
Role of Inflammatory Cytokines in Rheumatoid Arthritis and Development of Atherosclerosis: A Review



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Objective
2.2. Data Sources
2.3. Keywords Used in Article Selection
2.4. Article Selection: Inclusion and Exclusion Criteria
2.5. Limitations
2.6. Study Design
2.7. Review Period
3. Results
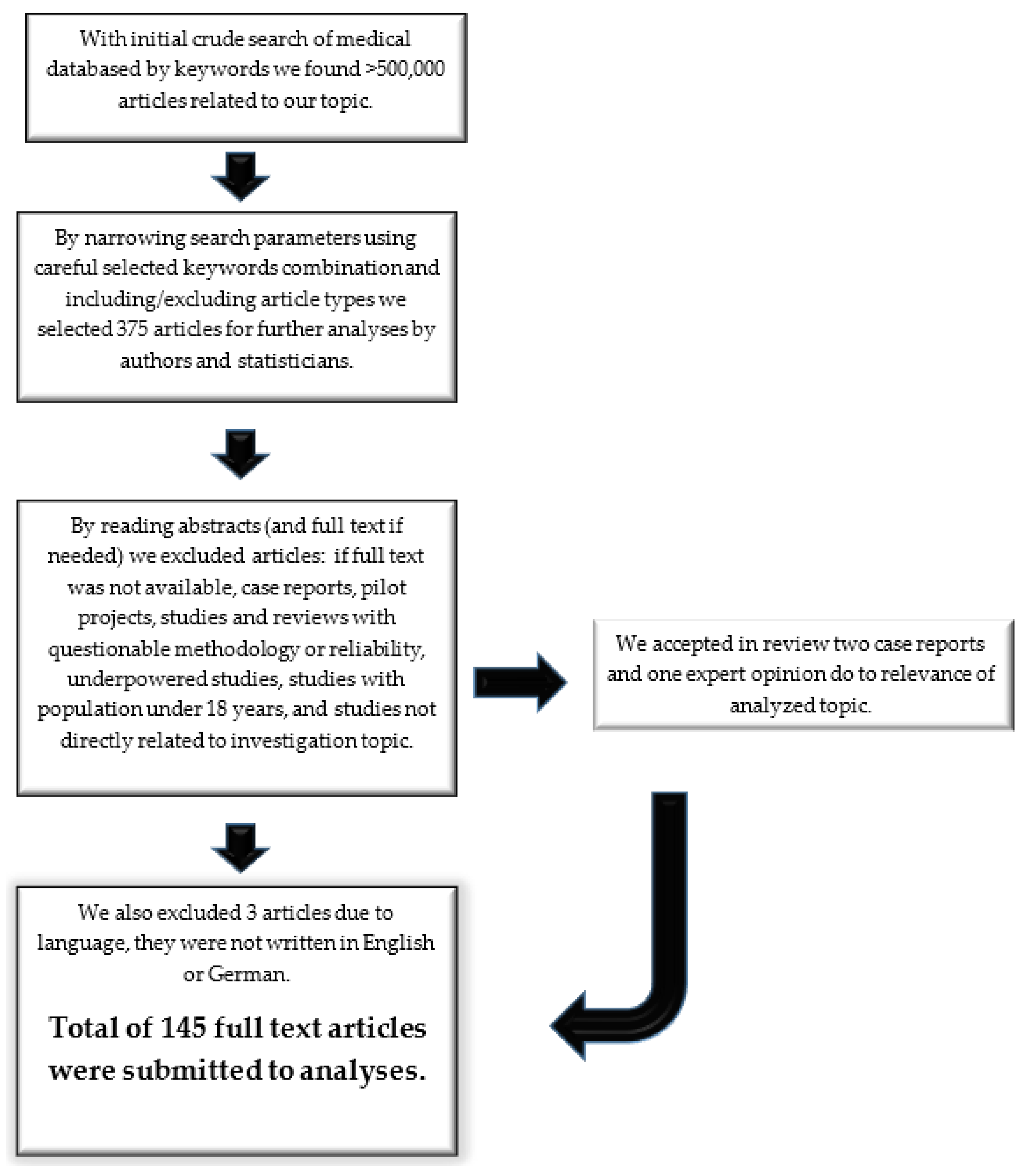
3.1. Article Selection Process
3.2. Analysis Results
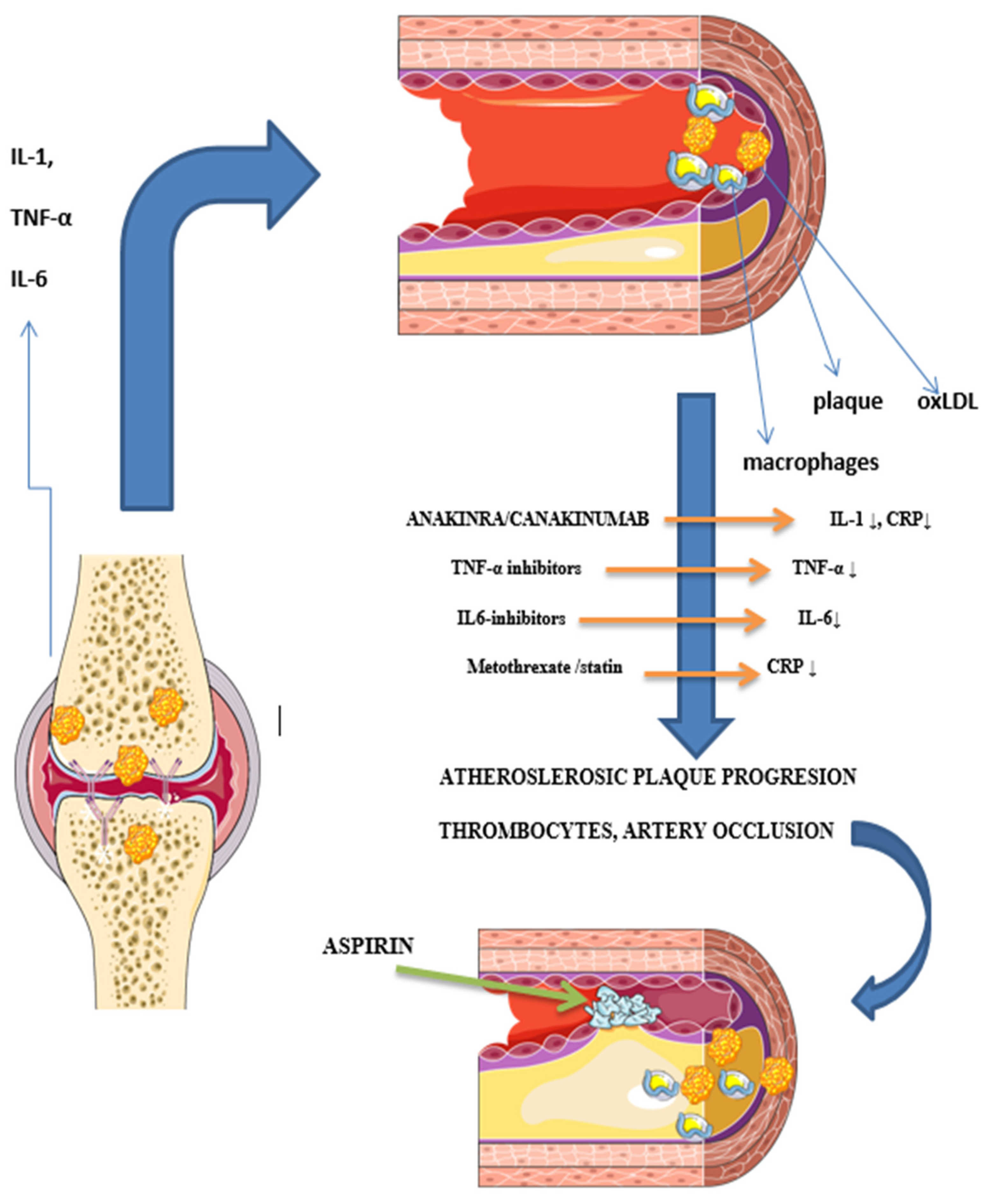
3.2.1. Chronic Inflammation
3.2.2. Influence of Medications
NSAIDs
Glucocorticoids
Classical DMARDs
Biologic Agents
Small Molecule Inhibitors of Janus Kinase
Statins
4. Discussion
4.1. Chronic Inflammation
4.2. Influence of Medications
4.2.1. NSAIDs
4.2.2. Glucocorticoids
4.2.3. Classical DMARDs
4.2.4. Biologic Agents
4.2.5. Small Molecule Inhibitors of JAK
4.2.6. Statins role in CVD prevention
4.3. The Role of Inflammatory Markers in RA Activity and CV Risk Assessment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitchell, D.M.; Spitz, P.W.; Young, D.Y.; Bloch, D.A.; McShane, D.J.; Fries, J.F. Survival, prognosis, and causes of death in rheumatoid-arthritis. Arthritis Rheum. 1986, 29, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Coulton, B.L.; Symmons, D.P.M. Long-term outcome of threating rheumatoid arthritis—Results after 20 years. Lancet 1987, 1, 1108–1111. [Google Scholar] [CrossRef]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; Mcshane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The american rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef]
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet 2001, 358, 903–911. [Google Scholar] [CrossRef]
- Symmons, D.P. Epidemiology of rheumatoid arthritis: Determinants of onset, persistence and outcome. Best Pract. Res. Clin. Rheumatol. 2002, 16, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Hirano, F.; Kihara, M.; Yokoyama, W.; Yamazaki, H.; Harada, S.; Nanki, T.; Koike, R.; Miyasaka, N.; Harigai, M. High prevalence of cardiovascular comorbidities in patients with rheumatoid arthritis from a population-based cross-sectional study of a Japanese health insurance database. Mod. Rheumatol. 2016, 26, 522–528. [Google Scholar] [CrossRef]
- Koivuniemi, R.; Paimela, L.; Leirisalo-Repo, M. Causes of death in patients with rheumatoid arthritis from 1971 to 1991 with special reference to autopsy. Clin. Rheumatol. 2009, 28, 1443–1447. [Google Scholar] [CrossRef]
- Maradit-Kremers, H.; Nicola, P.J.; Crowson, C.S.; Ballman, K.V.; Gabriel, S.E. Cardiovascular death in rheumatoid arthritis: A population-based study. Arthritis Rheum. 2005, 52, 722–732. [Google Scholar] [CrossRef]
- Sokka, T.; Abelson, B.; Pincus, T. Mortality in rheumatoid arthritis: 2008 update. Clin. Exp. Rheumatol. 2009, 26, S35–S61. [Google Scholar]
- Meune, C.; Touzé, E.; Trinquart, L.; Allanore, Y. High risk of clinical cardiovascular events in rheumatoid arthritis: Levels of associations of myocardial infarction and stroke through a systematic review and meta-analysis. Arch. Cardiovasc. Dis. 2010, 103, 253–261. [Google Scholar] [CrossRef]
- Meune, C.; Touzé, E.; Trinquart, L.; Allanore, Y. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: A systematic review and meta-analysis of cohort studies. Rheumatology 2009, 48, 1309–1313. [Google Scholar] [CrossRef]
- Aviña-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008, 59, 1690–1697. [Google Scholar] [CrossRef]
- Meyer, P.W.; Anderson, R.; Ker, J.A.; Ally, M.T. Rheumatoid arthritis and risk of cardiovascular disease. Cardiovasc. J. Afr. 2018, 29, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Siebert, S.; Lyall, D.M.; Mackay, D.F.; Porter, D.; McInnes, I.B.; Sattar, N.; Pell, J.P. Characteristics of rheumatoid arthritis and its association with major comorbid conditions: Cross-sectional study of 502 649 UK Biobank participants. RMD Open 2016, 2, e000267. [Google Scholar] [CrossRef]
- Van Doornum, S.; McColl, G.; Wicks, I.P. Accelerated atherosclerosis—An extraarticular feature of rheumatoid arthritis? Arthritis Rheum. 2002, 46, 862–873. [Google Scholar] [CrossRef]
- Visseren, F.L.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [PubMed]
- Panoulas, V.F.; Metsios, G.S.; Pace, A.V.; John, H.; Treharne, G.J.; Banks, M.J.; Kitas, G.D. Hypertension in rheumatoid arthritis. Rheumatology 2008, 4, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Innala, L.; Sjöberg, C.; Möller, B.; Ljung, L.; Smedby, T.; Södergren, A.; Magnusson, S.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S. Co-morbidity in patients with early rheumatoid arthritis—Inflammation matters. Thromb. Haemost. 2016, 18, 33. [Google Scholar] [CrossRef]
- Grosso, G.; Erba, G.; Valena, C.; Riva, M.; Betelli, M.; Allevi, E.; Bonomi, F.; Barbarossa, S.; Ricci, M.; Facchetti, R.; et al. THU0139 Cardiovascular Risk Factor Profile in an Italian Cohort of Patients with Rheumatoid Arthritis: Results of a Three Year Follow-up. Ann. Rheum. Dis. 2015, 74, 244. [Google Scholar] [CrossRef]
- Gherghe, A.M.; Dougados, M.; Combe, B.; Landewé, R.; Mihai, C.; Berenbaum, F.; Mariette, X.; Wolterbeek, R.; van der Heijde, D. Cardiovascular and selected comorbidities in early arthritis and early spondyloarthritis, a comparative study: Results from the ESPOIR and DESIR cohorts. RMD Open 2015, 1, e000128. [Google Scholar] [CrossRef]
- Boyer, J.-F.; Gourraud, P.-A.; Cantagrel, A.; Davignon, J.-L.; Constantin, A. Traditional cardiovascular risk factors in rheumatoid arthritis: A meta-analysis. Jt. Bone Spine 2011, 78, 179–183. [Google Scholar] [CrossRef]
- Heliövaara, M.; Aho, K.; Aromaa, A.; Knekt, P.; Reunanen, A. Smoking and risk of rheumatoid arthritis. J. Rheumatol. 1993, 20, 1830–1835. [Google Scholar]
- La Hoz, J.C.-D.; Amaya-Amaya, J.; Molano-González, N.; Gutiérrez-Infante, F.; Anaya, J.M.; Rojas-Villarraga, A. FRI0055 The influence of cigarette smoking on disease activity and joint erosions in rheumatoid arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2013, 72, A387. [Google Scholar] [CrossRef]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Harris, H.E.; Ulfgren, A.K.; Rantapää-Dahlqvist, S.; et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006, 54, 38–46. [Google Scholar] [CrossRef]
- Welsing, P.M.J.; Van Gestel, A.M.; Swinkels, H.L.; Kiemeney, L.A.L.M.; Van Riel, P.L.C.M. The relationship between disease activity, joint destruction, and functional capacity over the course of rheumatoid arthritis. Arthritis Rheum. 2001, 44, 2009–2017. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. Task Force 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts). Eur. J. Prev. Cardiol. 2016, 23, NP1–NP96. [Google Scholar]
- Baghdadi, L.R.; Woodman, R.J.; Shanahan, E.M.; Mangoni, A.A. The Impact of Traditional Cardiovascular Risk Factors on Cardiovascular Outcomes in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0117952. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Li, H.; Li, X. Diabetes mellitus risk factors in rheumatoid arthritis: A systematic review and meta-analysis. Clin. Exp. Rheumatol. 2015, 33, 115–121. [Google Scholar] [PubMed]
- Guin, A.; Sinhamahapatra, P.; Misra, S.; Mazumder, S.R.C.; Chatterjee, S.; Ghosh, A. Incidence and effect of insulin resistance on progression of atherosclerosis in rheumatoid arthritis patients of long disease duration. Biomed. J. 2019, 42, 394–402. [Google Scholar] [CrossRef]
- Dougados, M.; Soubrier, M.; Antunez, A.; Balint, P.; Balsa, A.; Buch, M.H.; Casado, G.; Detert, J.; El-Zorkany, B.; Emery, P.; et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: Results of an international, cross-sectional study (COMORA). Ann. Rheum. Dis. 2020, 73, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos-Kalinoglou, A.; Metsios, G.S.; Koutedakis, Y.; Nevill, A.M.; Douglas, K.M.; Jamurtas, A.; van Zanten, J.J.C.S.V.; Labib, M.; Kitas, G.D. Redefining overweight and obesity in rheumatoid arthritis patients. Ann. Rheum. Dis. 2007, 66, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.L.; Lanna, C.C.D.; Rocha, M.P.; Ribeiro, A.L.P.; Telles, R.W. Recognition and control of hypertension, diabetes, and dyslipidemia in patients with rheumatoid arthritis. Rheumatol. Int. 2018, 38, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Beinsberger, J.; Heemskerk, J.W.; Cosemans, J.M. Chronic arthritis and cardiovascular disease: Altered blood parameters give rise to a prothrombotic propensity. Semin. Arthritis Rheum. 2014, 44, 345–352. [Google Scholar] [CrossRef]
- Nowak, B.; Madej, M.; Łuczak, A.; Małecki, R.; Wiland, P. Disease Activity, Oxidized-LDL Fraction and Anti-Oxidized LDL Antibodies Influence Cardiovascular Risk in Rheumatoid Arthritis. Adv. Clin. Exp. Med. 2016, 25, 43–50. [Google Scholar] [CrossRef]
- Giles, J.T.; Wasko, M.C.M.; Chung, C.P.; Szklo, M.; Blumenthal, R.S.; Kao, A.; Bokhari, S.; Zartoshti, A.; Stein, C.M.; Bathon, J.M. Exploring the Lipid Paradox Theory in Rheumatoid Arthritis: Associations of Low Circulating Low-Density Lipoprotein Concentration With Subclinical Coronary Atherosclerosis. Arthritis Rheumatol. 2011, 71, 1426–1436. [Google Scholar] [CrossRef]
- McGrath, C.M.; Young, S.P. Lipid and Metabolic Changes in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2015, 17, 57. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Mechanisms of Disease: The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, J.; Lau, J.; Wang, S.; Taneja, V.; Matteson, E.L.; Vassallo, R. Mechanisms of lung disease development in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019, 15, 581–596. [Google Scholar] [CrossRef]
- Londei, M.; Savill, C.M.; Verhoef, A.; Brennan, F.; Leech, Z.A.; Duance, V.; Maini, R.N.; Feldmann, M. Persistence of collagen type II-specific T-cell clones in the synovial membrane of a patient with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 1987, 86, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Glant, T.T.; Radacs, M.; Nagyeri, G.; Olasz, K.; Laszlo, A.; Boldizsar, F.; Hegyi, A.; Finnegan, A.; Mikecz, K. Proteoglycan-induced arthritis and recombinant human proteoglycan aggrecan G1 domain-induced arthritis in BALB/c mice resembling two subtypes of rheumatoid arthritis. Arthritis Rheum. 2011, 63, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Verheijden, G.F.M.; Rijnders, A.W.M.; Bos, E.; Roo, C.J.J.C.-D.; van Staveren, C.J.; Miltenburg, A.M.M.; Meijerink, J.H.; Elewaut, D.; de Keyser, F.; Veys, E.; et al. Human cartilage glycoprotein-39 as a candidate autoantigen in rheumatoid arthritis. Arthritis Rheum. 2011, 40, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 23. [Google Scholar] [CrossRef]
- Burmester, G.R.; Feist, E.; Dörner, T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 77–88. [Google Scholar] [CrossRef]
- Carbone, F.; Bonaventura, A.; Liberale, L.; Paolino, S.; Torre, F.; Dallegri, F.; Montecucco, F.; Cutolo, M. Atherosclerosis in Rheumatoid Arthritis: Promoters and Opponents. Clin. Rev. Allergy Immunol. 2020, 58, 1–14. [Google Scholar] [CrossRef]
- Mewar, D.; Coote, A.; Moore, D.J.; Marinou, I.; Keyworth, J.; Dickson, M.C.; Montgomery, D.S.; Binks, M.H.; Wilson, A.G. Independent associations of anti-cyclic citrullinated peptide antibodies and rheumatoid factor with radiographic severity of rheumatoid arthritis. Thromb. Haemost. 2006, 8, R128. [Google Scholar] [CrossRef]
- Sokolove, J.; Bromberg, R.; Deane, K.D.; Lahey, L.J.; Derber, L.A.; Chandra, P.E.; Edison, J.D.; Gilliland, W.R.; Tibshirani, R.J.; Norris, J.M.; et al. Autoantibody Epitope Spreading in the Pre-Clinical Phase Predicts Progression to Rheumatoid Arthritis. PLoS ONE 2012, 7, e35296. [Google Scholar] [CrossRef]
- López-Longo, F.J.; Oliver-Miñarro, D.; de la Torre, I.; de Rábago, E.G.-D.; Sánchez-Ramón, S.; Rodríguez-Mahou, M.; Paravisini, A.; Monteagudo, I.; González, C.-M.; GarCía-Castro, M.; et al. Association between anti-cyclic citrullinated peptide antibodies and ischemic heart disease in patients with rheumatoid arthritis. Arthritis Rheum. 2009, 61, 419–424. [Google Scholar] [CrossRef]
- Sokolove, J.; Brennan, M.J.; Sharpe, O.; Lahey, L.J.; Kao, A.H.; Krishnan, E.; Edmundowicz, D.; Lepus, C.M.; Wasko, M.C.; Robinson, W.H. Brief Report: Citrullination Within the Atherosclerotic Plaque: A Potential Target for the Anti-Citrullinated Protein Antibody Response in Rheumatoid Arthritis. Arthritis Rheum. 2013, 65, 1719–1724. [Google Scholar] [CrossRef]
- Geraldino-Pardilla, L.; Zartoshti, A.; Ozbek, A.B.; Giles, J.T.; Weinberg, R.; Kinkhabwala, M.; Bokhari, S.; Bathon, J.M. Arterial Inflammation Detected With F-18-Fluorodeoxyglucose-Positron Emission Tomography in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 30–39. [Google Scholar] [CrossRef]
- DeMizio, D.J.; Geraldino-Pardilla, L.B. Autoimmunity and Inflammation Link to Cardiovascular Disease Risk in Rheumatoid Arthritis. Rheumatol. Ther. 2020, 7, 19–33. [Google Scholar] [CrossRef]
- Liang, K.P.; Maradit-Kremers, H.; Crowson, C.S.; Snyder, M.R.; Therneau, T.M.; Roger, V.L.; Gabriel, S.E. Autoantibodies and the Risk of Cardiovascular Events. J. Rheumatol. 2009, 36, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, J.H.; AB van Nies, J.; Chipping, J.; Marshall, T.; Mil, A.H.v.d.H.-V.; Symmons, D.P.; Verstappen, S.M. Rheumatoid factor and anti-citrullinated protein antibody positivity, but not level, are associated with increased mortality in patients with rheumatoid arthritis: Results from two large independent cohorts. Thromb. Haemost. 2014, 16, 1–8. [Google Scholar] [CrossRef]
- McCoy, S.S.; Crowson, C.S.; Maradit-Kremers, H.; Therneau, T.M.; Roger, V.L.; Matteson, E.L.; Gabriel, S.E. Longterm outcomes and treatment after myocardial infarction in patients with rheumatoid arthritis. J. Rheumatol. 2013, 40, 605–610. [Google Scholar] [CrossRef]
- Mackey, R.H.; Kuller, L.H.; Deane, K.D.; Walitt, B.T.; Chang, Y.F.; Holers, V.M.; Robinson, W.H.; Tracy, R.P.; Hlatky, M.A.; Eaton, C.B.; et al. Rheumatoid Arthritis, Anti-Cyclic Citrullinated Peptide Positivity, and Cardiovascular Disease Risk in the Women’s Health Initiative. Arthritis Rheumatol. 2015, 67, 2311–2322. [Google Scholar] [CrossRef]
- Innala, L.; Möller, B.; Ljung, L.; Magnusson, S.; Smedby, T.; Södergren, A.; Öhman, M.-L.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: A five year prospective study. Thromb. Haemost. 2010, 13, R131. [Google Scholar] [CrossRef]
- Pawlik, A.; Ostanek, L.; Brzosko, I.; Brzosko, M.; Masiuk, M.; Machalinski, B.; Gawronska-Szklarz, B. The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Thromb. Haemost. 2003, 5, R210–R213. [Google Scholar] [CrossRef]
- Winchester, R.; Giles, J.T.; Nativ, S.; Downer, K.; Zhang, H.-Z.; Bag-Ozbek, A.; Zartoshti, A.; Bokhari, S.; Bathon, J.M. Association of Elevations of Specific T Cell and Monocyte Subpopulations in Rheumatoid Arthritis with Subclinical Coronary Artery Atherosclerosis. Arthritis Rheumatol. 2016, 68, 92–102. [Google Scholar] [CrossRef]
- Nakajima, T.; Goek, O.; Zhang, X.Y.; Kopecky, S.L.; Frye, R.L.; Goronzy, J.J.; Weyand, C.M. De novo expression of killer immunoglobulin-like receptors and signaling proteins regulates the cytotoxic function of CD4 T cells in acute coronary syndromes. Circ. Res. 2003, 93, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, I.E.; Baruah, P.; Finlayson, C.J.; Loftus, I.M.; Antunes, R.F.; Lim, P.; Bunce, N.; Kaski, J.C. High Levels of Costimulatory Receptors OX40 and 4-1BB Characterize CD4 (+) CD28 (null) T Cells in Patients with Acute Coronary Syndrome. Circ. Res. 2010, 110, 857–869. [Google Scholar] [CrossRef]
- López-Mejías, R.; Castañeda, S.; González-Juanatey, C.; Corrales, A.; Ferraz-Amaro, I.; Genre, F.; Remuzgo-Martínez, S.; Rodriguez-Rodriguez, L.; Blanco, R.; Llorca, J.; et al. Cardiovascular risk assessment in patients with rheumatoid arthritis: The relevance of clinical, genetic and serological markers. Autoimmun. Rev. 2016, 15, 1013–1030. [Google Scholar] [CrossRef]
- Liuzzo, G.; Biasucci, L.M.; Brugaletta, S.; Digianuario, G.; Pinnelli, M.; Giubilato, G.; Giubilato, S.; Colafrancesco, V.; Rebuzzi, A.G.; Crea, F. An unusual population of T-lymphocytes, (CD4+CD28null) T-cells, is associated with the recurrence of acute coronary events in patients with unstable angina. Circulation 2005, 112, U586. [Google Scholar]
- Libby, P. Inflammatory Mechanisms: The Molecular Basis of Inflammation and Disease. Nutr. Rev. 2007, 65, 140–146. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.L.; Shah, T.; Sofat, R.; Guo, Y.R.; Chung, C.; Peasey, A.; Ster, R.P.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [PubMed]
- Kaptoge, S.; Seshasai, S.R.K.; Jørgensen, T.; Danesh, J.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef]
- van den Oever, I.A.M.; Sattar, N.; Nurmohamed, M.T. Thromboembolic and cardiovascular risk in rheumatoid arthritis: Role of the haemostatic system. Ann. Rheum. Dis. 2014, 73, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Small, H.Y.; Migliarino, S.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Hypertension: Focus on autoimmunity and oxidative stress. Free Radic. Biol. Med. 2018, 125, 104–115. [Google Scholar] [CrossRef]
- Peters, M.J.L.; Nurmohamed, M.T.; van Eijk, I.C.; Verkleij, C.J.N.; Marx, P.F. Thrombin-activatable fibrinolysis inhibitor and its relation with inflammation in rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 1232–1233. [Google Scholar] [CrossRef] [PubMed]
- Habets, K.L.; Trouw, L.A.; Levarht, E.N.; Korporaal, S.J.; Habets, P.A.; de Groot, P.; Huizinga, T.W.; Toes, R.E. Anti-citrullinated protein antibodies contribute to platelet activation in rheumatoid arthritis. Thromb. Haemost. 2015, 17, 209. [Google Scholar] [CrossRef]
- Zhou, Z.W.; Chen, H.M.; Ju, H.X.; Sun, M.Z.; Jin, H. Platelet indices in patients with chronic inflammatory arthritis: A systematic review and meta-analysis. Platelets 2020, 31, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Agca, R.; Hopman, L.H.; Laan, K.J.; van Halm, V.P.; Peters, M.J.; Smulders, Y.M.; Dekker, J.M.; Nijpels, G.; Stehouwer, C.D.; Voskuyl, A.E.; et al. Cardiovascular Event Risk in Rheumatoid Arthritis Compared with Type 2 Diabetes: A 15-year Longitudinal Study. J. Rheumatol. 2020, 47, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Cugno, M.; Marzano, A.V.; Asero, R.; Tedeschi, A. Activation of blood coagulation in chronic urticaria: Pathophysiological and clinical implications. Intern. Emerg. Med. 2010, 5, 97–101. [Google Scholar] [CrossRef]
- Choy, E.; Ganeshalingam, K.; Semb, A.G.; Szekanecz, Z.; Nurmohamed, M. Cardiovascular risk in rheumatoid arthritis: Recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology 2014, 53, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Roger, V.L.; Fitz-Gibbon, P.D.; Therneau, T.M.; Gabriel, S.E. Lipid paradox in rheumatoid arthritis: The impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann. Rheum. Dis. 2011, 70, 482–487. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Goodson, N.J.; Brookhart, A.M.; Symmons, D.P.M.; Silman, A.J.; Solomon, D.H. Non-steroidal anti-inflammatory drug use does not appear to be associated with increased cardiovascular mortality in patients with inflammatory polyarthritis: Results from a primary care based inception cohort of patients. Ann. Rheum. Dis. 2009, 68, 367–372. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Gonzalez-Juanatey, C.; Martin, J. Rheumatoid Arthritis: A Disease Associated with Accelerated Atherogenesis. Semin. Arthritis Rheum. 2005, 35, 8–17. [Google Scholar] [CrossRef]
- Kerola, A.M.; Kerola, T.; Kauppi, M.J.; Kautiainen, H.; Virta, L.J.; Puolakka, K.; Nieminen, T.V. Cardiovascular comorbidities antedating the diagnosis of rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 1826–1829. [Google Scholar] [CrossRef]
- Södergren, A.; Karp, K.; Boman, K.; Eriksson, C.; Lundström, E.; Smedby, T.; Söderlund, L.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S. Atherosclerosis in early rheumatoid arthritis: Very early endothelial activation and rapid progression of intima media thickness. Arthritis Res. Ther. 2010, 12, R158. [Google Scholar] [CrossRef]
- González-Gay, M.A.; González-Juanatey, C.; Miranda-Filloy, J.A.; García-Unzueta, M.T.; Llorca, J. Lack of association between flow-mediated endothelium-dependent vasodilatation and biomarkers of endothelial dysfunction in patients with severe rheumatoid arthritis. Rheumatol. Int. 2012, 32, 4071–4072. [Google Scholar] [CrossRef] [PubMed]
- Maga, M.; Laczak, P.; Kaczmarczyk, P.; Wandzilak, M.; Maga, P. Images in Vascular Medicine: Successful endovascular treatment of psoriasis-induced critical limb ischemia. Vasc. Med. 2021, 26, 350–351. [Google Scholar] [CrossRef]
- Di Minno, M.N.D.; Ambrosino, P.; Lupoli, R.; Di Minno, A.; Tasso, M.; Peluso, R.; Tremoli, E. Clinical assessment of endothelial function in patients with rheumatoid arthritis: A meta-analysis of literature studies. Eur. J. Intern. Med. 2015, 26, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Tasso, M.; Lupoli, R.; Di Minno, A.; Baldassarre, D.; Tremoli, E.; Di Minno, M.N.D. Non-invasive assessment of arterial stiffness in patients with rheumatoid arthritis: A systematic review and meta-analysis of literature studies. Ann. Med. 2015, 47, 457–467. [Google Scholar] [CrossRef]
- Paccou, J.; Renard, C.; Liabeuf, S.; Kamel, S.; Fardellone, P.; Massy, Z.A.; Brazier, M.; Mentaverri, R. Coronary and Abdominal Aorta Calcification in Rheumatoid Arthritis: Relationships with Traditional Cardiovascular Risk Factors, Disease Characteristics, and Concomitant Treatments. J. Rheumatol. 2014, 41, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.; Henry, D. Cardiovascular risk and inhibition of cyclooxygenase—A systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase. JAMA—J. Am. Med. Assoc. 2006, 296, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Kearney, P.M.; Baigent, C.; Godwin, J.; Halls, H.; Emberson, J.R.; Patrono, C. Faculty Opinions recommendation of Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ-Br. Med. J. 2006, 332, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Roubille, C.; Richer, V.; Starnino, T.; McCourt, C.; McFarlane, A.; Fleming, P.; Siu, S.; Kraft, J.; Lynde, C.; Pope, J.; et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 480–489. [Google Scholar] [CrossRef]
- Cabassi, A.; Tedeschi, S.; Perlini, S.; Verzicco, I.; Volpi, R.; Gonzi, G.; Del, S. Canale Non-steroidal anti-inflammatory drug effects on renal and cardiovascular function: From physiology to clinical practice. Eur. J. Prev. Cardiol. 2020, 27, 850–867. [Google Scholar] [CrossRef]
- Gargiulo, G.; Capodanno, D.; Longo, G.; Capranzano, P.; Tamburino, C. Updates on NSAIDs in patients with and without coronary artery disease: Pitfalls, interactions and cardiovascular outcomes. Expert Rev. Cardiovasc. Ther. 2014, 12, 1185–1203. [Google Scholar] [CrossRef]
- Gasparyan, A.Y.; Ayvazyan, L.; Cocco, G.; Kitas, G.D. Adverse Cardiovascular Effects of Antirheumatic Drugs: Implications for Clinical Practice and Research. Curr. Pharm. Des. 2012, 18, 1543–1555. [Google Scholar] [CrossRef]
- Szeto, C.-C.; Sugano, K.; Wang, J.-G.; Fujimoto, K.; Whittle, S.; Modi, G.K.; Chen, C.-H.; Park, J.-B.; Tam, L.-S.; Vareesangthip, K.; et al. Non-steroidal anti-inflammatory drug (NSAID) therapy in patients with hypertension, cardiovascular, renal or gastrointestinal comorbidities: Joint APAGE/APLAR/APSDE/APSH/APSN/PoA recommendations. Gut 2020, 69, 617–629. [Google Scholar] [CrossRef]
- Zheng, L.; Du, X. Non-steroidal Anti-inflammatory Drugs and Hypertension. Cell Biochem. Biophys. 2014, 69, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, B.; Aldington, S.; Weatherall, M.; Shirtcliffe, P.; Beasley, R. Risk of cardiovascular events and celecoxib: A systematic review and meta-analysis. J. R. Soc. Med. 2006, 99, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Helin-Salmivaara, A.; Virtanen, A.; Vesalainen, R.; Grönroos, J.M.; Klaukka, T.; Idänpään-Heikkilä, J.E.; Huupponen, R. NSAID use and the risk of hospitalization for first myocardial infarction in the general population: A nationwide case-control study from Finland. Eur. Heart J. 2006, 27, 1657–1663. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schneeweiss, S.; Solomon, D.H.; Wang, P.S.; Rassen, J.; Brookhart, M.A. Simultaneous assessment of short-term gastrointestinal benefits and cardiovascular risks of selective cyclooxygenase 2 inhibitors and nonselective nonsteroidal anti-inflammatory drugs—An instrumental variable analysis. Arthritis Rheum. 2006, 54, 3390–3398. [Google Scholar] [CrossRef]
- Fabule, J.; Adebajo, A. Comparative evaluation of cardiovascular outcomes in patients with osteoarthritis and rheumatoid arthritis on recommended doses of nonsteroidal anti-inflammatory drugs. Ther. Adv. Musculoskelet. Dis. 2014, 6, 111–130. [Google Scholar] [CrossRef]
- del Rincón, I.; Battafarano, D.F.; Restrepo, J.F.; Erikson, J.M.; Escalante, A. Glucocorticoid Dose Thresholds Associated With All-Cause and Cardiovascular Mortality in Rheumatoid Arthritis. Arthritis Rheumatol. 2014, 66, 264–272. [Google Scholar] [CrossRef]
- Soubrier, M.; Chamoux, N.B.; Tatar, Z.; Couderc, M.; Dubost, J.J.; Mathieu, S. Cardiovascular risk in rheumatoid arthritis. Jt. Bone Spine 2014, 81, 298–302. [Google Scholar] [CrossRef]
- Ravindran, V.; Rachapalli, S.; Choy, E.H. Safety of medium- to long-term glucocorticoid therapy in rheumatoid arthritis: A meta-analysis. Rheumatology 2009, 48, 807–811. [Google Scholar] [CrossRef]
- Ruyssen-Witrand, A.; Fautrel, B.; Saraux, A.; Le Loët, X.; Pham, T. Cardiovascular risk induced by low-dose corticosteroids in rheumatoid arthritis: A systematic literature review. Jt. Bone Spine 2011, 78, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Agca, R.; Heslinga, S.C.; Rollefstad, S.; Heslinga, M.; McInnes, B.; Peters, M.J.L.; Kvien, T.K.; Dougados, M.; Radner, H.; Atzeni, F.; et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann. Rheum. Dis. 2017, 76, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S.; Bernatsky, S.; Hudson, M. Antirheumatic drug use and the risk of acute myocardial infarction. Arthritis Rheum. 2006, 55, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Rempenault, C.; Combe, B.; Barnetche, T.; Gaujoux-Viala, C.; Lukas, C.; Morel, J.; Hua, C. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2018, 77, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Widdifield, J.; Abrahamowicz, M.; Paterson, J.M.; Huang, A.; Thorne, J.C.; Pope, J.E.; Kuriya, B.; Beauchamp, M.-E.; Bernatsky, S. Associations Between Methotrexate Use and the Risk of Cardiovascular Events in Patients with Elderly-onset Rheumatoid Arthritis. J. Rheumatol. 2019, 46, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Westlake, S.L.; Colebatch, A.N.; Baird, J.; Curzen, N.; Kiely, P.; Quinn, M.; Choy, E.; Ostor, A.J.K.; Edwards, C.J. Tumor necrosis factor antagonists and the risk of cardiovascular disease in patients with rheumatoid arthritis: A systematic literature review. Rheumatology 2011, 50, 518–531. [Google Scholar] [CrossRef]
- Micha, R.; Imamura, F.; von Ballmoos, M.W.; Solomon, D.H.; Hernán, M.A.; Ridker, P.M.; Mozaffarian, D. Systematic Review and Meta-Analysis of Methotrexate Use and Risk of Cardiovascular Disease. Am. J. Cardiol. 2011, 108, 1362–1370. [Google Scholar] [CrossRef]
- Morris, S.J.; Wasko, M.C.M.; Antohe, J.L.; Sartorius, J.A.; Kirchner, H.L.; Dancea, S.; Bili, A. Hydroxychloroquine use associated with improvement in lipid profiles in rheumatoid arthritis patients. Arthritis Care Res. 2011, 63, 530–534. [Google Scholar] [CrossRef]
- Li, H.-Z.; Xu, X.-H.; Lin, N.; Lu, H.-D. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2019, 78, e21. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Wang, X.; Lee, Y.Y.; Shahbazian, A.; Navarro-Millán, I.; Yang, S.; Chen, L.; Cofield, S.S.; Moreland, L.W.; O’Dell, J.; et al. Association of Triple Therapy With Improvement in Cholesterol Profiles Over Two-Year Followup in the Treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis Rheumatol. 2016, 68, 577–586. [Google Scholar] [CrossRef]
- Solomon, D.H.; Avorn, J.; Katz, J.N.; Weinblatt, M.E.; Setoguchi, S.; Levin, R.; Schneeweiss, S. Immunosuppressive medications and hospitalization for cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum. 2006, 54, 3790–3798. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.; Breedveld, F.C.; Buch, M.; Burmester, G.; Dougados, M.; Emery, P.; Gaujoux-Viala, C.; Gossec, L.; Nam, J.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann. Rheum. Dis. 2014, 73, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Curtis, J.R.; Saag, K.G.; Lii, J.; Chen, L.; Harrold, L.R.; Herrinton, L.J.; Graham, D.J.; Kowal, M.K.; Kuriya, B.; et al. Cardiovascular Risk in Rheumatoid Arthritis: Comparing TNF-α Blockade with Nonbiologic DMARDs. Am. J. Med. 2013, 126, 730.e9–730.e17. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E. Effects of TNF alpha inhibitors on adiposity and other cardiovascular risk factors: Implications for the cardiovascular prognosis in patients with rheumatoid arthritis. Expert Opin. Drug Saf. 2015, 14, 525–532. [Google Scholar] [CrossRef]
- Barnabe, C.; Martin, B.-J.; Ghali, W.A. Systematic review and meta-analysis: Anti-tumor necrosis factor α therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res. 2011, 63, 522–529. [Google Scholar] [CrossRef]
- Jacobsson, L.T.H.; Turesson, C.; Gülfe, A.; Kapetanovic, M.C.; Petersson, I.F.; Saxne, T.; Geborek, P. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J. Rheumatol. 2005, 32, 1213–1218. [Google Scholar]
- Ljung, L.; Rantapää-Dahlqvist, S.; Jacobsson, L.T.H.; Askling, J. Response to biological treatment and subsequent risk of coronary events in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 2087–2094. [Google Scholar] [CrossRef]
- Karpouzas, G.A.; Ormseth, S.R.; Hernandez, E.; Budoff, M.J. Impact of Cumulative Inflammation, Cardiac Risk Factors, and Medication Exposure on Coronary Atherosclerosis Progression in Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 400–408. [Google Scholar] [CrossRef]
- Singh, S.; Fumery, M.; Singh, A.G.; Singh, N.; Prokop, L.J.; Dulai, P.S.; Sandborn, W.J.; Curtis, J.R. Comparative Risk of Cardiovascular Events With Biologic and Synthetic Disease-Modifying Antirheumatic Drugs in Patients With Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Arthritis Care Res. 2020, 72, 561–576. [Google Scholar] [CrossRef]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T.; Investigators, A. Randomized, double-blind, placebo-controlled, trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure—Results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar]
- Leporini, C.; Russo, E.; D`angelo, S.; Arturi, F.; Tripepi, G.; Peluso, R.; Grembiale, R.D.; Olivieri, I.; De Sarro, G.; Ursini, F. Insulin-Sensiting Effects of Tumor Necrosis Factor Alpha Inhibitors in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Rev. Recent Clin. Trials 2018, 13, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Castagné, B.; Viprey, M.; Martin, J.; Schott, A.-M.; Cucherat, M.; Soubrier, M. Cardiovascular safety of tocilizumab: A systematic review and network meta-analysis. PLoS ONE 2019, 14, e0220178. [Google Scholar] [CrossRef] [PubMed]
- Divonne, M.d.L.F.; Gottenberg, J.E.; Salliot, C. Safety of biologic DMARDs in RA patients in real life: A systematic literature review and meta-analyses of biologic registers. Jt. Bone Spine 2017, 84, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.; Tsoi, M.; Cheung, B. SAT0099 Effect of TNF Inhibitors on Subclinical Atherosclerosis in Patients with Rheumatoid Arthritis: A Meta-Analysis. Ann. Rheum. Dis. 2015, 74, 685. [Google Scholar] [CrossRef]
- Daien, C.I.; Duny, Y.; Barnetche, T.; Daures, J.P.; Combe, B.; Morel, J. Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: A systematic review with meta-analysis. Ann. Rheum. Dis. 2012, 71, 862–868. [Google Scholar] [CrossRef]
- Zhao, Q.W.; Hong, D.S.; Zhang, Y.; Sang, Y.L.; Yang, Z.H.; Zhang, X.G. Association Between Anti-TNF Therapy for Rheumatoid Arthritis and Hypertension A Meta-Analysis of Randomized Controlled Trials. Medicine 2015, 94, e731. [Google Scholar] [CrossRef]
- Kim, S.K.; Kwak, S.G.; Choe, J.Y. Association between biologic disease modifying antirheumatic drugs and incident hypertension in patients with rheumatoid arthritis Results from prospective nationwide KOBIO Registry. Medicine 2020, 99, e19415. [Google Scholar] [CrossRef]
- Desai, R.J.; Solomon, D.H.; Schneeweiss, S.; Danaei, G.; Liao, K.P.; Kim, S.C. Tumor Necrosis Factor-α Inhibitor Use and the Risk of Incident Hypertension in Patients with Rheumatoid Arthritis. Epidemiology 2016, 27, 414–422. [Google Scholar] [CrossRef]
- Jin, Y.; Kang, E.H.; Brill, G.; Desai, R.J.; Kim, S.C. Cardiovascular (CV) Risk after Initiation of Abatacept versus TNF Inhibitors in Rheumatoid Arthritis Patients with and without Baseline CV Disease. J. Rheumatol. 2018, 45, 1240–1248. [Google Scholar] [CrossRef]
- Schiff, M.H.; Kremer, J.M.; Jahreis, A.; Vernon, E.; Isaacs, J.D.; van Vollenhoven, R.F. Integrated safety in tocilizumab clinical trials. Thromb. Haemost. 2011, 13, R141. [Google Scholar] [CrossRef]
- Fleischmann, R.; Lin, Y.; John, G.S.; van der Heijde, D.; Qiu, C.; Gomez-Reino, J.J.; Maldonado-Cocco, J.A.; Stanislav, M.; Seriolo, B.; Burmester, G.R. SAT0125 Long-Term Safety with Sarilumab Plus Conventional Synthetic Disease-Modifying Antirheumatic Drugs and Sarilumab Monotherapy in Rheumatoid Arthritis: An Integrated Analysis with 9000 Patient-Years of Follow-Up. Ann. Rheum. Dis. 2019, 78, 1130–1131. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Lekakis, J.P.; Nikolaou, M.; Paraskevaidis, I.; Andreadou, I.; Kaplanoglou, T.; Katsimbri, P.; Skarantavos, G.; Soucacos, P.N.; Kremastinos, D.T. Inhibition of Interleukin-1 by Anakinra Improves Vascular and Left Ventricular Function in Patients With Rheumatoid Arthritis. Circulation 2008, 117, 2662–2669. [Google Scholar] [CrossRef] [PubMed]
- van Vollenhoven, R.F.; Emery, P.; O Bingham, C.; Keystone, E.C.; Fleischmann, R.M.; E Furst, D.; Tyson, N.; Collinson, N.; Lehane, P.B. Long-term safety of rituximab in rheumatoid arthritis: 9.5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann. Rheum. Dis. 2013, 72, 1496–1502. [Google Scholar] [CrossRef]
- Day, A.L.; Singh, J.A. Cardiovascular Disease Risk in Older Adults and Elderly Patients with Rheumatoid Arthritis: What Role Can Disease-Modifying Antirheumatic Drugs Play in Cardiovascular Risk Reduction? Drugs Aging 2019, 36, 493–510. [Google Scholar] [CrossRef]
- McInnes, I.B.; Thompson, L.; Giles, J.T.; Bathon, J.M.; E Salmon, J.; Beaulieu, A.D.; E Codding, C.; Carlson, T.H.; Delles, C.; Lee, J.S.; et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann. Rheum. Dis. 2015, 74, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Suzuki, M.; Nakamura, H.; Toyoizumi, S.; Zwillich, S.H. Tofacitinib Study Investigators Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res. 2011, 63, 1150–1158. [Google Scholar] [CrossRef]
- Taylor, P.C.; Weinblatt, M.E.; Burmester, G.R.; Rooney, T.P.; Witt, S.; Walls, C.D.; Issa, M.; Salinas, C.A.; Saifan, C.; Zhang, X.; et al. Cardiovascular Safety During Treatment With Baricitinib in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1042–1055. [Google Scholar] [CrossRef]
- Souto, A.; Salgado, E.; Maneiro, J.R.; Mera, A.; Carmona, L.; Gómez-Reino, J.J. Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: A systematic review and meta-analysis. Arthritis Rheumatol. 2015, 67, 117–127. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, F.; Yun, H.; Chen, L.; Muntner, P.; Levitan, E.B.; Safford, M.M.; Kent, S.T.; Osterman, M.T.; Lewis, J.D.; et al. Comparative effects of biologics on cardiovascular risk among older patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1813–1818. [Google Scholar] [CrossRef]
- Myasoedova, E.; Karmacharya, P.; Garcia, A.D.; Davis, J.; Murad, M.H.; Crowson, C. Effect of statin use on the risk of rheumatoid arthritis: A systematic review and meta-analysis. Semin Arthritis Rheum. 2020, 50, 1348–1356. [Google Scholar] [CrossRef]
- Soulaidopoulos, S.; Nikiphorou, E.; Dimitroulas, T.; Kitas, G.D. The Role of Statins in Disease Modification and Cardiovascular Risk in Rheumatoid Arthritis. Front. Med. 2018, 5, 24. [Google Scholar] [CrossRef]
- Danninger, K.; Hoppe, U.C.; Pieringer, H. Do statins reduce the cardiovascular risk in patients with rheumatoid arthritis? Int. J. Rheum. Dis. 2014, 17, 606–611. [Google Scholar] [CrossRef]
- Xing, B.; Yin, Y.-F.; Zhao, L.-D.; Wang, L.; Zheng, W.-J.; Chen, H.; Wu, Q.-J.; Tang, F.-L.; Zhang, F.-C.; Shan, G.; et al. Effect of 3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Inhibitor on Disease Activity in Patients with Rheumatoid Arthritis A Meta-Analysis. Medicine 2015, 94, e572. [Google Scholar] [CrossRef] [PubMed]
- Marnell, L.; Mold, C.; Du Clos, T.W. C-reactive protein: Ligands, receptors and role in inflammation. Clin. Immunol. 2005, 117, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Nakada, T.-A.; Hattori, N.; Takahashi, W.; Takahashi, N.; Aizimu, T.; Yoshida, M.; Morizane, T.; Oda, S. Interleukin-6 as a diagnostic marker for infection in critically ill patients: A systematic review and meta-analysis. Am. J. Emerg. Med. 2019, 37, 260–265. [Google Scholar] [CrossRef]
- Shimamoto, K.; Ito, T.; Ozaki, Y.; Amuro, H.; Tanaka, A.; Nishizawa, T.; Son, Y.; Inaba, M.; Nomura, S. Serum Interleukin 6 Before and After Therapy with Tocilizumab Is a Principal Biomarker in Patients with Rheumatoid Arthritis. J. Rheumatol. 2013, 40, 1074–1081. [Google Scholar] [CrossRef]
- Rincón-López, E.M.; Gómez, M.L.N.; Matos, T.H.-S.; Aguilera-Alonso, D.; Moreno, E.D.; Saavedra-Lozano, J.; García, B.S.; Sebastián, M.d.M.S.; Morín, M.G.; Bieler, C.B.; et al. Interleukin 6 as a marker of severe bacterial infection in children with sickle cell disease and fever: A case–control study. BMC Infect. Dis. 2021, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Inam Illahi, M.; Amjad, S.; Alam, S.M.; Ahmed, S.T.; Fatima, M.; Shahid, M.A. Serum tumor necrosis fac-tor-alpha as a competent biomarker for evaluation of disease activity in early rheumatoid arthritis. Cureus 2021, 13, e15314. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bedeković, D.; Bošnjak, I.; Šarić, S.; Kirner, D.; Novak, S. Role of Inflammatory Cytokines in Rheumatoid Arthritis and Development of Atherosclerosis: A Review. Medicina 2023, 59, 1550. https://doi.org/10.3390/medicina59091550
Bedeković D, Bošnjak I, Šarić S, Kirner D, Novak S. Role of Inflammatory Cytokines in Rheumatoid Arthritis and Development of Atherosclerosis: A Review. Medicina. 2023; 59(9):1550. https://doi.org/10.3390/medicina59091550
Chicago/Turabian StyleBedeković, Dražen, Ivica Bošnjak, Sandra Šarić, Damir Kirner, and Srđan Novak. 2023. "Role of Inflammatory Cytokines in Rheumatoid Arthritis and Development of Atherosclerosis: A Review" Medicina 59, no. 9: 1550. https://doi.org/10.3390/medicina59091550
APA StyleBedeković, D., Bošnjak, I., Šarić, S., Kirner, D., & Novak, S. (2023). Role of Inflammatory Cytokines in Rheumatoid Arthritis and Development of Atherosclerosis: A Review. Medicina, 59(9), 1550. https://doi.org/10.3390/medicina59091550