
Bioinformatic Analysis for Influential Core Gene Identification and Prognostic Significance in Advanced Serous Ovarian Carcinoma


Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Data Source and Cluster Analysis
2.2. Screening for DEGs
2.3. GO Analysis of DEGs
2.4. PPI Network Analysis
2.5. DEG Survival Analysis
2.6. Data Source for Analysis of Association with Clinicopathological Parameters
2.7. Statistical Analysis
3. Results
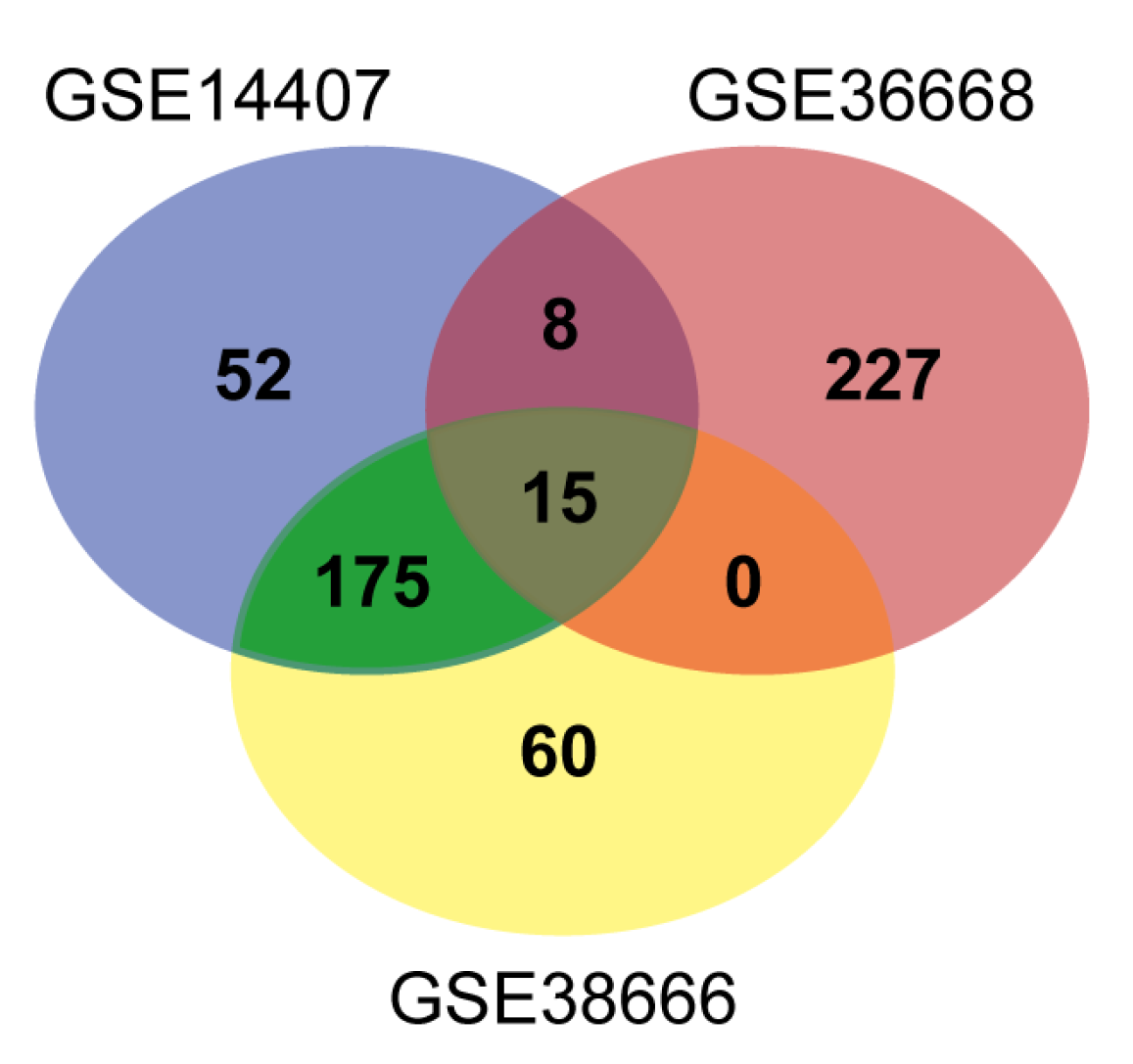
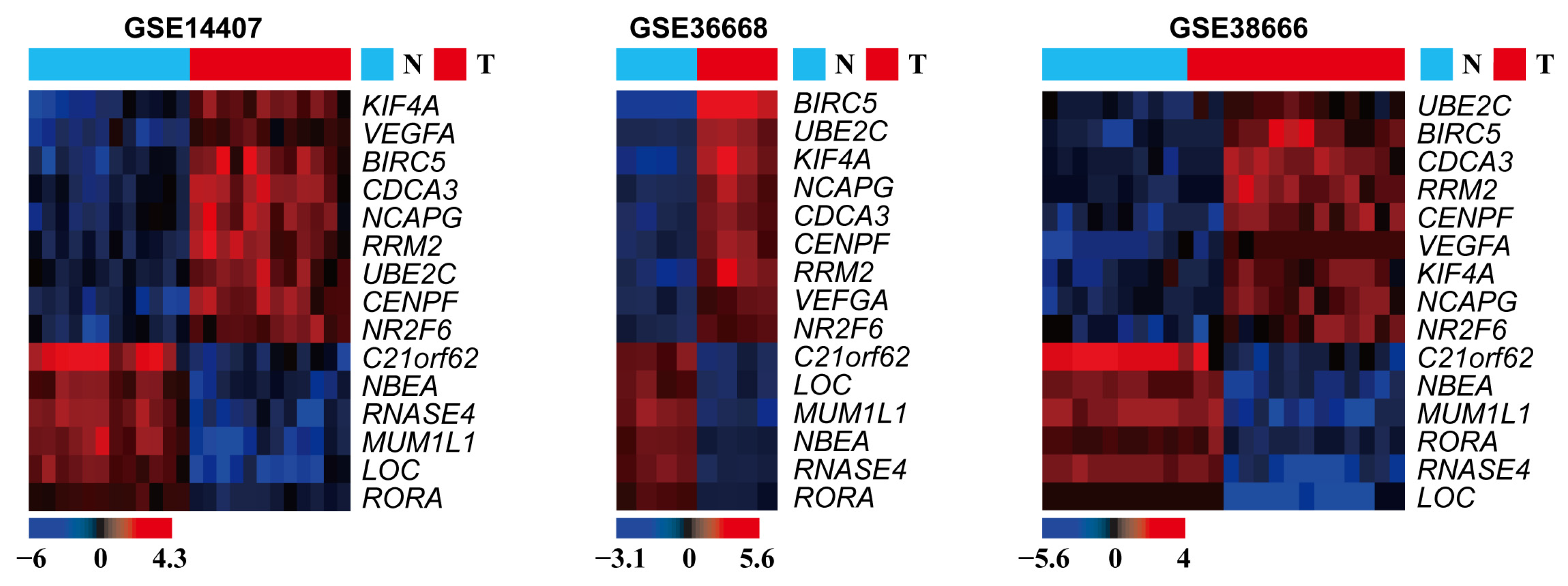
3.1. DEG Identification
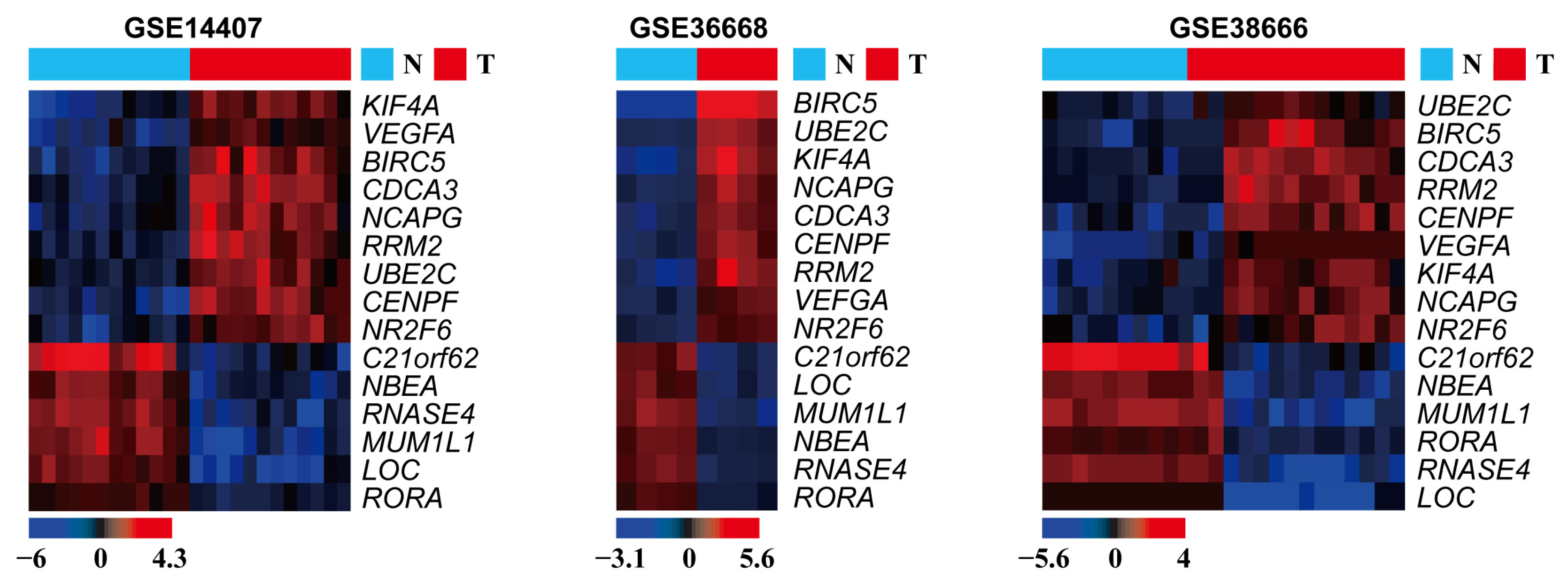
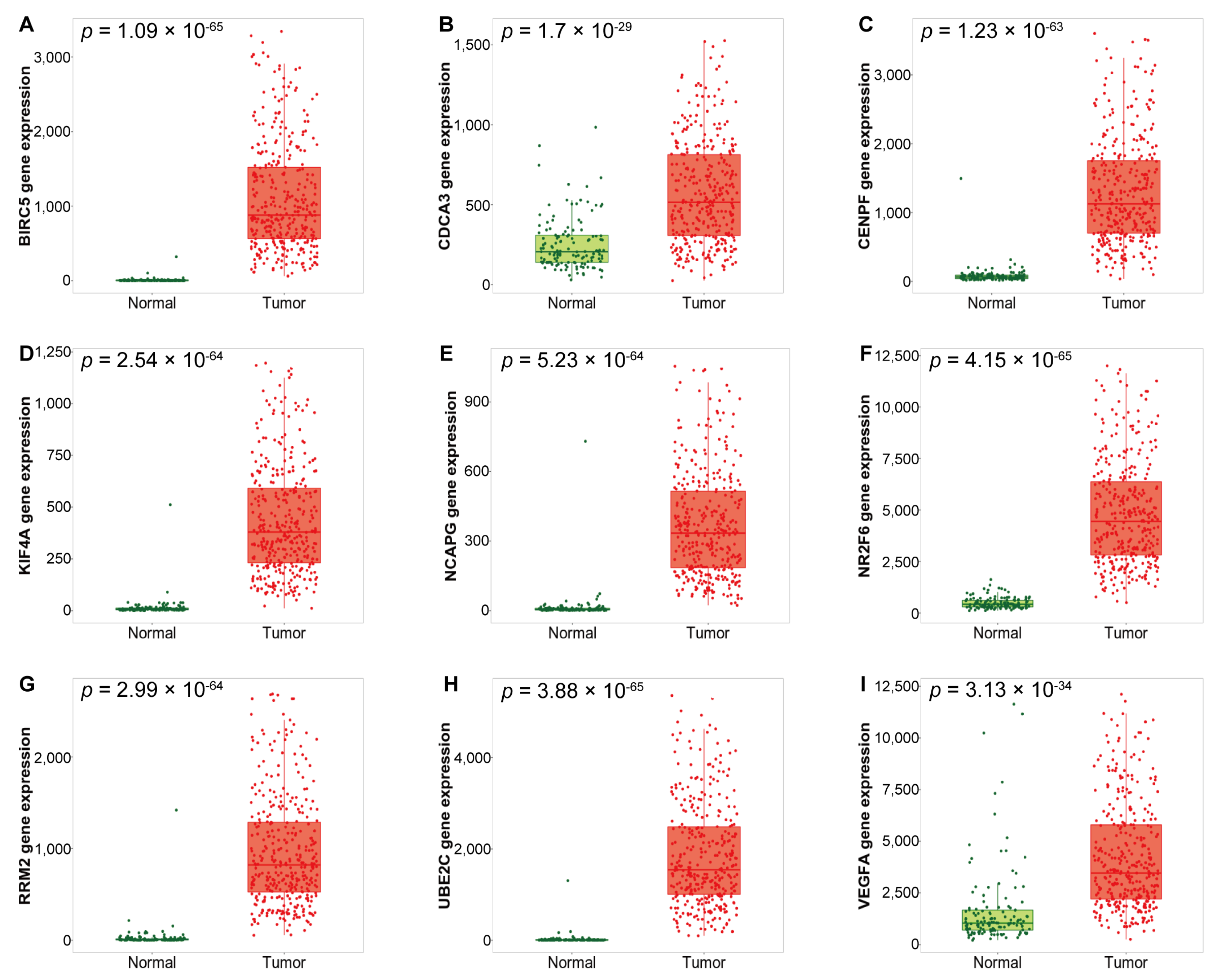
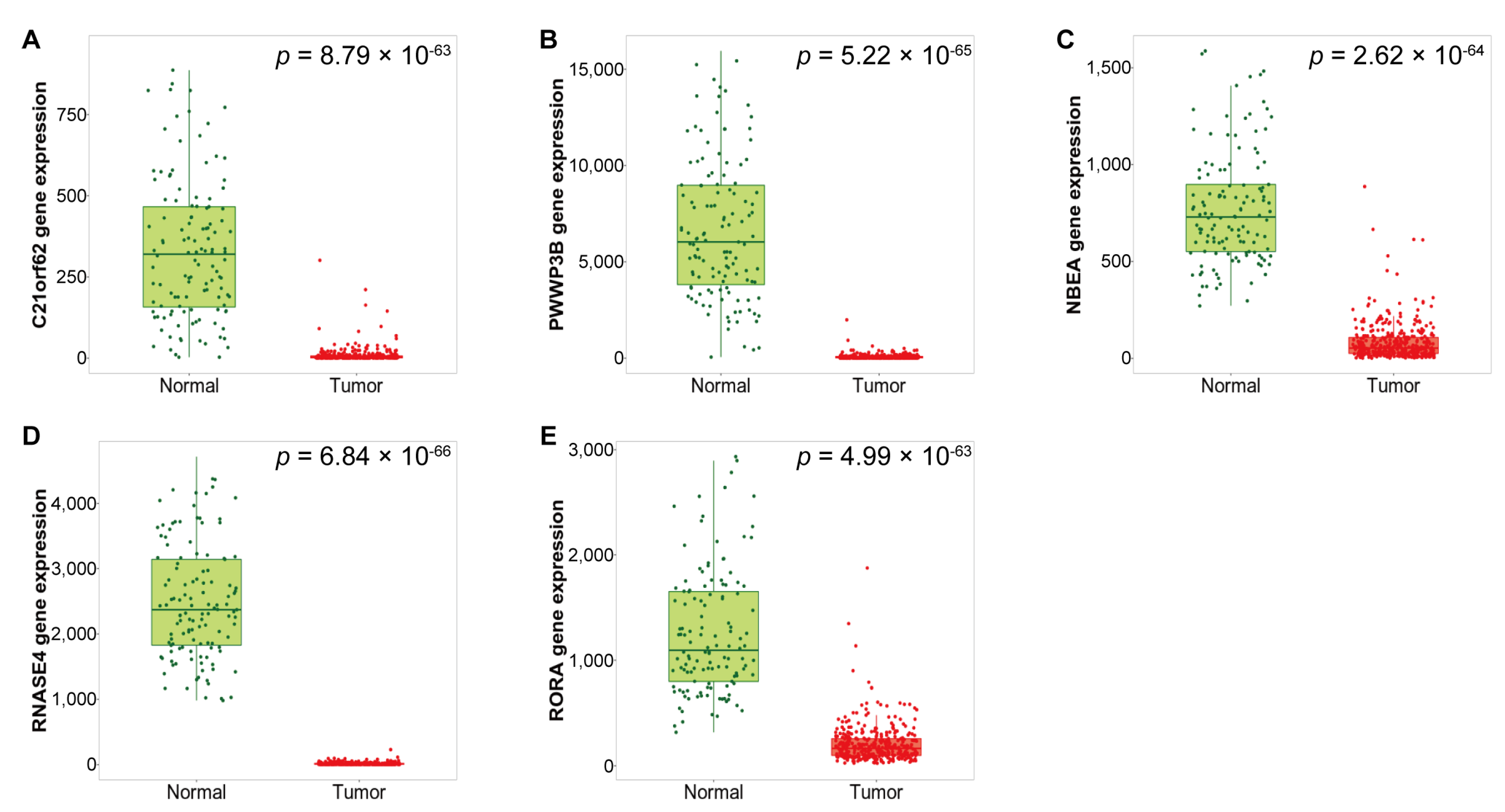
3.2. Expression of the Identified DEGs between Normal and Serous Ovarian Cancer Tissues
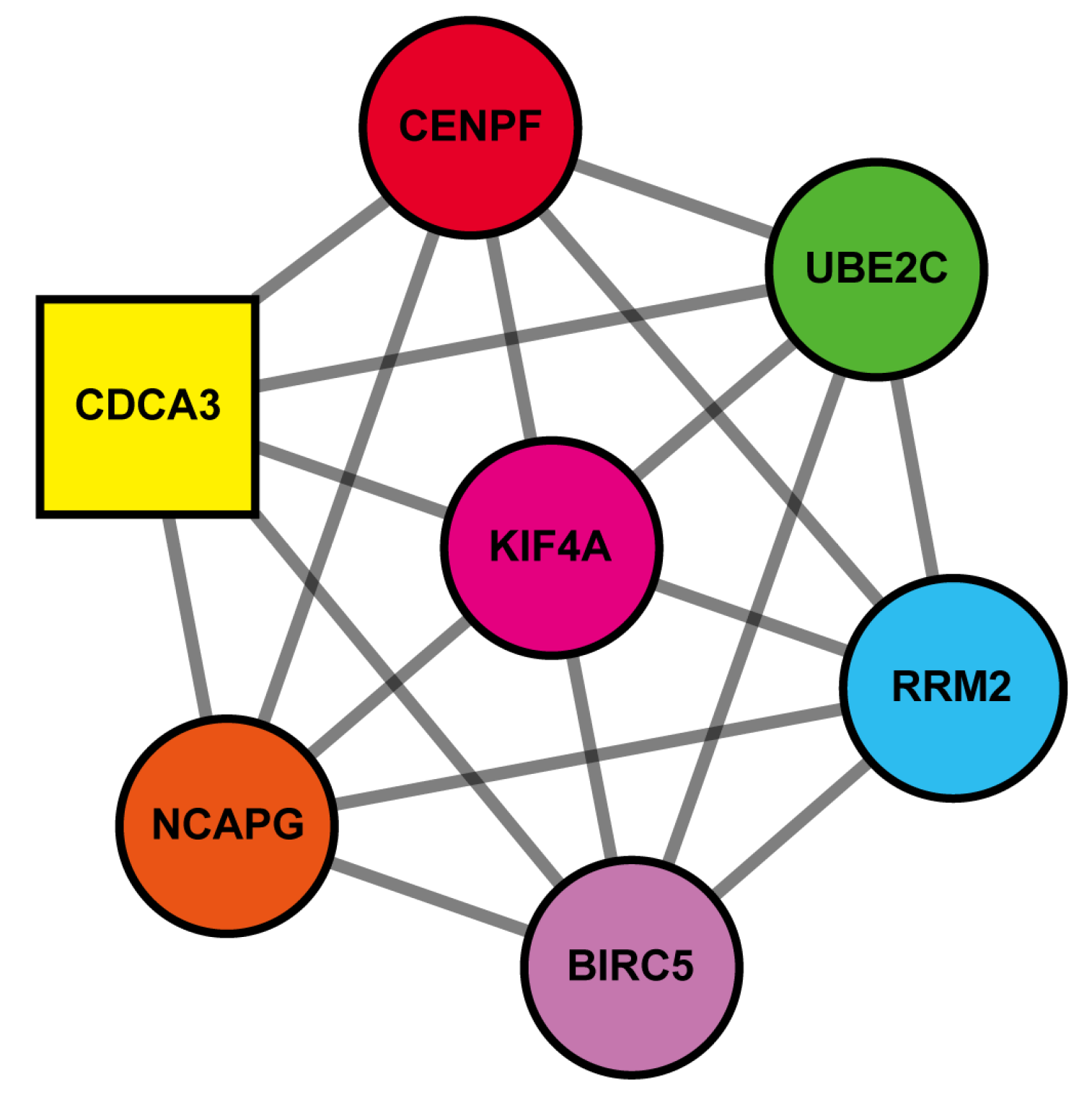
3.3. PPI Network Construction
3.4. Clustered Genes Are Mainly Involved in Cell Cycle Regulation
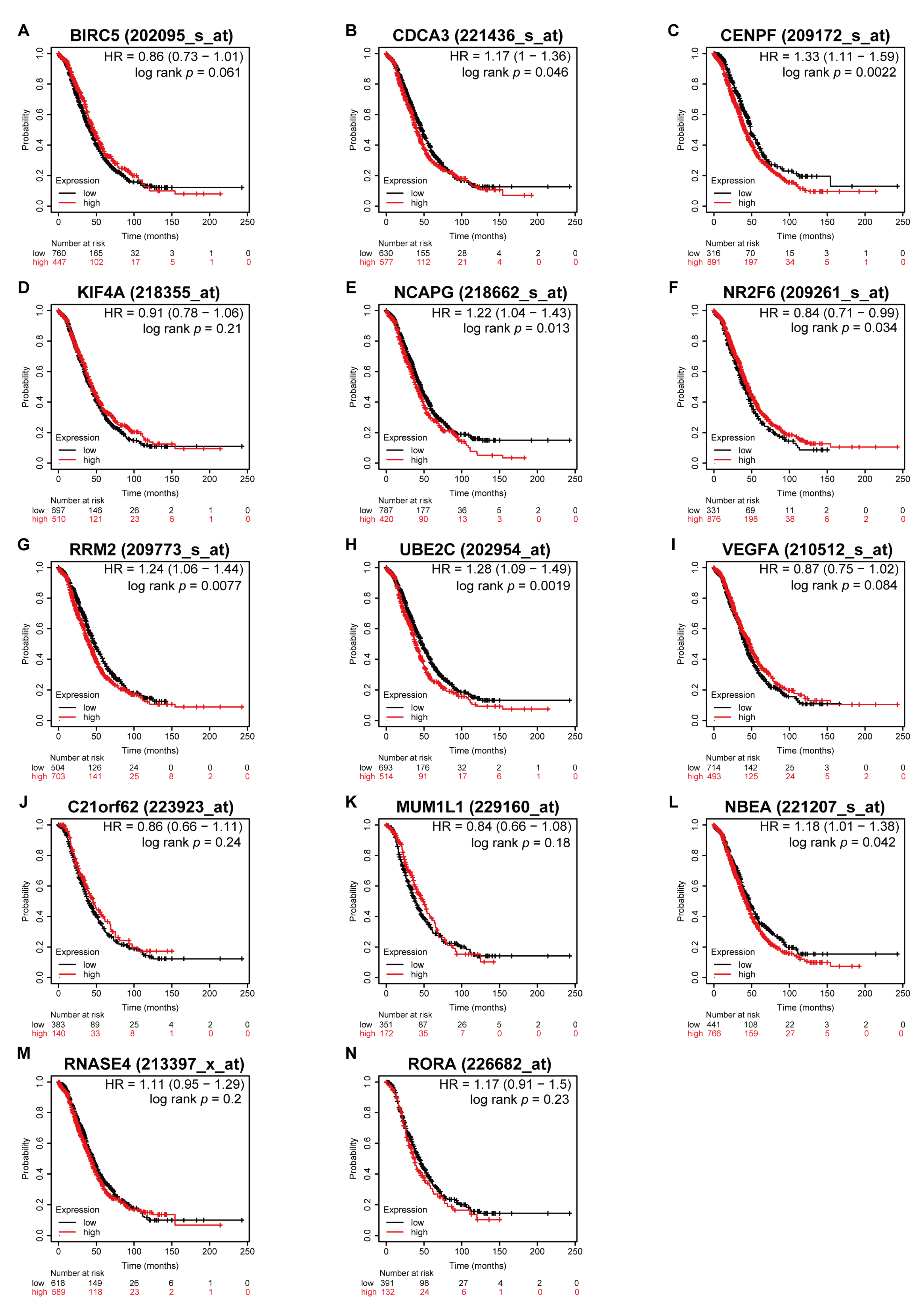
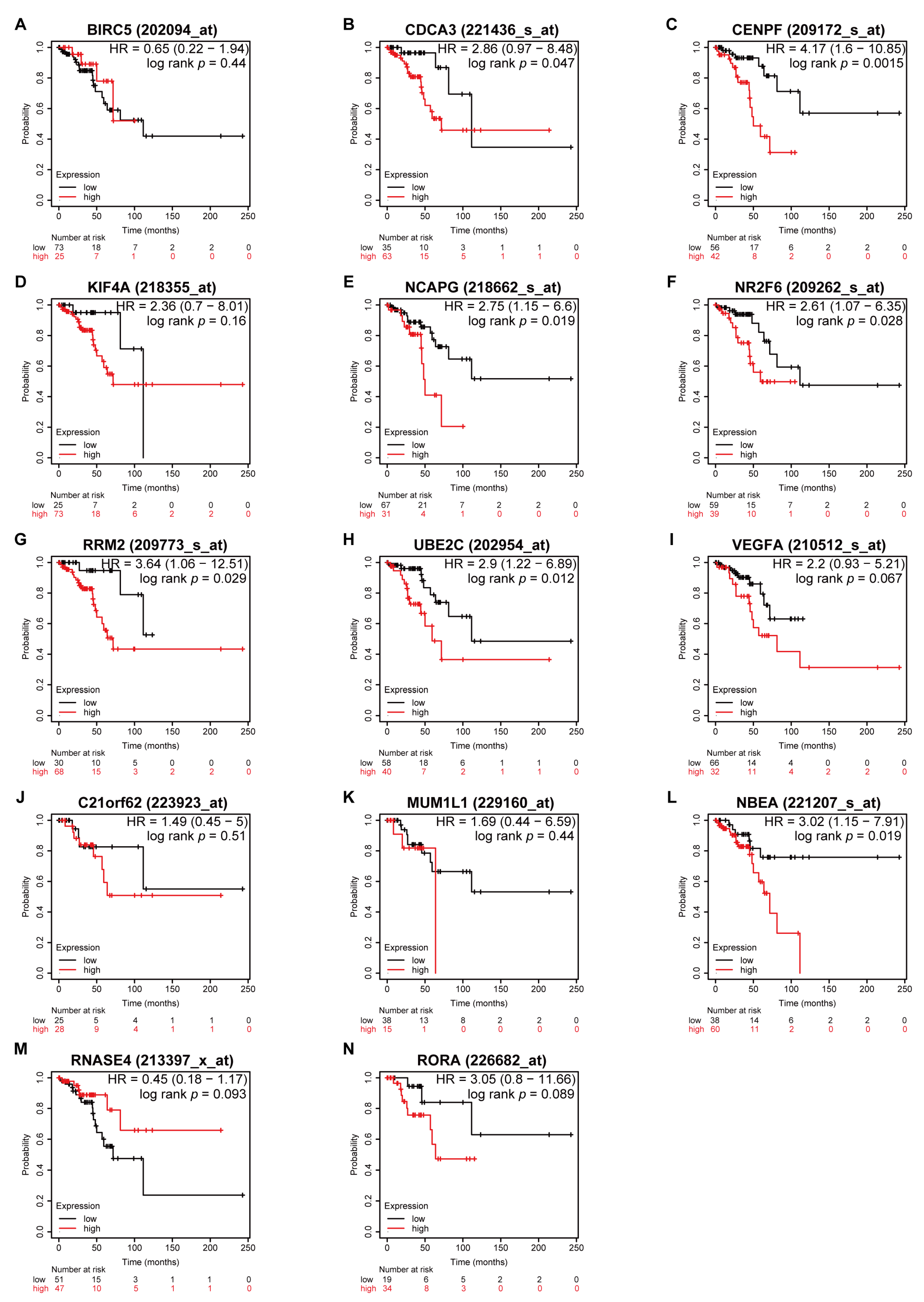
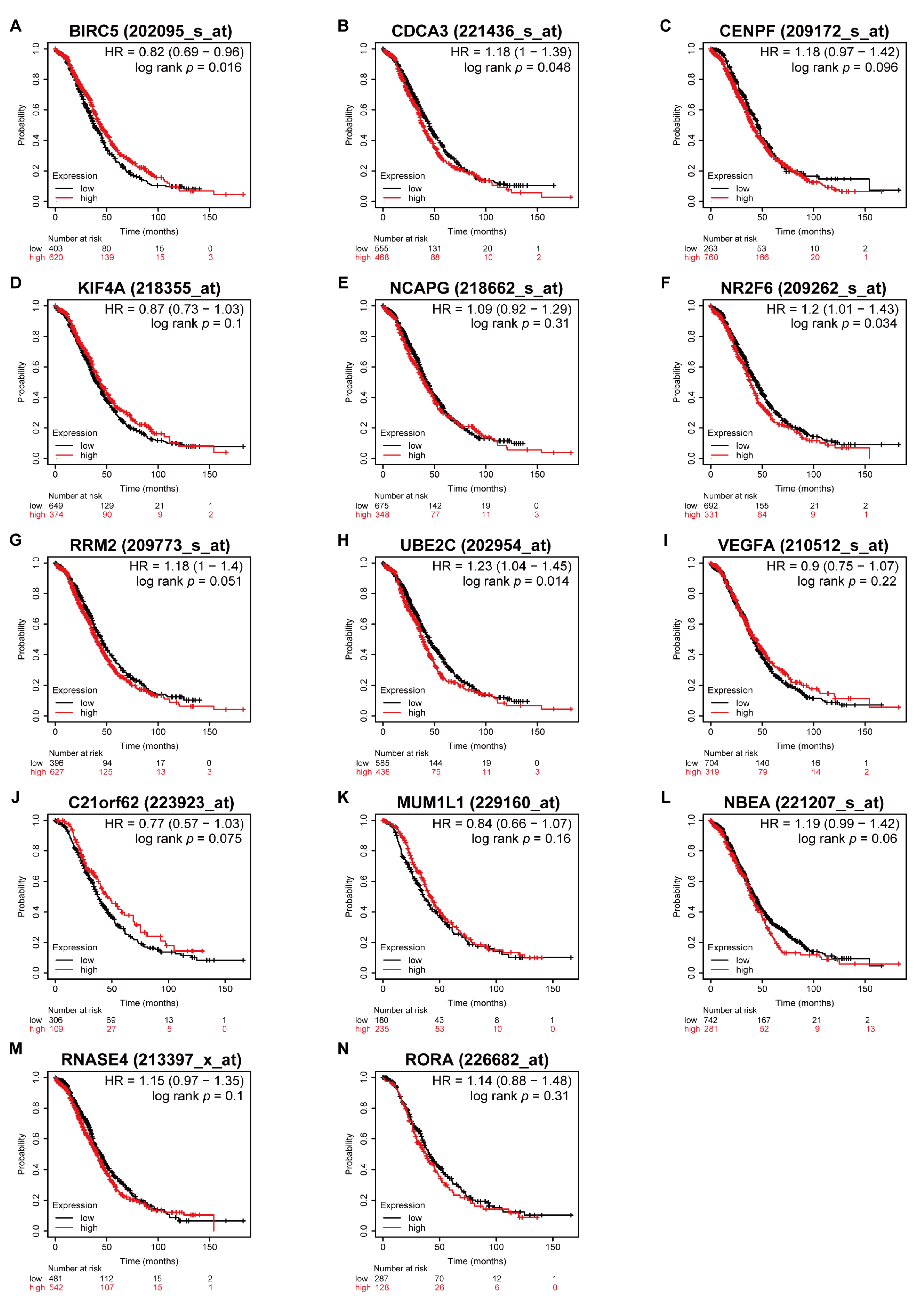
3.5. Higher Expression of CDCA3 and UBE2C Associated with Poor OS in All Stages
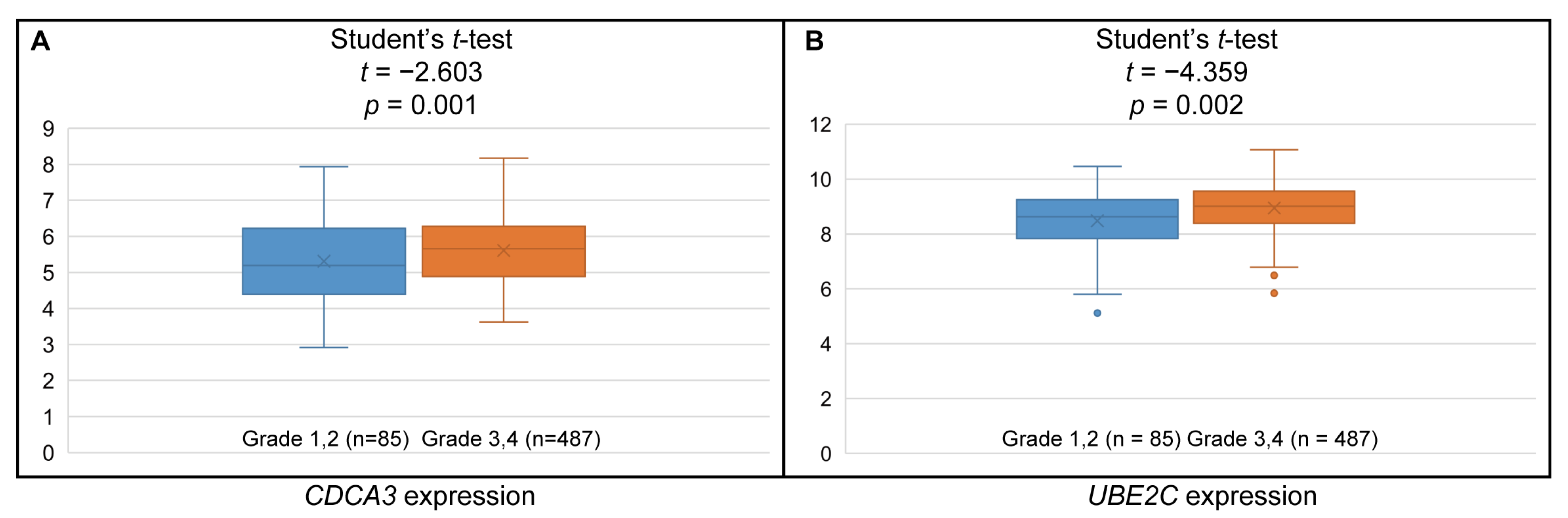
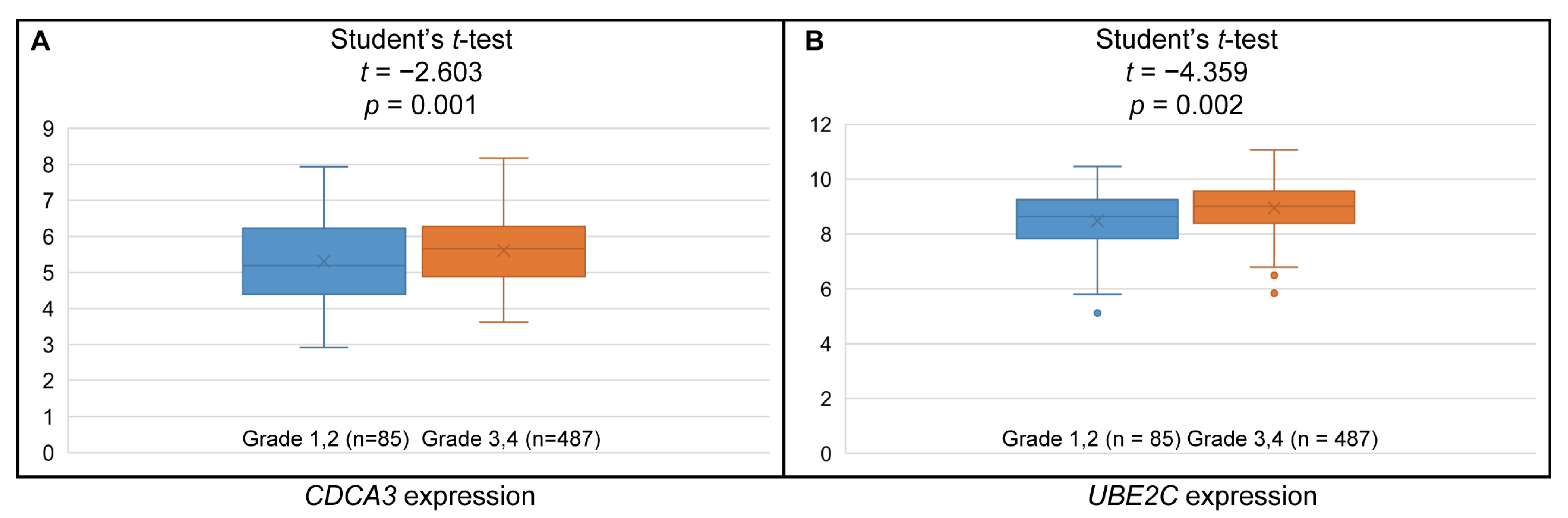
3.6. Higher CDCA3 and UBE2C Expression Associated with Higher Histologic Grades
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Schink, J.C. Current initial therapy of stage III and IV ovarian cancer: Challenges for managed care. Semin. Oncol. 1999, 26, 2–7. [Google Scholar] [PubMed]
- Scaletta, G.; Plotti, F.; Luvero, D.; Capriglione, S.; Montera, R.; Miranda, A.; Lopez, S.; Terranova, C.; De Cicco Nardone, C.; Angioli, R. The role of novel biomarker HE4 in the diagnosis, prognosis and follow-up of ovarian cancer: A systematic review. Expert Rev. Anticancer 2017, 17, 827–839. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, S.; Kim, Y.T.; Lim, M.C.; Lee, B.; Jung, K.W.; Kim, J.W.; Park, S.Y.; Won, Y.J. Changes in ovarian cancer survival during the 20 years before the era of targeted therapy. BMC Cancer 2018, 18, 601. [Google Scholar] [CrossRef] [PubMed]
- Papp, E.; Hallberg, D.; Konecny, G.E.; Bruhm, D.C.; Adleff, V.; Noe, M.; Kagiampakis, I.; Palsgrove, D.; Conklin, D.; Kinose, Y.; et al. Integrated Genomic, Epigenomic, and Expression Analyses of Ovarian Cancer Cell Lines. Cell Rep. 2018, 25, 2617–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Gu, Z.Y.; Li, Q.; Liu, Q.H.; Yang, X.Y.; Zhang, J.J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J. Ovarian Res. 2019, 12, 35. [Google Scholar] [CrossRef]
- Zheng, M.J.; Li, X.; Hu, Y.X.; Dong, H.; Gou, R.; Nie, X.; Liu, Q.; Ying-Ying, H.; Liu, J.J.; Lin, B. Identification of molecular marker associated with ovarian cancer prognosis using bioinformatics analysis and experiments. J. Cell. Physiol. 2019, 234, 11023–11036. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Bowen, N.J.; Walker, L.D.; Matyunina, L.V.; Logani, S.; Totten, K.A.; Benigno, B.B.; McDonald, J.F. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med. Genom. 2009, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Elgaaen, B.V.; Olstad, O.K.; Sandvik, L.; Odegaard, E.; Sauer, T.; Staff, A.C.; Gautvik, K.M. ZNF385B and VEGFA are strongly differentially expressed in serous ovarian carcinomas and correlate with survival. PLoS ONE 2012, 7, e46317. [Google Scholar] [CrossRef]
- Lili, L.N.; Matyunina, L.V.; Walker, L.D.; Benigno, B.B.; McDonald, J.F. Molecular profiling predicts the existence of two functionally distinct classes of ovarian cancer stroma. BioMed Res. Int. 2013, 2013, 846387. [Google Scholar] [CrossRef] [Green Version]
- de Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [Green Version]
- Saldanha, A.J. Java Treeview--extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [Green Version]
- Bartha, A.; Gyorffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Estes, J.M.; Leath, C.A., 3rd; Straughn, J.M., Jr.; Rocconi, R.P.; Kirby, T.O.; Huh, W.K.; Barnes, M.N., 3rd. Bowel resection at the time of primary debulking for epithelial ovarian carcinoma: Outcomes in patients treated with platinum and taxane-based chemotherapy. J. Am. Coll Surg 2006, 203, 527–532. [Google Scholar] [CrossRef]
- Salani, R.; Backes, F.J.; Fung, M.F.; Holschneider, C.H.; Parker, L.P.; Bristow, R.E.; Goff, B.A. Posttreatment surveillance and diagnosis of recurrence in women with gynecologic malignancies: Society of Gynecologic Oncologists recommendations. Am. J. Obs. Gynecol 2011, 204, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.G.; Stewart, M.E.; Altman, D.G.; Gabra, H.; Smyth, J.F. A prognostic model for ovarian cancer. Br. J. Cancer 2001, 85, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Eltabbakh, G.H.; Belinson, J.L.; Kennedy, A.W.; Biscotti, C.V.; Casey, G.; Tubbs, R.R.; Blumenson, L.E. p53 overexpression is not an independent prognostic factor for patients with primary ovarian epithelial cancer. Cancer 1997, 80, 892–898. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Q.; Zhao, G.; Ke, B.; Ma, G.; Liu, G.L.; Liang, H.; Liu, L.R.; Hao, X.S. Overexpression of UBE2C correlates with poor prognosis in gastric cancer patients. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhi, X.; Shen, X.; Chen, C.; Yuan, L.; Dong, X.; Zhu, C.; Yao, L.; Chen, M. Depletion of UBE2C reduces ovarian cancer malignancy and reverses cisplatin resistance via downregulating CDK1. Biochem. Biophys. Res. Commun. 2020, 523, 434–440. [Google Scholar] [CrossRef]
- Ayad, N.G.; Rankin, S.; Murakami, M.; Jebanathirajah, J.; Gygi, S.; Kirschner, M.W. Tome-1, a trigger of mitotic entry, is degraded during G1 via the APC. Cell 2003, 113, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.N.; Burgess, J.T.; He, Y.; Gately, K.; Snell, C.; Zhang, S.D.; Hooper, J.D.; Richard, D.J.; O’Byrne, K.J. Expression of CDCA3 Is a Prognostic Biomarker and Potential Therapeutic Target in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1071–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yin, W.; Cao, W.; Chen, P.; Bian, L.; Ni, Q. CDCA3 is a potential prognostic marker that promotes cell proliferation in gastric cancer. Oncol. Rep. 2019, 41, 2471–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | GEO Dataset | Samples | Reference |
|---|---|---|---|
| GPL570 | GSE14407 | 12 Normal, 12 Cancer | Bowen et al. [13] |
| GPL570 | GSE36668 | 4 Normal, 4 Cancer | Elgaaen et al. [14] |
| GPL570 | GSE38666 | 12 Normal, 18 Cancer | Lili et al. [15] |
| Gene Names | Log2FC | Adjusted p-Value | ||||
|---|---|---|---|---|---|---|
| GSE14407 | GSE36668 | GSE38666 | GSE14407 | GSE36668 | GSE38666 | |
| BIRC5 | 3.784515 | 13.83283 | 3.792818 | 4.58 × 10−6 | 2.45 × 10−4 | 3.34 × 10−8 |
| CDCA3 | 3.40002 | 3.542135 | 3.543772 | 9.12 × 10−7 | 2.72 × 10−3 | 2.20 × 10−9 |
| CENPF | 3.904451 | 3.450972 | 3.934447 | 1.69 × 10−7 | 3.17 × 10−3 | 2.20 × 10−9 |
| KIF4A | 3.533572 | 5.173891 | 3.746773 | 4.76 × 10−6 | 2.84 × 10−3 | 1.70 × 10−8 |
| NCAPG | 3.233566 | 3.533272 | 3.092676 | 5.15 × 10−6 | 3.17 × 10−3 | 1.26 × 10−7 |
| RRM2 | 3.212708 | 4.504844 | 3.215252 | 3.24 × 10−6 | 3.17 × 10−3 | 2.23 × 10−8 |
| UBE2C | 2.539233 | 3.816594 | 2.699924 | 7.37 × 10−6 | 2.56 × 10−3 | 1.26 × 10−7 |
| VEGFA | 2.86746 | 2.906894 | 2.647287 | 2.89 × 10−6 | 2.84 × 10−3 | 4.33 × 10−7 |
| NR2F6 | 2.950696 | 2.75339 | 3.029833 | 1.85 × 10−5 | 2.72 × 10−3 | 1.63 × 10−7 |
| Gene Names | Log2FC | Adjusted p-Value | ||||
|---|---|---|---|---|---|---|
| GSE14407 | GSE36668 | GSE38666 | GSE14407 | GSE36668 | GSE38666 | |
| C21orf62 | −3.784515 | −13.83283 | −3.792818 | 4.58 × 10−6 | 2.45 × 10−4 | 3.34 × 10−8 |
| MUM1L1 | −3.40002 | −3.542135 | −3.543772 | 9.12 × 10−7 | 2.72 × 10−3 | 2.20 × 10−9 |
| NBEA | −3.904451 | −3.450972 | −3.934447 | 1.69 × 10−7 | 3.17 × 10−3 | 2.20 × 10−9 |
| RNASE4 | −3.533572 | −5.173891 | −3.746773 | 4.76 × 10−6 | 2.84 × 10−3 | 1.70 × 10−8 |
| RORA | −3.233566 | −3.533272 | −3.092676 | 5.15 × 10−6 | 3.17 × 10−3 | 1.26 × 10−7 |
| LOC101930363 /LOC101928349 /LOC100507387 /FAM153C /FAM153A /FAM153B | −4.26894 | −3.17681 | −4.17039 | 4.56 × 10−7 | 0.003168 | 8.62 × 10−9 |
| Gene Names | Fold Change | p-Value |
|---|---|---|
| BIRC5 | 157.01 | 1.09 × 10−65 |
| CDCA3 | 2.58 | 1.7 × 10−29 |
| CENPF | 18.82 | 1.23 × 10−63 |
| KIF4A | 34.68 | 2.54 × 10−64 |
| NCAPG | 29.63 | 5.23 × 10−64 |
| NR2F6 | 11.6 | 4.15 × 10−65 |
| RRM2 | 41.23 | 2.99 × 10−64 |
| UBE2C | 101.05 | 3.88 × 10−65 |
| VEGFA | 2.61 | 3.13 × 10−34 |
| Gene Names | Fold Change | p-Value |
|---|---|---|
| C21orf62 | 0.02 | 8.79 × 10−63 |
| MUM1L1(PWWP3B) | 0.01 | 5.22 × 10−65 |
| NBEA | 0.11 | 2.62 × 10−64 |
| RNASE4 | 0.01 | 6.84 × 10−66 |
| RORA | 0.15 | 4.99 × 10−63 |
| Category | Term | Gene Names | p-Value |
|---|---|---|---|
| GO_BP | GO:0051301 Cell division | BIRC5, CDCA3, CENPF, NCAPG, UBE2C | 5.5 × 10−5 |
| GO:0031536 Positive regulation of exit from mitosis | BIRC5, UBE2C | 3.9 × 10−3 | |
| GO:0000278 Mitotic nuclear division | BIRC5, CDCA3, CENPF | 1.1 × 10−2 | |
| GO:0016567 Protein ubiquitination | BIRC5, CDCA3, UBE2C | 2.2 × 10−2 | |
| GO:0030522 Intracellular receptor signaling pathway | RORA, NR2F6 | 2.5 × 10−2 | |
| GO:0043401 Steroid hormone mediated signaling pathway | RORA, NR2F6 | 3.7 × 10−2 | |
| GO:0043154 Negative regulation of cysteine–type endopeptidase activity involved in apoptotic process | BIRC5, VEGFA | 4.4 × 10−2 | |
| GO_CC | Cytosol | BIRC5, CDCA3, CENPF, KIF4A, NBEA, NCAPG, RRM2, UBE2C | 2.2 × 10−3 |
| Midbody | BIRC5, CENPF, KIF4A | 3.1 × 10−3 | |
| Nucleoplasm | RORA, BIRC5, CENPF, KIF4A, NR2F6, RRM2, UBE2C | 5.1 × 10−3 | |
| Spindle microtubule | BIRC5, KIF4A | 2.9 × 10−2 | |
| Chromosome, centromeric region | BIRC5, CENPF | 3.7 × 10−2 | |
| GO_MF | RNA polymerase II transcription factor activity, ligand activated sequence–specific DNA | RORA, NR2F6 | 2.3 × 10−2 |
| Steroid hormone receptor activity | RORA, NR2F6 | 3.6 × 10−2 | |
| Protein binding | RORA, BIRC5, CDCA3, CENPF, KIF4A, NCAPG, NR2F6, RRM2, UBE2C, VEGFA | 4.4 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Kim, K.-B.; Lee, J.-H.; Kim, S. Bioinformatic Analysis for Influential Core Gene Identification and Prognostic Significance in Advanced Serous Ovarian Carcinoma. Medicina 2021, 57, 933. https://doi.org/10.3390/medicina57090933
Song C, Kim K-B, Lee J-H, Kim S. Bioinformatic Analysis for Influential Core Gene Identification and Prognostic Significance in Advanced Serous Ovarian Carcinoma. Medicina. 2021; 57(9):933. https://doi.org/10.3390/medicina57090933
Chicago/Turabian StyleSong, Changho, Kyoung-Bo Kim, Jae-Ho Lee, and Shin Kim. 2021. "Bioinformatic Analysis for Influential Core Gene Identification and Prognostic Significance in Advanced Serous Ovarian Carcinoma" Medicina 57, no. 9: 933. https://doi.org/10.3390/medicina57090933
APA StyleSong, C., Kim, K.-B., Lee, J.-H., & Kim, S. (2021). Bioinformatic Analysis for Influential Core Gene Identification and Prognostic Significance in Advanced Serous Ovarian Carcinoma. Medicina, 57(9), 933. https://doi.org/10.3390/medicina57090933