Ohira–Bestmann reagent 21 was applied to the aldehyde derivative 12a (vide infra) and afforded the desired alkyne 8a in quantitative yield. After acidic treatment of 8a, the alkyne derivative 8 was obtained in 68% yield.
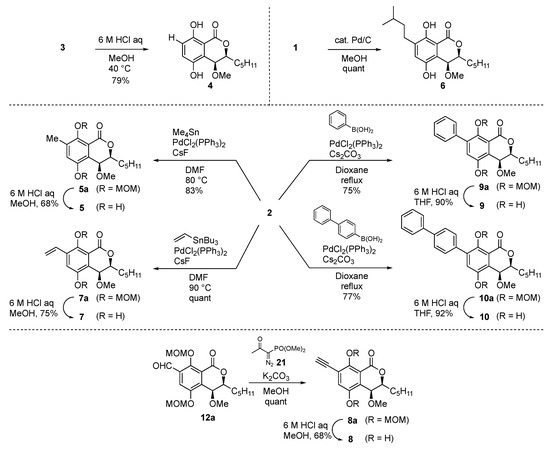
Scheme 1.
Synthesis of the hydrocarbon derivatives (type A).
With type A derivatives in hand, we turned our attention to preparing type B derivatives having heteroatom-containing side chains (Scheme 2). For the introduction of an alkyl group containing heteroatoms, we chose the styrene derivative 7a as a starting point. Ozonolysis of the alkene moiety of 7a afforded the diMOM-protected benzaldehyde 12a in excellent yield. Acidic treatment of 12a gave the desired deprotected benzaldehyde derivative 12 in 77%. On the other hand, reduction of the aldehyde moiety of 12a with sodium borohydride to give the benzyl alcohol 11a and the deprotection furnished the hydroxymethyl derivative 11 in moderate yield. To introduce a nitrogen group at the benzyl position of 11a, the primary alcohol moiety was converted to a mesyl group (22) and a nucleophilic substitution reaction with sodium azide afforded diMOM-protected azide 13a in good yield. Derivative 13a was treated with aqueous 6 M HCl in MeOH to furnish the desired dihydroxy azide derivative 13. We then tried to convert the azide into an amine functionality. After several attempts, we found that addition of triethylamine was crucial to keep the reaction clean and we succeeded to get 14a. Then, deprotection of the diMOM group gave the desired aminomethyl derivative 14.
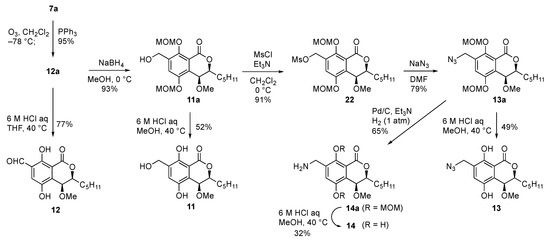
Scheme 2.
Synthesis of the derivatives having heteroatom-containing side chains (type B).
Next, a nitration reaction was conducted with non-substituted derivative 3 by adding HNO3 in AcOH to afford monoMOM-protected nitro derivative 15a as a crude product; then it was deprotected under acidic condition to give the nitro derivative 15 (Scheme 3). After that, hydrogenation with Adam’s catalyst produced the aniline derivative 16 from 15.
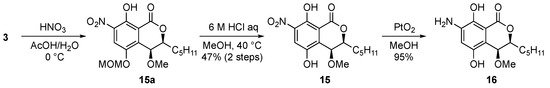
Scheme 3.
Synthesis of nitro and aniline derivatives.
Finally, we tried to synthesize the halogenated derivatives (Scheme 4). Chloro and iodo groups were introduced to treat 3 with N-chlorosuccinimide and N-iodosuccinimide in DMF to afford the chloro derivative 18a and the iodo derivative 20a, respectively. The diMOM groups of 18a and 20a were then deprotected under acidic conditions to afford the desired 18 and 20. Bromo derivative 19 was obtained from 2 in 97% yield by acid treatment to cleave the diMOM group. However, despite several efforts to introduce fluorine to the aromatic ring from 3, we could not get the desired fluoro derivative 17. We also tried the Sandmeyer reaction with 16 but did not obtain the desired 17.
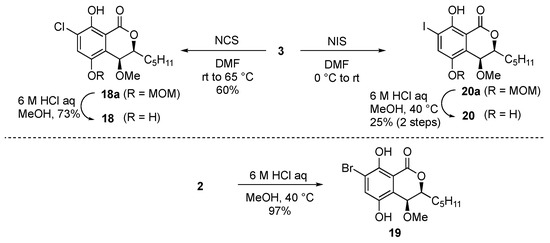
Scheme 4.
Synthesis of halogenated derivatives (type C).
2.2. Antimicrobial Evaluation of Synthesized Derivatives
After the initially set derivatives of eurotiumide A were synthesized, the first antimicrobial activity screening was conducted against the Gram-positive MSSA and MRSA as well as the Gram-negative P. gingivalis in 10 µM solutions of the synthesized derivatives to narrow down the promising antimicrobial candidates. The results are depicted in Figure 3. (+/−)-Eurotiumide A (1) exhibited mild antimicrobial activity against MSSA at this concentration (Figure 3a). While most of the derivatives did not show antimicrobial activity against this strain, the isopentyl derivative 6 and the iodo derivative 20 exhibited more potent antimicrobial activity than 1. Next, we tested the same screening against MRSA (Figure 3b). Most of the derivatives that displayed good activity against MSSA showed no antimicrobial activity against MRSA. Even natural product 1 and the iodo derivative 20 also did not show good antimicrobial activity against MRSA. Surprisingly, only the isopentyl derivative 6, which was a reduced derivative of 1, was found to have good antimicrobial activity against MRSA. We also conducted antimicrobial screening against P. gingivalis (Figure 3c). Unlike the case with S. aureus, many derivatives, specifically eurotiumide A (1), isopentyl derivative 6, vinyl derivative 7, aniline derivative 16, and three halogenated derivatives (18, 19, 20), were effective against P. gingivalis.
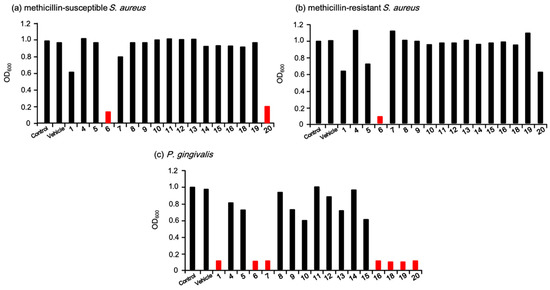
Figure 3.
Initial screening of antimicrobial activity against (a) methicillin-susceptible S. aureus, (b) methicillin-resistant S. aureus, and (c) P. gingivalis. The terminal concentration was 10 µM.
Since we acquired promising agents against all three strains, we determined the IC50 values of these candidates (Table 1). The IC50 values of the isopentyl derivative 6 and the iodo derivative 20 against MSSA were 5.6 µM (2.0 µg/mL) and 9.0 µM (3.7 µg/mL), respectively. Moreover, the IC50 value of 6 against MRSA was 4.3 µM (1.5 µg/mL), which is the same level of activity against MSSA. The IC50 values of these seven candidates (1, 6, 7, 16, 18, 19, and 20) against P. gingivalis ranged from 2.0 to 7.0 µM. We also checked the cytotoxicity of three compounds (1, 6, and 20) against the A549 cell line, and these three compounds were non-toxic in 10 µM.

Table 1.
The IC50 values (µM) of the selected side chain derivatives against methicillin-susceptible S. aureus (MSSA), methicillin-resistant S. aureus (MRSA), and P. gingivalis. Vancomycin (VCM) was used as a positive control against MSSA and MRSA. Cefcapene pivoxyl (CFPN-PI) was used as a positive control against P. gingivalis.
In this study, we discovered that the isopentyl derivative 6, which is a one-point modified compound of natural product 1, and the iodo derivative 20 have superior antimicrobial activity to 1 against MSSA and P. gingivalis. Although 20 did not exhibit good efficacy against MRSA, 6 was found to maintain antimicrobial activity against these three strains, including MRSA. These results indicate that S. aureus is sensitive to changes in the side chain of the aromatic ring and that MRSA can distinguish the subtle difference between prenyl and isopentyl moieties. Moreover, the weak antimicrobial activity of 1 against MRSA suggests a binding affinity between 1 and the penicillin binding protein 2’ [26], which is the main resistance mechanism of MRSA against antibiotics. The inhibition of cell wall synthesis seems to be the mode of action of 1, although a more detailed study is needed to clarify the mode of action of 6 and 20. On the other hand, we found that several compounds having alkyl and halogenated side chains well suppressed the increase in P. gingivalis.
3. Materials and Methods
3.1. Preparation of Eurotiumide A Derivatives.
3.1.1. General Procedure
All the reactions were carried out in a round-bottomed flask with an appropriate number of necks and side arms connected to a three-way stopcock and/or a rubber septum cap under an argon atmosphere. All vessels were first evacuated by rotary pump and then flushed with argon prior to use. Solutions and solvents were introduced by hypodermic syringe through a rubber septum. During the reaction, the vessel was kept under a positive pressure of argon. Dry THF was freshly prepared by distillation from benzophenone ketyl before use. Anhydrous CH2Cl2, DMF, ethanol, MeCN, methanol, pyridine, and toluene were purchased from Kanto Chemical Co. Inc. Infrared (IR) spectra were recorded on a JASCO FT/IR-4100 spectrophotometer using a 5 mm KBr plate. Wavelengths of maximum absorbance are quoted in cm−1. 1H-NMR spectra were recorded on a JEOL ECA–400 (400 MHz), Bruker AV-400N (400 MHz), and Bruker AV–500 (500 MHz) in CDCl3. Chemical shifts are reported in parts per million (ppm), and signals are expressed as singlet (s), doublet (d), triplet (t), multiplet (m), broad (br), and overlapped. 13C-NMR spectra were recorded on a JEOL ECA–400 (100 MHz), Bruker AV–400N (100 MHz), and Bruker AV–500 (125 MHz) in CDCl3. Chemical shifts are reported in parts per million (ppm) (see Supplementary Materials). High resolution mass (HRMS) spectra were recorded on a Thermo Scientific Exactive. All melting points were measured with a Yanaco MP-500D. Analytical thin layer chromatography (TLC) was performed using 0.25 mm E. Merck Silica gel (60F-254) plates. Reaction components were visualized phosphomolybdic acid or ninhydrin or p-anisaldehyde in 10% sulfuric acid in ethanol. Kanto Chem. Co. Silica Gel 60N (particle size 0.040–0.050 mm) was used for column chromatography.
3.1.2. Synthesis of (3S,4S)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (4)
To a solution of bromo compound 3 (10.0 mg, 30.8 µmol) in MeOH (2.3 mL) was added 6 M aqueous HCl (0.77 mL) at 0 °C. After stirring for 30 min at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by preparative thin layer chromatography (PTLC) (EtOAc:n-hexane = 3:7) to give non-substituted derivative 4 (6.8 mg, 79%) as a white solid. m.p. 120–121 °C; 1H-NMR (400 MHz, CDCl3) δ 10.62 (1H, s), 7.06 (1H, d, J = 9.0 Hz), 6.91 (1H, d, J = 9.0 Hz), 5.89 (1H, br-s), 4.77 (1H, d, J = 2.7 Hz), 4.50 (1H, ddd, J = 2.7, 5.4, 8.3 Hz), 3.40 (3H, s), 1.95 (1H, m), 1.85 (1H, m), 1.70–1.50 (1H, overlapped), 1.46 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (100 MHz, CDCl3) δ 169.0, 156.2, 145.7, 125.1, 121.7, 118.8, 107.6, 81.4, 69.8, 56.8, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3219, 2955, 2924, 2860, 1661, 1586, 1471, 1293, 1204, 905 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H20O5Na)+ 303.1208, found 303.1200.
3.1.3. Synthesis of (3S,4S)-5,8-dihydroxy-7-isopentyl-4-methoxy-3-pentylisochroman-1-one (6)
To a solution of eurotiumide A (1) (1.6 mg, 4.6 µmol) in MeOH (0.23 mL) was added Pd/C (1.6 mg, 100 w/w%) at room temperature. After stirring for 1.5 h under hydrogen atmosphere (balloon), the reaction mixture was passed through Celite and the organic solvent was removed under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 2:3) to give isopentyl derivative 6 (1.4 mg, 88%) as a white wax. 1H-NMR (500 MHz, CDCl3) δ 10.91 (1H, s), 6.93 (1H, s), 5.62 (1H, br-s), 4.74 (1H, d, J = 2.5 Hz), 4.48 (1H, ddd, J = 2.6, 5.4, 8.6 Hz), 3.38 (3H, s), 2.62 (2H, m), 1.95 (1H, m), 1.85 (1H, m), 1.65–1.50 (2H, overlapped), 1.50–1.40 (3H, overlapped), 1.40–1.30 (4H, overlapped), 0.95 (6H, d, J = 6.3 Hz), 0.90 (3H, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.4, 154.7, 145.0, 133.6, 124.8, 118.6, 106.8, 81.4, 69.9, 56.6, 38.4, 31.6, 29.8, 29.7, 27.9, 27.5, 14.9, 22.5, 14.0.; IR (KBr) 3290, 2956, 2927, 2870, 1761, 1445, 1171, 807 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H31O5)+ 351.2171, found 351.2177.
3.1.4. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-7-methyl-3-pentylisochroman-1-one (5a)
To a solution of bromo compound 3 (40.0 mg, 89.4 µmol) and CsF (16.3 mg, 107 µmol) in degassed DMF (0.45 mL) were added Me4Sn (15 µL, 107 µmol) and PdCl2(PPh3)2 (6.3 mg, 8.94 µmol) at room temperature. After stirring for 50 min at 80 °C, the reaction was quenched by adding water. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 3:7) to give diMOM-protected methyl derivative 5a (28.5 mg, 83%) as a yellow amorphous. 1H-NMR (400 MHz, CDCl3) δ 7.255 (1H, s), 5.21 (2H, s), 5.10 (1H, d, J = 6.8 Hz), 5.07 (1H, d, J = 6.8 Hz), 4.59 (1H, d, J = 1.5 Hz), 4.26 (1H, ddd, J = 1.5, 5.9, 7.5 Hz), 3.60 (3H, s), 3.50 (3H, s), 3.30 (3H, s), 2.39 (3H, s), 2.02 (1H, m), 1.81 (1H, m), 1.70–1.50 (1H, overlapped), 1.43 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.4, 152.3, 149.8, 135.7, 126.3, 121.3, 118.7, 101.5, 95.0, 80.9, 68.2, 57.5, 56.7, 56.4, 31.6, 30.6, 24.9, 22.6, 17.6, 14.0.; IR (KBr) 2958, 2927, 2858, 2828, 1728, 1478, 1153 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H31O7)+ 383.2070, found 383.2069.
3.1.5. (3S,4S)-5,8-dihydroxy-4-methoxy-7-methyl-3-pentylisochroman-1-one (5)
To a solution of diMOM-protected methyl derivative 5a (10.0 mg, 26.0 µmol) in MeOH (2.0 mL) was added 6 M aqueous HCl (0.65 mL) at 0 °C. After stirring for 1 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give methyl derivative 5 (5.2 mg, 68%) as a yellow solid. m.p. 113 °C; 1H-NMR (400 MHz, CDCl3) δ 10.89 (1H, s), 6.93 (1H, s), 5.59 (1H, br-s), 4.75 (1H, d, J = 2.7 Hz), 4.48 (1H, ddd, J = 2.7, 5.4, 8,3 Hz), 3.37 (3H, s), 2.25 (3H, s), 1.93 (1H, m), 1.84 (1H, m), 1.70-1.50 (1H, overlapped), 1.45 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.6 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.4, 154.9, 144.9, 128.7, 125.8, 118.6, 106.6, 81.4, 69.8, 56.5, 31.6, 29.8, 24.9, 22.5, 15.8, 14.0.; IR (KBr) 3340, 2957, 2928, 2859, 1682, 1654, 1604, 1296, 1172 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C16H22O5Na)+ 317.1365, found 317.1350.
3.1.6. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentyl-7-vinylisochroman-1-one (7a)
To a solution of bromo compound 3 (200 mg, 0.447 mmol) and CsF (135.8 mg, 0.894 mmol) in degassed DMF (2.2 mL) were added tributylvinyltin (0.26 mL, 0.894 mmol) and PdCl2(PPh3)2 (62.8 mg, 89.0 µmol) at room temperature. After stirring for 1 h at 80 °C, the reaction was quenched by adding water. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 3:7) to give diMOM-protected vinyl derivative 7a (185.1 mg, quant) as a yellow solid. m.p. 63–64 °C; 1H-NMR (500 MHz, CDCl3) δ 7.56 (1H, s), 7.14 (1H, dd, J = 11.1, 17.7 Hz), 5.76 (1H, d, J = 17.7 Hz), 5.40 (1H, d, J = 11.1 Hz), 5.24 (2H, s), 5.08 (1H, d, J = 6.3 Hz), 5.05 (1H, d, J = 6.3 Hz), 4.60 (1H, d, J = 1.3 Hz), 4.26 (1H, ddd, J = 1.3, 5.8, 7.4 Hz), 3.58 (3H, s), 3.50 (3H, s), 3.31 (3H, s), 2.03 (1H, m), 1.81 (1H, m), 1.56 (1H, m), 1.43 (1H, m), 1.40–1.25 (4H, overlapped), 0.90 (3H, t, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.0, 150.7, 150.2, 134.9, 131.3, 128.5, 119.7, 116.7, 116.0, 101.5, 95.2, 80.8, 68.3, 57.9, 56.8, 56.4, 31.6, 30.6, 24.9, 22.5, 14.0.; IR (KBr) 2953, 2931, 2861, 2829, 1730, 1471, 1426, 1155, 929 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C21H31O7)+ 395.2070, found 395.2078.
3.1.7. (3S,4S)-5,8-dihydroxy-4-methoxy-3-pentyl-7-vinylisochroman-1-one (7)
To a solution of diMOM-protected methyl derivative 7a (13.7 mg, 34.7 µmol) in MeOH (2.6 mL) was added 6 M aqueous HCl (0.87 mL) at 0 °C. After stirring for 3 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give vinyl derivative 7 (8.5 mg, 75%) as a yellow wax. 1H-NMR (500 MHz, CDCl3) δ 11.10 (1H, s), 7.23 (1H, s), 7.01 (1H, dd, J = 11.4, 17.7 Hz), 5.82 (1H, br-s), 5.80 (1H, d, J = 18.0 Hz), 5.37 (1H, d, J = 11.0 Hz), 4.77 (1H, br-s), 4.50 (1H, br-s), 3.40 (3H, s), 1.95 (1H, m), 1.85 (1H, m), 1.58 (1H, m), 1.45 (1H,m), 1.40–1.25 (4H, overlapped), 0.90 (3H, br-s); 13C-NMR (125 MHz, CDCl3) δ 169.3, 153.9, 145.4, 129.8, 128.0, 121.4, 120.9, 116.5, 107.7, 81.5, 69.7, 56.8, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3311, 2956, 2930, 2859, 1659, 1438, 1171 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C17H22O5Na)+ 329.1365, found 329.1368.
3.1.8. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentyl-7-phenylisochroman-1-one (9a)
Bromo compound 3 (10.0 mg, 22.4 µmol), Cs2CO3 (21.9 mg, 67.1 µmol), phenylboronic acid (5.5 mg, 44.7 µM), and PdCl2(PPh3)2 (3.1 mg, 44.7 µmol) were dissolved in degassed dioxane (0.22 mL) at room temperature. After stirring for 1 h under reflux condition, the reaction was quenched by adding saturated aqueous NH4Cl. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 3:7) to give diMOM-protected phenyl derivative 9a (7.4 mg, 75%) as a white wax. 1H-NMR (500 MHz, CDCl3) δ 7.55 (1H, d, J = 7.6 Hz), 7.50-7.38 (3H, overlapped), 7.36 (1H, dd, J = 7.3 Hz), 5.25 (2H, s), 4.80 (2H, s), 4.66 (1H, s), 4.33 (1H, t, J = 7.0 Hz), 3.50 (3H, s), 3.37 (3H, s), 2.92 (3H, s), 2.06 (1H, m), 1.85 (1H, m), 1.70–1.50 (1H, overlapped), 1.50–1.25 (5H, overlapped), 0.92 (3H, br-s); 13C-NMR (125 MHz, CDCl3) δ 162.1, 150.5, 150.0, 139.5, 137.9, 129.8, 128.3, 128.1, 127.7, 121.0, 119.9, 101.0, 95.1, 80.8, 68.3, 57.1, 56.4, 31.6, 30.6, 24.9, 22.5, 14.0.; IR (KBr) 2956, 2927, 2859, 2828, 1728, 1467, 1152, 1008, 932 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C25H32O7Na)+ 467.2046, found 467.2043.
3.1.9. (3S,4S)-5,8-dihydroxy-4-methoxy-3-pentyl-7-phenylisochroman-1-one (9)
To a solution of diMOM-protected methyl derivative 9a (7.4 mg, 16.8 µmol) in THF (1.0 mL) was added 6 M aqueous HCl (0.50 mL) at 0 °C. After stirring for 6 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give phenyl derivative 9 (6.0 mg, 90%) as a yellow solid. m.p. 173–174 °C; 1H-NMR (400 MHz, CDCl3) δ 11.21 (1H, s), 7.58 (2H, d, J = 7.3 Hz), 7.44 (2H, t, J = 7.3 Hz), 7.38 (1H, d, J = 7.6 Hz), 7.13 (1H, s), 5.76 (1H, br-s), 4.82 (1H, d, J = 2.7 Hz), 4.55 (1H, ddd, J = 2.7, 5.1, 8.3 Hz), 3.44 (3H, s), 1.98 (1H, m), 1.89 (1H, m), 1.70–1.40 (2H, overlapped), 1.40–1.25 (4H, overlapped), 0.92 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.5, 153.7, 145.4, 136.2, 131.8, 129.2, 128.3, 127.9, 125.5, 121.1, 107.8, 81.6, 69.6, 56.9, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3307, 2955, 2928, 2859, 1650, 1425, 1295, 1194 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C21H25O5)+ 357.1702, found 357.1707.
3.1.10. (3S,4S)-7-([1,1’-biphenyl]-4-yl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (10a)
Bromo compound 3 (20.0 mg, 44.7 µmol), Cs2CO3 (21.9 mg, 67.1 µmol), 4-biphenylboronic acid (5.5 mg, 44.7 µmol), and PdCl2(PPh3)2 (3.2 mg, 4.47 µmol) were dissolved in degassed dioxane (0.23 mL) at room temperature. After stirring for 1 h under reflux condition, the reaction was quenched by adding saturated aqueous NH4Cl. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give diMOM-protected biphenyl derivative 10a (18.0 mg, 88%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 7.74–7.60 (6H, overlapped), 7.53–7.40 (3H, overlapped), 7.38 (1H, t, J = 7.3 Hz), 5.28 (2H, s), 4.85 (1H, d, J = 7.0 Hz), 4.84 (1H, d, J = 7.0 Hz), 4.68 (1H, d, J = 1.3 Hz), 4.35 (1H, ddd, J = 1.3, 6.0, 7.6 Hz), 3.51 (3H, s), 3.38 (3H, s), 2.99 (3H, s), 2.08 (1H, m), 1.86 (1H, m), 1.70-1.50 (1H, overlapped), 1.46 (1H, m), 1.40–1.25 (4H, overlapped), 0.92 (3H, t, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.1, 150.6, 150.0, 140.5, 140.4, 139.1, 136.8, 130.2, 128.9, 128.1, 127.5, 127.0, 126.9, 120.9, 120.0, 101.1, 95.1, 80.8, 68.3, 57.2, 56.9, 56.4, 31.6, 30.6, 24.9, 22.5, 14.0.; IR (KBr) 2956, 2927, 2858, 2827, 1728, 1467, 1152, 1007, 931 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C31H37O7)+ 521.2539, found 521.2539.
3.1.11. (3S,4S)-7-([1,1’-biphenyl]-4-yl)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (10)
To a solution of diMOM-protected biphenyl derivative 10a (12.9 mg, 24.8 µmol) in THF (1.7 mL) was added 6 M aqueous HCl (0.83 mL) at 0 °C. After stirring for 17 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give biphenyl derivative 10 (9.9 mg, 92%) as a yellow solid. m.p. 181–182 °C; 1H-NMR (400 MHz, CDCl3) δ 11.28 (1H, s), 7.67 (4H, s), 7.64 (2H, d, J = 7.3 Hz), 7.46 (2H, t, J = 7.3 Hz), 7.37 (1H, t, J = 7.3 Hz), 7.19 (1H, s), 5.75 (1H, br-s), 4.84 (1H, d, J = 2.7 Hz), 4.56 (1H, ddd, J = 2.7, 5.4, 8.3 Hz), 3.46 (3H, s), 1.98 (1H, m), 1.89 (1H, m), 1.70–1.50 (2H, overlapped), 1.45–1.25 (4H, overlapped), 0.92 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.4, 153.8, 145.5, 140.7, 135.2, 131.4, 129.6, 128.8, 127.5,127.15, 127.07, 125.3, 121.0, 107.9, 81.5, 69.8, 56.9, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3283, 2954, 2929, 2863, 1668, 1595, 1295, 1220, 772 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C27H28O5Na)+ 455.1834, found 455.1831.
3.1.12. (3S,4S)-7-ethynyl-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (8a)
To a solution of aldehyde 12a (5.4 mg, 13.6 µmol) in MeOH (0.14 mL) were added K2CO3 (5.7 mg, 40.9 µmol) and Ohira–Bestmann reagent (3.9 mg, 20.4 µmol) at room temperature. After stirring for 40 min at the same temperature, the mixture was concentrated under reduced pressure. The residue was purified with column chromatography (EtOAc:n-hexane = 1:4 to 1:1) to give diMOM alkyne derivative 8a (6.3 mg, quant) as a yellow oil. 1H-NMR (500 MHz, CDCl3) δ 7.52 (1H, s), 5.27 (1H, d, J = 6.0 Hz), 5.22 (2H, s), 5.17 (1H, d, J = 6.0 Hz), 4.59 (1H, d, J = 1.3 Hz), 4.27 (1H, ddd, J = 1.3, 5.8, 7.4 Hz), 3.65 (3H, s), 3.49 (3H, s), 3.32 (3H, s), 2.05 (1H, m), 1.82 (1H, m), 1.65–1.50 (1H, overlapped), 1.42 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 161.2, 154.6, 149.5, 130.0, 123.5, 120.4, 120.1, 101.0, 95.2, 82.7, 80.7, 79.3, 68.3, 58.1, 57.0, 56.5, 31.6, 30.5, 24.8, 22.5, 14.0.; IR (KBr) 3260, 2954, 2932, 2861, 2830, 1730, 1155, 1012, 931 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C21H29O7)+ 393.1913, found 393.1903.
3.1.13. (3S,4S)-7-ethynyl-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (8)
To a solution of diMOM alkyne derivative 8a (6.3 mg, 13.6 µmol) in MeOH (1.2 mL) was added 6 M aqueous HCl (0.40 mL) at room temperature. After stirring for 24 h at the same temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with column chromatography (EtOAc:n-hexane = 1:4 to 1:1) to give alkyne derivative 8 (3.3 mg, 67%) as a yellow solid. m.p. 132–133 °C; 1H-NMR (500 MHz, CDCl3) δ 11.20 (1H, s), 7.22 (1H, s), 6.03 (1H, br-s), 4.76 (1H, d, J = 2.5 Hz), 4.51 (1H, ddd, J = 2.5, 5.1, 8.2 Hz), 3.40 (3H, s), 3.39 (1H, s), 1.94 (1H, m), 1.84 (1H, m), 1.70–1.50 (1H, overlapped), 1.45 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.6, 157.3, 145.1, 127.8, 123.4, 112.6, 108.0, 83.2, 81.4, 77.7, 69.7, 57.0, 31.5, 29.7, 24.8, 22.5, 14.0.; IR (KBr) 3294, 2956, 2930, 2859, 1679, 1434, 1172 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C17H21O5)+ 305.1389, found 305.1391.
3.1.14. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-1-oxo-3-pentylisochromane-7-carbaldehyde (12a)
A stirred solution of 7a (185.1 mg, 0.469 mmol) in CH2Cl2 (10.0 mL) was cooled to −78 °C and a stream of ozone was passed through it for 30 min. At this time, ozone gas was bubbled into the reaction mixture until the color of the reaction mixture turned to blue. After completion of the reaction, the mixture was purged with oxygen gas for 30 min before being treated with PPh3 (246.2 mg, 0.939 mmol) and allowed to warm to room temperature. After stirring at the same temperature for 12 h, the mixture was concentrated under reduced pressure and the resultant mixture was purified with column chromatography (EtOAc:n-hexane = 1:4 to 2:3) to give diMOM benzaldehyde derivative 12a (177.4 mg, 95%) as a white solid. m.p. 38–39 °C; 1H-NMR (400 MHz, CDCl3) δ 10.42 (1H, s), 7.83 (1H, s), 5.29 (2H, s), 5.2 (2H, s), 4.65 (1H, d, J = 1.0 Hz), 4.29 (1H, J = 1.0, 5.6, 8.3 Hz), 3.59 (3H, s), 3.50 (3H, s), 3.35 (3H, s), 2.06 (1H, m), 1.83 (1H, m), 1.70-1.50 (1H, overlapped), 1.44 (1H, m), 1.40-1.30 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 189.9, 161.4, 156.6, 150.6, 135.8, 132.5, 120.8, 116.9, 103.0, 95.4, 81.0, 68.7, 58.4, 57.8, 57.0, 31.9, 30.8, 25.2, 22.8, 14.3.; IR (KBr) 2957, 2929, 2859, 2829, 1730, 1691, 1379, 1155, 930 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H29O8)+ 397.1862, found 397.1866.
3.1.15. (3S,4S)-5,8-dihydroxy-4-methoxy-1-oxo-3-pentylisochromane-7-carbaldehyde (12)
To a solution of diMOM aldehyde derivative 12a (10.0 mg, 25.2 µmol) in THF (1.9 mL) was added 6 M aqueous HCl (0.63 mL) at 0 °C. After stirring for 4 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 2:3) to give benzaldehyde derivative 12 (6.0 mg, 77%) as a pale yellow solid. m.p. 170 °C (dec); 1H-NMR (400 MHz, CDCl3) δ 11.33 (1H, s), 10.47 (1H, s), 7.70 (1H, d, J = 1.5 Hz), 6.62 (1H, br-s), 4.75 (1H, d, J = 2.2 Hz), 4.49 (1H, ddd, J = 2.2, 5.6, 8.0 Hz), 3.43 (3H, s), 2.03 (1H, s), 1.88 (1H, m), 1.61 (1H, m), 1.48 (1H, m), 1.42–1.30 (4H, overlapped), 0.92 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 189.0, 168.8, 158.9, 146.0, 131.3, 124.9, 121.5, 110.2, 82.2, 69.2, 57.9, 31.9, 30.3, 25.1, 22.8, 14.3.; IR (KBr) 3444, 3169, 2953, 2940, 2920, 1676, 1455, 1395, 1299 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C16H21O6)+ 309.1338, found 309.1342.
3.1.16. (3S,4S)-7-(hydroxymethyl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (11a)
To a solution of diMOM aldehyde derivative 12a (20.0 mg, 50.5 µmol) in MeOH (0.25 mL) was added NaBH4 (2.1 mg, 55.5 µmol) at 0 °C. After stirring for 15 min at the same temperature, the reaction was quenched by adding water at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:1) to give diMOM hydroxymethyl derivative 11a (18.6 mg, 93%) as a white wax. 1H-NMR (400 MHz, CDCl3) δ 7.46 (1H, s), 5.25 (1H, d, J = 6.8 Hz), 5.24 (1H, d, J = 6.8 Hz), 5.15 (2H, s), 4.72 (1H, dd, J = 6.4, 12.5 Hz), 4.62 (1H, d, J = 1.2 Hz), 4.58 (1H, dd, J = 7.8, 12.5 Hz), 4.25 (1H, ddd, J = 1.2, 5.8, 8.0 Hz), 3.64 (3H, s), 3.55 (1H, t, J = 6.8 Hz), 3.50 (3H, s), 3.31 (3H, s), 2.05 (1H, m), 1.83 (1H, m), 1.65-1.50 (1H, overlapped), 1.43 (1H, m), 1.42–1.30 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.4, 152.7, 150.7, 138.7, 128.9, 120.8, 119.3, 102.2, 95.4, 81.2, 68.4, 61.4, 57.8, 57.2, 56.8, 31.9, 30.9, 25.2, 22.9, 14.4.; IR (KBr) 3443, 2957, 2928, 2859, 2828, 1724, 1153, 1012 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H31O8)+ 399.2019, found 399.2017.
3.1.17. (3S,4S)-5,8-dihydroxy-7-(hydroxymethyl)-4-methoxy-3-pentylisochroman-1-one (11)
To a solution of diMOM hydroxymethyl derivative 11a (7.2 mg, 24.1 µmol) in MeOH (1.8 mL) was added 6 M aqueous HCl (0.45 mL) at 0 °C. After stirring for 4 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:1) to give hydroxymethyl derivative 11 (3.9 mg, 52%) as a white solid. m.p. 143–145 °C; 1H-NMR (400 MHz, CDCl3) δ 10.99 (1H, s), 7.12 (1H, s), 6.03 (1H, br-s), 4.74 (1H, d, J = 2.4 Hz), 4.72 (2H, br-s), 4.48 (1H, ddd, J = 2.4, 5.2, 8.0 Hz), 3.38 (3H, s), 2.53 (1H, br-s), 1.96 (1H, m), 1.86 (1H, m), 1.70–1.50 (1H, overlapped), 1.46 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.5, 154.2, 145.8, 130.8, 123.8, 121.4, 107.8, 82.1, 69.8, 61.2, 57.2, 31.9, 30.2, 25.2, 22.8, 14.3.; IR (KBr) 2951, 2921, 2854, 1682, 1440, 1302 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C16H23O6)+ 311.1495, found 311.1498.
3.1.18. ((3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-1-oxo-3-pentylisochroman-7-yl)methylmethanesulfonate (22)
To a solution of diMOM hydroxymethyl derivative 11a (7.2 mg, 24.1 µmol) in CH2Cl2 (0.47 mL) were added Et3N (10.8 µL, 77.5 µmol) and MsCl (6.0 µL, 77.5 µmol) at 0 °C. After stirring for 40 min at the same temperature, the reaction was quenched by adding water at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 2:3) to give diMOM mesylated derivative 22 (30.4 mg, 91%) as a white wax. 1H-NMR (400 MHz, CDCl3) δ 7.51 (1H, s), 5.45 (1H, d, J = 12.0 Hz), 5.37 (1H, d, J = 12.2 Hz), 5.25 (2H, s), 5.14 (1H, d, J = 6.6 Hz), 5.12 (1H, d, J = 6.6 Hz), 4.62 (1H, d, J = 1.4 Hz), 4.27 (1H, ddd, J = 1.2, 5.6, 7.8 Hz), 3.59 (3H, s), 3.50 (3H, s), 3.33 (3H, s), 3.07 (3H, s), 2.03 (1H, m), 1.82 (1H, m), 1.58 (1H, m), 1.44 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (100 MHz, CDCl3) δ 161.9, 152.2, 150.5, 131.3, 130.5, 120.3, 119.7, 102.8, 95.5, 81.2, 68.6, 66.9, 58.1, 57.4, 56.9, 38.2, 31.9, 30.9, 25.2, 22.8, 14.3.; IR (KBr) 2958, 2930, 2860, 1829, 1681, 1440, 1358, 1175, 933 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C21H32O10SNa)+ 499.1614, found 499.1616.
3.1.19. (3S,4S)-7-(azidomethyl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (13a)
To a solution of diMOM mesylated derivative 22 (5.3 mg, 11.1 µmol) in DMF (55 µL) was added NaN3 (0.79 mg, 12.1 µmol) at room temperature. After stirring for 6 h at the same temperature, the reaction was quenched by adding water at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give diMOM azide derivative 13a (3.7 mg, 79%) as a pale-yellow oil. 1H-NMR (500 MHz, CDCl3) δ 7.44 (1H, s), 5.26 (1H, d, J = 6.9 Hz), 5.25 (1H, d, J = 6.9 Hz), 5.13 (1H, d, J = 6.9 Hz), 5.11 (1H, d, J = 6.9 Hz), 4.65 (1H, d, J = 14.5 Hz), 4.62 (1H, d, J = 1.3 Hz), 4.53 (1H, d, J = 14.5 Hz), 4.27 (1H, ddd, J = 1.3, 5.7, 7.3 Hz), 3.60 (3H, s), 3.51 (3H, s), 3.32 (3H, s), 2.04 (1H, m), 1.82 (1H, m), 1.65–1.50 (1H, overlapped), 1.43 (1H, m), 1.40–1.30 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.2, 152.1, 150.5, 133.5, 129.1, 119.7, 119.5, 102.6, 95.5, 81.2, 68.6, 57.9, 57.3, 56.8, 50.2, 31.9, 30.9, 25.2, 22.9, 14.4.; IR (KBr) 2957, 2928, 2858, 2829, 2105, 1729, 1153, 1009 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H30N3O7)+ 424.2084, found 424.2085.
3.1.20. (3S,4S)-7-(azidomethyl)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (13)
To a solution of diMOM azide derivative 13a (8.3 mg, 19.6 µmol) in MeOH (1.5 mL) was added 6 M aqueous HCl (0.49 mL) at room temperature. After stirring for 4 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give nitro derivative 13 (3.1 mg, 49%) as a white solid. m.p. 98–99 °C; 1H-NMR (400 MHz, CDCl3) δ 10.98 (1H, s), 7.10 (1H, s), 5.81 (1H, br-s), 4.78 (1H, d, J = 2.9 Hz), 4.52 (1H, ddd, J = 2.9, 5.4, 8.5 Hz), 4.45 (1H, d, J = 14.4 Hz), 4.42 (1H, d, J = 14.4 Hz), 3.41 (3H, s), 1.93 (1H, m), 1.86 (1H, m), 1.70-1.50 (1H, overlapped), 1.47 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.1, 154.4, 145.7, 126.2, 124.6, 121.8, 108.0, 81.7, 70.4, 57.2, 49.3, 31.9, 30.0, 25.2, 22.8, 14.3.; IR (KBr) 2959, 2924, 2857, 2108, 1654, 1441, 1293, 1170 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C16H22N3O5)+ 336.1559, found 336.1563.
3.1.21. (3S,4S)-7-(aminomethyl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (14a)
To a solution of diMOM azide derivative 13a (3.3 mg, 7.8 µmol) in MeOH (0.78 mL) was added Et3N (0.10 mL, 7.35 mmol) and Pd/C (1.6 mg, 1.5 µmol) at room temperature. After stirring for 1 h at the same temperature, the mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was purified with PTLC (MeOH:CH2Cl2 = 1:9) to give diMOM amine derivative 14a (2.0 mg, 65%) as brown oil. 1H-NMR (400 MHz, CDCl3) δ 7.49 (1H, s), 5.26 (2H, s), 5.16 (1H, d, J = 7.2 Hz), 5.07 (1H, d, J = 6.8 Hz), 4.61 (1H, d, J = 1.2 Hz), 4.27 (1H, ddd, J = 1.2, 6.0, 7.6 Hz), 4.00 (2H, s), 3.61 (3H, s), 3.50 (3H, s), 3.32 (3H, s), 2.59 (1H, br-s), 2.03 (1H, m), 1.82 (1H, m), 1.57 (1H, m), 1.43 (1H, m), 1.40-1.25 (1H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.6, 152.6, 150.5, 128.1, 120.0, 119.0, 102.4, 95.4, 81.2, 68.5, 57.9, 57.2, 56.8, 42.5, 32.0, 30.9, 30.0, 25.2, 22.9, 14.4.; IR (KBr) 2957, 2925, 2857, 2827, 1726, 1470, 1153, 1005 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H32NO7)+ 398.2179, found 398.2178.
3.1.22. (3S,4S)-7-(aminomethyl)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (14)
To a solution of diMOM amine derivative 14a (4.4 mg, 11.1 µmol) in MeOH (0.83 mL) was added 6 M aqueous HCl (0.28 mL) at 0 °C. After stirring for 5 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with the mixture of MeOH and CH2Cl2 (MeOH:CH2Cl2 = 1:4) (×4) and the combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified with PTLC (MeOH:CHCl3 saturated with NH3 = 1:9) to give amiomethyl derivative 14 (1.1 mg, 32%) as brown solid. m.p. 78–80 °C; 1H-NMR (400 MHz, CDCl3) δ 6.98 (1H, s), 4.59 (1H, d, J = 1.8 Hz), 4.35 (1H, ddd, J = 1.8, 6.0, 8.0 Hz), 3.97 (1H, d, J = 13.3 Hz), 3.88 (1H, d, J = 13.3 Hz), 3.19 (3H, s), 1.98 (1H, m), 1.83 (1H, m), 1.56 (1H, m), 1.43 (1H, m), 1.40–1.25 (4H, overlapped), 0.90 (3H, t, J = 7.0 Hz) ; 13C-NMR (125 MHz, CDCl3) δ 169.9, 154.2, 146.2, 130.0, 125.8, 122.9, 108.1, 82.8, 68.5, 56.9, 42.3, 31.9, 30.6, 25.1, 22.8, 14,3.; IR (KBr) 2956, 2921, 2857, 1676, 1441, 1171 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C16H23NO5Na)+ 332.1474, found 332.1474.
3.1.23. ((3S,4S)-5,8-dihydroxy-4-methoxy-7-nitro-3-pentylisochroman-1-one (15)
To a solution of 3 (28.9 mg, 89.1 µmol) in AcOH (0.50 mL) was added the mixture of AcOH and 70% HNO3 (0.80 mL:0.20 mL) at 0 °C. After stirring for 10 min at the same temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was pathed through SiO2 plug and the resultant mixture of monoMOM nitro derivative 15a was used for the next reaction without further purification. To a solution of 15a mixture in MeOH (7.5 mL) was added 6 M aqueous HCl (2.4 mL) at 0 °C. After stirring for 5 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:1) to give nitro derivative 15 (21.5 mg, 74%) as a yellow solid. m.p. 158-159; 1H-NMR (400 MHz, CDCl3) δ 11.89 (1H, s), 7.78 (1H, s), 6.80 (1H, br-s), 4.82 (1H, d, J = 2.6 Hz), 4.55 (1H, ddd, J = 2.6, 5.2, 8.3 Hz), 3.46 (3H, s), 1.96 (1H, m), 1.86 (1H, m), 1.59 (1H, m), 1.47 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 167.5, 150.4, 144.9, 137.6, 129.4, 119.7, 110.7, 81.0, 70.3, 57.6 , 31.4, 29.4, 24.7, 22.4, 14.0; IR (KBr) 3416, 2962, 2927, 2857, 1679, 1445, 1261, 1018, 800 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C15H20NO7)+ 326.1240, found 326.1224.
3.1.24. (3S,4S)-7-amino-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (16)
To a solution of nitro derivative 15 (5.0 mg, 15.4 µmol) in THF (0.62 mL) and MeOH (80 µL) was added PtO2 (0.3 mg, 1.54 µmol) at room temperature. After stirring for 1.5 h at the same temperature under hydrogen atmosphere (1 atm), the mixture was passed through a membrane filter to remove PtO2. The mixture was concentrated under reduced pressure and the residue was purified with PTLC (EtOAc:n-hexane = 3:7, developed by three times) to give nitro derivative 16 (4.3 mg, 95%) as a yellow solid. m.p. 118–119 °C; 1H-NMR (500 MHz, CDCl3) δ 10.72 (1H, s), 6.45 (1H, s), 5.68 (1H, br-s), 4.67 (1H, d, J = 2.5 Hz), 4.46 (1H, ddd, J = 2.5, 5.5, 8.3 Hz), 4.05 (1H, br-s), 3.32 (3H, s), 1.94 (1H, m), 1.84 (1H, m), 1.75–1.50 (1H, overlapped), 1.45 (1H, m), 1.40–1.25 (4H, overlapped), 0.90 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.8, 145.9, 144.5, 137.2, 109.8, 108.4, 106.8, 82.4, 69.1, 56.1, 31.6, 30.1, 24.9, 22.5, 14.0; IR (KBr) 3378, 2957, 2926, 2858, 1681, 1464, 1217, 1171 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H21NO5Na)+ 318.1317, found 318.1321.
3.1.25. (3S,4S)-7-chloro-8-hydroxy-4-methoxy-5-(methoxymethoxy)-3-pentylisochroman-1-one (18a)
To a solution of 3 (5.0 mg, 15.4 µmol) in DMF (0.18 mL) was added the solution of N-chlorosuccinimide (4.1 mg, 30.8 µmol) in DMF (31 µL) at room temperature. After stirring for 5 h at 65 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give monoMOM chloro derivative 18a (3.3 mg, 60%) as a brown solid. m.p. 79–81 °C; 1H-NMR (400 MHz, CDCl3) δ 11.23 (1H, s), 7.55 (1H, s), 5.18 (1H, d, J = 7.0 Hz), 5.16 (1H, d, J = 7.0 Hz), 4.59 (1H, d, J = 1.7 Hz), 4.39 (1H, ddd, J = 1.7, 6.0, 8.0 Hz), 3.50 (3H, s), 3.30 (3H, s), 2.07 (1H, m), 1.86 (1H, m), 1.70-1.50 (1H, overlapped), 1.47 (1H, m), 1.45-1.25 (4H, overlapped), 0.92 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.7, 152.8, 146.3, 125.1, 123.6, 123.0, 109.0, 95.7, 82.7, 67.4, 56.8, 56.4, 31.5, 30.4, 24.7, 22.5, 14.0.; IR (KBr) 2955, 2927, 2853, 2826, 1681, 1453, 1433, 1206 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C17H23O6ClNa)+ 381.1081, found 381.1088.
3.1.26. (3S,4S)-7-chloro-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (18)
To a solution of monoMOM chloro derivative 18a (3.3 mg, 9.20 µmol) in MeOH (0.69 mL) was added 6 M aqueous HCl (0.23 mL) at 0 °C. After stirring for 2 h at 40 °C, the reaction was quenched by adding saturated NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give chloro derivative 18 (2.1 mg, 73%) as a brown solid. m.p. 119-120 °C; 1H-NMR (400 MHz, CDCl3) δ 11.17 (1H, br-s), 7.34 (1H, s), 6.34 (1H, br-s), 4.82 (1H, br-s), 4.59 (1H, ddd, J = 2.8, 5.6, 8.4 Hz), 3.48 (3H, s), 2.03 (1H, m), 1.93 (1H, m), 1.64 (1H, m), 1.53 (1H, m), 1.51–1.35 (4H, overlapped), 0.98 (3H, t, J = 7.2 Hz); 13C-NMR (100 MHz, CDCl3) δ 168.7, 152.1, 145.6, 124.9, 122.8, 121.1, 108.5, 81.8, 69.6, 57.0, 31.5, 29.8, 24.8, 22.5, 14.0.; IR (KBr) 3282, 2958, 2929, 2860, 1681, 1437, 1198 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C15H20O5Cl)+ 315.0999, found 315.0998.
3.1.27. (3S,4S)-7-bromo-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (19)
To a solution of bromo derivative 2 (11.0 mg, 24.6 µmol) in MeOH (1.8 mL) was added 6 M aqueous HCl (0.62 mL) at 0 °C. After stirring for 3.5 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give bromo derivative 19 (8.6 mg, 97%) as a white solid. m.p. 132 °C; 1H-NMR (400 MHz, CDCl3) δ 11.26 (1H, s), 7.36 (1H, s), 6.00 (1H, br-s), 4.76 (1H, d, J = 2.7 Hz), 4.52 (1H, ddd, J = 2.7, 5.1, 8.3 Hz), 3.41 (3H, s), 1.95 (1H, m), 1.86 (1H, m), 1.70-1.50 (2H, overlapped), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.4, 153.0, 145.8, 127.9, 121.5, 111.6, 108.2, 81.4, 70.0, 57.0, 31.5, 29.6, 24.8, 22.5, 14.0.; IR (KBr) 3296, 2955, 2930, 2859, 1679, 1432, 1197 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H19O5BrNa)+ 381.0314, found 381.0322.
3.1.28. (3S,4S)-5,8-dihydroxy-7-iodo-4-methoxy-3-pentylisochroman-1-one (20)
To a solution of 3 (12.6 mg, 38.8 µmol) in DMF (0.35 mL) was added the solution of N-iodosuccinimide (17.5 mg, 77.6 µmol) in DMF (50 µL) at room temperature. After stirring for 3 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with CH2Cl2 (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was pathed through SiO2 plug and the resultant mixture of monoMOM iodo derivative 20a was used for the next reaction without further purification. To a solution of crude mixture of 20a in MeOH (0.83 mL) was added 6 M aqueous HCl (0.30 mL) at 0 °C. After stirring for 5 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give iodo derivative 20 (4.0 mg, 87%) as a pale-yellow oil. m.p. 109–110 °C; 1H-NMR (500 MHz, CDCl3) δ 11.44 (1H, s), 7.57 (1H, s), 6.11 (1H, br-s), 4.51 (1H, ddd, J = 2.8, 5.4, 8.5 Hz), 3.40 (3H, s), 1.94 (1H, m), 1.85 (1H, m), 1.75–1.50 (4H, overlapped), 1.45 (1H, m), 1.40–1.30 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.3, 155.3, 146.3, 133.8, 122.6, 107.1, 85.5, 81.5, 69.8, 56.9, 31.5, 29.7, 24.8, 22.5, 14.0; IR (KBr) 3293, 2977, 298, 2857, 1674, 1427, 1197 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H19O5Ina)+ 429.0175, found 429.0174.
3.2. Bactericidal Assay
Methicillin-susceptible Staphylococcus aureus (MSSA) ATCC25923 and methicillin-resistant Staphylococcus aureus (MRSA) ATCC 33,591 were aerobically incubated at 37 °C in Luria–Bertani medium (LB, Nippon Becton Dickinson Company, Tokyo, Japan). Porphyromonas gingivalis W83 was anaerobically incubated at 37 °C in Gifu anaerobic medium (GAM, Nissui, Tokyo, Japan). Each culture (20 µL) prepared to an optical density of 1.5 at 600 nm were appropriately incubated with various concentrations of synthesized compounds in 200 µL of culture medium at 37 °C for 24 h in 96-well plate (Thermo scientific, MA, USA). Compounds were dissolved in DMSO (Wako, Osaka, Japan). The degree of turbidity in the broth culture was measured at absorbance 600 nm using microplate reader (Thermo scientific, MA, USA).
3.3. Cellular Toxicity
Human lung adenocarcinoma epithelial cell line A549 cells were cultured at 37 °C in growth medium (DMEM with 10% fetal bovine serum) in 5% CO2, and then seeded into 96-well plates at a density of 1 × 105 cells/mL. Once the cells reached 80%–90% confluence, they were treated with or without 10 µM of various compounds at 37 °C for 12 h. Next, 10 μL Cell Counting Kit-8 (Dojindo Molecular Technologies, Kumamoto, Japan) solution was added to each well, and the plate was incubated for 2 h at 37 °C. Cell viability was determined by measuring the absorbance at 450 nm using a fluorimeter (Varioscan, Thermo, USA).
4. Conclusions
We constructed a chemical library of the side-chain derivatives of eurotiumide A, which is a dihydroisocoumarin-type marine natural product. The antimicrobial evaluation of these compounds was conducted against MSSA, MRSA, and P. gingivalis. We discovered several compounds to be effective against these strains; among them, the isopentyl derivative 6 is especially more active against all three strains than 1. Continuous research to clarify the modes of action of these derivatives is under way in our laboratory.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/18/2/92/s1, 1H- and 13C-NMR charts of all new compounds.
Author Contributions
A.N. conceived and designed this research and analyzed the experimental data; H.S., T.N., M.H., S.N., and S.K. (Shuhei Kameyama) prepared compounds and collected their spectral data; S.K. (Sangita Karanjit) checked the experimental data; Y.F., N.H., G.K. and M.O. evaluated the antimicrobial activity; A.N., M.O. and K.N. wrote the paper; all of the authors reviewed and approved the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Nos. 17K08365 (A.N.), 18H02657 (M.O.), JP19H02851 (K.N.), and JP16H01156 (K.N.), as well as the Kurita Water and Environment Foundation. We also acknowledge Tokushima University for their financial support of the Research Clusters program of Tokushima University (No. 1802001).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ma, B.; Forney, L.; Ravel, J. Vaginal microbiology: Rethinking health and disease. Annu. Rev. Microbiol. 2012, 66, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Parker, E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef] [PubMed]
- MCKenney, P.T.; Palmer, E.G. From hype to hope: The gut microbiome in Health and Disease. Cell 2015, 163, 1326–1332. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Petersen, O. The Human Intestinal Microbiome in Health and Disease. N. Eng. J. M. 2016, 375, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.H.; Piters, W.A.A.S.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 13, 259–270. [Google Scholar]
- Lewis, K. Platforms for Antibiotic Discovery. Nat. Rev. Drug Discovery 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Taru, Y.; Palmer, E.G. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef]
- Brown, E.D.; Wright, G.D. Antibacterial Drug Discovery in the Resistance Era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Fleming, A. On the antibacterial action of cultures of a penicillium with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Chu, D.T.W.; Plattner, J.J.; Katz, L. New Directions in Antibacterial Research. J. Med. Chem. 1996, 39, 3853–3874. [Google Scholar] [CrossRef]
- Saleem, M.; Nazir, M.; Ali, M.S.; Hussain, H.; Lee, Y.S.; Riaz, N.; Jabbar, A. Antimicrobial natural products: An update on future antibiotic drug candidates. Nat. Prod. Rep. 2010, 27, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Bologa, C.G.; Ursu, O.; Oprea, T.I.; Melancon, C.E., III; Tegos, G.P. Emerging trends in the discovery of natural product antibacterials. Curr. Opin. Pharmacol. 2013, 13, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Robertson, A.A.B.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef]
- Szychowski, J.; Truchon, J.-F.; Bennani, Y.L. Natural Products in Medicine: Transformational Outcome of Synthetic Chemistry. J. Med. Chem. 2014, 57, 9292–9308. [Google Scholar] [CrossRef] [PubMed]
- Sclinke, C.; Martins, T.; Queiroz, S.C.; Melo, I.S.; Reyes, F.G.R. Antibacterial Compounds from Marine Bacteria, 2010–2015. J. Nat. Prod. 2017, 80, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Masscheletin, J.; Henner, M.; Challis, G.L. Antibiotics from Gram-negative bacteria: A comprehensive overview and selected biosynthetic highlights. Nat. Prod. Rep. 2017, 34, 712–783. [Google Scholar] [CrossRef]
- Von Nussbaum, F.; Brands, M.; Hizen, B.; Weigand, S.; Habich, D. Antibacterial natural products in medicinal chemistry-exodus or revival? Angew, Chem. Int. Ed. 2006, 45, 5072–5129. [Google Scholar] [CrossRef]
- Clardy, J.; Fischbach, M.A.; Walsh, C.T. New antibiotics from bacterial natural product. Nat. Biotechnol. 2006, 24, 1541–1550. [Google Scholar] [CrossRef]
- Abouelhassan, Y.; Garrison, A.T.; Yang, H.; Riveros, A.C.; Burch, G.M.; Huigens, R.W., III. Recent Progress in Natural-Product-Inspired Progra,s Aimed To Address Antibiotic Resistance and Tolerance. J. Med. Chem. 2019, 62, 7618–7642. [Google Scholar] [CrossRef]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Ali, J.; Rafiq, Q.A.; Ratcliffe, E. Antimicrobial Resistance Mechanisms and Potential Synthetic Treatments. Futur. Sci. OA 2018, 4, FSO290. [Google Scholar] [CrossRef]
- Francino, M.P. Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shao, C.-L.; Wang, K.-L.; Xu, Y.; She, Z.-G.; Wang, C.-Y. Dihydroisocoumarin derivatives with antifouling activities from a gorgonian-derived Eurotium sp. fungus. Tetrahedron 2014, 70, 9132–9138. [Google Scholar] [CrossRef]
- Nakayama, A.; Sato, H.; Karanjit, S.; Hayashi, N.; Oda, M.; Namba, K. Asymmetric Total Syntheses and Structure Revisions of Eurotiumide A and Eurotiumide B, and Their Evaluation as Natural Fluorescent Probes. Eur. J. Org. Chem. 2018. [Google Scholar] [CrossRef]
- Nakayama, A.; Sato, H.; Nagano, S.; Karanjit, S.; Imagawa, H.; Namba, K. Asymmetric Total Syntheses and Structure Elucidations of (+)-Eurtiumide F and (+)-Eurotiumide G. Chem. Pharm. Bull. 2019, 67, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Ito, T.; Hiramatsu, K. A new class of genetic element, Staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 1549–1555. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).