Pyrenosetins A–C, New Decalinoylspirotetramic Acid Derivatives Isolated by Bioactivity-Based Molecular Networking from the Seaweed-Derived Fungus Pyrenochaetopsis sp. FVE-001




Abstract
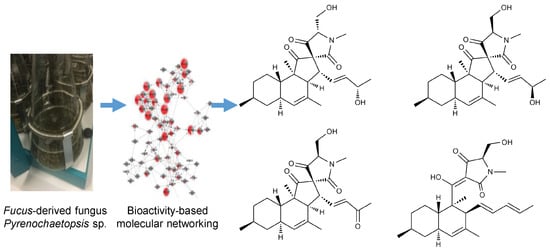
1. Introduction
2. Results
2.1. Strain Isolation and Identification
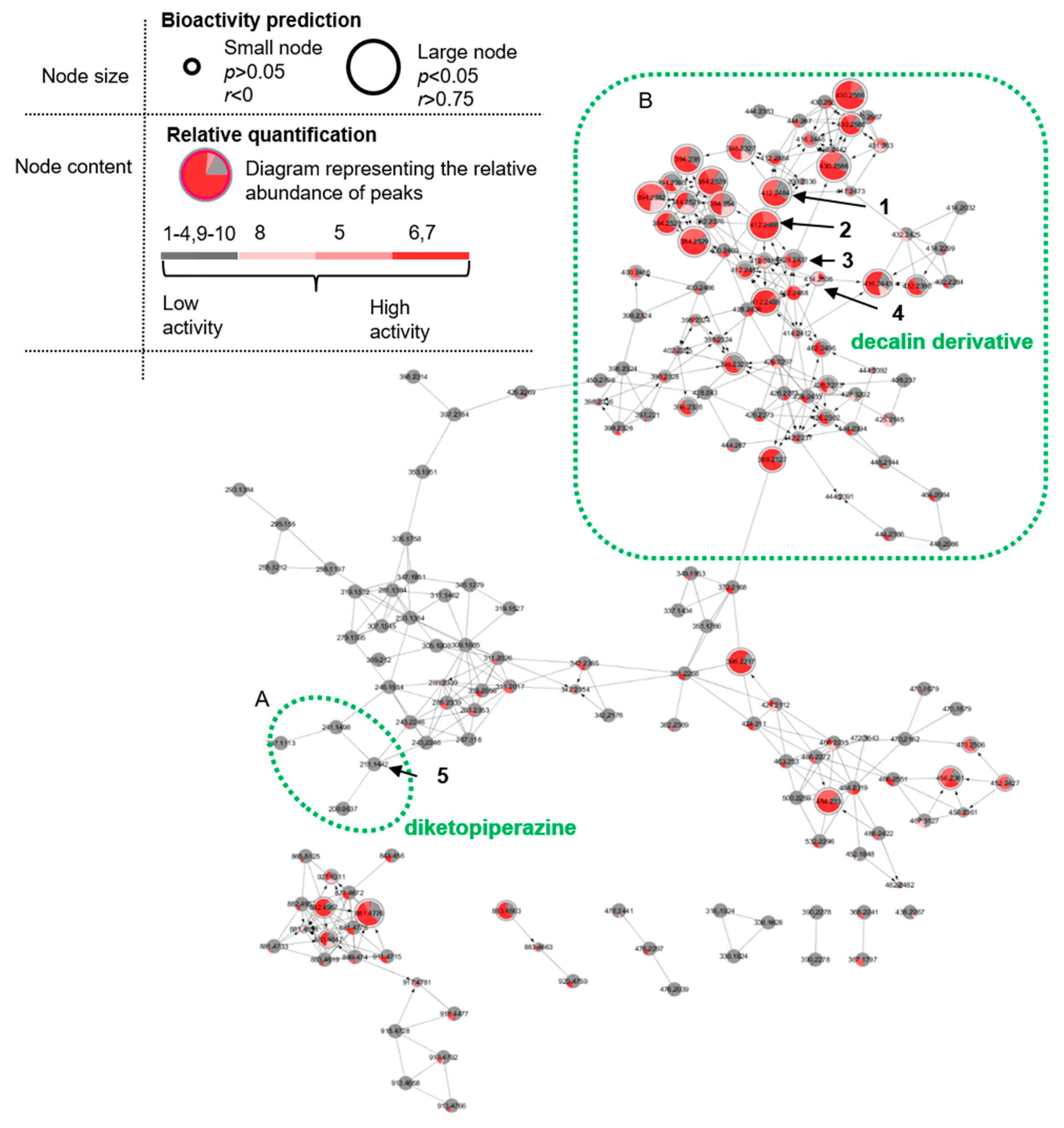
2.2. Cultivation, Extraction, Bioactivity Test and Molecular Networking
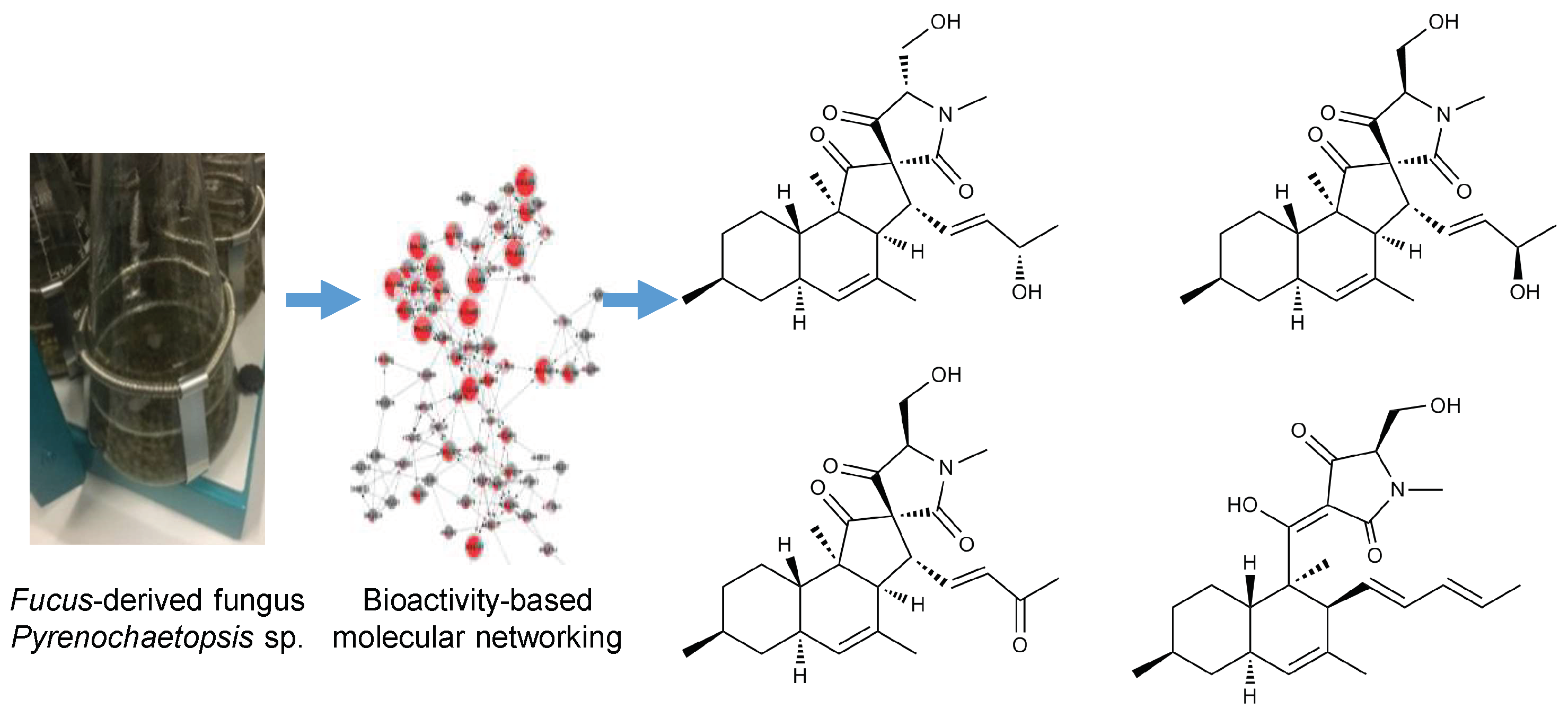
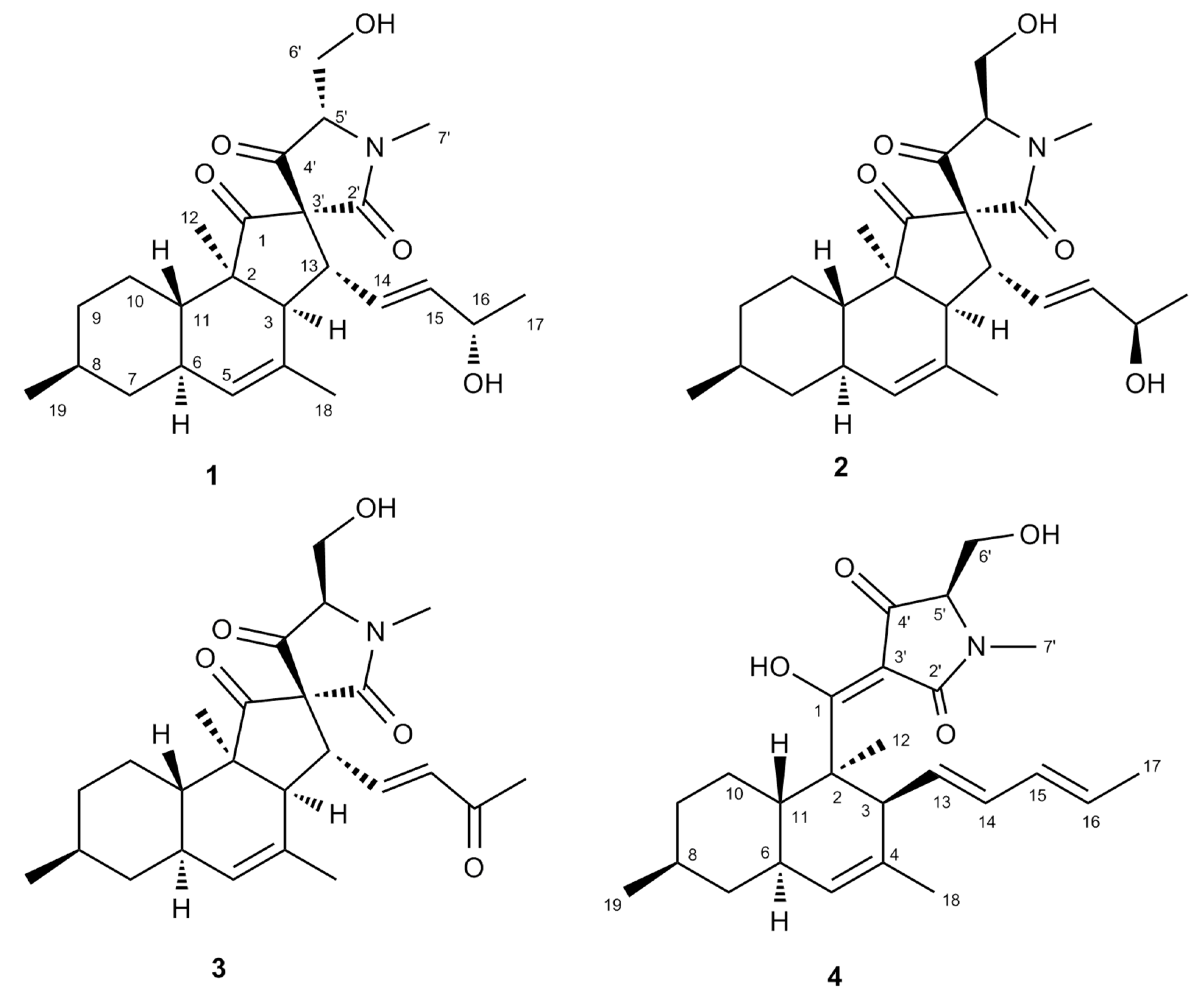
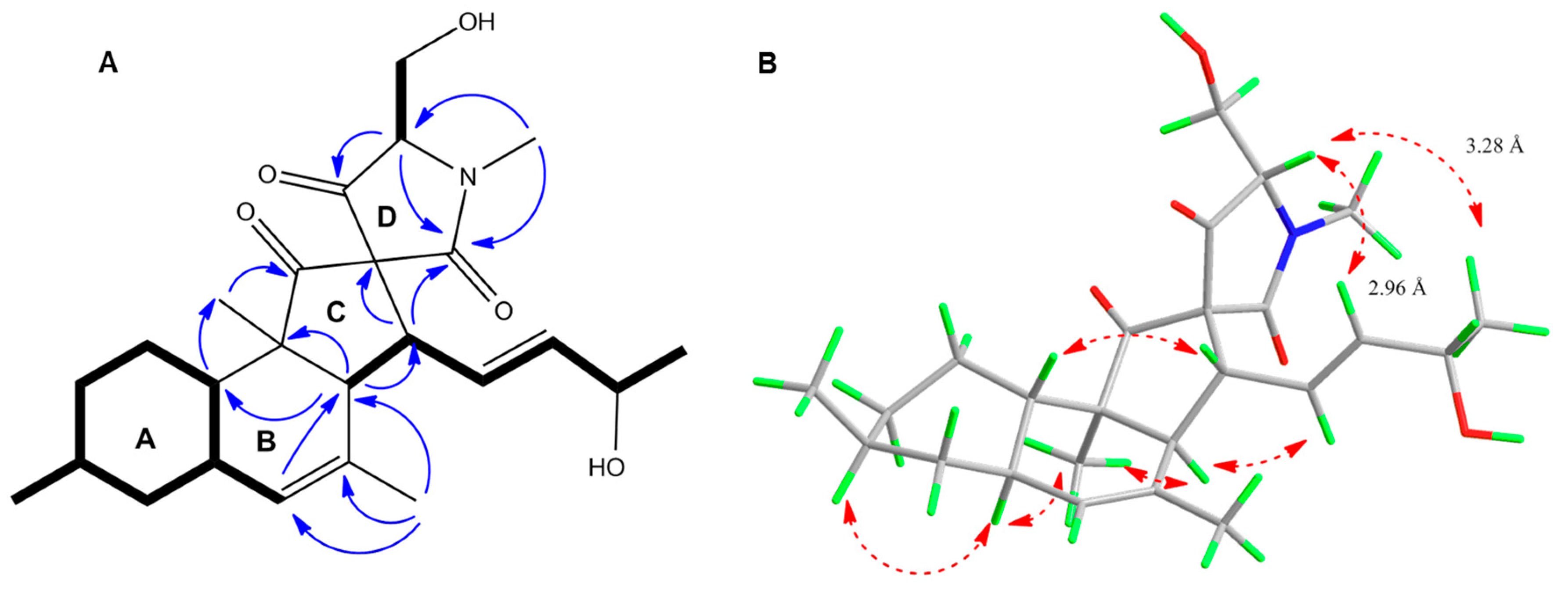
2.3. Purification and Structure Elucidation
2.4. Biological Activity of Compounds 1–4
3. Discussion
4. Material and Methods
4.1. General Procedures
4.2. Strain Identification and Cultivation
4.3. Extraction and Isolation
4.4. UHPLC-QToF-MS/MS Analysis and Molecular Networking
4.5. Mosher’s Esterification
4.5.1. Preparation of 16-(S) MTPA Ester 6 and 16-(R)-MTPA Ester 7
4.5.2. Preparation of 16-(S) MTPA Ester 8 and 16-(R)-MTPA Ester 9
4.6. Bioactivity Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed–bacteria interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; Jensen, P.R.; Cheng, X.C. Halimide, a Cytotoxic Marine Natural Product, and Derivatives Thereof. U.S. Patent US6358957B1, 30 May 2000. [Google Scholar]
- Cimino, P.J.; Huang, L.; Du, L.; Wu, Y.; Bishop, J.; Dalsing-Hernandez, J.; Kotlarczyk, K.; Gonzales, P.; Carew, J.; Nawrocki, S. Plinabulin, an inhibitor of tubulin polymerization, targets KRAS signaling through disruption of endosomal recycling. Biomed. Rep. 2019, 10, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, A.; Schulz, B.; Mitchell, J.I. Molecular detection of ascomycetes associated with Fucus serratus. Mycol. Res. 2004, 107, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, A.; Schoch, C.L.; Spatafora, J.W.; Kohlmeyer, J.; Draeger, S.; Mitchell, J.I. Detection and identification of fungi intimately associated with the brown seaweed Fucus serratus. Appl. Environ. Microbiol. 2008, 74, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Flewelling, A.J.; Johnson, J.A.; Gray, C.A. Isolation and bioassay screening of fungal endophytes from North Atlantic marine macroalgae. Bot. Mar. 2013, 56, 287–297. [Google Scholar] [CrossRef]
- Osterhage, C.; König, G.M.; Jones, P.G.; Wright, A.D. 5-Hydroxyramulosin, a new natural product produced by Phoma tropica, a marine-derived fungus isolated from the alga Fucus spiralis. Planta Med. 2002, 68, 1052–1054. [Google Scholar] [CrossRef]
- Santiago, C.; Fitchett, C.; Munro, M.H.G.; Jalil, J.; Santhanam, J. Cytotoxic and antifungal activities of 5-hydroxyramulosin, a compound produced by an endophytic fungus isolated from Cinnamomum mollisimum. Evid. Based Complement. Altern. Med. 2012, 2012, 689310. [Google Scholar] [CrossRef]
- Abdel-Lateff, A.; Fisch, K.M.; Wright, A.D.; Konig, G.M. A new antioxidant isobenzofuranone derivative from the algicolous marine fungus Epicoccum sp. Planta Med. 2003, 69, 831–834. [Google Scholar]
- Quinn, R.A.; Nothias, L.F.; Vining, O.; Meehan, M.; Esquenazi, E.; Dorrestein, P.C. Molecular networking as a drug discovery, drug metabolism, and precision medicine strategy. Trends Pharmacol. Sci. 2017, 38, 143–154. [Google Scholar] [CrossRef]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef]
- Fan, B.; Parrot, D.; Blümel, M.; Labes, A.; Tasdemir, D. Influence of OSMAC-based cultivation in metabolome and anticancer activity of fungi associated with the brown alga Fucus vesiculosus. Mar. Drugs 2019, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Olivon, F.; Allard, P.M.; Koval, A.; Righi, D.; Genta-Jouve, G.; Neyts, J.; Apel, C.; Pannecouque, C.; Nothias, L.F.; Cachet, X.; et al. Bioactive natural products prioritization using massive multi-informational molecular networks. ACS Chem. Biol. 2017, 12, 2644–2651. [Google Scholar] [CrossRef]
- Nothias, L.F.; Nothias-Esposito, M.; da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-based molecular networking for the discovery of drug leads in natural product bioassay-guided fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef]
- Parrot, D.; Blümel, M.; Utermann, C.; Chianese, G.; Krause, S.; Kovalev, A.; Gorb, S.N.; Tasdemir, D. Mapping the surface microbiome and metabolome of brown seaweed Fucus vesiculosus by amplicon sequencing, integrated metabolomics and imaging techniques. Sci. Rep. 2019, 9, 1061. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Hayashi, M.; Murakami, Y.; Araki, Y.; Otsuka, Y.; Kashiwagi, T.; Shimamura, T.; Ukeda, H. Development of LC-MS/MS analysis of cyclic dipeptides and its application to tea extract. Biosci. Biotechnol. Biochem. 2016, 80, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Nogawa, T.; Kato, N.; Shimizu, T.; Okano, A.; Futamura, Y.; Takahashi, S.; Osada, H. Wakodecalines A and B, new decaline metabolites isolated from a fungus Pyrenochaetopsis sp. RK10-F058. J. Antibiot. 2017, 71, 123–128. [Google Scholar] [CrossRef]
- Singh, S.B.; Zink, D.L.; Goetz, M.A.; Dombrowski, A.W.; Polishook, J.D.; Hazuda, D.J. Equisetin and a novel opposite stereochemical homolog phomasetin, two fungal metabolites as inhibitors of HIV-1 integrase. Tetrahedron Lett. 1998, 39, 2243–2246. [Google Scholar] [CrossRef]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef]
- Papizadeh, M.; Soudi, M.R.; Amini, L.; Wijayawardene, N.N.; Hyde, K.D. Pyrenochaetopsis tabarestanensis (Cucurbitariaceae, Pleosporales), a new species isolated from rice farms in north Iran. Phytotaxa 2017, 297, 15–28. [Google Scholar] [CrossRef]
- de Gruyter, J.; Woudenberg, J.H.C.; Aveskamp, M.M.; Verkley, G.J.M.; Groenewald, J.Z.; Crous, P.W. Systematic reappraisal of species in Phoma section Paraphoma, Pyrenochaeta and Pleurophoma. Mycologia 2010, 102, 1066–1081. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Nogawa, T.; Takita, R.; Kinugasa, K.; Kanai, M.; Uchiyama, M.; Osada, H.; Takahashi, S. Control of the stereochemical course of [4+2] cycloaddition during trans-decalin formation by Fsa2-family enzymes. Angew. Chem. Int. Ed. 2018, 57, 9754–9758. [Google Scholar] [CrossRef] [PubMed]
- Fox Ramos, A.E.; Evanno, L.; Poupon, E.; Champy, P.; Beniddir, M.A. Natural products targeting strategies involving molecular networking: Different manners, one goal. Nat. Prod. Rep. 2019, 36, 960–980. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kusari, S.; Spiteller, M. Natural products containing ‘decalin’ motif in microorganisms. Nat. Prod. Rep. 2014, 31, 1175–1201. [Google Scholar] [CrossRef]
- Jang, J.H.; Asami, Y.; Jang, J.P.; Kim, S.O.; Moon, D.O.; Shin, K.S.; Hashizume, D.; Muroi, M.; Saito, T.; Oh, H. Fusarisetin A, an acinar morphogenesis inhibitor from a soil fungus, Fusarium sp. FN080326. J. Am. Chem. Soc. 2011, 133, 6865–6867. [Google Scholar] [CrossRef]
- Zhao, D.; Han, X.; Wang, D.; Liu, M.; Gou, J.; Peng, Y.; Liu, J.; Li, Y.; Cao, F.; Zhang, C. Bioactive 3-decalinoyltetramic acids derivatives from a marine-derived strain of the fungus Fusarium equiseti D39. Front. Microbiol. 2019, 10, 1285–1294. [Google Scholar] [CrossRef]
- Yamada, T.; Tanaka, A.; Nehira, T.; Nishii, T.; Kikuchi, T. Altercrasins A–E, decalin derivatives, from a sea-urchin-derived Alternaria sp.: Isolation and structural analysis including stereochemistry. Mar. Drugs 2019, 17, 218. [Google Scholar] [CrossRef]
- Liu, S.Z.; Yan, X.; Tang, X.X.; Lin, J.G.; Qiu, Y.K. New bis-alkenoic acid derivatives from a marine-derived fungus Fusarium solani H915. Mar. Drugs 2018, 16, 483. [Google Scholar] [CrossRef]
- Kato, N.; Nogawa, T.; Hirota, H.; Jang, J.H.; Takahashi, S.; Ahn, J.S.; Osada, H. A new enzyme involved in the control of the stereochemistry in the decalin formation during equisetin biosynthesis. Biochem. Biophys. Res. Commun. 2015, 460, 210–215. [Google Scholar] [CrossRef]
- Waddell, P.J.; Steel, M.A. General time-reversible distances with unequal rates across sites: Mixing Γ and inverse gaussian distributions with invariant ites. Mol. Phylogenet. Evol. 1997, 8, 398–414. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 4 b |
|---|---|---|---|---|
| δH, Mult (J in Hz) | δH, Mult (J in Hz) | δH, Mult (J in Hz) | δH, Mult (J in Hz) | |
| 1 | - | - | - | - |
| 2 | - | - | - | - |
| 3 | 2.57, d (11.4) | 2.73, d (11.4) | 2.66, d (11.3) | 3.17, br d (9.4) |
| 4 | - | - | - | - |
| 5 | 5.24, br s | 5.22, br s | 5.28, br.s | 5.22, br s |
| 6 | 1.82, m | 1.82, m | 1.83, m | 1.84, m |
| 7 | 1.78, m | 1.80, m | 1.82, m | 1.82, m |
| 0.86, m | 0.84, m | 0.88, m | 0.86, m | |
| 8 | 1.43, m | 1.44, m | 1.44, m | 1.51, m |
| 9 | 1.72, m | 1.73, m | 1.73, m | 1.76, m |
| 1.01, m | 0.93, m | 0.99, m | 1.04, m | |
| 10 | 1.42, m | 1.40, m | 1.41, m | 1.97, m |
| 1.04, m | 1.07, qd (12.8,3.4) | 1.04, m | 1.06, m | |
| 11 | 1.63, m | 1.42, m | 1.64, td (11.0, 2.7) | 1.64 m |
| 12 | 0.98, s | 1.00, s | 1.01, s | 1.38, br s |
| 13 | 3.44, dd (11.4, 8.6) | 3.26, dd (11.4, 9.4) | 3.57, dd (11.4, 9.8) | 5.27, dd (13.6, 10.5) |
| 14 | 5.75, dd (15.4, 8.6) | 5.97, dd (15.3, 9.4) | 6.85, dd (15.9, 9.8) | 5.78, dd (14.6, 10.5) |
| 15 | 5.71, dd (15.4 4.8) | 5.50, dd (15.3, 8.0) | 6.18, d (15.9) | 5.91, t (12.8) |
| 16 | 4.24, m | 4.18, m | - | 5.56, dq (14.2, 6.8) |
| 17 | 1.19, d (6.4) | 1.19, d (6.2) | 2.22, s | 1.67, d (6.8) |
| 18 | 1.73, br s | 1.69, br s | 1.68, br s | 1.55, t (1.9) |
| 19 | 0.90, d (6.5) | 0.91, d (6.5) | 0.90, d (6.2) | 0.91, d (6.5) |
| 2′ | - | - | - | - |
| 3′ | - | - | - | - |
| 4′ | - | - | - | - |
| 5′ | 3.57, dd (4.8, 2.6) | 3.94, dd (2.7, 1.9) | 3.61, dd (4.9, 2.7) | 3.61, t (2.7) |
| 6′ | 4.09, dd (12.2, 2.6) | 4.08, m | 4.10, m | 3.87, m |
| 3.92, dd (12.3, 4.8) | 3.86, dd (12.4, 2.7) | 3.94, m | 3.81, m | |
| 7′ | 3.10, s | 3.07, s | 3.11, s | 2.97, brs |
| 6′-OH | 2.74, m |
| Position | 1 a | 2 a | 3 a | 4 b |
|---|---|---|---|---|
| δC | δC | δC | δC | |
| 1 | 213.3 (C) | 209.8 (C) | 212.1 (C) | 197.6 (C) |
| 2 | 54.8 (C) | 54.1 (C) | 54.7 (C) | 49.9 (C) |
| 3 | 53.6 (CH) | 52.8 (CH) | 53.6 (CH) | 50.2 (CH) |
| 4 | 131.8 (C) | 132.3 (C) | 130.9 (C) | 132.4 (C) |
| 5 | 128.1 (CH) | 127.6 (CH) | 128.8 (CH) | 127.1 (CH) |
| 6 | 37.6 (CH) | 37.6 (CH) | 37.6 (CH) | 40.1 (CH) |
| 7 | 41.9 (CH2) | 42.0 (CH2) | 41.8 (CH2) | 43.1 (CH2) |
| 8 | 33.0 (CH) | 32.9 (CH) | 32.9 (CH) | 34.3 (CH) |
| 9 | 35.3 (CH2) | 35.3 (CH2) | 35.2 (CH2) | 36.6 (CH2) |
| 10 | 25.3 (CH2) | 25.3 (CH2) | 25.2 (CH2) | 29.0 (CH2) |
| 11 | 37.4 (CH) | 38.0 (CH) | 37.4 (CH) | 40.6 (CH) |
| 12 | 15.0 (CH3) | 15.2 (CH3) | 15.2 (CH3) | 14.2 (CH3) |
| 13 | 51.5 (CH) | 51.0 (CH) | 50.6 (CH) | 131.6 (CH) |
| 14 | 127.3 (CH) | 130.5 (CH) | 144.4 (CH) | 133.4 (CH) |
| 15 | 138.8 (CH) | 137.8 (CH) | 133.9 (CH) | 132.1 (CH) |
| 16 | 67.9 (CH) | 69.0 (CH) | 197.6 (C) | 129.2 (CH) |
| 17 | 23.5 (CH3) | 22.9 (CH3) | 27.6 (CH3) | 18.1 (CH3) |
| 18 | 24.2 (CH3) | 23.9 (CH3) | 23.7 (CH3) | 22.5 (CH3) |
| 19 | 22.4 (CH3) | 22.4 (CH3) | 22.4 (CH3) | 22.7 (CH3) |
| 2′ | 168.6 (C) | 168.6 (C) | 167.8 (C) | 178.1 (C) |
| 3′ | 73.1 (C) | 73.8 (C) | 72.7 (C) | 101.6 (C) |
| 4′ | 207.2 (C) | 205.0 (C) | 206.4 (C) | 191.5 (C) |
| 5′ | 69.8 (CH) | 69.4 (CH) | 69.8 (CH) | 68.8 (CH) |
| 6′ | 60.3 (CH2) | 58.3 (CH2) | 60.3 (CH2) | 59.6 (CH2) |
| 7′ | 28.3 (CH3) | 27.7 (CH3) | 28.5 (CH3) | 27.4 (CH3) |
| Compound | A-375 Cells | HaCaT Cells |
|---|---|---|
| 1 | 2.8 (±0.0) | 4.2 (±0.0) |
| 2 | 6.3 (±0.0) | 35.0 (±0.0) |
| 3 | 140.3 (±0.9) | 142.9 (±1.4) |
| 4 | 37.3 (±0.1) | 45.0 (±0.2) |
| Positive control | 0.6 (±0.0) | 22.1 (±2.9) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, B.; Dewapriya, P.; Li, F.; Blümel, M.; Tasdemir, D. Pyrenosetins A–C, New Decalinoylspirotetramic Acid Derivatives Isolated by Bioactivity-Based Molecular Networking from the Seaweed-Derived Fungus Pyrenochaetopsis sp. FVE-001. Mar. Drugs 2020, 18, 47. https://doi.org/10.3390/md18010047
Fan B, Dewapriya P, Li F, Blümel M, Tasdemir D. Pyrenosetins A–C, New Decalinoylspirotetramic Acid Derivatives Isolated by Bioactivity-Based Molecular Networking from the Seaweed-Derived Fungus Pyrenochaetopsis sp. FVE-001. Marine Drugs. 2020; 18(1):47. https://doi.org/10.3390/md18010047
Chicago/Turabian StyleFan, Bicheng, Pradeep Dewapriya, Fengjie Li, Martina Blümel, and Deniz Tasdemir. 2020. "Pyrenosetins A–C, New Decalinoylspirotetramic Acid Derivatives Isolated by Bioactivity-Based Molecular Networking from the Seaweed-Derived Fungus Pyrenochaetopsis sp. FVE-001" Marine Drugs 18, no. 1: 47. https://doi.org/10.3390/md18010047
APA StyleFan, B., Dewapriya, P., Li, F., Blümel, M., & Tasdemir, D. (2020). Pyrenosetins A–C, New Decalinoylspirotetramic Acid Derivatives Isolated by Bioactivity-Based Molecular Networking from the Seaweed-Derived Fungus Pyrenochaetopsis sp. FVE-001. Marine Drugs, 18(1), 47. https://doi.org/10.3390/md18010047