
Cardiac Amyloidosis-Challenging Diagnosis and Unclear Clinical Picture

 , , ,
, , ,  and
and 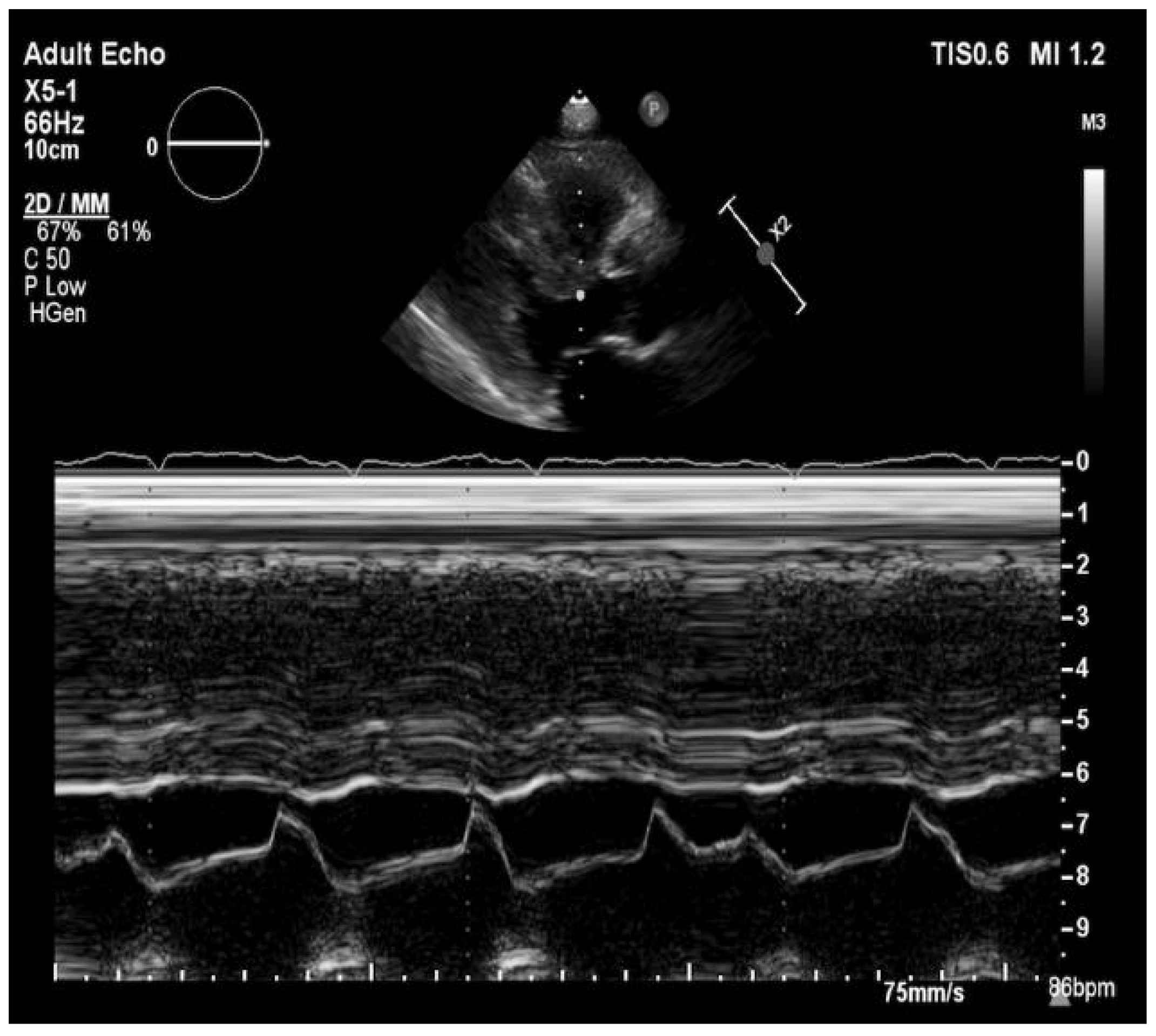{kind=link}
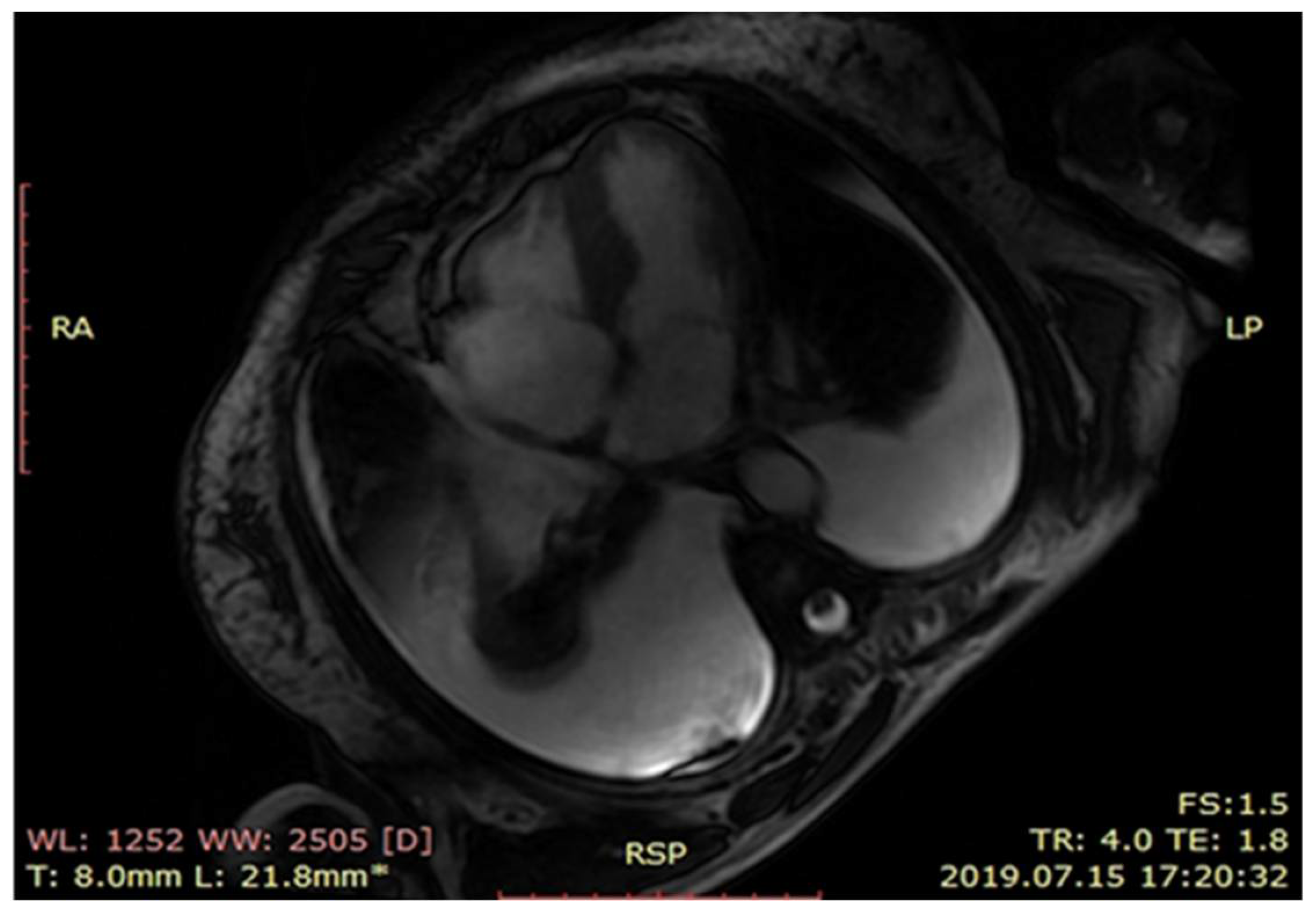{kind=link}
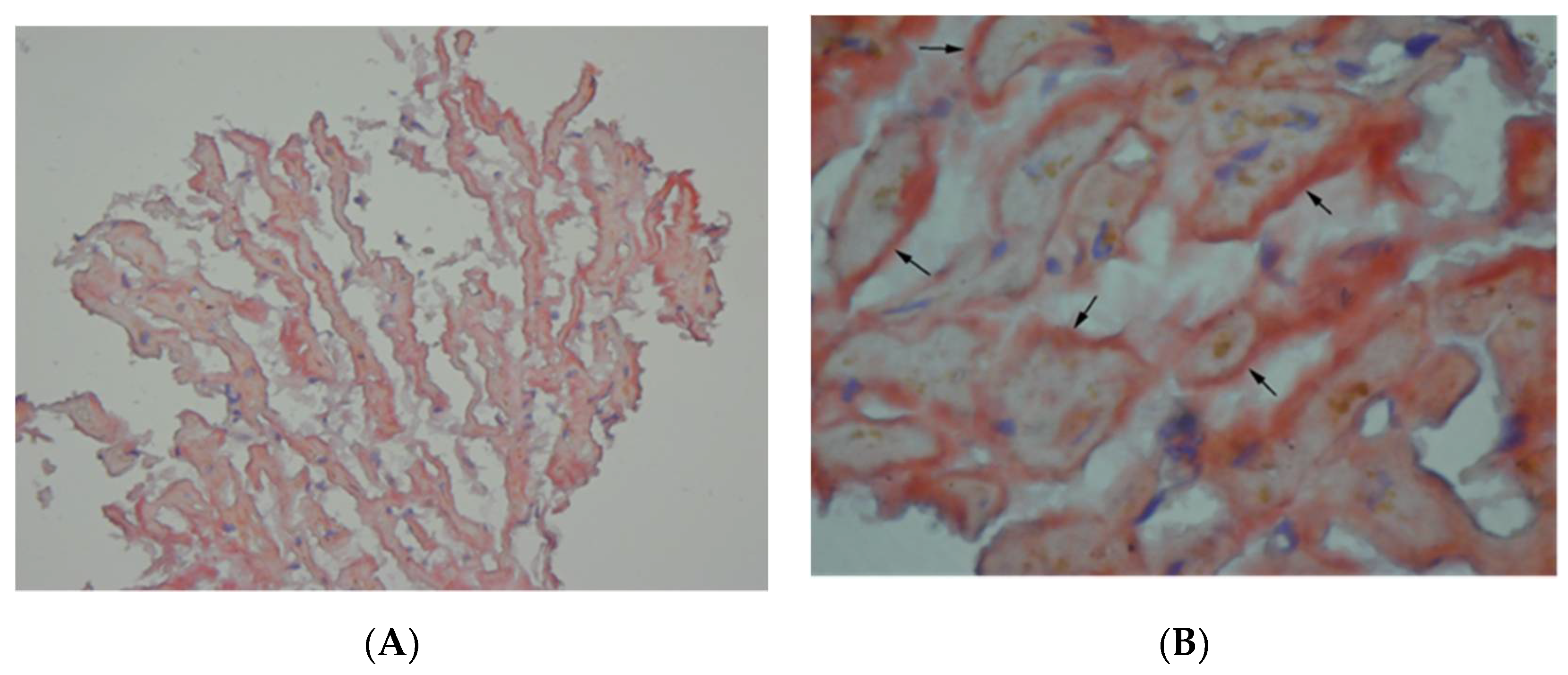{kind=link}
Abstract
:1. Introduction
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ash, S.; Shorer, E.; Bs, D.R.; Vo, M.; Bs, J.G.; Golamari, R.; Jain, R.; Jain, R. Cardiac amyloidosis—A review of current literature for the practicing physician. Clin. Cardiol. 2021, 44, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Bistola, V.; Parissis, J.; Foukarakis, E.; Valsamaki, P.N.; Anastasakis, A.; Koutsis, G.; Efthimiadis, G.; Kastritis, E. Practical recommendations for the diagnosis and management of transthyretin cardiac amyloidosis. Hear. Fail. Rev. 2021, 1–19. [Google Scholar] [CrossRef]
- Holcman, K.; Kostkiewicz, M.; Podolec, P.; Rubiś, P. Amyloidoza serca—Właściwe rozpoznanie i nowe terapie na horyzoncie. Folia Cardiol. 2019, 14, 616–624. [Google Scholar] [CrossRef]
- Oerlemans, M.I.F.J.; Rutten, K.H.G.; Minnema, M.C.; Raymakers, R.A.P.; Asselbergs, F.W.; De Jonge, N. Cardiac amyloidosis: The need for early diagnosis. Neth. Hear. J. 2019, 27, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonderman, D.; Pölzl, G.; Ablasser, K.; Agis, H.; Aschauer, S.; Auer-Grumbach, M.; Binder, C.; Dörler, J.; Duca, F.; Ebner, C.; et al. Diagnosis and treatment of cardiac amyloidosis: An interdisciplinary consensus statement. Wien. Klin. Wochenschr. 2020, 132, 742–761. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.; Fontana, M.; Martinez-Naharro, A.; Quarta, C.C.; Whelan, C.J.; Petrie, A.; Rowczenio, D.M.; Gilbertson, J.A.; Hutt, D.F.; Rezk, T.; et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation 2019, 140, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-González, P.; Cozar-Santiago, M.D.P.; Maceira, A.M. Cardiac Amyloidosis Detected Using 18 F-florbetapir PET/CT. Revista Española de Cardiología 2016, 69, 1215. [Google Scholar] [CrossRef] [PubMed]
- Sivrikoz, I.A.; Çavuşoğlu, Y. Cardiac Scintigraphy-Centered Diagnostic Process in Transthyretin Cardiac Amyloidosis. Turk. Kardiyol. Dernegi Arsivi-Archives Turk. Soc. Cardiol. 2020, 48, 10. [Google Scholar] [CrossRef]
- Mollee, P.; Renaut, P.; Gottlieb, D.; Goodman, H. How to diagnose amyloidosis. Intern. Med. J. 2014, 44, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Italian Group of Nuclear Cardiology (GICN); Genovesi, D.; Milan, E.; Acampa, W.; Giubbini, R.; Cuocolo, A.; Marzullo, P. Cardiac amyloidosis. Clin. Transl. Imaging 2019, 7, 21–32. [Google Scholar] [CrossRef]
- Barroso, F.A.; Judge, D.P.; Ebede, B.; Leslie, A.; Stewart, M.; Amass, L.; Sultan, M.B. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: Results up to 6 years. Amyloid 2017, 24, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Tojo, K.; Morita, H.; Koyama, J.; Ikeda, S.-I. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2014, 22, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Dispenzieri, A.; Sher, T. Pathophysiology and treatment of cardiac amyloidosis. Nat. Rev. Cardiol. 2015, 12, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Stern, L.K.; Kittleson, M.M. Updates in Cardiac Amyloidosis Diagnosis and Treatment. Curr. Oncol. Rep. 2021, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozak, S.; Ulbrich, K.; Migacz, M.; Szydło, K.; Mizia-Stec, K.; Holecki, M. Cardiac Amyloidosis-Challenging Diagnosis and Unclear Clinical Picture. Medicina 2021, 57, 450. https://doi.org/10.3390/medicina57050450
Kozak S, Ulbrich K, Migacz M, Szydło K, Mizia-Stec K, Holecki M. Cardiac Amyloidosis-Challenging Diagnosis and Unclear Clinical Picture. Medicina. 2021; 57(5):450. https://doi.org/10.3390/medicina57050450
Chicago/Turabian StyleKozak, Sylwia, Krzysztof Ulbrich, Maciej Migacz, Krzysztof Szydło, Katarzyna Mizia-Stec, and Michał Holecki. 2021. "Cardiac Amyloidosis-Challenging Diagnosis and Unclear Clinical Picture" Medicina 57, no. 5: 450. https://doi.org/10.3390/medicina57050450
APA StyleKozak, S., Ulbrich, K., Migacz, M., Szydło, K., Mizia-Stec, K., & Holecki, M. (2021). Cardiac Amyloidosis-Challenging Diagnosis and Unclear Clinical Picture. Medicina, 57(5), 450. https://doi.org/10.3390/medicina57050450