

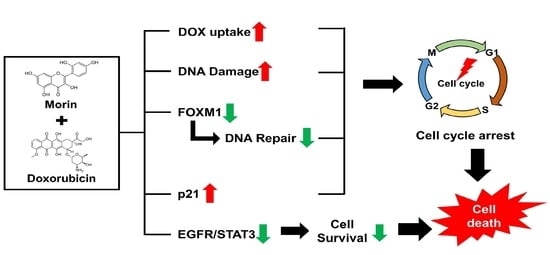
Morin Sensitizes MDA-MB-231 Triple-Negative Breast Cancer Cells to Doxorubicin Cytotoxicity by Suppressing FOXM1 and Attenuating EGFR/STAT3 Signaling Pathways

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Morin/Dox Co-Treatment Increased the Cytotoxic Effect on MDA-MB-231 TNBC Cells
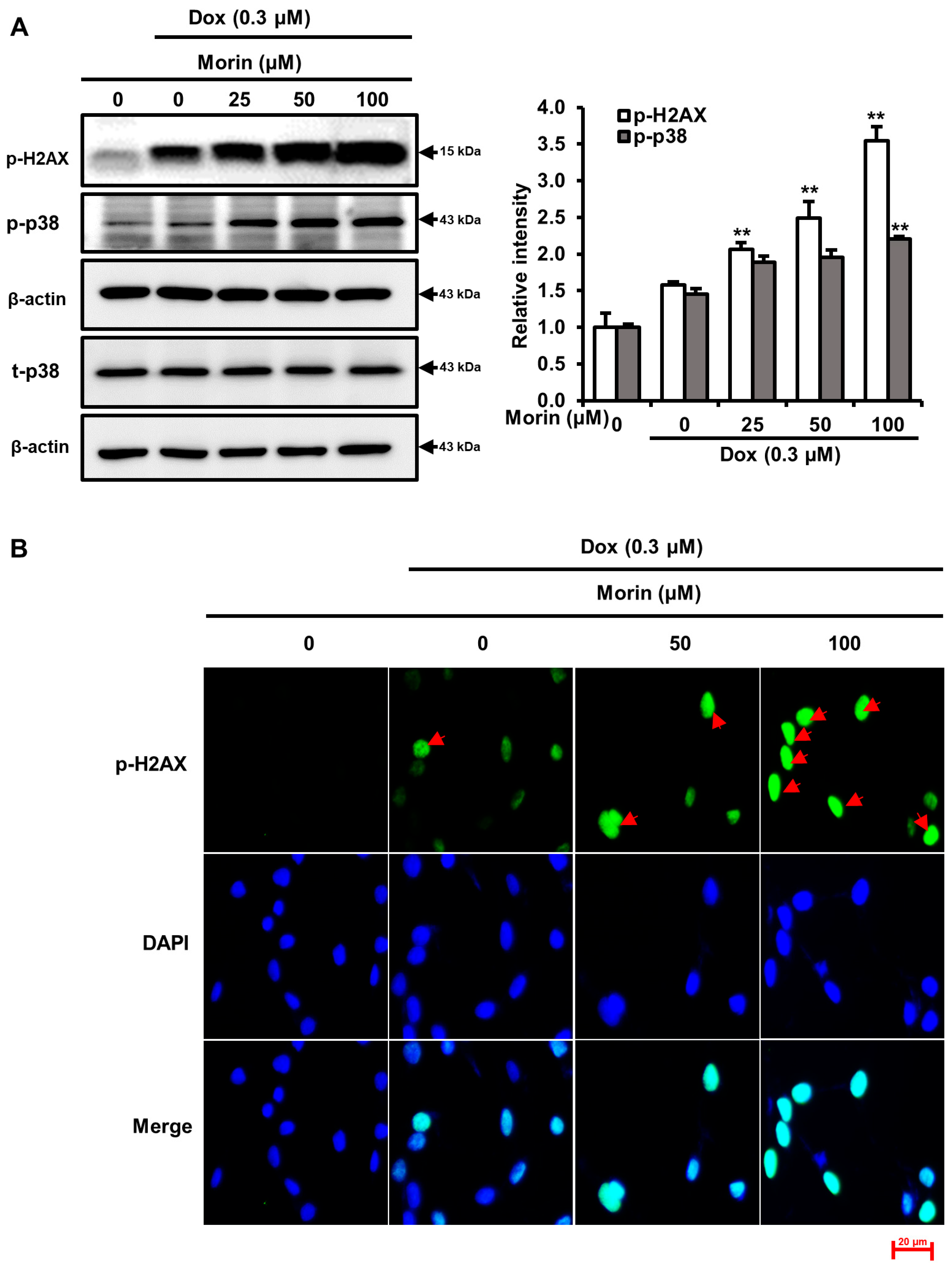
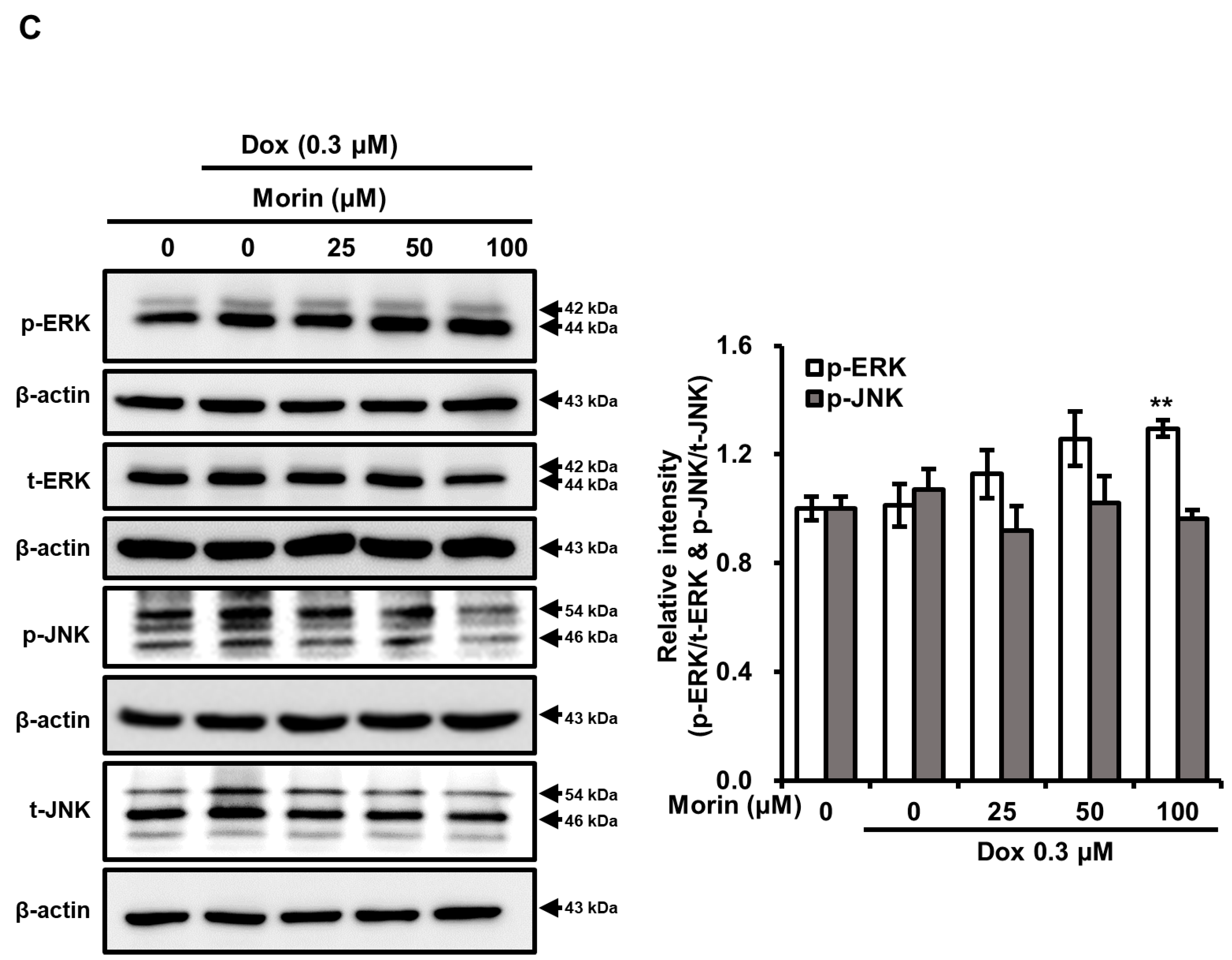
2.2. Morin/Dox Co-Treatment Increased DNA Damage and p38 Activation
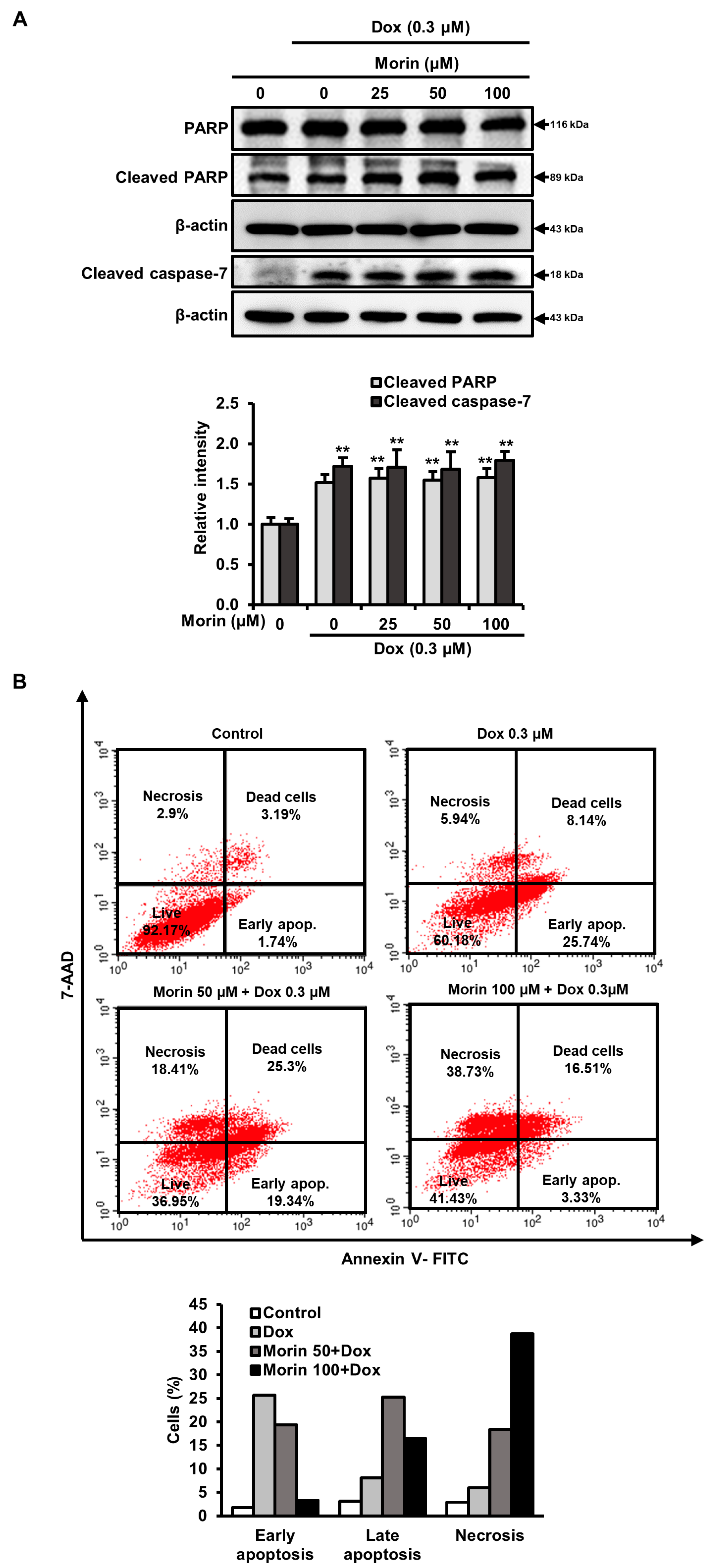
2.3. Morin/Dox Co-Treatment Promoted Necrotic Cell Death
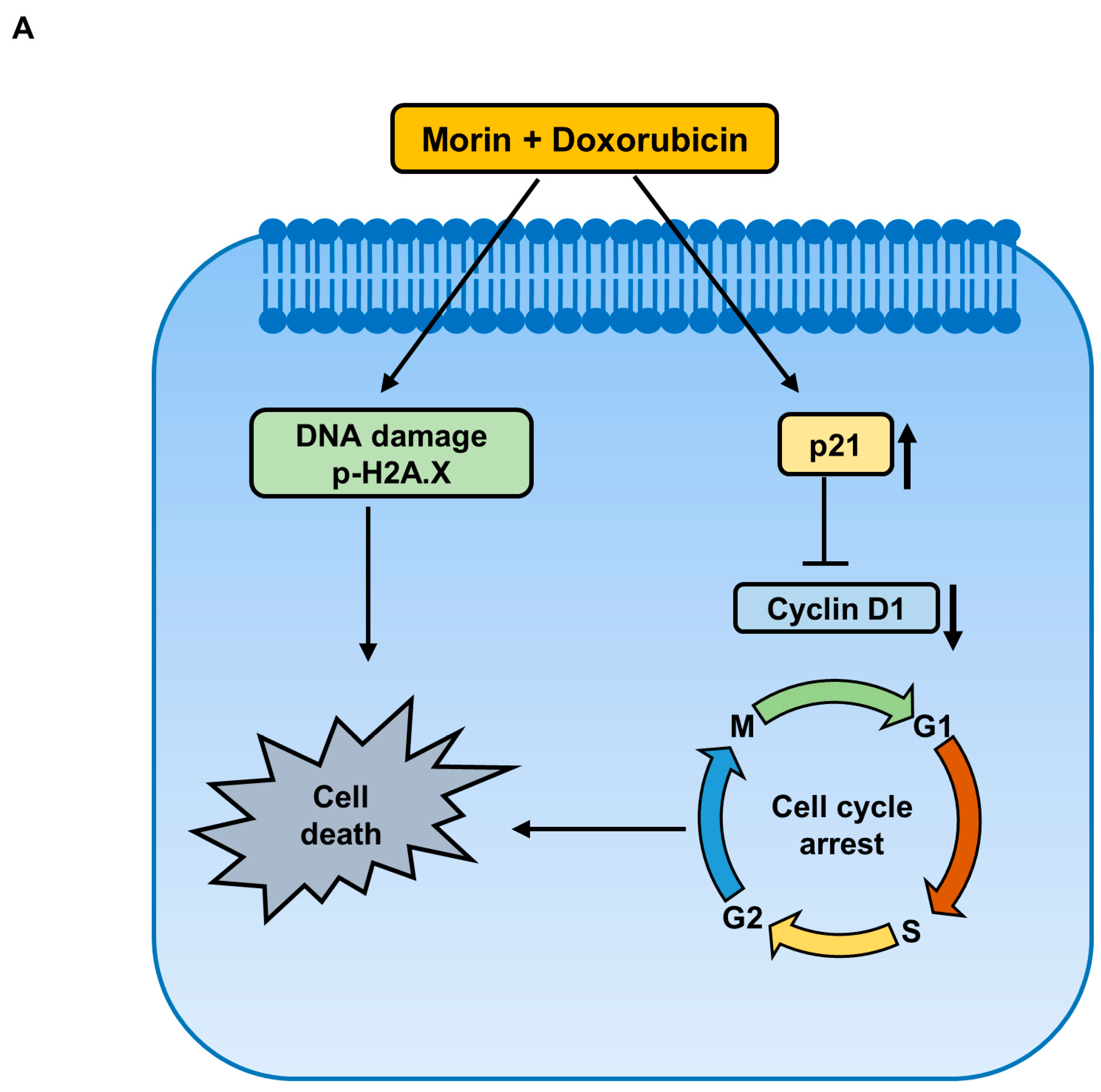
2.4. Morin/Dox Co-Treatment Induced Cell Death by Suppressing the FOXM1 Pathway
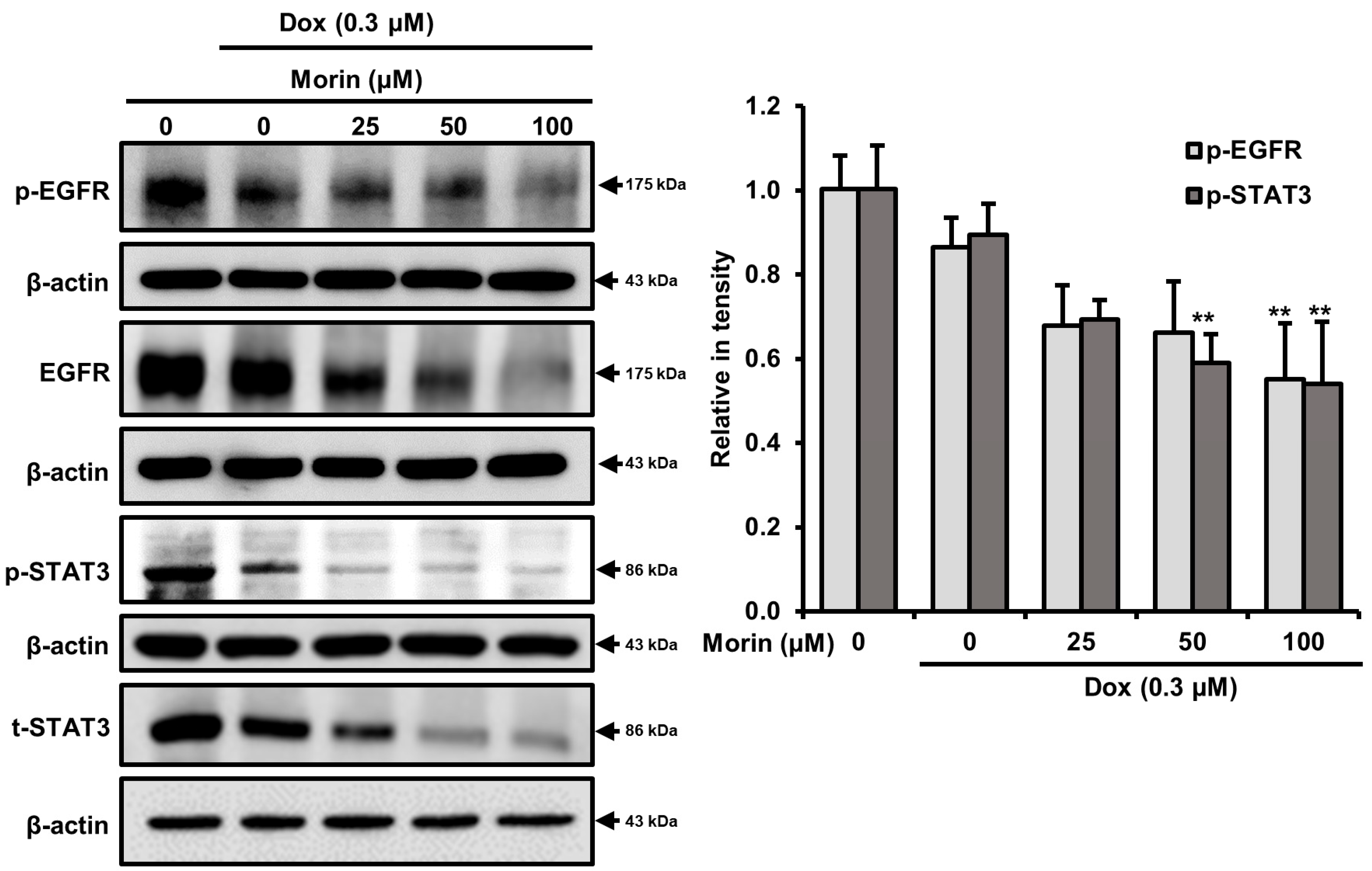
2.5. Morin/Dox Co-Treatment Induced Cell Death by Inhibiting the EGFR/STAT3 Pathway
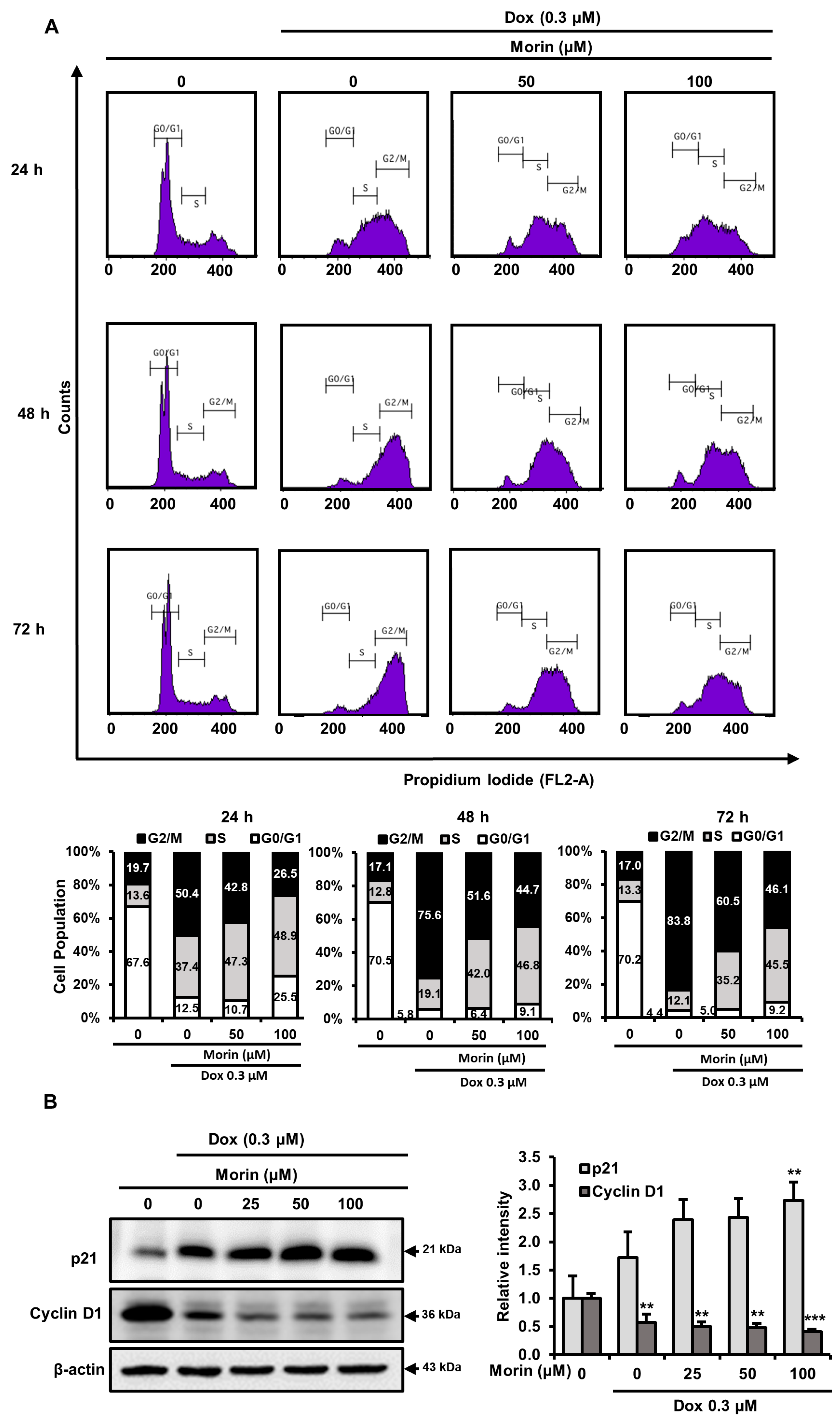
2.6. Morin/Dox Co-Treatment Disrupted Cell Cycle Progression
2.7. Morin/Dox Co-Treatment Affected the Expressions of Cell Cycle Regulators
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cytotoxicity Assay
4.4. Doxorubicin Accumulation Assay
4.5. Combination Index Determinations
4.6. Cell Cycle Analysis
4.7. Western Blotting
4.8. Immunofluorescence Staining
4.9. Analysis of Apoptotic and Necrotic Cells
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shuhendler, A.J.; O’Brien, P.J.; Rauth, A.M.; Wu, X.Y. On the synergistic effect of doxorubicin and mitomycin C against breast cancer cells. Drug Metab. Drug Interact 2007, 22, 201–233. [Google Scholar] [CrossRef] [PubMed]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Wright, G.; Bryant, H.; Wiggins, L.A.; Dal Zotto, V.L.; Schuler, M.; Malozzi, C.; Cohen, M.V.; Gassman, N.R. Cytoprotective Effect of Vitamin D on Doxorubicin-Induced Cardiac Toxicity in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 7439. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Wang, L.; Agyin, J.; Tang, Y.; Lin, S.; Yeh, I.T.; De, K.; Sun, L.-Z. Doxorubicin in Combination with a Small TGFβ Inhibitor: A Potential Novel Therapy for Metastatic Breast Cancer in Mouse Models. PLoS ONE 2010, 5, e10365. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Lee, J.S.; Hong, E.K. Hericium erinaceus enhances doxorubicin-induced apoptosis in human hepatocellular carcinoma cells. Cancer Lett. 2010, 297, 144–154. [Google Scholar] [CrossRef]
- Abad, M.N.; Calabuig-Fariñas, S.; de Mena, M.L.; de Bremond, M.J.G.S.; González, C.G.; Martínez, S.T.; García-García, J.Á.; González-Cruz, V.I.; Herrero, C.C. Update on systemic treatment in early triple negative breast cancer. Adv. Med. Oncol. 2021, 13, 1758835920986749. [Google Scholar]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Ciocan-Cartita, C.A.; Jurj, A.; Zanoaga, O.; Cojocneanu, R.; Pop, L.-A.; Moldovan, A.; Moldovan, C.; Zimta, A.A.; Raduly, L.; Pop-Bica, C.; et al. New insights in gene expression alteration as effect of doxorubicin drug resistance in triple negative breast cancer cells. J. Exp. Clin. Cancer Res. 2020, 39, 241. [Google Scholar] [CrossRef]
- Kuzu, M.; Kandemir, F.M.; Yildirim, S.; Kucukler, S.; Caglayan, C.; Turk, E. Morin attenuates doxorubicin-induced heart and brain damage by reducing oxidative stress, inflammation and apoptosis. Biomed. Pharmacother. 2018, 106, 443–453. [Google Scholar]
- Injac, R.; Strukelj, B. Recent advances in protection against doxorubicin-induced toxicity. Technol. Cancer Res. Treat. 2008, 7, 497–516. [Google Scholar] [CrossRef]
- Tyagi, A.K.; Agarwal, C.; Chan, D.C.; Agarwal, R. Synergistic anti-cancer effects of silibinin with conventional cytotoxic agents doxorubicin, cisplatin and carboplatin against human breast carcinoma MCF-7 and MDA-MB468 cells. Oncol. Rep. 2004, 11, 493–499. [Google Scholar] [CrossRef]
- Kapadia, G.J.; Rao, G.S.; Ramachandran, C.; Iida, A.; Suzuki, N.; Tokuda, H. Synergistic cytotoxicity of red beetroot (Beta vulgaris L.) extract with doxorubicin in human pancreatic, breast and prostate cancer cell lines. J. Complement. Integr. Med. 2013, 10, 113–122. [Google Scholar] [CrossRef]
- Tsao, R.; Yang, R.; Young, J.C. Antioxidant isoflavones in osage orange, Maclura pomifera (Raf.) Schneid. J. Agric. Food Chem. 2003, 51, 6445–6451. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, W.I.; Kim, S.Y.; Cho, S.W.; Nam, H.S.; Lee, S.H.; Cho, M.K. Flavonoid morin inhibits proliferation and induces apoptosis of melanoma cells by regulating reactive oxygen species, Sp1 and Mcl-1. Arch. Pharmacal Res. 2019, 42, 531–542. [Google Scholar] [CrossRef]
- Kapoor, R.; Kakkar, P. Protective role of morin, a flavonoid, against high glucose induced oxidative stress mediated apoptosis in primary rat hepatocytes. PLoS ONE 2012, 7, e41663. [Google Scholar] [CrossRef]
- Gupta, S.C.; Phromnoi, K.; Aggarwal, B.B. Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells through activation of protein tyrosine phosphatase SHP1. Biochem. Pharmacol. 2013, 85, 898–912. [Google Scholar] [CrossRef]
- Jin, H.; Lee, W.S.; Eun, S.Y.; Jung, J.H.; Park, H.-S.; Kim, G.; Choi, Y.H.; Ryu, C.H.; Jung, J.M.; Hong, S.C. Morin, a flavonoid from Moraceae, suppresses growth and invasion of the highly metastatic breast cancer cell line MDA-MB-231 partly through suppression of the Akt pathway. Int. J. Oncol. 2014, 45, 1629–1637. [Google Scholar] [CrossRef]
- Li, B.; Jin, X.; Meng, H.; Hu, B.; Zhang, T.; Yu, J.; Chen, S.; Guo, X.; Wang, W.; Jiang, W. Morin promotes prostate cancer cells chemosensitivity to paclitaxel through miR-155/GATA3 axis. Oncotarget 2017, 8, 47849. [Google Scholar] [CrossRef]
- Chung, S.S.; Oliva, B.; Dwabe, S.; Vadgama, J.V. Combination treatment with flavonoid morin and telomerase inhibitor MST-312 reduces cancer stem cell traits by targeting STAT3 and telomerase. Int. J. Oncol. 2016, 49, 487–498. [Google Scholar] [CrossRef]
- Popp, H.D.; Brendel, S.; Hofmann, W.-K.; Fabarius, A. Immunofluorescence Microscopy of γH2AX and 53BP1 for Analyzing the Formation and Repair of DNA Double-strand Breaks. J. Vis. Exp. JoVE 2017, 129, 56617. [Google Scholar]
- Eom, H.-J.; Choi, J. p38 MAPK Activation, DNA Damage, Cell Cycle Arrest and Apoptosis As Mechanisms of Toxicity of Silver Nanoparticles in Jurkat T Cells. Environ. Sci. Technol. 2010, 44, 8337–8342. [Google Scholar] [CrossRef] [PubMed]
- Synowiec, E.; Hoser, G.; Bialkowska-Warzecha, J.; Pawlowska, E.; Skorski, T.; Blasiak, J. Doxorubicin differentially induces apoptosis, expression of mitochondrial apoptosis-related genes, and mitochondrial potential in BCR-ABL1-expressing cells sensitive and resistant to imatinib. BioMed Res. Int. 2015, 2015, 673512. [Google Scholar] [CrossRef] [PubMed]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W.F. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim. Biophys. Acta 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Lee, K.S.; Nam, K.S. The association of changes in RAD51 and survivin expression levels with the proton beam sensitivity of Capan-1 and Panc-1 human pancreatic cancer cells. Int. J. Oncol. 2019, 54, 744–752. [Google Scholar] [CrossRef]
- Yuan, J.; Yan, R.; Krämer, A.; Eckerdt, F.; Roller, M.; Kaufmann, M.; Strebhardt, K. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene 2004, 23, 5843–5852. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, X.; Li, X.L. Expression and clinical significance of STAT3, P-STAT3, and VEGF-C in small cell lung cancer. Asian Pac. J. Cancer Prev. 2012, 13, 2873–2877. [Google Scholar] [CrossRef]
- Kim, H.S.; Cho, H.J.; Cho, H.J.; Park, S.J.; Park, K.W.; Chae, I.H.; Oh, B.H.; Park, Y.B.; Lee, M.M. The essential role of p21 in radiation-induced cell cycle arrest of vascular smooth muscle cell. J. Mol. Cell. Cardiol. 2004, 37, 871–880. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef]
- Yap, T.A.; Omlin, A.; De Bono, J.S. Development of therapeutic combinations targeting major cancer signaling pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef]
- Hong, C.E.; Park, A.K.; Lyu, S.Y. Synergistic anticancer effects of lectin and doxorubicin in breast cancer cells. Mol. Cell. Biochem. 2014, 394, 225–235. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Mizutani, H.; Tada-Oikawa, S.; Hiraku, Y.; Kojima, M.; Kawanishi, S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci. 2005, 76, 1439–1453. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Morgan, M.J.; Kim, Y.-S. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp. Mol. Med. 2022, 54, 1695–1704. [Google Scholar] [CrossRef]
- Günther, C.; Neumann, H.; Neurath, M.F.; Becker, C. Apoptosis, necrosis and necroptosis: Cell death regulation in the intestinal epithelium. Gut 2013, 62, 1062–1071. [Google Scholar] [CrossRef]
- Kim, M.-N.; Ahn, E.-Y.; Park, S.E.; Hossain, M.A.; Kim, M.Y.; Moon, J.-O.; Kim, N.D.; Yoon, J.-H. Morin inhibits the growth of murine hepatoma cells via cell cycle arrest and induction of apoptosis. Korean J. Cancer Prev. 2010, 15, 190–197. [Google Scholar]
- Tan, Y.; Raychaudhuri, P.; Costa, R.H. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell. Biol. 2007, 27, 1007–1016. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, X.; Wang, X.; Ge, H.-Y.; Wang, X.-y.; Garfield, D.; Yang, P.; Song, Y.-l.; Bai, C.-x. FoxM1 mediated resistance to gefitinib in non-smallcell lung cancer cells. Acta Pharmacol. Sin. 2012, 33, 675–681. [Google Scholar] [CrossRef]
- Zhang, N.; Wu, X.; Yang, L.; Xiao, F.; Zhang, H.; Zhou, A.; Huang, Z.; Huang, S. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51. Clin. Cancer Res. 2012, 18, 5961–5971. [Google Scholar] [CrossRef]
- Leung, T.W.; Lin, S.S.; Tsang, A.C.; Tong, C.S.; Ching, J.C.; Leung, W.Y.; Gimlich, R.; Wong, G.G.; Yao, K.M. Over-expression of FoxM1 stimulates cyclin B1 expression. FEBS Lett. 2001, 507, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, Q.; Xie, Y.; Qiao, X.; Zhang, S.; Wang, Y.; Yang, Y.; Zhang, B. Identification of FOXM1 as a specific marker for triple-negative breast cancer. Int. J. Oncol. 2019, 54, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Gartel, A.L. Suppression of FOXM1 Sensitizes Human Cancer Cells to Cell Death Induced by DNA-Damage. PLoS ONE 2012, 7, e31761. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-Y.; Jung, S.Y.; Jennings, N.B.; Rodriguez-Aguayo, C.; Peng, G.; Lee, S.-R.; Kim, S.B.; Kim, K.; Leem, S.-H.; Lin, S.-Y.; et al. FOXM1 mediates Dox resistance in breast cancer by enhancing DNA repair. Carcinogenesis 2012, 33, 1843–1853. [Google Scholar] [CrossRef]
- Androic, I.; Krämer, A.; Yan, R.; Rödel, F.; Gätje, R.; Kaufmann, M.; Strebhardt, K.; Yuan, J. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer 2008, 8, 391. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Guo, X.G.; Bai, X.P. Epidermal growth factor receptor-related DNA repair and radiation-resistance regulatory mechanisms: A mini-review. Asian Pac. J. Cancer Prev. 2012, 13, 4879–4881. [Google Scholar] [CrossRef]
- Chua, C.Y.; Liu, Y.; Granberg, K.J.; Hu, L.; Haapasalo, H.; Annala, M.J.; Cogdell, D.E.; Verploegen, M.; Moore, L.M.; Fuller, G.N.; et al. IGFBP2 potentiates nuclear EGFR–STAT3 signaling. Oncogene 2016, 35, 738–747. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, H.; Zhou, Z.; Tian, Y.; Cao, X.; Cheng, G.; Liu, Q. Blocking of the EGFR-STAT3 signaling pathway through afatinib treatment inhibited the intrahepatic cholangiocarcinoma. Exp. Ther. Med. 2018, 15, 4995–5000. [Google Scholar] [CrossRef]
- Pan, Z.; Zhang, X.; Yu, P.; Chen, X.; Lu, P.; Li, M.; Liu, X.; Li, Z.; Wei, F.; Wang, K.; et al. Cinobufagin Induces Cell Cycle Arrest at the G2/M Phase and Promotes Apoptosis in Malignant Melanoma Cells. Front. Oncol. 2019, 9, 853. [Google Scholar] [CrossRef]
- Choi, Y.H.; Zhang, L.; Lee, W.H.; Park, K.Y. Genistein-induced G2/M arrest is associated with the inhibition of cyclin B1 and the induction of p21 in human breast carcinoma cells. Int. J. Oncol. 1998, 13, 391–396. [Google Scholar] [CrossRef]
- Ling, Y.H.; el-Naggar, A.K.; Priebe, W.; Perez-Soler, R. Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Mol. Pharm. 1996, 49, 832–841. [Google Scholar]
- Suzuki, M.; Hosaka, Y.; Matsushima, H.; Goto, T.; Kitamura, T.; Kawabe, K. Butyrolactone I induces cyclin B1 and causes G2/M arrest and skipping of mitosis in human prostate cell lines. Cancer Lett. 1999, 138, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zuryń, A.; Litwiniec, A.; Gackowska, L.; Pawlik, A.; Grzanka, A.A.; Grzanka, A. Expression of cyclin A, B1 and D1 after induction of cell cycle arrest in the Jurkat cell line exposed to doxorubicin. Cell Biol. Int. 2012, 36, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, S.; Kwon, Y.S.; Lee, M.G.; Lee, K.S.; Nam, K.S. Cell cycle arrest-mediated cell death by morin in MDA-MB-231 triple-negative breast cancer cells. Pharm. Rep. 2021, 73, 1315–1327. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Zhang, X.; Min, K.-W.; Wimalasena, J.; Baek, S.J. Cyclin D1 degradation and p21 induction contribute to growth inhibition of colorectal cancer cells induced by epigallocatechin-3-gallate. J. Cancer Res. Clin. Oncol. 2012, 138, 2051–2060. [Google Scholar] [CrossRef]
- Sim, K.H.; Shu, M.-S.; Kim, S.; Kim, J.-Y.; Choi, B.-H.; Lee, Y.J. Cilostazol Induces Apoptosis and Inhibits Proliferation of Hepatocellular Carcinoma Cells by Activating AMPK. Biotechnol. Bioprocess Eng. 2021, 26, 776–785. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay methodsynergy quantification method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, S.; Lee, M.-G.; Kim, S.-Y.; Lee, K.-S.; Nam, K.-S. Morin Sensitizes MDA-MB-231 Triple-Negative Breast Cancer Cells to Doxorubicin Cytotoxicity by Suppressing FOXM1 and Attenuating EGFR/STAT3 Signaling Pathways. Pharmaceuticals 2023, 16, 672. https://doi.org/10.3390/ph16050672
Maharjan S, Lee M-G, Kim S-Y, Lee K-S, Nam K-S. Morin Sensitizes MDA-MB-231 Triple-Negative Breast Cancer Cells to Doxorubicin Cytotoxicity by Suppressing FOXM1 and Attenuating EGFR/STAT3 Signaling Pathways. Pharmaceuticals. 2023; 16(5):672. https://doi.org/10.3390/ph16050672
Chicago/Turabian StyleMaharjan, Sushma, Min-Gu Lee, So-Young Kim, Kyu-Shik Lee, and Kyung-Soo Nam. 2023. "Morin Sensitizes MDA-MB-231 Triple-Negative Breast Cancer Cells to Doxorubicin Cytotoxicity by Suppressing FOXM1 and Attenuating EGFR/STAT3 Signaling Pathways" Pharmaceuticals 16, no. 5: 672. https://doi.org/10.3390/ph16050672
APA StyleMaharjan, S., Lee, M.-G., Kim, S.-Y., Lee, K.-S., & Nam, K.-S. (2023). Morin Sensitizes MDA-MB-231 Triple-Negative Breast Cancer Cells to Doxorubicin Cytotoxicity by Suppressing FOXM1 and Attenuating EGFR/STAT3 Signaling Pathways. Pharmaceuticals, 16(5), 672. https://doi.org/10.3390/ph16050672