
Sindbis Virus Platform Provides an Oncolytic-Virus-Mediated and Immunotherapeutic Strategy to Overcome the Challenging Microenvironment of Pancreatic Cancer


Abstract
1. Introduction
2. Results
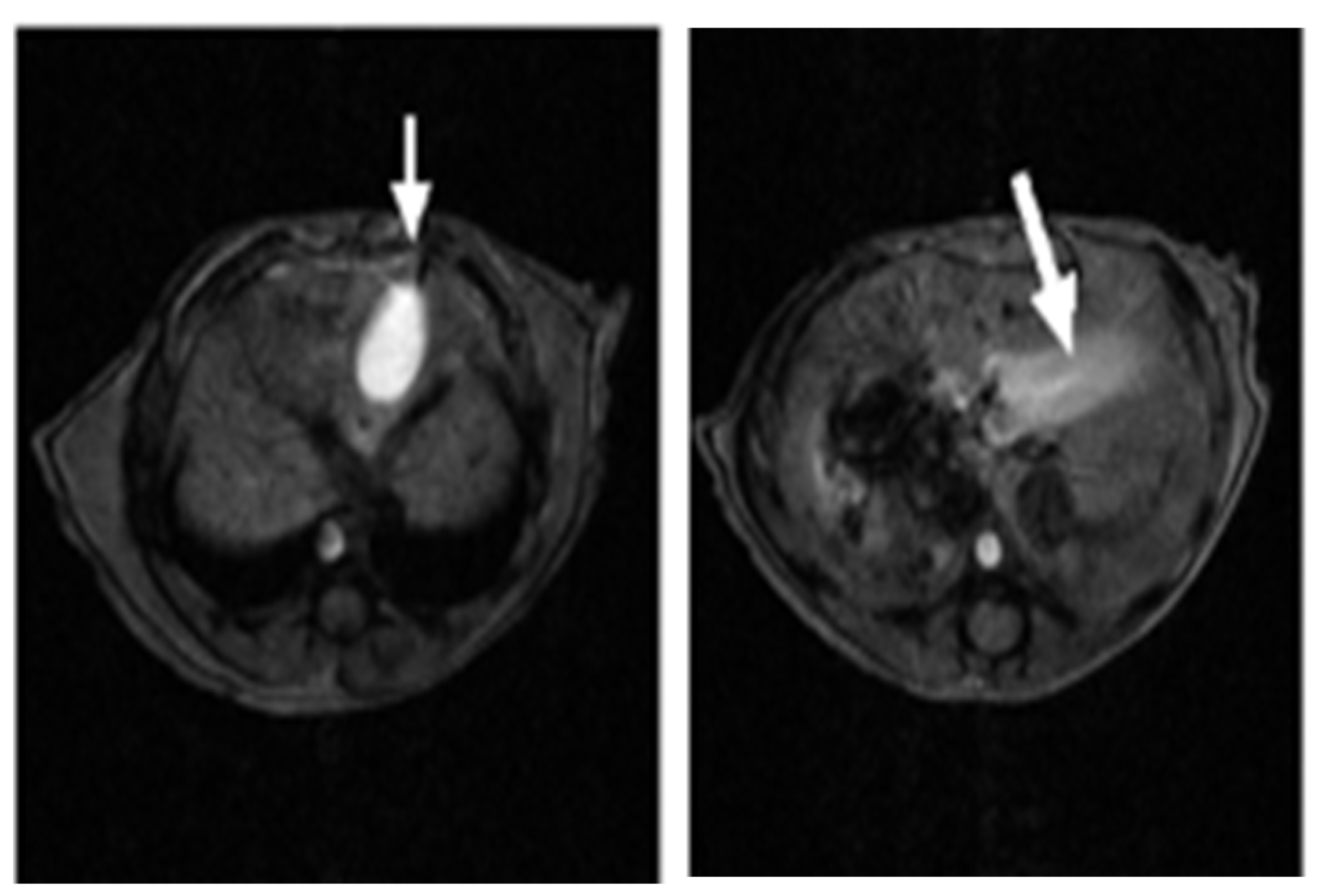
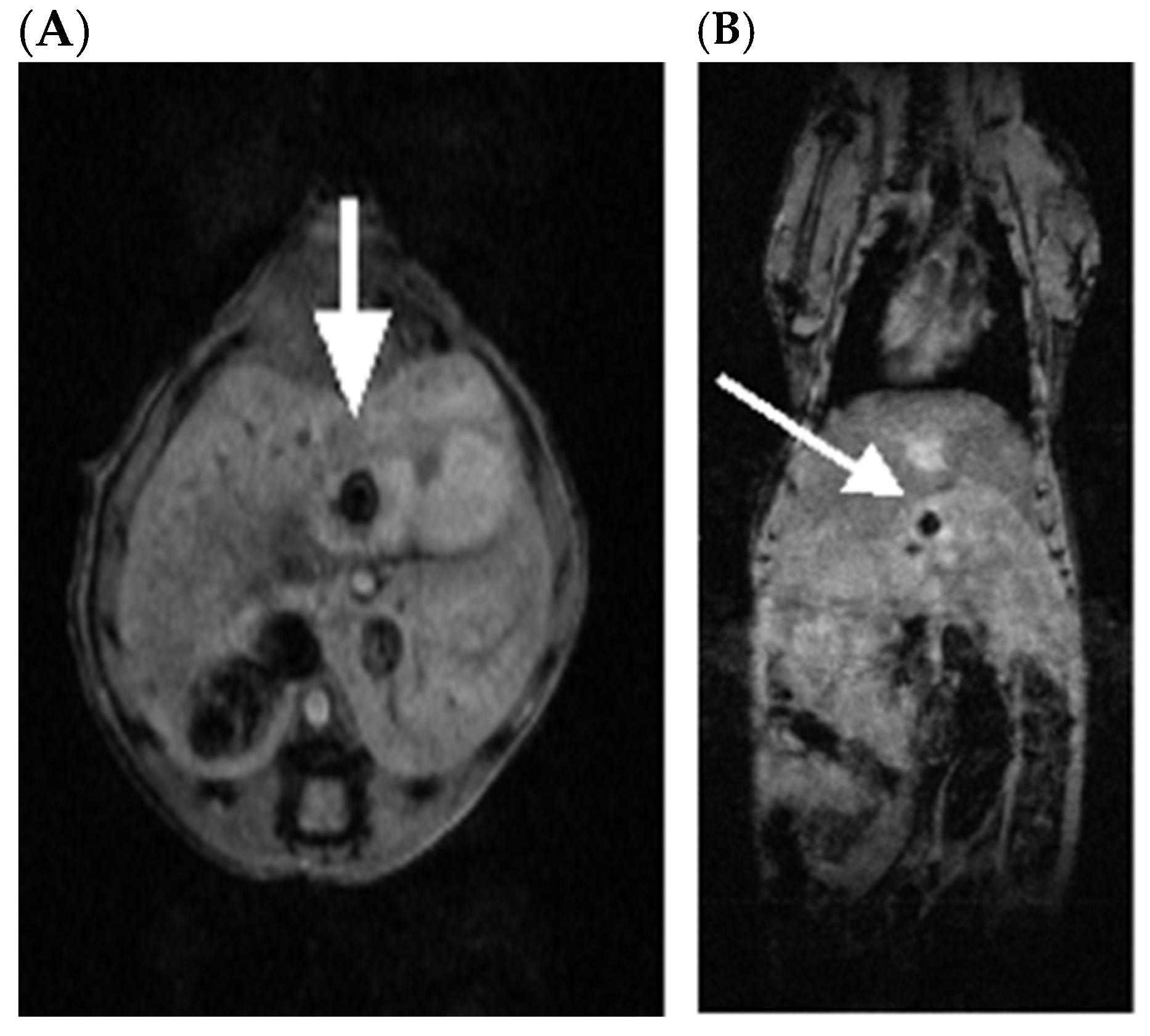
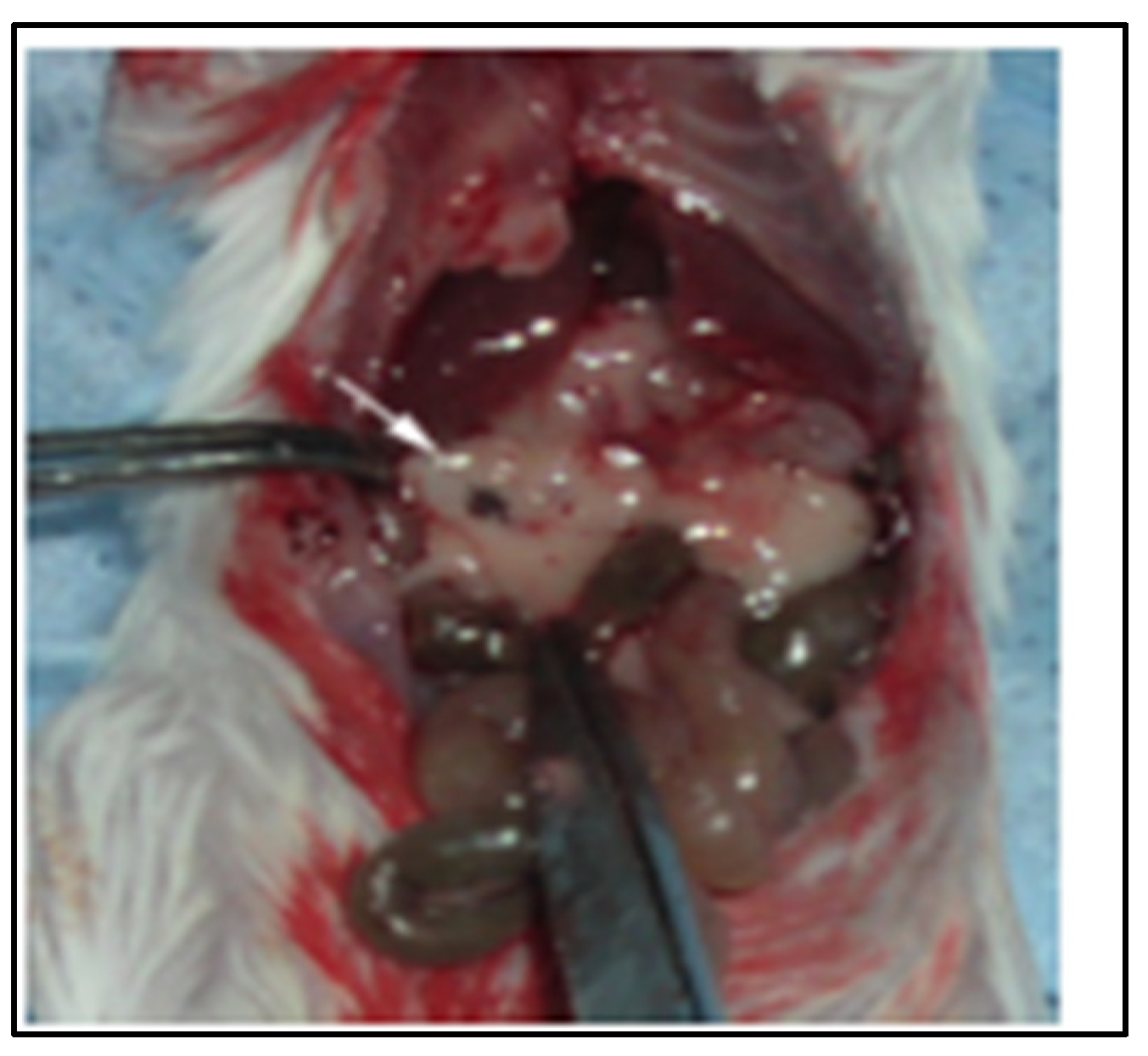
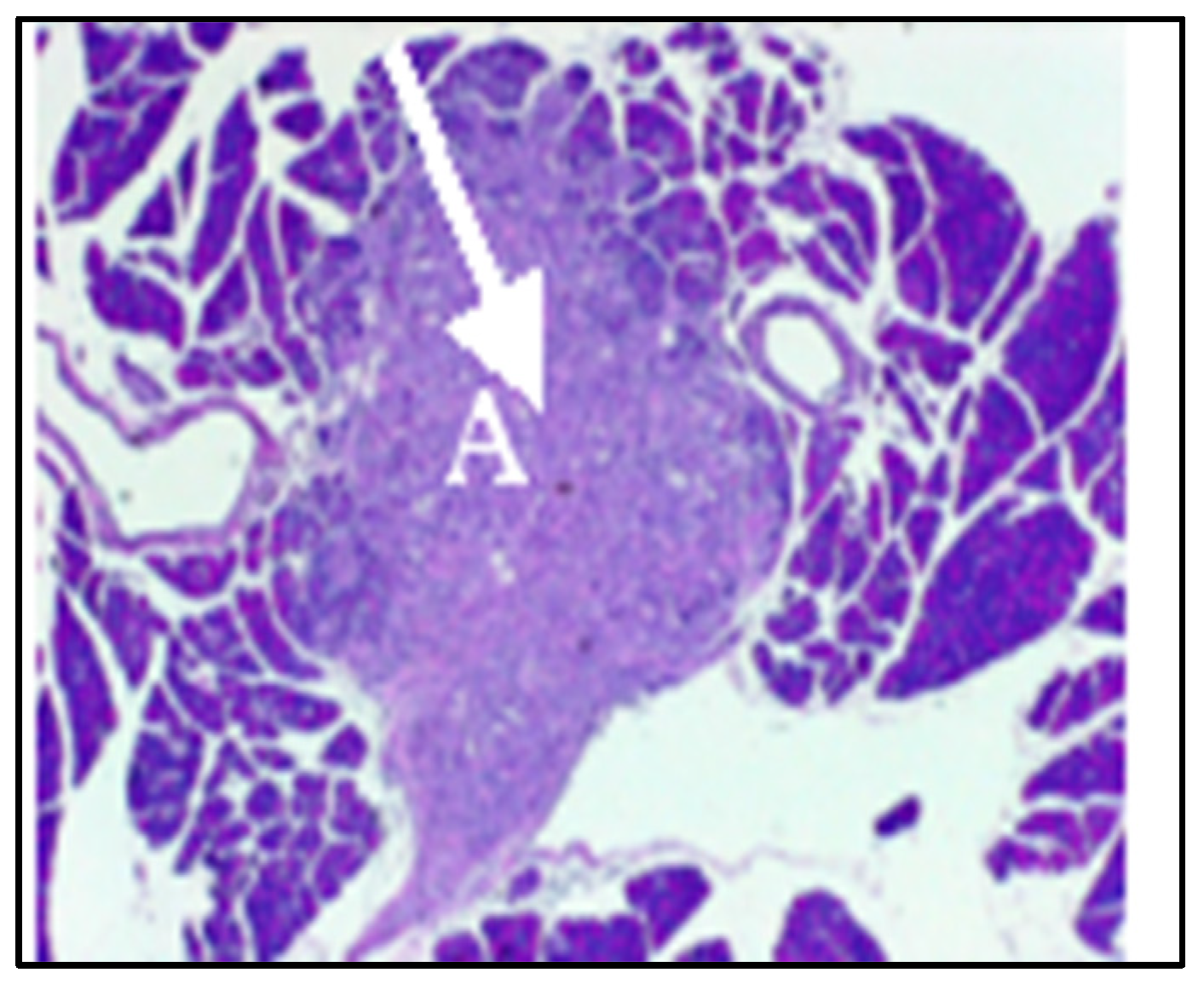
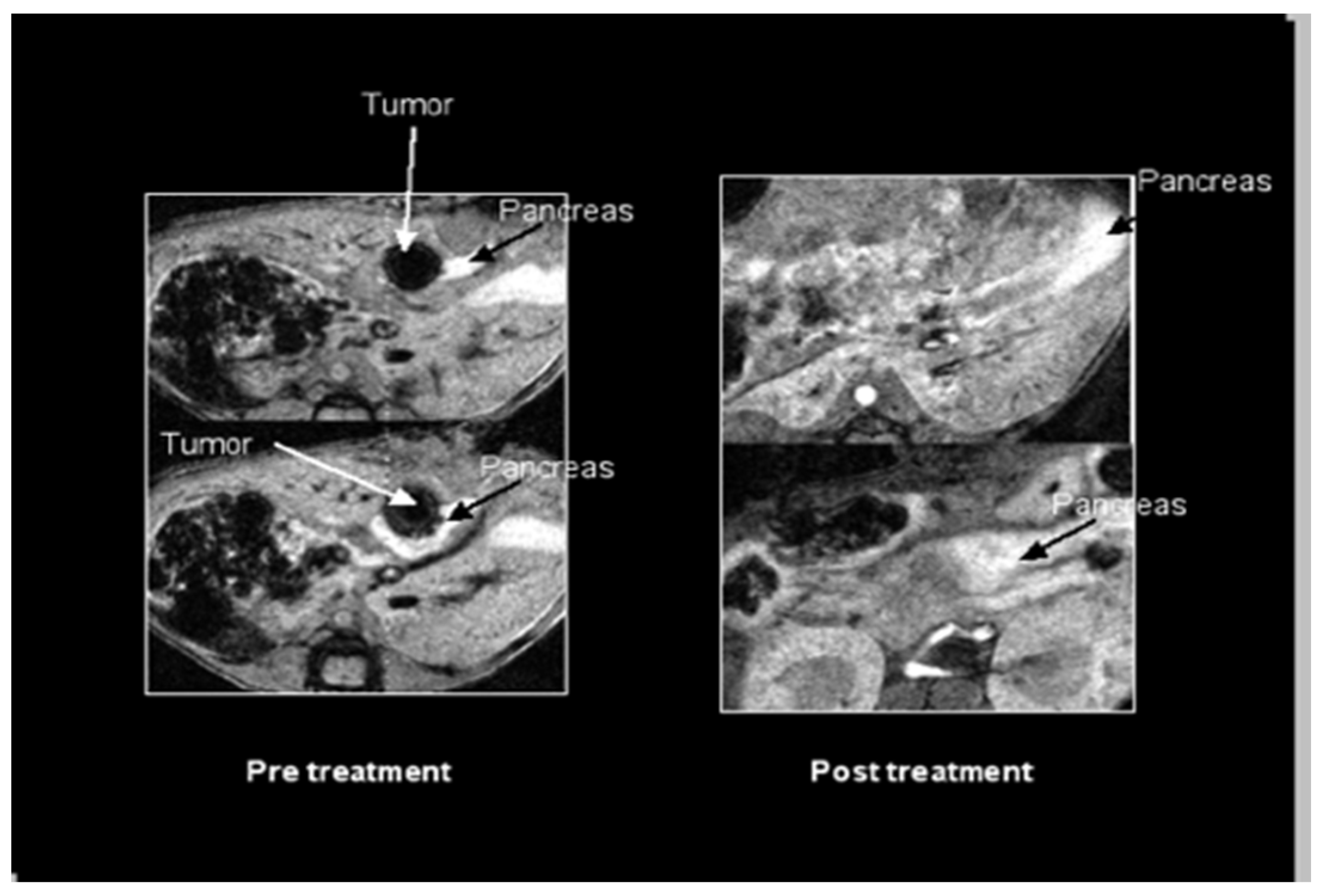
2.1. SV Vectors Can Generate Regression of Human Pancreatic Tumors in an SCID Mouse Model
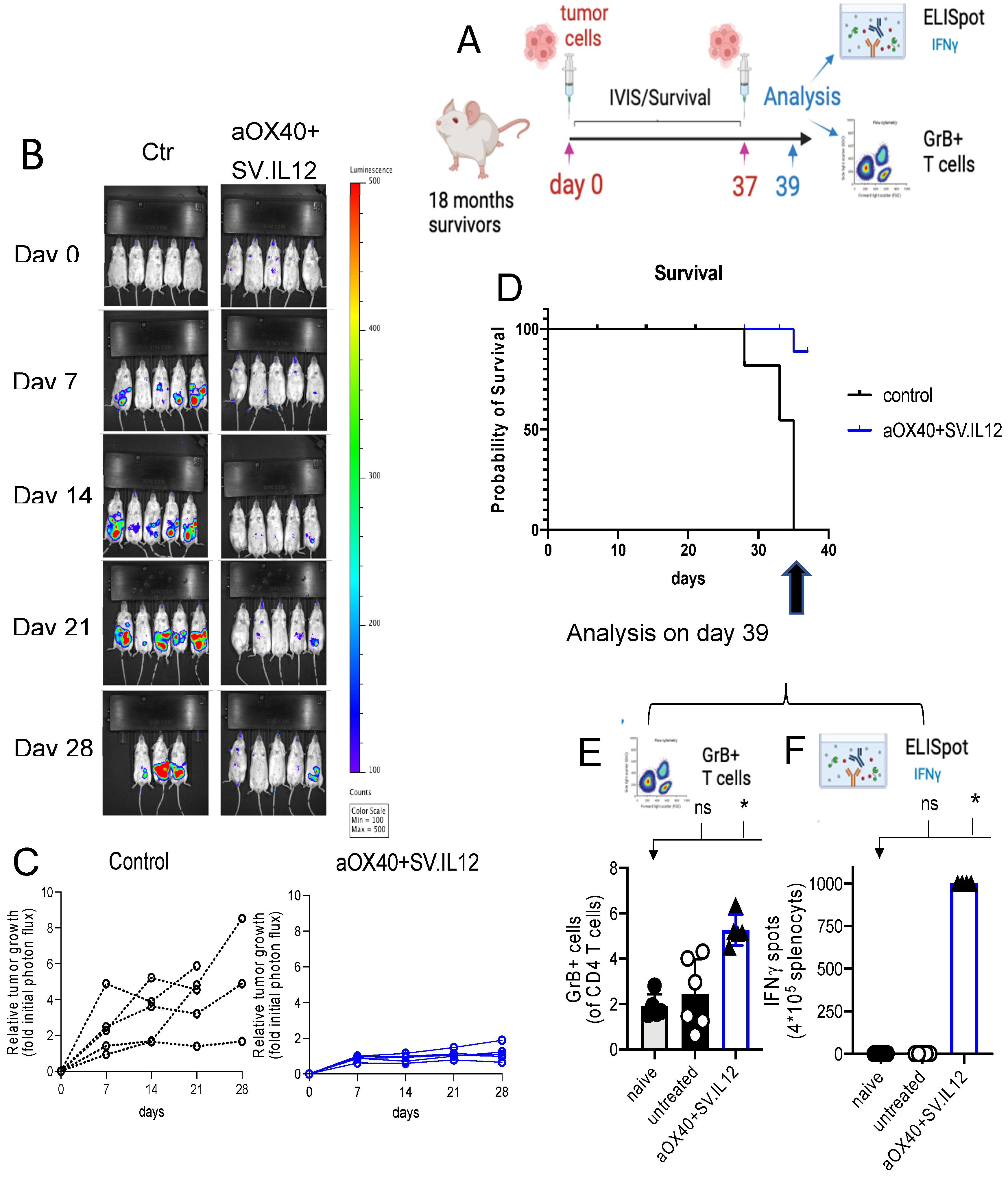
2.2. SV.IL-12 Can Reduce Metastasis in a Pan02 Syngeneic Mouse Model
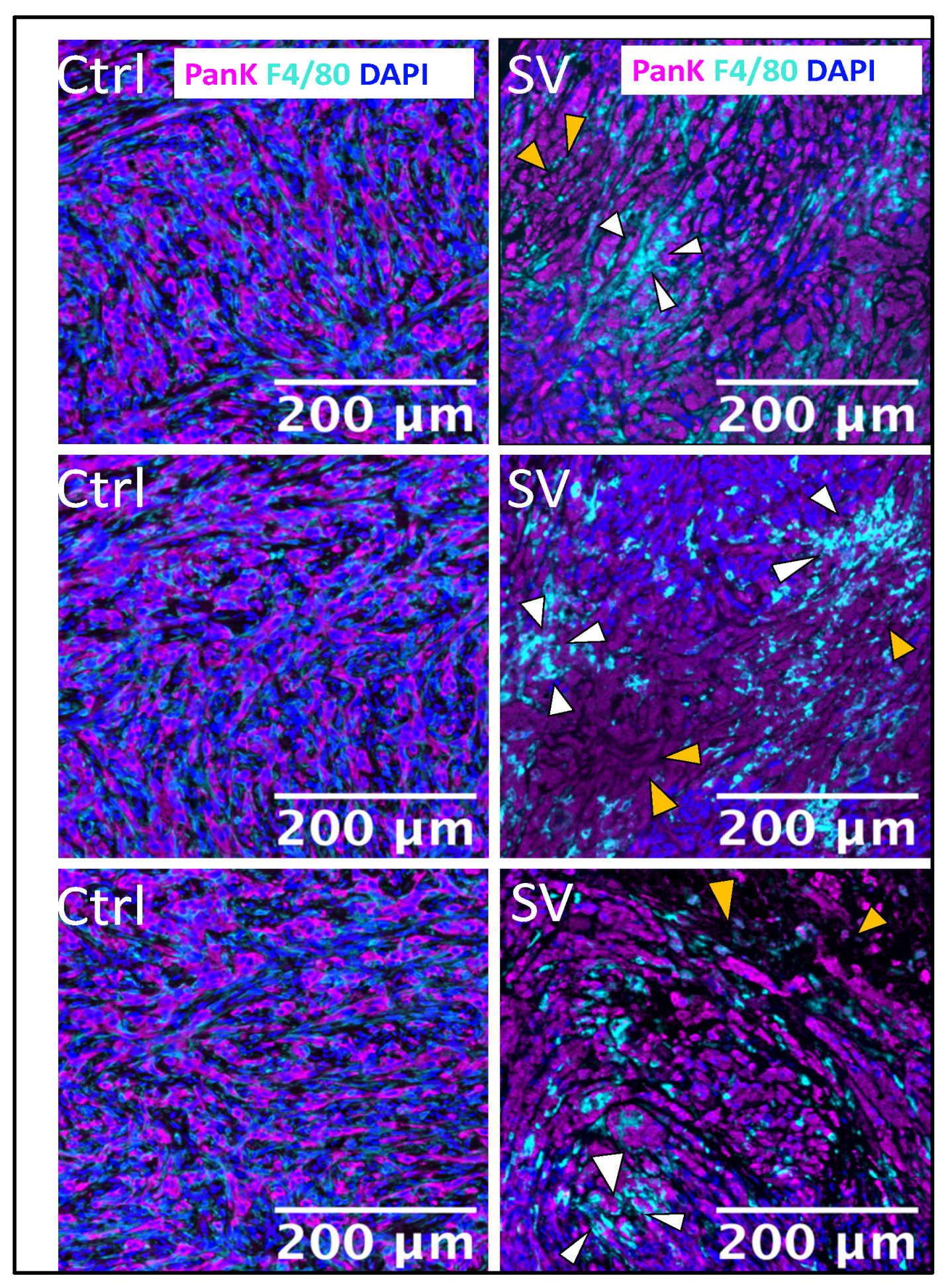
2.3. Effect of SV Vector on PDAC Models with Immunosuppressive TME
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. SV Production
4.3. Animal Experiments and Tumor Models
4.4. Histochemistry and Multiplex Immunofluorescence (MIF)
4.5. Flow Cytometry Analysis of Granzyme B on CD4 T Cells and Elispot Analysis of IFNγ
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SV | Sindbis Viral Vector |
| PDAC | Pancreatic ductal carcinoma |
| TME | Tumor microenvironment |
| TNFR | Tumor necrosis factor receptor |
| PanK | Pan cytokeratin |
| TAA | Tumor-associated antigens |
References
- Opp, S.; Hurtado, A.; Pampeno, C.; Lin, Z.; Meruelo, D. Potent and Targeted Sindbis Virus Platform for Immunotherapy of Ovarian Cancer. Cells 2022, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Pampeno, C.; Opp, S.; Hurtado, A.; Meruelo, D. Sindbis Virus Vaccine Platform: A Promising Oncolytic Virus-Mediated Approach for Ovarian Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 2925. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Ryan, D.P. Pancreatic Cancer: A Time to Change. Ann. Surg. 2020, 271, 1003–1004. [Google Scholar] [CrossRef]
- Guo, S.; Contratto, M.; Miller, G.; Leichman, L.; Wu, J. Immunotherapy in pancreatic cancer: Unleash its potential through novel combinations. World J. Clin. Oncol. 2017, 8, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Ebelt, N.D.; Zamloot, V.; Manuel, E.R. Targeting desmoplasia in pancreatic cancer as an essential first step to effective therapy. Oncotarget 2020, 11, 3486–3488. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Bayne, L.J. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol. 2013, 25, 200–205. [Google Scholar] [CrossRef]
- Ullman, N.A.; Burchard, P.R.; Dunne, R.F.; Linehan, D.C. Immunologic Strategies in Pancreatic Cancer: Making Cold Tumors Hot. J. Clin. Oncol. 2022, 40, 2789–2805. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef]
- Scharping, N.E.; Delgoffe, G.M. Tumor Microenvironment Metabolism: A New Checkpoint for Anti-Tumor Immunity. Vaccines 2016, 4, 46. [Google Scholar] [CrossRef] [PubMed]
- Scherwitzl, I.; Hurtado, A.; Pierce, C.M.; Vogt, S.; Pampeno, C.; Meruelo, D. Systemically Administered Sindbis Virus in Combination with Immune Checkpoint Blockade Induces Curative Anti-tumor Immunity. Mol. Ther. Oncolytics 2018, 9, 51–63. [Google Scholar] [CrossRef]
- Scherwitzl, I.; Opp, S.; Hurtado, A.M.; Pampeno, C.; Loomis, C.; Kannan, K.; Yu, M.; Meruelo, D. Sindbis Virus with Anti-OX40 Overcomes the Immunosuppressive Tumor Microenvironment of Low-Immunogenic Tumors. Mol. Ther. Oncolytics 2020, 17, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Bansal-Pakala, P.; Halteman, B.S.; Cheng, M.H.; Croft, M. Costimulation of CD8 T cell responses by OX40. J. Immunol. 2004, 172, 4821–4825. [Google Scholar] [CrossRef]
- Gramaglia, I.; Weinberg, A.D.; Lemon, M.; Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998, 161, 6510–6517. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shi, B.M.; Xie, F.; Fu, Z.Y.; Chen, Y.J.; An, J.N.; Ma, Y.; Liu, C.P.; Zhang, X.K.; Zhang, X.G. Enhancement of CD4(+) T cell response and survival via coexpressed OX40/OX40L in Graves’ disease. Mol. Cell. Endocrinol. 2016, 430, 115–124. [Google Scholar] [CrossRef]
- Zander, R.A.; Obeng-Adjei, N.; Guthmiller, J.J.; Kulu, D.I.; Li, J.; Ongoiba, A.; Traore, B.; Crompton, P.D.; Butler, N.S. PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity. Cell Host Microbe 2015, 17, 628–641. [Google Scholar] [CrossRef]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef]
- Song, A.; Tang, X.; Harms, K.M.; Croft, M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. J. Immunol. 2005, 175, 3534–3541. [Google Scholar] [CrossRef]
- Tahiliani, V.; Hutchinson, T.E.; Abboud, G.; Croft, M.; Salek-Ardakani, S. OX40 Cooperates with ICOS To Amplify Follicular Th Cell Development and Germinal Center Reactions during Infection. J. Immunol. 2017, 198, 218–228. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, X.; Lan, P.; Li, J.; Dou, Y.; Chen, W.; Ishii, N.; Chen, S.; Xia, B.; Chen, K.; et al. OX40 Costimulation Inhibits Foxp3 Expression and Treg Induction via BATF3-Dependent and Independent Mechanisms. Cell Rep. 2018, 24, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, S.; Kim, S.; Chung, E.Y.; Homma, Y.; Guan, X.; Jimenez, V.; Ma, X. Interleukin-12: An update on its immunological activities, signaling and regulation of gene expression. Curr. Immunol. Rev. 2005, 1, 119–137. [Google Scholar] [CrossRef]
- Tseng, J.C.; Levin, B.; Hirano, T.; Yee, H.; Pampeno, C.; Meruelo, D. In vivo antitumor activity of Sindbis viral vectors. J. Natl. Cancer Inst. 2002, 94, 1790–1802. [Google Scholar] [CrossRef]
- Tseng, J.C.; Levin, B.; Hurtado, A.; Yee, H.; Perez de Castro, I.; Jimenez, M.; Shamamian, P.; Jin, R.; Novick, R.P.; Pellicer, A.; et al. Systemic tumor targeting and killing by Sindbis viral vectors. Nat. Biotechnol. 2004, 22, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Venticinque, L.; Meruelo, D. Sindbis viral vector induced apoptosis requires translational inhibition and signaling through Mcl-1 and Bak. Mol. Cancer 2010, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, A.; Tseng, J.C.; Meruelo, D. Gene therapy that safely targets and kills tumor cells throughout the body. Rejuvenation Res. 2006, 9, 36–44. [Google Scholar] [CrossRef]
- Tseng, J.C.; Hurtado, A.; Yee, H.; Levin, B.; Boivin, C.; Benet, M.; Blank, S.V.; Pellicer, A.; Meruelo, D. Using sindbis viral vectors for specific detection and suppression of advanced ovarian cancer in animal models. Cancer Res. 2004, 64, 6684–6692. [Google Scholar] [CrossRef]
- Lundstrom, K. Alphaviruses in Cancer Therapy. Front. Mol. Biosci. 2022, 9, 864781. [Google Scholar] [CrossRef]
- Lundstrom, K. Alphaviruses in Immunotherapy and Anticancer Therapy. Biomedicines 2022, 10, 2263. [Google Scholar] [CrossRef]
- Pampeno, C.; Hurtado, A.; Opp, S.; Meruelo, D. Channeling the Natural Properties of Sindbis Alphavirus for Targeted Tumor Therapy. Int. J. Mol. Sci. 2023, 24, 4948. [Google Scholar] [CrossRef]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Zimmerman, O.; Holmes, A.C.; Kafai, N.M.; Adams, L.J.; Diamond, M.S. Entry receptors—The gateway to alphavirus infection. J. Clin. Invest. 2023, 133, e165307. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, D.; Maehara, N.; Kuba, K.; Mizumoto, K.; Tanaka, M.; Matsumoto, K.; Nakamura, T. Inhibition of growth, invasion, and metastasis of human pancreatic carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res. 2001, 61, 7518–7524. [Google Scholar]
- Corbett, T.H.; Roberts, B.J.; Leopold, W.R.; Peckham, J.C.; Wilkoff, L.J.; Griswold, D.P., Jr.; Schabel, F.M., Jr. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984, 44, 717–726. [Google Scholar] [PubMed]
- Greco, S.H.; Tomkötter, L.; Vahle, A.K.; Rokosh, R.; Avanzi, A.; Mahmood, S.K.; Deutsch, M.; Alothman, S.; Alqunaibit, D.; Ochi, A.; et al. TGF-β Blockade Reduces Mortality and Metabolic Changes in a Validated Murine Model of Pancreatic Cancer Cachexia. PLoS ONE 2015, 10, e0132786. [Google Scholar] [CrossRef]
- Pham, T.N.D.; Shields, M.A.; Spaulding, C.; Principe, D.R.; Li, B.; Underwood, P.W.; Trevino, J.G.; Bentrem, D.J.; Munshi, H.G. Preclinical Models of Pancreatic Ductal Adenocarcinoma and Their Utility in Immunotherapy Studies. Cancers 2021, 13, 440. [Google Scholar] [CrossRef] [PubMed]
- Suklabaidya, S.; Dash, P.; Das, B.; Suresh, V.; Sasmal, P.K.; Senapati, S. Experimental models of pancreatic cancer desmoplasia. Lab. Invest. 2018, 98, 27–40. [Google Scholar] [CrossRef]
- Torres, M.P.; Rachagani, S.; Souchek, J.J.; Mallya, K.; Johansson, S.L.; Batra, S.K. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: Applications in diagnosis and therapy. PLoS ONE 2013, 8, e80580. [Google Scholar] [CrossRef]
- Yu, M.; Scherwitzl, I.; Opp, S.; Tsirigos, A.; Meruelo, D. Molecular and metabolic pathways mediating curative treatment of a non-Hodgkin B cell lymphoma by Sindbis viral vectors and anti-4-1BB monoclonal antibody. J. Immunother. Cancer 2019, 7, 185. [Google Scholar] [CrossRef]
- Ishihara, J.; Ishihara, A.; Potin, L.; Hosseinchi, P.; Fukunaga, K.; Damo, M.; Gajewski, T.F.; Swartz, M.A.; Hubbell, J.A. Improving Efficacy and Safety of Agonistic Anti-CD40 Antibody Through Extracellular Matrix Affinity. Mol. Cancer Ther. 2018, 17, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Thoidingjam, S.; Bhatnagar, A.R.; Sriramulu, S.; Siddiqui, F.; Nyati, S. Optimizing Pancreatic Cancer Therapy: The Promise of Immune Stimulatory Oncolytic Viruses. Int. J. Mol. Sci. 2024, 25, 9912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Chen, K.; Qian, L.; Wang, P. Oncolytic virotherapy against the tumor microenvironment and its potential in pancreatic cancer. J. Cancer Res. Ther. 2022, 18, 1247–1255. [Google Scholar] [CrossRef]
- Bhatnagar, A.R.; Siddiqui, F.; Khan, G.; Pompa, R.; Kwon, D.; Nyati, S. Long-Term Follow-Up of Phase I Trial of Oncolytic Adenovirus-Mediated Cytotoxic and Interleukin-12 Gene Therapy for Treatment of Metastatic Pancreatic Cancer. Biomedicines 2024, 12, 1065. [Google Scholar] [CrossRef]
- Musher, B.L.; Rowinsky, E.K.; Smaglo, B.G.; Abidi, W.; Othman, M.; Patel, K.; Jawaid, S.; Jing, J.; Brisco, A.; Leen, A.M.; et al. LOAd703, an oncolytic virus-based immunostimulatory gene therapy, combined with chemotherapy for unresectable or metastatic pancreatic cancer (LOKON001): Results from arm 1 of a non-randomised, single-centre, phase 1/2 study. Lancet Oncol. 2024, 25, 488–500. [Google Scholar] [CrossRef]
- Liu, S.; Li, F.; Ma, Q.; Du, M.; Wang, H.; Zhu, Y.; Deng, L.; Gao, W.; Wang, C.; Liu, Y.; et al. OX40L-Armed Oncolytic Virus Boosts T-cell Response and Remodels Tumor Microenvironment for Pancreatic Cancer Treatment. Theranostics 2023, 13, 4016–4029. [Google Scholar] [CrossRef]
- Ma, Y.; Li, J.; Wang, H.; Chiu, Y.; Kingsley, C.V.; Fry, D.; Delaney, S.N.; Wei, S.C.; Zhang, J.; Maitra, A.; et al. Combination of PD-1 Inhibitor and OX40 Agonist Induces Tumor Rejection and Immune Memory in Mouse Models of Pancreatic Cancer. Gastroenterology 2020, 159, 306–319.e312. [Google Scholar] [CrossRef]
- Choi, Y.; Chang, J. Viral vectors for vaccine applications. Clin. Exp. Vaccine Res. 2013, 2, 97–105. [Google Scholar] [CrossRef]
- Gardner, J.P.; Frolov, I.; Perri, S.; Ji, Y.; MacKichan, M.L.; zur Megede, J.; Chen, M.; Belli, B.A.; Driver, D.A.; Sherrill, S.; et al. Infection of human dendritic cells by a sindbis virus replicon vector is determined by a single amino acid substitution in the E2 glycoprotein. J. Virol. 2000, 74, 11849–11857. [Google Scholar] [CrossRef]
- Granot, T.; Yamanashi, Y.; Meruelo, D. Sindbis viral vectors transiently deliver tumor-associated antigens to lymph nodes and elicit diversified antitumor CD8+ T-cell immunity. Mol. Ther. 2014, 22, 112–122. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, G.H.; Johnston, R.E. Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J. Virol. 2000, 74, 914–922. [Google Scholar] [CrossRef]
- Osada, T.; Morse, M.A.; Hobeika, A.; Lyerly, H.K. Novel recombinant alphaviral and adenoviral vectors for cancer immunotherapy. Semin. Oncol. 2012, 39, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Pushko, P.; Parker, M.; Ludwig, G.V.; Davis, N.L.; Johnston, R.E.; Smith, J.F. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: Expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 1997, 239, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, Y.; Vajdy, M.; Lian, Y.; Perri, S.; Greer, C.E.; Legg, H.S.; Galli, G.; Saletti, G.; Otten, G.R.; Rappuoli, R.; et al. Lack of interference with immunogenicity of a chimeric alphavirus replicon particle-based influenza vaccine by preexisting antivector immunity. Clin. Vaccine Immunol. 2012, 19, 991–998. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Levine, B. Sindbis virus vector system for functional analysis of apoptosis regulators. Methods Enzymol. 2000, 322, 492–508. [Google Scholar] [CrossRef]
- Sane, J.; Kurkela, S.; Lokki, M.L.; Miettinen, A.; Helve, T.; Vaheri, A.; Vapalahti, O. Clinical Sindbis alphavirus infection is associated with HLA-DRB1*01 allele and production of autoantibodies. Clin. Infect. Dis. 2012, 55, 358–363. [Google Scholar] [CrossRef]
- Strauss, J.H.; Wang, K.S.; Schmaljohn, A.L.; Kuhn, R.J.; Strauss, E.G. Host-cell receptors for Sindbis virus. Arch. Virol. Suppl. 1994, 9, 473–484. [Google Scholar] [CrossRef]
- Bredenbeek, P.J.; Frolov, I.; Rice, C.M.; Schlesinger, S. Sindbis virus expression vectors: Packaging of RNA replicons by using defective helper RNAs. J. Virol. 1993, 67, 6439–6446. [Google Scholar] [CrossRef]
- Frolov, I.; Hoffman, T.A.; Prágai, B.M.; Dryga, S.A.; Huang, H.V.; Schlesinger, S.; Rice, C.M. Alphavirus-based expression vectors: Strategies and applications. Proc. Natl. Acad. Sci. USA 1996, 93, 11371–11377. [Google Scholar] [CrossRef]
- Hurtado, A.; Tseng, J.C.; Boivin, C.; Levin, B.; Yee, H.; Pampeno, C.; Meruelo, D. Identification of amino acids of Sindbis virus E2 protein involved in targeting tumor metastases in vivo. Mol. Ther. 2005, 12, 813–823. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | No Vector Treatment | Sindbis-IL-12 |
|---|---|---|
| Primary tumor | ++++ | + |
| Liver | ++ | + |
| Spleen | +++ | + |
| Perirectum | ++ | None |
| Peritoneal seeding | + | None |
| Lymph nodes | ++ | None |
| Periaortic | ++ | + |
| Omental | + | + |
| Mesenteric | ++ | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opp, S.; Pampeno, C.; Hurtado, A.; Meruelo, D. Sindbis Virus Platform Provides an Oncolytic-Virus-Mediated and Immunotherapeutic Strategy to Overcome the Challenging Microenvironment of Pancreatic Cancer. Pharmaceuticals 2025, 18, 725. https://doi.org/10.3390/ph18050725
Opp S, Pampeno C, Hurtado A, Meruelo D. Sindbis Virus Platform Provides an Oncolytic-Virus-Mediated and Immunotherapeutic Strategy to Overcome the Challenging Microenvironment of Pancreatic Cancer. Pharmaceuticals. 2025; 18(5):725. https://doi.org/10.3390/ph18050725
Chicago/Turabian StyleOpp, Silvana, Christine Pampeno, Alicia Hurtado, and Daniel Meruelo. 2025. "Sindbis Virus Platform Provides an Oncolytic-Virus-Mediated and Immunotherapeutic Strategy to Overcome the Challenging Microenvironment of Pancreatic Cancer" Pharmaceuticals 18, no. 5: 725. https://doi.org/10.3390/ph18050725
APA StyleOpp, S., Pampeno, C., Hurtado, A., & Meruelo, D. (2025). Sindbis Virus Platform Provides an Oncolytic-Virus-Mediated and Immunotherapeutic Strategy to Overcome the Challenging Microenvironment of Pancreatic Cancer. Pharmaceuticals, 18(5), 725. https://doi.org/10.3390/ph18050725