Precise Structure and Anticoagulant Activity of Fucosylated Glycosaminoglycan from Apostichopus japonicus: Analysis of Its Depolymerized Fragments

 and
and
Abstract
1. Introduction
2. Results and Discussion
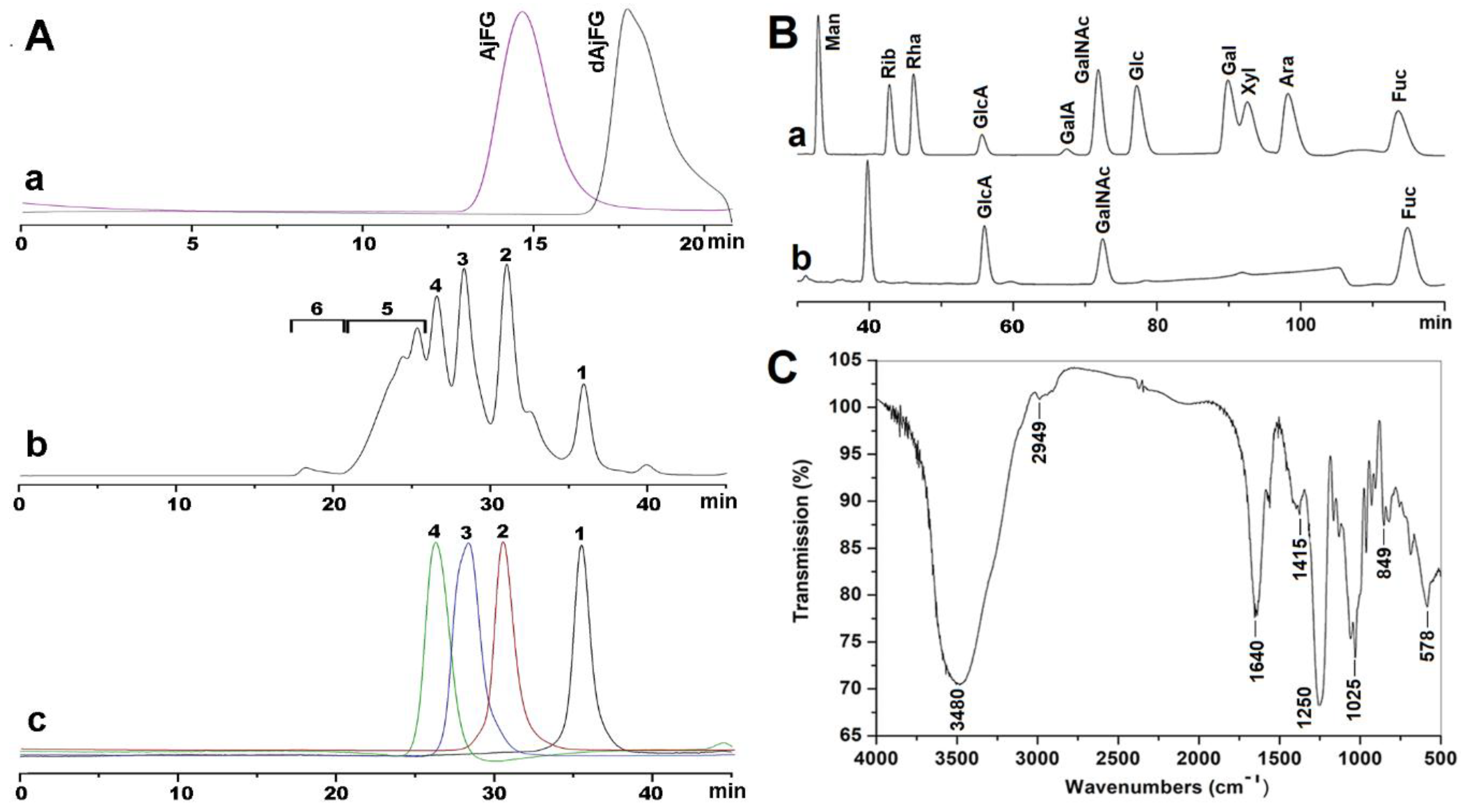
2.1. Extraction, Isolation, and Purification of AjFG
2.2. Analysis of Physicochemical Properties
2.3. Preparation of Oligosaccharides with Various dp
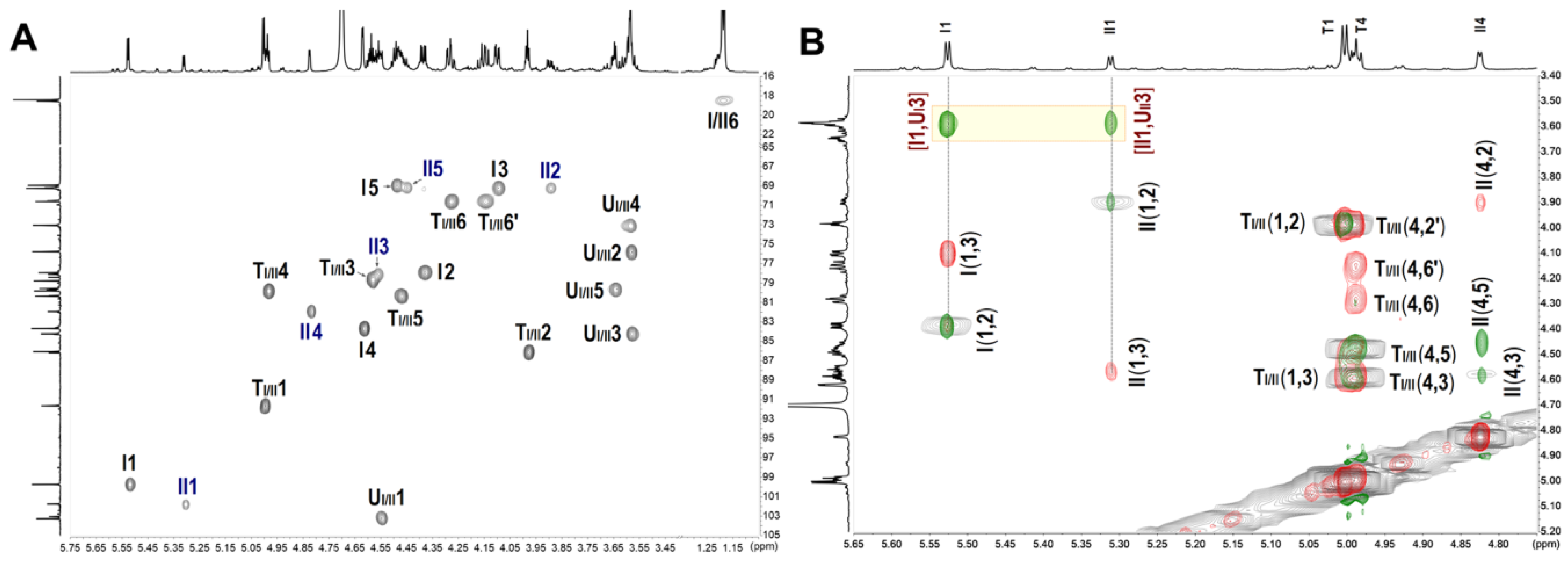
2.4. NMR and ESI-Q-TOF-MS Analysis
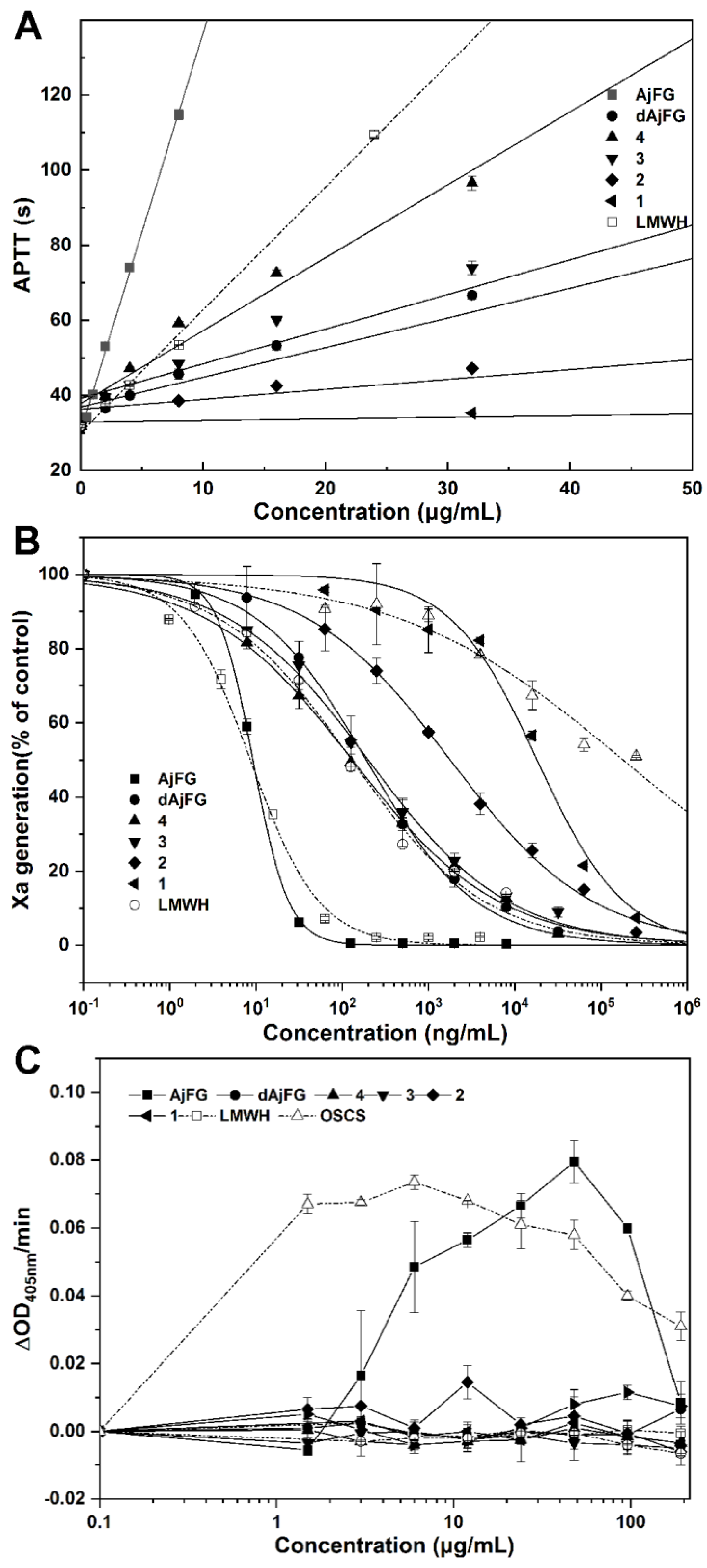
2.5. Analysis of the Anticoagulant Activity
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Extraction, Isolation and Purification of AjFG
3.3. Determination of Physicochemical Properties
3.4. Preparation and Isolation of Oligosaccharides of AjFG
3.5. NMR and ESI-Q-TOF-MS Analysis
3.6. Determination of Anticoagulant Activities In Vitro
3.7. Human Factor XII Activation and Platelet Aggregation Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Ma, H.; Mai, K.; Zhang, W.; Liufu, Z.; Xu, W. Interaction of dietary Bacillus subtilis and fructooligosaccharide on the growth performance, non-specific immunity of sea cucumber, Apostichopus japonicus. Fish Shellfish Immunol. 2010, 29, 204–211. [Google Scholar] [CrossRef]
- Bordbar, S.; Anwar, F.; Saari, N. High-value components and bioactives from sea cucumbers for functional foods—A review. Mar. Drugs 2011, 9, 1761–1805. [Google Scholar] [CrossRef] [PubMed]
- Buyue, Y.; Sheehan, J.P. Fucosylated chondroitin sulfate inhibits plasma thrombin generation via targeting of the factor IXa heparin-binding exosite. Blood 2009, 114, 3092–3100. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Holothurian fucosylated chondroitin sulfate. Mar. Drugs 2014, 12, 232–254. [Google Scholar] [CrossRef]
- Wu, M.Y.; Wen, D.D.; Gao, N.; Xiao, C.; Yang, L.; Xu, L.; Lian, W.; Peng, W.L.; Jiang, J.M.; Zhao, J.H. Anticoagulant and antithrombotic evaluation of native fucosylated chondroitin sulfates and their derivatives as selective inhibitors of intrinsic factor Xase. Eur. J. Med. Chem. 2015, 92, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.Z.; Chen, J.D.; Lin, K.Z. An acidic mucopolysaccharide isolated from Stichopus japonicus Selenka and some of its physical and chemical properties. Yao Xue Xue Bao 1980, 15, 263–270. [Google Scholar]
- Kariya, Y.; Watabe, S.; Hashimoto, K.; Yoshida, K. Occurence of chondroitin sulfate E in glycosaminoglycan islated from the body wall of sea cucumber Stichopus japanicus. J. Biol. Chem. 1990, 265, 5081–5085. [Google Scholar]
- Yoshida, K.; Minami, Y.; Nemoto, H.; Numata, K.; Yamanaka, E. Structure of DHG, a depolymerized glycosaminoglycan from sea cucumber, Stichopus japonicus. Tetrahedron Lett. 1992, 33, 4959–4962. [Google Scholar] [CrossRef]
- Kariya, Y.; Watabe, S.; Kyogashima, M.; Ishihara, M.; Ishii, T. Structure of fucose branches in the glycosaminoglycan from the body wall of the sea cucumber Stichopus japonicus. Carbohydr. Res. 1997, 297, 273–279. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.H.; Jiang, T.F.; Lv, Z.H. Novel branch patterns and anticoagulant activity of glycosaminoglycan from sea cucumber Apostichopus japonicus. Int. J. Biol. Macromol. 2015, 72, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Ustyuzhanina, N.E.; Bilan, M.I.; Dmitrenok, A.S.; Tsvetkova, E.A.; Shashkov, A.S.; Stonik, V.A.; Nifantiev, N.E.; Usov, A.I. Structural characterization of fucosylated chondroitin sulfates from sea cucumbers Apostichopus japonicus and Actinopyga mauritiana. Carbohydr. Polym. 2016, 153, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Mou, J.J.; Li, Q.; Qin, X.H.; Yang, J. Structural comparison, antioxidant and anti-inflammatory properties of fucosylated chondroitin sulfate of three edible sea cucumbers. Carbohydr. Polym. 2018, 185, 41–47. [Google Scholar] [CrossRef]
- Cui, C.; Cui, N.S.; Wang, P.; Song, S.L.; Liang, H.; Ji, A.G. Neuroprotective effect of sulfated polysaccharide isolated from sea cucumber Stichopus japonicus on 6-OHDA-induced death in SH-SY5Y through inhibition of MAPK and NF-κB and activation of PI3K/Akt signaling pathways. Biochem. Biophys. Res. Commun. 2016, 470, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, Z.L.; Zhang, M.S.; Meng, X.M.; Xia, X.K.; Yuan, W.P.; Xue, F.; Liu, C.H. Antioxidant and antihyperlipidemic activities of polysaccharides from sea cucumber Apostichopus japonicus. Carbohydr. Polym. 2012, 90, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Kitazato, K.; Takamatsu, J.; Saito, H. Antithrombotic and anticoagulant activity of depolymerized fragment of the glycosaminoglycan extracted from Stichopus japonicus Selenka. Thromb. Haemost. 1991, 65, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Y.; Wu, M.Y.; Xiao, C.; Yang, L.; Zhou, L.T.; Gao, N.; Li, Z.; Chen, J.; Chen, J.C.; Liu, J.K.; et al. Discovery of an intrinsic tenase complex inhibitor: Pure nonasaccharide from fucosylated glycosaminoglycan. Proc. Natl. Acad. Sci. USA 2015, 112, 8284–8289. [Google Scholar] [CrossRef]
- Fonseca, R.J.; Sucupira, I.D.; Oliveira, S.N.; Santos, G.R.; Mourao, P.A. Improved anticoagulant effect of fucosylated chondroitin sulfate orally administered as gastro-resistant tablets. Thromb. Haemost. 2017, 117, 662–670. [Google Scholar]
- Xu, L.; Gao, N.; Xiao, C.; Lin, L.S.; Purcell, S.W.; Wu, M.Y.; Zhao, J.H. Modulating the degree of fucosylation of fucosylated chondroitin sulfate enhances heparin cofactor II-dependent thrombin inhibition. Eur. J. Med. Chem. 2018, 154, 133–143. [Google Scholar] [CrossRef]
- Shang, F.N.; Gao, N.; Yin, R.H.; Lin, L.S.; Xiao, C.; Zhou, L.T.; Li, Z.; Purcell, S.W.; Wu, M.Y.; Zhao, J.H. Precise structures of fucosylated glycosaminoglycan and its oligosaccharides as novel intrinsic factor Xase inhibitors. Eur. J. Med. Chem. 2018, 148, 423–435. [Google Scholar] [CrossRef]
- Yin, R.H.; Zhou, L.T.; Gao, N.; Li, Z.; Zhao, L.Y.; Shang, F.N.; Wu, M.Y.; Zhao, J.H. Oligosaccharides from depolymerized fucosylated glycosaminoglycan: Structures and minimum size for intrinsic factor Xase complex inhibition. J. Biol. Chem. 2018, 293, 14089–14099. [Google Scholar] [CrossRef] [PubMed]
- Mourao, P.A.S.; Pereira, M.S.; Pavo, M.S.G.; Mulloy, B.; Tollefsen, D.M.; Mowinckel, M.C.; Abildgaard, U. Structure and anticoagulant activity of a fucosylated chondroitin sulfate from echinoderm. J. Biol. Chem. 1996, 271, 23973–23984. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Qin, Y.J.; Guan, R.W.; Zheng, W.Q.; Liu, J.K.; Zhao, J.H. Digestibility of fucosylated glycosaminoglycan from sea cucumber and its effects on digestive enzymes under simulated salivary and gastrointestinal conditions. Carbohydr. Polym. 2018, 186, 217–225. [Google Scholar] [CrossRef]
- Rabenstein, D.L. Heparin and heparan sulfate: Structure and function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.P.; Mulloy, B.; Mourão, P.A.S. Structure of a fucose-branched chondroitin sulfate from sea cucumber. J. Biol. Chem. 1991, 266, 13530–13536. [Google Scholar] [PubMed]
- Luo, L.; Wu, M.Y.; Xu, L.; Lian, W.; Xiang, J.Y.; Lu, F.; Gao, N.; Xiao, C.; Wang, S.M.; Zhao, J.H. Comparison of physicochemical characteristics and anticoagulant activities of polysaccharides from three sea cucumbers. Mar. Drugs 2013, 11, 399–417. [Google Scholar] [CrossRef]
- Shang, F.N.; Mou, R.R.; Zhang, Z.D.; Gao, N.; Lin, L.S.; Li, Z.; Wu, M.Y.; Zhao, J.H. Structural analysis and anticoagulant activities of three highly regular fucan sulfates as novel intrinsic factor Xase inhibitors. Carbohydr. Polym. 2018, 195, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.J.C.; Santos, G.R.C.; Mourão, P.A.S. Effects of polysaccharides enriched in 2,4-disulfated fucose units on coagulation, thrombosis and bleeding: Practical and conceptual implications. Thromb. Haemost. 2009, 102, 829–836. [Google Scholar] [CrossRef]
- Fonseca, R.J.C.; Oliveira, S.N.M.C.G.; Pomin, V.H.; Mecawi, A.S.; Araujo, I.G.; Mourão, P.A.S. Effects of oversulfated and fucosylated chondroitin sulfates on coagulation. Challenges for the study of anticoagulant polysaccharides. Thromb. Haemost. 2010, 103, 994–1004. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Weïwer, M.; Li, B.Y.; Kemp, M.M.; Daman, T.H.; Linhardt, R.J. Oversulfated chondroitin sulfate: Impact of a heparin impurity, associated with adverse clinical events, on low-molecular-weight heparin preparation. J. Med. Chem. 2008, 51, 5498–5501. [Google Scholar] [CrossRef]
- Kishimoto, T.K.; Viswanathan, K.; Ganguly, T.; Elankumaran, S.; Smith, S.; Pelzer, K.; Lansing, J.C.; Sriranganathan, N.; Zhao, G.; Galcheva-Gargova, Z.; et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N. Engl. J. Med. 2008, 358, 2457–2467. [Google Scholar] [CrossRef]
- Li, J.Z.; Lian, E.C.Y. Mechanism of rabbit platelet agglutination induced by acidic mucopolysaccharide extracted from Stichopus japonicus Selenka. Thromb. Haemost. 1988, 59, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Lian, E.C.Y. Aggregation of human platelets by acidic mucopolysaccharide extracted from Stichopus japonicus Selenka. Thromb. Haemost. 1988, 59, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Walke, E.N. Depolymerized holothurian glycosaminoglycan and heparin inhibit the intrinsic tenase complex by a common antithrombin-independent mechanism. Blood 2006, 107, 3876–3882. [Google Scholar] [CrossRef] [PubMed]
- Nagase, B.H.; Enjyoji, K.; Minamiguchi, K.; Kitazato, K.T.; Kitazato, K.; Saito, H. Depolymerized holothurian glycosaminoglycan with novel anticoagulant actions: Antithrombin III- and heparin cofactor II-independent inhibition of factor. Blood 1995, 85, 1527–1534. [Google Scholar] [PubMed]
- Yuan, Q.X.; Xie, Y.F.; Wang, W.; Yan, Y.H.; Ye, H.; Jabbar, S.; Zeng, X.X. Extraction optimization, characterization and antioxidant activity in vitro of polysaccharides from mulberry (Morus alba L.) leaves. Carbohydr. Polym. 2015, 128, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Gennaro, U. A conductimetric method for the determination of sulphate and carboxyl groups in heparin and other mucopolysaccharides. Carbohydr. Res. 1975, 39, 168–176. [Google Scholar] [CrossRef]
- Gao, N.; Wu, M.Y.; Liu, S.; Lian, W.; Li, Z.; Zhao, J.H. Preparation and characterization of O-acylated fucosylated chondroitin sulfate from sea cucumber. Mar. Drugs 2012, 10, 1647–1661. [Google Scholar] [CrossRef]
- Xiao, C.; Lian, W.; Zhou, L.T.; Gao, N.; Xu, L.; Chen, J.; Wu, M.Y.; Peng, W.L.; Zhao, J.H. Interactions between depolymerized fucosylated glycosaminoglycan and coagulation proteases or inhibitors. Thromb. Res. 2016, 146, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Born, G.V.R. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| δH (ppm) | Coupling Constant in Hz | δC (ppm) | δH (ppm) | Coupling Constant in Hz | δC (ppm) | ||||||
| TI | H-1 | 5.00 | J(1,2) = 4.41 | C-1 | 91.7 | TII | H-1 | 5.00 | J(1,2) = 4.41 | C-1 | 91.6 |
| H-2 | 3.98 | J(2,3) = 4.52 | C-2 | 86.1 | H-2 | 3.99 | J(2,3) = 4.52 | C-2 | 86.2 | ||
| H-3 | 4.59 | J(3,4) = 5.20 | C-3 | 78.8 | H-3 | 4.60 | J(3,4) = 5.06 | C-3 | 78.4 | ||
| H-4 | 4.99 | J(4,5) = 5.10 | C-4 | 79.8 | H-4 | 4.99 | J(2,3) = 5.13 | C-4 | 79.7 | ||
| H-5 | 4.48 | J(5,6′) = 9.10 | C-5 | 80.4 | H-5 | 4.48 | J(5,6′) = 9.10 | C-5 | 80.3 | ||
| H-6 | 4.29 | J(5,6) = 2.50 | C-6 | 70.6 | H-6 | 4.27 | J(5,6) = 2.47 | C-6 | 70.5 | ||
| H-6′ | 4.15 | J(6,6′) =11.29 | H-6′ | 4.16 | J(6,6′) = 11.26 | ||||||
| UI | H-1 | 4.56 | J(1,2) = 7.74 | C-1 | 103.3 | UII | H-1 | 4.55 | J(1,2) = 7.44 | C-1 | 103.1 |
| H-2 | 3.59 | J(2,3) = 8.29 | C-2 | 75.8 | H-2 | 3.59 | J(2,3) = 8.37 | C-2 | 75.8 | ||
| H-3 | 3.60 | J(3,4) = 7.55 | C-3 | 84.2 | H-3 | 3.57 | J(3,4) = 7.78 | C-3 | 84.3 | ||
| H-4 | 3.60 | J(4,5) = 9.73 | C-4 | 73.1 | H-4 | 3.62 | J(4,5) = 9.38 | C-4 | 73.1 | ||
| H-5 | 3.65 | C-5 | 79.6 | H-5 | 3.65 | C-5 | 79.7 | ||||
| C-6 | 178.4 | C-6 | 178.4 | ||||||||
| I | H-1 | 5.53 | J(1,2) = 3.92 | C-1 | 99.7 | II | H-1 | 5.31 | J(1,2) = 3.98 | C-1 | 101.8 |
| H-2 | 4.39 | J(2,3) = 10.58 | C-2 | 77.9 | H-2 | 3.90 | J(2,3) = 10.46 | C-2 | 69.2 | ||
| H-3 | 4.10 | J(3,4) = 2.96 | C-3 | 69.2 | H-3 | 4.57 | J(3,4) = 2.95 | C-3 | 78.1 | ||
| H-4 | 4.62 | C-4 | 83.7 | H-4 | 4.83 | C-4 | 81.9 | ||||
| H-5 | 4.50 | J(5,6) = 6.80 | C-5 | 68.9 | H-5 | 4.46 | J(5,6) = 6.62 | C-5 | 69.2 | ||
| H-6 | 1.19 | C-6 | 18.4 | H-6 | 1.19 | C-6 | 18.6 | ||||
| Sample | Mw (kDa) | APTT (μg/mL) a | Anti-FXase (IC50, ng/mL) b |
|---|---|---|---|
| AjFG | 76.4 | 3.06 | 9.20 |
| dAjFG | 3.90 | 26.5 | 189 |
| 4 | 3.49 | 10.3 | 131 |
| 3 | 2.63 | 20.1 | 200 |
| 2 | 1.77 | >128 | >1000 |
| 1 | 0.93 | >128 | >1000 |
| LMWH | 4.50 | 11.3 | 128 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, R.; Peng, Y.; Zhou, L.; Zheng, W.; Liu, X.; Wang, P.; Yuan, Q.; Gao, N.; Zhao, L.; Zhao, J. Precise Structure and Anticoagulant Activity of Fucosylated Glycosaminoglycan from Apostichopus japonicus: Analysis of Its Depolymerized Fragments. Mar. Drugs 2019, 17, 195. https://doi.org/10.3390/md17040195
Guan R, Peng Y, Zhou L, Zheng W, Liu X, Wang P, Yuan Q, Gao N, Zhao L, Zhao J. Precise Structure and Anticoagulant Activity of Fucosylated Glycosaminoglycan from Apostichopus japonicus: Analysis of Its Depolymerized Fragments. Marine Drugs. 2019; 17(4):195. https://doi.org/10.3390/md17040195
Chicago/Turabian StyleGuan, Ruowei, Yuan Peng, Lutan Zhou, Wenqi Zheng, Xixi Liu, Pin Wang, Qingxia Yuan, Na Gao, Longyan Zhao, and Jinhua Zhao. 2019. "Precise Structure and Anticoagulant Activity of Fucosylated Glycosaminoglycan from Apostichopus japonicus: Analysis of Its Depolymerized Fragments" Marine Drugs 17, no. 4: 195. https://doi.org/10.3390/md17040195
APA StyleGuan, R., Peng, Y., Zhou, L., Zheng, W., Liu, X., Wang, P., Yuan, Q., Gao, N., Zhao, L., & Zhao, J. (2019). Precise Structure and Anticoagulant Activity of Fucosylated Glycosaminoglycan from Apostichopus japonicus: Analysis of Its Depolymerized Fragments. Marine Drugs, 17(4), 195. https://doi.org/10.3390/md17040195