
RNAi Screening in Tumor Cells Identifies Artificial microRNAs That Improve Oncolytic Virus Replication

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
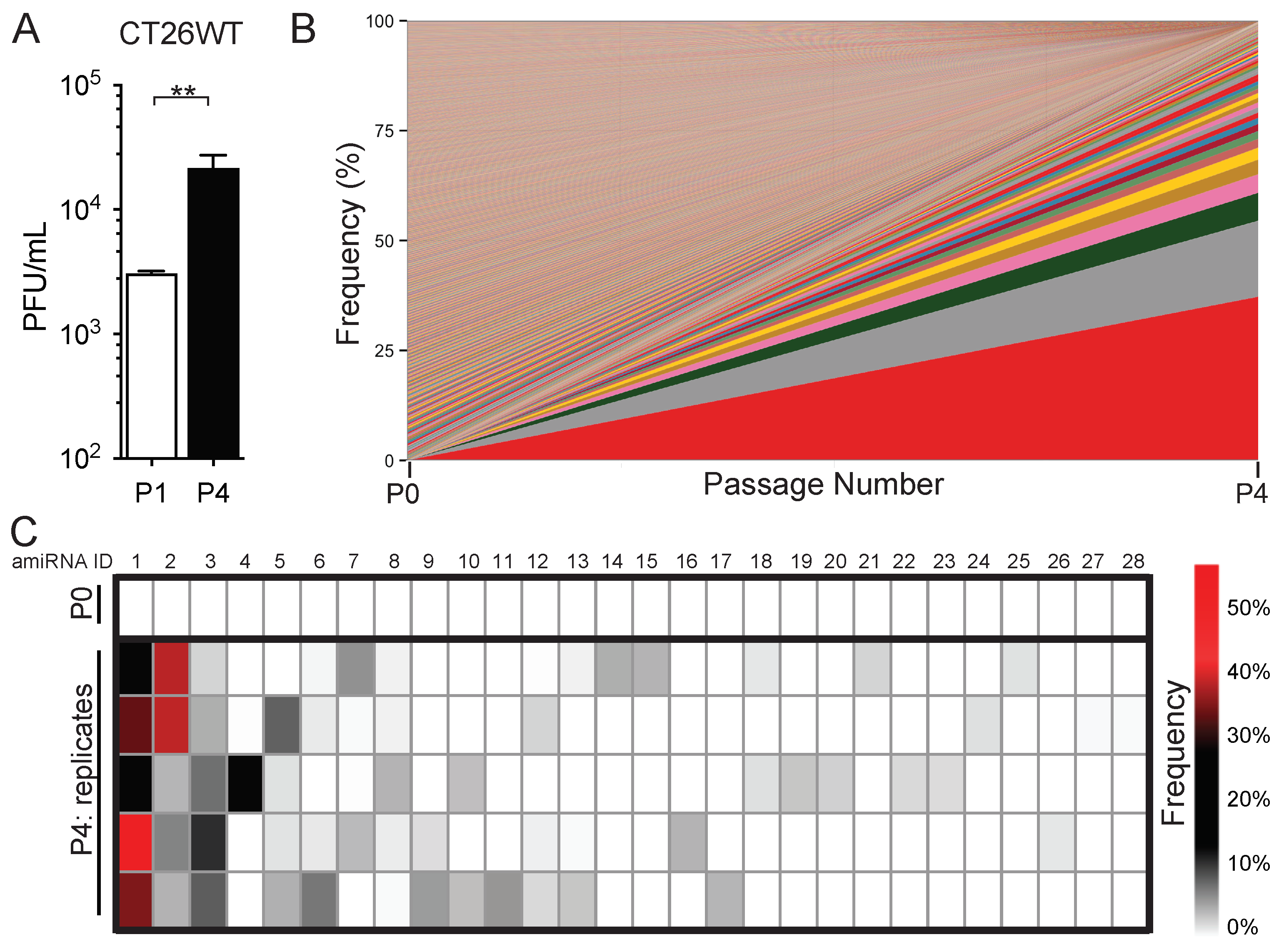
2.1. In Vitro Serial Passage of SINV–amiRNA Library in Tumor Cells Selects for Specific amiRNAs
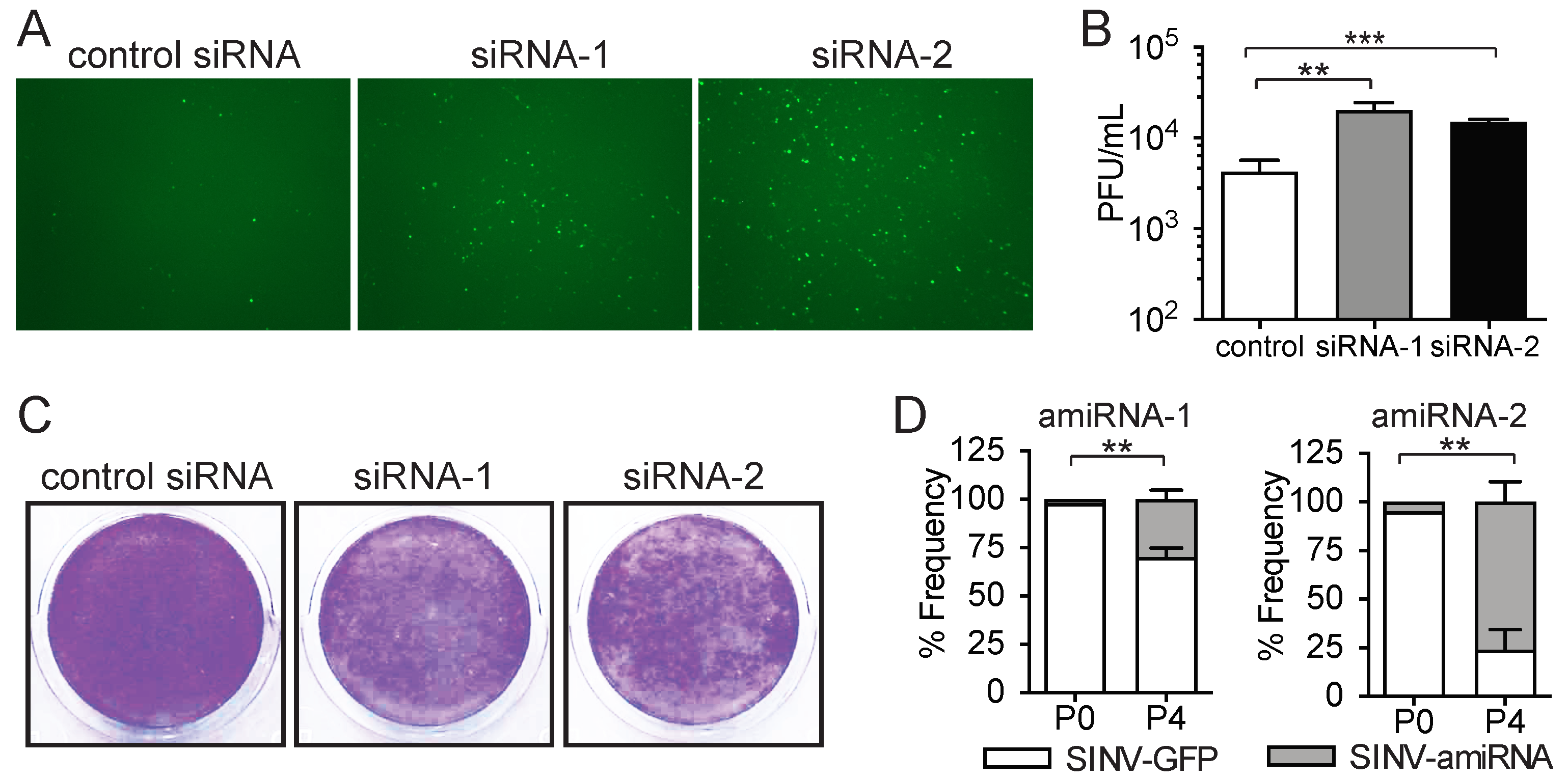
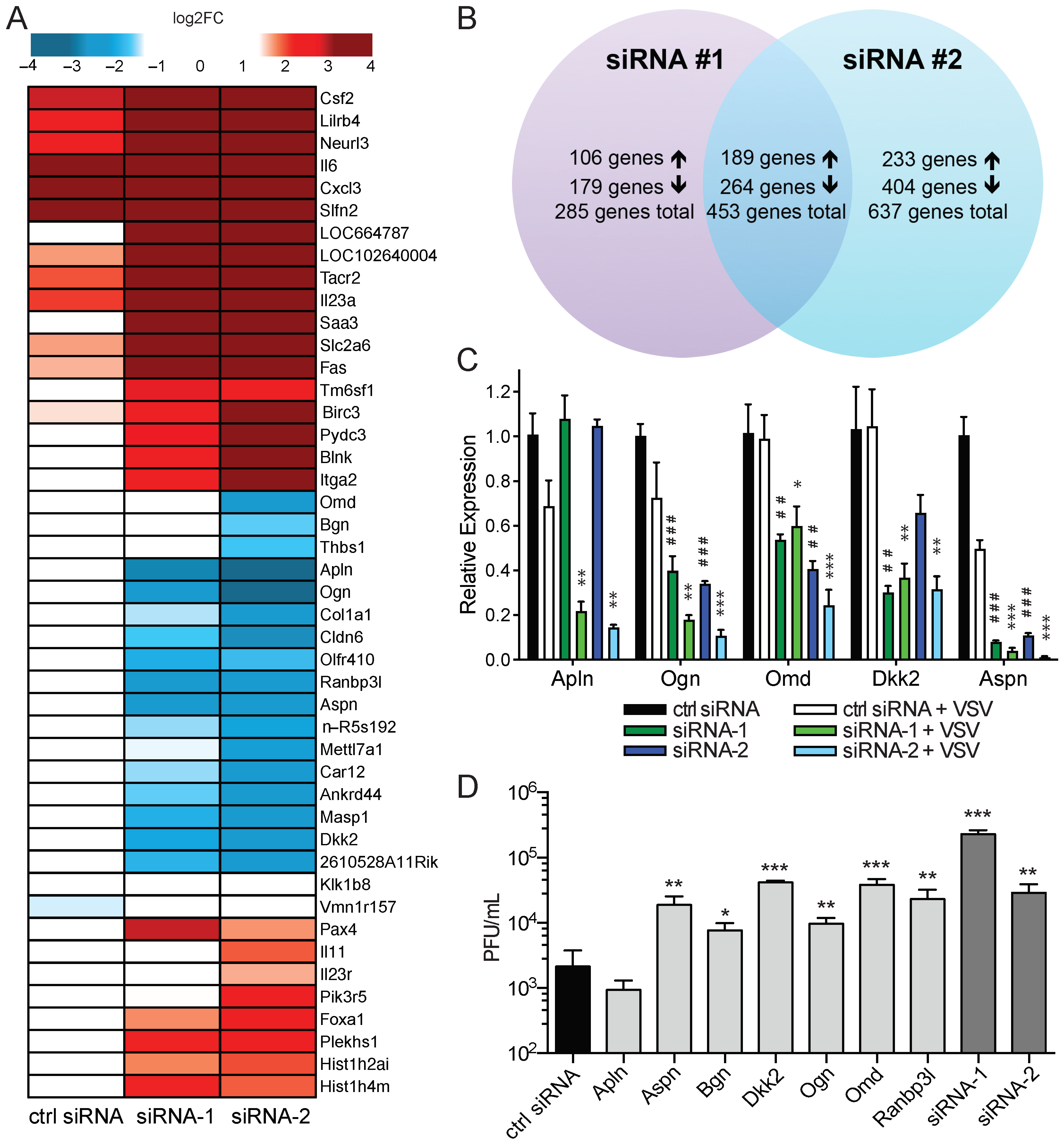
2.2. Enriched amiRNA Sequences Increase SINV Replication in Tumor Cells
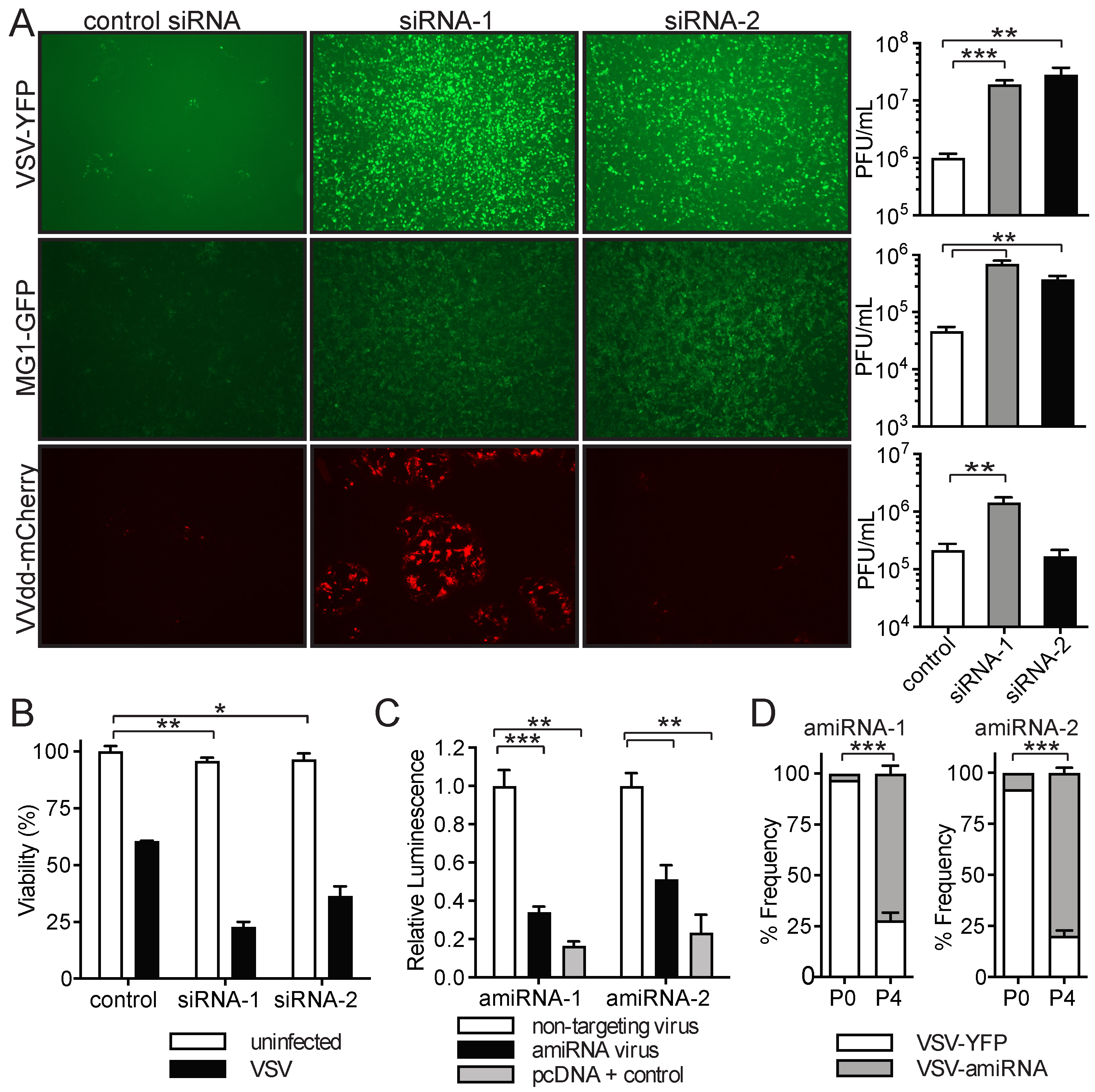
2.3. amiRNA-1 and amiRNA-2 Enhance Replication and Tumor Cell Killing of Other OVs
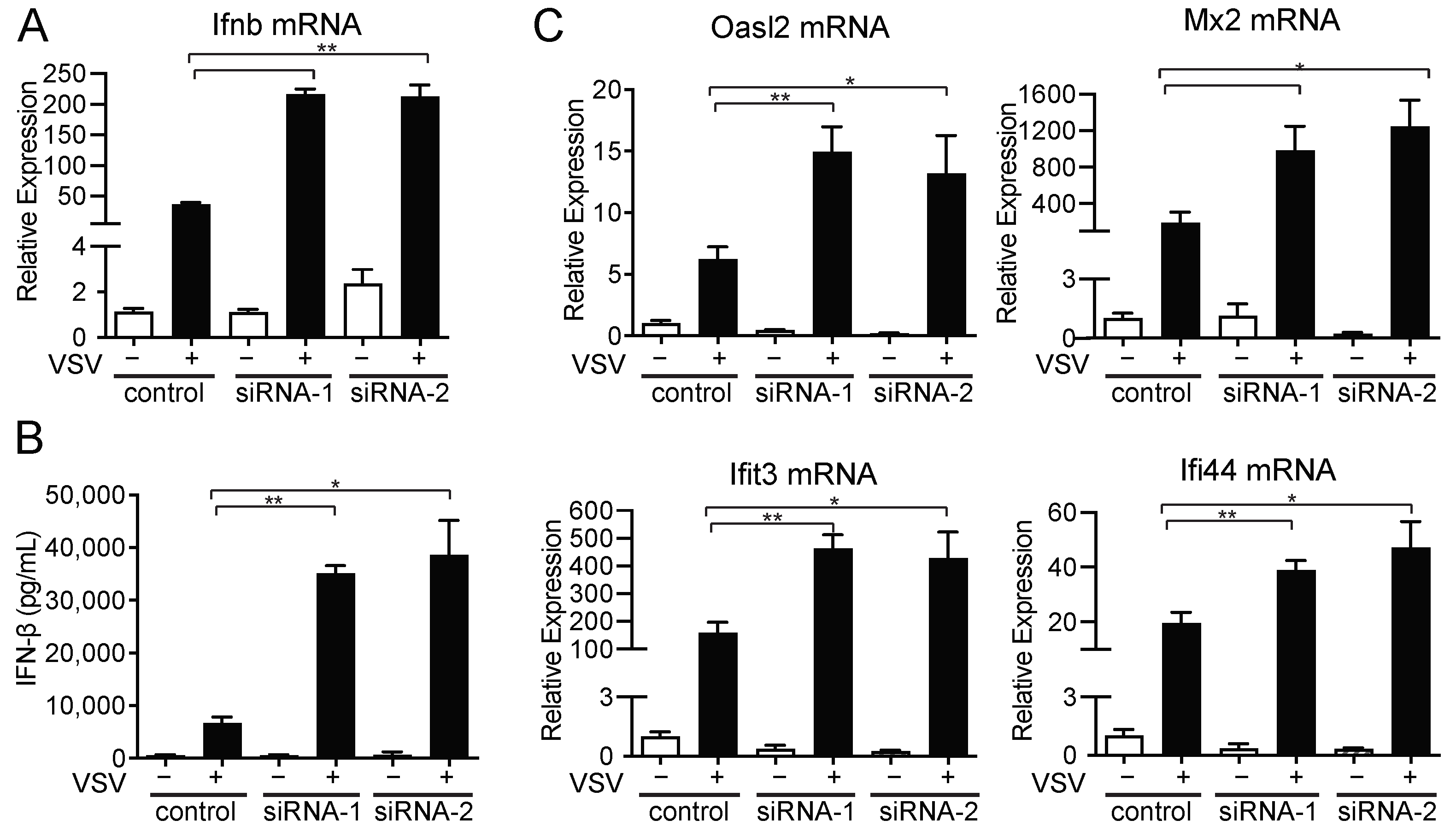
2.4. amiRNA-1 and amiRNA-2 Enhance VSV Replication in an IFN-Independent Manner
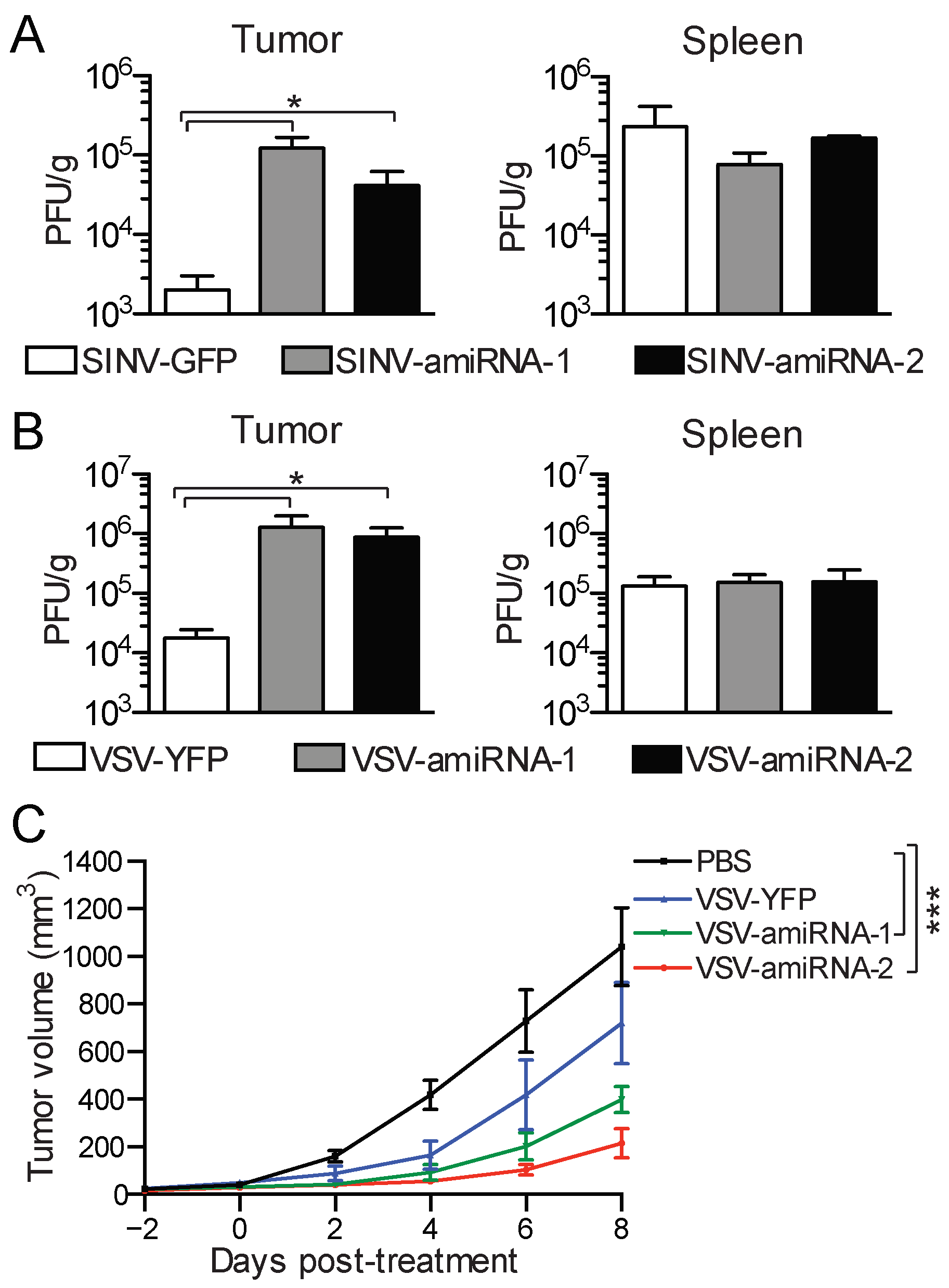
2.5. amiRNA-Expressing Viruses Demonstrate Enhanced Replication and Efficacy In Vivo
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Viruses
4.3. SINV–amiRNA Library Screen
4.4. Sequencing of SINV–amiRNA Library
4.5. Cell Viability Assay
4.6. Coomassie Blue Staining
4.7. siRNA Transfection
4.8. Quantification of Infection in Cultured Cells, Tumors, and Spleens
4.9. Dual-Luciferase Reporter Assay
4.10. Virus Competition Assay
4.11. Quantitative PCR
4.12. ELISA
4.13. Microarray
4.14. Mouse and Tumor Models
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with Oncolytic Viruses: Progress and Challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. Incorporation of the B18R Gene of Vaccinia Virus into an Oncolytic Herpes Simplex Virus Improves Antitumor Activity. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Le Bœuf, F.; Batenchuk, C.; Vähä-Koskela, M.; Breton, S.; Roy, D.; Lemay, C.; Cox, J.; Abdelbary, H.; Falls, T.; Waghray, G.; et al. Model-Based Rational Design of an Oncolytic Virus with Improved Therapeutic Potential. Nat. Commun. 2013, 4, 1974. [Google Scholar] [CrossRef]
- Li, W.; Turaga, R.C.; Li, X.; Sharma, M.; Enadi, Z.; Dunham Tompkins, S.N.; Hardy, K.C.; Mishra, F.; Tsao, J.; Liu, Z.R.; et al. Overexpression of Smac by an Armed Vesicular Stomatitis Virus Overcomes Tumor Resistance. Mol. Ther. Oncolytics 2019, 14, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S.O.; Rogulski, K.R.; Paielli, D.L.; Gilbert, J.D.; Kim, J.H. A Novel Three-Pronged Approach to Kill Cancer Cells Selectively: Concomitant Viral, Double Suicide Gene, and Radiotherapy. Hum. Gene Ther. 1998, 9, 1323–1333. [Google Scholar] [CrossRef]
- Bourgeois-Daigneault, M.; Roy, D. Oncolytic Vesicular Stomatitis Virus Expressing Interferon-γ Has Enhanced Therapeutic Activity. Mol. Ther. Oncolytics 2016, 3, 16001. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.Y. An Engineered Oncolytic Virus Expressing PD-L1 Inhibitors Activates Tumor Neoantigen-Specific T Cell Responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Nakao, S.; Arai, Y.; Tasaki, M.; Yamashita, M.; Murakami, R.; Kawase, T.; Amino, N.; Nakatake, M.; Kurosaki, H.; Mori, M.; et al. Intratumoral Expression of IL-7 and IL-12 Using an Oncolytic Virus Increases Systemic Sensitivity to Immune Checkpoint Blockade. Sci. Transl. Med. 2020, 12, 7992. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Amatruda, T.; Reid, T.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; Nemunaitis, J.; Zloza, A.; Wolf, M.; et al. Systemic versus Local Responses in Melanoma Patients Treated with Talimogene Laherparepvec from a Multi-Institutional Phase II Study. J. Immunother. Cancer 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the Clinic: Talimogene Laherparepvec (T-VEC), a First-in-Class Intratumoral Oncolytic Viral Therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor-Encoding, Second-Generation Oncolytic Herpesvirus in Patients with Unresectable Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical Landscape of Oncolytic Virus Research in 2020. J. Immunother. Cancer 2020, 8, 1486. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; tenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV Strains with Defects in Their Ability to Shutdown Innate Immunity Are Potent Systemic Anti-Cancer Agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Obuchi, M.; Fernandez, M.; Barber, G.N. Development of Recombinant Vesicular Stomatitis Viruses That Exploit Defects in Host Defense to Augment Specific Oncolytic Activity. J. Virol. 2003, 77, 8843–8856. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting Tumor-Specific Defects in the Interferon Pathway with a Previously Unknown Oncolytic Virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Isenmann, M.; Stoddart, M.J.; Schmelzeisen, R.; Gross, C.; Della Bella, E.; Rothweiler, R.M. Basic Principles of RNA Interference: Nucleic Acid Types and In Vitro Intracellular Delivery Methods. Micromachines 2023, 14, 1321. [Google Scholar] [CrossRef]
- Varble, A.; Benitez, A.A.; Schmid, S.; Sachs, D.; Shim, J.V.; Rodriguez-Barrueco, R.; Panis, M.; Crumiller, M.; Silva, J.M.; Sachidanandam, R.; et al. An in Vivo RNAi Screening Approach to Identify Host Determinants of Virus Replication. Cell Host Microbe 2013, 14, 346–356. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A Diverse Range of Gene Products Are Effectors of the Type I Interferon Antiviral Response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Li, J.; Ding, S.C.; Cho, H.; Chung, B.C.; Gale, M.; Chanda, S.K.; Diamond, M.S. A Short Hairpin RNA Screen of Interferon-Stimulated Genes Identifies a Novel Negative Regulator of the Cellular Antiviral Response. mBio 2013, 4, e00385-13. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Bozym, R.; Morosky, S.A.; Hanna, S.L.; Mukherjee, A.; Tudor, M.; Kim, K.S.; Cherry, S. Comparative RNAi Screening Reveals Host Factors Involved in Enterovirus Infection of Polarized Endothelial Monolayers. Cell Host Microbe 2011, 9, 70–82. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of Host Proteins Required for HIV Infection through a Functional Genomic Screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A Genome-Wide Genetic Screen for Host Factors Required for Hepatitis C Virus Propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [Google Scholar] [CrossRef]
- Panda, D.; Das, A.; Dinh, P.X.; Subramaniam, S.; Nayak, D.; Barrows, N.J.; Pearson, J.L.; Thompson, J.; Kelly, D.L.; Ladunga, I.; et al. RNAi Screening Reveals Requirement for Host Cell Secretory Pathway in Infection by Diverse Families of Negative-Strand RNA Viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 19036–19041. [Google Scholar] [CrossRef] [PubMed]
- Sessions, O.M.; Barrows, N.J.; Souza-Neto, J.A.; Robinson, T.J.; Hershey, C.L.; Rodgers, M.A.; Ramirez, J.L.; Dimopoulos, G.; Yang, P.L.; Pearson, J.L.; et al. Discovery of Insect and Human Dengue Virus Host Factors. Nature 2009, 458, 1047–1050. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Grönberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-Tumor Interactome Screen Reveals ER Stress Response Can Reprogram Resistant Cancers for Oncolytic Virus-Triggered Caspase-2 Cell Death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef]
- Wedge, M.E.; Jennings, V.A.; Crupi, M.J.F.; Poutou, J.; Jamieson, T.; Pelin, A.; Pugliese, G.; de Souza, C.T.; Petryk, J.; Laight, B.J.; et al. Virally Programmed Extracellular Vesicles Sensitize Cancer Cells to Oncolytic Virus and Small Molecule Therapy. Nature Commun. 2022, 13, 1898. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, J.-H.; Kwon, Y.-G.; Kim, E.-C.; Kim, N.K.; Choi, H.J.; Yun, C.-O. VEGF-Specific Short Hairpin RNA-Expressing Oncolytic Adenovirus Elicits Potent Inhibition of Angiogenesis and Tumor Growth. Mol. Ther. 2007, 15, 295–302. [Google Scholar] [CrossRef]
- Mao, L.; Zhang, J.; Liu, N.; Fan, L.; Yang, D.; Xue, B.; Shan, Y.; Zheng, J. Oncolytic Virus Carrying shRNA Targeting SATB1 Inhibits Prostate Cancer Growth and Metastasis. Tumor Biol. 2015, 36, 9073–9081. [Google Scholar] [CrossRef]
- Jennings, V.A.; Rumbold-Hall, R.; Migneco, G.; Barr, T.; Reilly, K.; Ingram, N.; St Hilare, I.; Heaton, S.; Alzamel, N.; Jackson, D.; et al. Enhancing Oncolytic Virotherapy by Extracellular Vesicle Mediated microRNA Reprograming of the Tumour Microenvironment. Front. Immunol. 2024, 15, 1500570. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.-K.; Kim, Y.J.; Hong, J.; Chang, H.-G.; Yoon, A.-R.; Yun, C.-O. ErbB3-Targeting Oncolytic Adenovirus Causes Potent Tumor Suppression by Induction of Apoptosis in Cancer Cells. Int. J. Mol. Sci. 2022, 23, 7127. [Google Scholar] [CrossRef]
- Wong, B.; Birtch, R.; Bergeron, A.; Ng, K.; Maznyi, G.; Spinelli, M.; Chen, A.; Landry, A.; Crupi, M.J.F.; Arulanandam, R.; et al. High Throughput Screen Identifies Lysosomal Acid Phosphatase 2 (ACP2) to Regulate IFN-1 Responses to Potentiate Oncolytic VSV∆51 Activity. Sci. Rep. 2024, 14, 28284. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, M.; Lu, Q.; Yu, B.; Yang, Y.; Li, B.; Peng, Z.; Li, Z. Oncolytic Adenovirus-Mediated Dual Knockdown of Survivin and OCT4 Improves Therapeutic Efficacy in Esophageal Cancer. Ann. Transl. Med. 2023, 11, 193. [Google Scholar] [CrossRef]
- Qiu, M.; Wei, R.; Zhang, Q.; Zhao, J.; Zhang, H.; Tan, J.; Qiao, W. ISG15 Depletion Enhances oHSV-1 Replication and Antitumor Efficacy in Oral Squamous Cell Carcinoma. Virology 2025, 606, 110504. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Liu, H.; Zhu, H.; Zhang, W.; Sun, D.; Han, X.; Liu, Y.; Luo, G. RCAd-LTH-shPD-L1, a Double-Gene Recombinant Oncolytic Adenovirus with Enhanced Antitumor Immunity, Increases Lymphocyte Infiltration and Reshapes the Tumor Microenvironment. J. Immunother. Cancer 2024, 12, e007171. [Google Scholar] [CrossRef]
- Rovira-Rigau, M.; Raimondi, G.; Marín, M.Á.; Gironella, M.; Alemany, R.; Fillat, C. Bioselection Reveals miR-99b and miR-485 as Enhancers of Adenoviral Oncolysis in Pancreatic Cancer. Mol. Ther. 2019, 27, 230–243. [Google Scholar] [CrossRef]
- Huang, P.-Y.; Guo, J.-H.; Hwang, L.-H. Oncolytic Sindbis Virus Targets Tumors Defective in the Interferon Response and Induces Significant Bystander Antitumor Immunity in Vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 298–305. [Google Scholar] [CrossRef]
- Pikor, L.A.; Bell, J.C.; Diallo, J.-S. Oncolytic Viruses: Exploiting Cancer’s Deal with the Devil. Trends Cancer 2015, 1, 266–277. [Google Scholar] [CrossRef]
- Scherwitzl, I.; Hurtado, A.; Pierce, C.M.; Vogt, S.; Pampeno, C.; Meruelo, D. Systemically Administered Sindbis Virus in Combination with Immune Checkpoint Blockade Induces Curative Anti-Tumor Immunity. Mol. Ther. Oncolytics 2018, 9, 51–63. [Google Scholar] [CrossRef]
- Pampeno, C.; Opp, S.; Hurtado, A.; Meruelo, D. Sindbis Virus Vaccine Platform: A Promising Oncolytic Virus-Mediated Approach for Ovarian Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 2925. [Google Scholar] [CrossRef]
- Tseng, J.-C.; Levin, B.; Hurtado, A.; Yee, H.; Perez de Castro, I.; Jimenez, M.; Shamamian, P.; Jin, R.; Novick, R.P.; Pellicer, A.; et al. Systemic Tumor Targeting and Killing by Sindbis Viral Vectors. Nat. Biotechnol. 2004, 22, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Uzawa, K.; Kasamatsu, A.; Shinozuka, K.; Sakuma, K.; Yamatoji, M.; Shiiba, M.; Shino, Y.; Shirasawa, H.; Tanzawa, H. Oncolytic Activity of Sindbis Virus in Human Oral Squamous Carcinoma Cells. Br. J. Cancer 2009, 101, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.; McManus, D.; Lefebvre, C.; Hu, K.; Falls, T.; Atkins, H.; Bell, J.C.; McCart, J.A.; Mahoney, D.; Stojdl, D.F. Identification of Genetically Modified Maraba Virus as an Oncolytic Rhabdovirus. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1440–1449. [Google Scholar] [CrossRef]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic Cancer Therapy with a Tumor-Selective Vaccinia Virus Mutant Lacking Thymidine Kinase and Vaccinia Growth Factor Genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar]
- Lavanya, M.; Cuevas, C.D.; Thomas, M.; Cherry, S.; Ross, S.R. siRNA Screen for Genes That Affect Junín Virus Entry Uncovers Voltage-Gated Calcium Channels as a Therapeutic Target. Sci. Transl. Med. 2013, 5, 204ra131. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martin, R.; Wang, G.; Brandão, B.B.; Zanotto, T.M.; Shah, S.; Patel, S.K.; Schilling, B.; Kahn, C.R. MicroRNA Sequence Codes for Small Extracellular Vesicle Release and Cellular Retention. Nature 2022, 601, 446–451. [Google Scholar] [CrossRef]
- Li, S.-D.; Chen, Y.-C.; Hackett, M.J.; Huang, L. Tumor-Targeted Delivery of siRNA by Self-Assembled Nanoparticles. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 163–169. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA Therapy by Tumor Selective Delivery with Ligand-Targeted Sterically Stabilized Nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef]
- Song, E.; Zhu, P.; Lee, S.-K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody Mediated in Vivo Delivery of Small Interfering RNAs via Cell-Surface Receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- Backes, S.; Shapiro, J.S.; Sabin, L.R.; Pham, A.M.; Reyes, I.; Moss, B.; Cherry, S.; tenOever, B.R. Degradation of Host microRNAs by Poxvirus Poly(A) Polymerase Reveals Terminal RNA Methylation as a Protective Antiviral Mechanism. Cell Host Microbe 2012, 12, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Belkowski, L.S.; Sen, G.C. Inhibition of Vesicular Stomatitis Viral mRNA Synthesis by Interferons. J. Virol. 1987, 61, 653–660. [Google Scholar] [CrossRef]
- Ryman, K.D.; Klimstra, W.B.; Nguyen, K.B.; Biron, C.A.; Johnston, R.E. Alpha/Beta Interferon Protects Adult Mice from Fatal Sindbis Virus Infection and Is an Important Determinant of Cell and Tissue Tropism. J. Virol. 2000, 74, 3366–3378. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Iozzo, R.V. Biological Functions of the Small Leucine-Rich Proteoglycans: From Genetics to Signal Transduction. J. Biol. Chem. 2008, 283, 21305–21309. [Google Scholar] [CrossRef]
- Schaefer, L.; Iozzo, R.V. Small Leucine-Rich Proteoglycans, at the Crossroad of Cancer Growth and Inflammation. Curr. Opin. Genet. Dev. 2012, 22, 56–57. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Li, M.Z.; Chang, K.; Ge, W.; Golding, M.C.; Rickles, R.J.; Siolas, D.; Hu, G.; Paddison, P.J.; Schlabach, M.R.; et al. Second-Generation shRNA Libraries Covering the Mouse and Human Genomes. Nat. Genet. 2005, 37, 1281–1288. [Google Scholar] [CrossRef]
- Cheng, E.H.; Levine, B.; Boise, L.H.; Thompson, C.B.; Hardwick, J.M. Bax-Independent Inhibition of Apoptosis by Bcl-XL. Nature 1996, 379, 554–556. [Google Scholar] [CrossRef]
- Roy, D.G.; Power, A.T.; Bourgeois-Daigneault, M.C.; Falls, T.; Ferreira, L.; Stern, A.; Tanese de Souza, C.; McCart, J.A.; Stojdl, D.F.; Lichty, B.D.; et al. Programmable Insect Cell Carriers for Systemic Delivery of Integrated Cancer Biotherapy. J. Control. Release Off. J. Control. Release Soc. 2015, 220, 210–221. [Google Scholar] [CrossRef]
- Lawson, N.D.; Stillman, E.A.; Whitt, M.A.; Rose, J.K. Recombinant Vesicular Stomatitis Viruses from DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 4477–4481. [Google Scholar] [CrossRef]
- Schnell, M.J.; Buonocore, L.; Whitt, M.A.; Rose, J.K. The Minimal Conserved Transcription Stop-Start Signal Promotes Stable Expression of a Foreign Gene in Vesicular Stomatitis Virus. J. Virol. 1996, 70, 2318–2323. [Google Scholar] [CrossRef]
- Roy, D. Improving the Delivery and Replication of Oncolytic Viruses. Ph.D. Thesis, Université d’Ottawa/University of Ottawa, Ottawa, ON, Canada, 2016. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klemets, H.; Bardoul, A.; Pelin, A.; Singaravelu, R.; Boileau, M.; Falls, T.; Petryk, J.; Bourgeois-Daigneault, M.-C.; Bell, J.C.; Roy, D.G. RNAi Screening in Tumor Cells Identifies Artificial microRNAs That Improve Oncolytic Virus Replication. Pharmaceuticals 2025, 18, 708. https://doi.org/10.3390/ph18050708
Klemets H, Bardoul A, Pelin A, Singaravelu R, Boileau M, Falls T, Petryk J, Bourgeois-Daigneault M-C, Bell JC, Roy DG. RNAi Screening in Tumor Cells Identifies Artificial microRNAs That Improve Oncolytic Virus Replication. Pharmaceuticals. 2025; 18(5):708. https://doi.org/10.3390/ph18050708
Chicago/Turabian StyleKlemets, Hannah, Angelina Bardoul, Adrian Pelin, Ragunath Singaravelu, Meaghan Boileau, Theresa Falls, Julia Petryk, Marie-Claude Bourgeois-Daigneault, John C. Bell, and Dominic G. Roy. 2025. "RNAi Screening in Tumor Cells Identifies Artificial microRNAs That Improve Oncolytic Virus Replication" Pharmaceuticals 18, no. 5: 708. https://doi.org/10.3390/ph18050708
APA StyleKlemets, H., Bardoul, A., Pelin, A., Singaravelu, R., Boileau, M., Falls, T., Petryk, J., Bourgeois-Daigneault, M.-C., Bell, J. C., & Roy, D. G. (2025). RNAi Screening in Tumor Cells Identifies Artificial microRNAs That Improve Oncolytic Virus Replication. Pharmaceuticals, 18(5), 708. https://doi.org/10.3390/ph18050708