3.1.12. General Procedure K
To a solution of the selected phenol (1 mmol) and K2CO3 (2.5 mmol per phenolic group) in acetone (30 mL), benzyl bromide was added (2.5 mmol per phenolic group). The mixture was stirred at room temperature for 12–24 h (until full conversion, reaction monitored by TLC). The reaction was then filtered, and the solvent concentrated under reduced pressure. Purification by flash chromatography afforded the desired compound.
Synthesis of 3,9-dihydroxy-6H-benzo[c]chromen-6-one (6)
3-Hydroxy-9-methoxy-6H-benzo[c]chromen-6-one (5)
Following general procedure A, 2-bromo-4-methoxybenzoic acid (3) (0.92 g, 4.00 mmol) was reacted and recrystallized from MeOH to afford 0.71 g (73%) of 5 as a pink solid. Mp = 272–274 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.43–7.90 (m, 2H), 7.64 (s, 1H), 7.26–6.21 (m, 3H), 3.95 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.81, 160.81, 132.04, 116.45, 112.67, 110.20, 105.17, 56.37.
3,9-dihydroxy-6H-benzo[c]chromen-6-one (6)
Following general procedure B, 5 (0.200 g, 0.82 mmol) was reacted and triturated with Et2O to afford 0.18 g (95%) of 6 as a grey solid. Mp > 350 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 10.27 (s, 1H), 8.02 (d, J = 8.7 Hz, 1H), 7.95 (d, J = 8.8 Hz, 1H), 7.42 (d, J = 2.3 Hz, 1H), 6.95 (dd, J = 8.7, 2.2 Hz, 1H), 6.79 (dd, J = 8.7, 2.4 Hz, 1H), 6.69 (d, J = 2.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 164.17, 160.78, 160.25, 152.86, 137.83, 132.89, 125.13, 116.93, 113.43, 111.07, 109.81, 106.60, 103.28; HRMS (ESI) m/z: 227.0356 [M − H]−, calculated for C12H8O4 227.0350.
Synthesis of 8,9-dihydroxy-6H-benzo[c]chromen-6-one (13)
2-Bromo-4,5-dimethoxybenzoic acid (8)
To a suspension of 3,4-dimethoxybenzoic acid (7) (5.00 g, 27.44 mmol) in HCl conc. (100.0 mL), bromine (4.82 g, 30.19 mmol) was added dropwise at room temperature. The reaction mixture was stirred for 7 h at room temperature. Water (100.0 mL) was then added, and the resulting precipitate was collected by filtration and recrystallized from MeOH to afford 6.1 g (85%) of 8 as a white solid. Mp = 185–187 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.35 (s, 1H), 7.19 (s, 1H), 3.81 (s, 3H), 3.76 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.91, 151.93, 148.01, 124.37, 117.18, 114.22, 112.83, 56.52, 56.14.
Methyl 2-bromo-4,5-dimethoxybenzoate (9)
Following general procedure C, 8 (5.00 g, 19.15 mmol) was reacted and crystallized from MeOH to afford 4.43 g (84%) of 9 as a white solid. Mp = 88–90 °C; 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 1H), 6.99 (s, 1H), 3.91–3.71 (m, 9H); 13C NMR (101 MHz, CDCl3) δ 165.85, 151.92, 147.64, 122.62, 116.82, 114.05, 113.84, 56.15, 56.00, 52.17, 50.13.
Methyl 4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylate (10)
Following general procedure D, 9 (0.50 g, 1.81 mmol), phenylboronic acid (0.24 g, 1.99 mmol), were reacted. The crude was purified with flash chromatography (pentane/EtOAc) to afford 0.27 g (55%) of 10 as a colorless oil. 1H-NMR (400 MHz, CDCl3): δ 7.43 (s, 1H), 7.25–7.45 (m, 5H), 6.80 (s, 1H), 3.94 (s, 3H), 3.90 (s, 3H), 3.60 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 167.88, 150.92, 147.46, 141.43, 137.12, 128.20, 127.59, 126.71, 121.67, 113.31, 112.60, 55.84, 55.78, 51.45.
4,5-Dimethoxy-[1,1′-biphenyl]-2-carboxylic acid (11)
Following general procedure E, 10 (0.27 g, 0.99 mmol) was reacted to afford 0.24 g (94%) of 11 as a white solid. Mp = 198–200 °C; 1H-NMR (CDCl3, 400 MHz): δ 7.54 (s, 1H), 7.20–7.44 (m, 5H), 6.78 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 172.75, 151.86, 147.63, 141.43, 138.67, 128.63, 127.87, 127.08, 120.35, 113.99, 113.56, 56.11, 56.08.
8,9-Dimethoxy-6H-benzo[c]chromen-6-one (12)
Following general procedure F, 11 (0.24 g, 0.92 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.19 g (90%) of 12 as a white powder. Mp = 203–205 °C; 1H-NMR (400 MHz, CDCl3,): δ 7.93 (dd, J = 1.5, 7.9 Hz, 1H), 7.73 (s, 1H), 7.41–7.47 (m, 2H), 7.28–7.39 (m, 2H), 4.08 (s, 3H), 3.99 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 161.03, 155.05, 150.96, 150.08, 129.86, 129.53, 124.31, 122.10, 118.02, 117.68, 114.46, 110.44, 102.61, 56.29, 56.28.
8,9-Dihydroxy-6H-benzo[c]chromen-6-one (13)
Following general procedure B, 12 (0.17 g, 0.66 mmol) was reacted and triturated with Et2O to afford 0.110 g (73%) of 13 as a white solid. Mp = 288–290 °C; 1H-NMR (400 MHz, DMSO-d6): δ 10.45 (br s, 1H), 10.22 (br s, 1H), 8.04 (d, J = 8.5 Hz, 1H), 7.60 (s, 1H), 7.55 (s, 1H), 7.44 (ddd, J = 1.5, 7.1, 8.5 Hz, 1H), 7.29–7.38 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 160.37, 153.77, 150.61, 147.80, 129.60, 128.58, 124.95, 123.10, 118.46, 117.44, 114.77, 112.98, 108.28, 40.58, 40.22; HRMS (ESI) m/z: 227.036 [M − H]−, calculated for C12H8O4 227.0350.
[1,1′-Biphenyl]-3,4,4′-triol (17)
3,4,4′-Trimethoxy-1,1′-biphenyl (16)
Following general procedure D, 3,4-dimethoxybenzeneboronic acid (14) (0.53 g, 2.94 mmol) and 4-bromoanisole (15) (0.50 g, 2.67 mmol) were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.57 g (87%) of 16 as a white solid. Mp = 104–106 °C; 1H NMR (400 MHz, CDCl3) δ 7.53–7.43 (m, 2H), 7.13–7.04 (m, 2H), 7.01–6.89 (m, 3H), 3.95 (s, 3H), 3.92 (s, 3H), 3.85 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 158.80, 149.09, 148.14, 133.96, 133.66, 127.84, 118.91, 114.15, 111.49, 110.17, 55.98, 55.91, 55.35.
[1,1′-Biphenyl]-3,4,4′-triol (17)
Following general procedure B, 16 (0.300 g, 1.23 mmol) was reacted and triturated with Et2O to afford 0.19 g (78%) of 17 as a white solid. Mp = 246–248 °C; 1H NMR (400 MHz, CD3OD) δ 7.40–7.24 (m, 2H), 6.97 (d, J = 2.2 Hz, 1H), 6.86 (dd, J = 8.2, 2.2 Hz, 1H), 6.82–6.72 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 155.90, 145.00, 143.85, 133.26, 132.70, 127.05, 117.51, 115.22, 114.98, 113.19; HRMS (ESI) m/z: 201.0564 [M − H]−, calculated for C12H9O3 201.0557.
8,9-dihydroxy-3-(methylamino)-6H-benzo[c]chromen-6-one (26)
Methyl 4′-((tert-butoxycarbonyl)(methyl)amino)-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylate (20)
Following general procedure D, 9 (250 mg, 0.9 mmol) and tert-butyl-N-methyl-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]carbamate (18) (0.25 mg, 1.0 mmol) were reacted and purified by flash chromatography (pentane/EtOAc) to afford 20 as a yellow oil (244 mg, 0.6 mmol, 67%). 1H NMR (400 MHz, DMSO-d6) δ 7.34–7.18 (m, 5H), 6.91 (s, 1H), 3.85 (s, 3H), 3.83 (s, 3H), 3.55 (s, 3H), 3.21 (s, 3H), 1.41 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 167.87, 153.70, 150.91, 147.48, 142.30, 137.50, 135.08, 128.43, 124.67, 121.89, 113.63, 112.67, 79.61, 55.75, 55.72, 51.63, 51.61, 36.98, 27.95.
4′-((tert-butoxycarbonyl)(methyl)amino)-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylic acid (22)
Following general procedure E (reaction conducted at 65 °C), 20 (180 mg, 0.5 mmol) was reacted to afford 0.17 g (99%) of 22 as a white solid. The compound was used in the next step without any purification. 1H NMR (400 MHz, DMSO-d6) δ 7.42 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.6 Hz, 2H), 6.99 (s, 1H), 6.72 (s, 1H), 3.75 (d, J = 4.2 Hz, 6H), 3.19 (s, 3H), 1.41 (s, 9H).
Tert-butyl (8,9-dimethoxy-6-oxo-6H-benzo[c]chromen-3-yl)(methyl)carbamate (24)
Following general procedure F, 22 (0.17 g, 0.49 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 24 as an orange solid 0.09 mg (55%). 1H NMR (400 MHz, DMSO-d6) δ 8.35 (d, J = 9.4 Hz, 1H), 7.81 (s, 1H), 7.60 (s, 1H), 7.38–7.33 (m, 2H), 4.04 (s, 3H), 3.91 (s, 3H), 3.27 (s, 3H), 1.43 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 160.00, 155.25, 153.33, 150.26, 149.72, 144.50, 129.41, 123.33, 121.20, 114.54, 112.90, 112.66, 109.82, 104.16, 80.29, 56.45, 55.77, 36.72, 27.91.
8,9-dihydroxy-3-(methylamino)-6H-benzo[c]chromen-6-one (26)
Following general procedure B, 24 (0.04 g, 0.1 mmol) was reacted and triturated with Et2O to afford 0.02 g (82%) of 25 as a brown solid. Mp = 279–281 °C; 1H NMR (400 MHz, CD3OD) δ 8.14 (d, J = 8.8 Hz, 1H), 7.64 (s, 1H), 7.56 (s, 1H), 7.35–7.20 (m, 2H), 3.09 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 162.04, 155.04, 152.47, 149.18, 139.42, 129.24, 125.76, 125.74, 120.13, 118.43, 115.58, 114.26, 111.23, 108.70, 37.11; HRMS (ESI): m/z: 258,0773 [M + H]+; calculated C14H12NO4: 258,0761.
3-Amino-8,9-dihydroxy-6H-benzo[c]chromen-6-one (28)
Methyl 4,5-dimethoxy-4′-nitro-[1,1′-biphenyl]-2-carboxylate (21)
Following general procedure D, 9 (3.080 g, 11.2 mmol) and 4-nitrobenzeneboronic acid (19) (2.056 g, 12.3 mmol), were reacted and purified by flash chromatography (pentane/EtOAc) to afford 1.92 g (54%) of 21 as a pale-orange solid. Mp = 134–136 °C; 1H-NMR (400 MHz, CDCl3) δ 8.26–8.23 (m, 2H), 7.52 (s, 1H), 7.45–7.41 (m, 2H), 6.73 (s, 1H), 3.98 (s, 3H), 3.93 (s, 3H), 3.65 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 167.22, 151.73, 148.94, 148.68, 147.02, 135.49, 129.64, 123.25, 121.59, 113.34, 113.24, 56.36, 56.33, 52.10.
4,5-Dimethoxy-4′-nitro-[1,1′-biphenyl]-2-carboxylic acid (23)
Following general procedure E, 21 (1.910 g, 6.02 mmol) was reacted to afford 1.79 g (98%) of 23 as a pale-brown solid. Mp = 266–268 °C; 1H-NMR (400 MHz, DMSO-d6) δ 12.66 (br s, 1H), 8.24–8.21 (m, 2H), 7.59–7.56 (m, 2H), 7.44 (s, 1H), 6.92 (s, 1H), 3.86 (s, 3H), 3.85 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 167.87, 150.89, 148.62, 148.11, 146.27, 133.98, 130.05, 122.85, 122.61, 113.71, 113.15, 55.84, 55.72.
8,9-Dimethoxy-3-nitro-6H-benzo[c]chromen-6-one (25)
Following general procedure G, 23 (1.090 g, 3.594 mmol) was reacted and triturated from CH2Cl2 to afford 0.9 g (84%) of 25 as a yellow solid. Mp = 322–324 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.73 (d, J = 8.7 Hz, 1H), 8.24–8.20 (m, 2H), 7.99 (s, 1H), 7.68 (s, 1H), 4.07 (s, 3H), 3.96 (s, 3H); 13C-NMR (201 MHz, DMSO-d6, 60 °C because of low solubility in DMSO) δ 158.88, 155.24, 151.31, 149.83, 147.06, 127.40, 124.71, 123.77, 118.56, 114.36, 112.21, 110.19, 105.50, 56.46, 55.88.
3-Amino-8,9-dimethoxy-6H-benzo[c]chromen-6-one (27)
Following general procedure H (conducted at 50 °C), 25 (1.410 g, 4.68 mmol) was reacted to afford to afford 1.16 g (92%) of 27 as a beige solid. Mp = 318–320 °C; 1H-NMR (400 MHz, DMSO-d6) δ 7.99 (d, J = 8.6 Hz, 1H), 7.57 (s, 1H), 7.49 (s, 1H), 6.61 (dd, J = 8.6, 2.2 Hz, 1H), 6.46 (d, J = 2.2 Hz, 1H), 5.79–5.77 (m, 2H), 3.99 (s, 3H), 3.85 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 160.62, 155.28, 152.20, 150.98, 148.07, 131.51, 124.28, 111.38, 110.66, 109.65, 106.18, 102.60, 99.59, 56.20, 55.60.
3-Amino-8,9-dihydroxy-6H-benzo[c]chromen-6-one (28)
Following general procedure B, 27 (0.040 g, 0.15 mmol) was reacted to afford 0.030 g (84%) of 28 as a white solid. Mp > 350 °C; 1H-NMR (400 MHz, CD3OD): δ 8.20 (d, J = 9.2 Hz, 1H, Ar-H), 7.64 (s, 1H, Ar-H), 7.59 (s, 1H, Ar-H), 7.33–7.39 (m, 2H, Ar-H); 13C NMR (101 MHz, CD3OD) δ 160.55, 153.65, 150.90, 147.86, 130.89, 127.72, 124.25, 119.16, 118.67, 114.16, 112.93, 111.74, 107.37; HRMS (ESI) m/z: 242.0462 [M − H]−, calculated for C13H8NO4 242.0459.
N-(8,9-dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)acetamide (33)
N-(8,9-dimethoxy-6-oxo-6H-benzo[c]chromen-3-yl)acetamide (29)
Following general procedure I, 27 (70.0 mg, 0.258 mmol) and acetyl chloride (83 µL, 1.162 mmol) were reacted to afford 0.06 g (71%) of 29 as a beige solid. Mp = 333–335 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 8.30 (d, J = 8.8 Hz, 1H), 7.76 (d, J = 2.1 Hz, 1H), 7.73 (s, 1H), 7.56 (s, 1H), 7.47 (dd, J = 8.7, 2.1 Hz, 1H), 4.02 (s, 3H), 3.89 (s, 3H), 2.09 (s, 3H); 13C NMR (101 MHz, dm DMSO-d6) δ 168.81, 160.12, 155.26, 150.73, 149.39, 140.64, 129.84, 123.95, 115.13, 112.82, 112.43, 109.77, 106.23, 103.73, 56.38, 55.75, 39.99, 24.15.
N-(8,9-dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)acetamide (33)
Following general procedure B, 29 (0.033 g, 0.11 mmol) was reacted to afford 0.023 g (77%) of 33 as a beige solid. Mp > 350 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 10.25 (s, 1H), 10.14 (s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 2.1 Hz, 1H), 7.52 (s, 1H), 7.50 (s, 1H), 7.45 (dd, J = 8.7, 2.1 Hz, 1H), 2.08 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 168.76, 160.14, 153.43, 150.45, 146.77, 140.12, 128.50, 123.08, 115.31, 114.28, 113.03, 111.68, 107.30, 106.32, 24.15; HRMS (ESI): m/z: 286,0712 [M + H]+; calculated for C15H12NO5: 286,071.
N-(8,9-dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)propionamide (34)
N-(8,9-dimethoxy-6-oxo-6H-benzo[c]chromen-3-yl)propionamide (30)
Following general procedure I, 27 (0.05 g, 0.184 mmol) and propionyl chloride (48 µL, 0.553 mmol) were reacted to afford 0.04 g (61%) of 30 as a beige solid. Mp = 283–285 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.37–8.21 (m, 1H), 7.77 (d, J = 2.3 Hz, 1H), 7.75–7.70 (m, 1H), 7.59–7.54 (m, 1H), 7.54–7.48 (m, 1H), 4.05–3.99 (m, 3H), 3.89 (t, J = 1.4 Hz, 3H), 2.42–2.33 (m, 2H), 1.16–1.05 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 172.47, 160.14, 155.26, 150.74, 149.37, 140.72, 129.86, 123.94, 115.17, 112.73, 112.40, 109.77, 106.22, 103.72, 56.37, 55.74, 29.61, 9.46.
N-(8,9-dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)propionamide (34)
Following general procedure B, 30 (0.022 g, 0.07 mmol) was reacted to afford 0.016 g (78%) of 34 as a beige solid. Mp > 350 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.71–9.79 (m, 3H), 7.96 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 2.1 Hz, 1H), 7.52 (s, 1H), 7.50 (s, 1H), 7.47 (dd, J = 8.7, 2.1 Hz, 1H), 2.37 (q, J = 7.5 Hz, 2H), 1.10 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 172.41, 160.14, 153.43, 150.46, 146.74, 140.18, 128.51, 123.05, 115.32, 114.26, 112.93, 111.64, 107.28, 106.31, 29.61, 9.52; HRMS (ESI): m/z: 300,0872 [M + H]+; calculated for C16H14NO5: 300,0874.
N-(8,9-dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)butyramide (35)
N-(8,9-dimethoxy-6-oxo-6H-benzo[c]chromen-3-yl)butyramide (31)
Following general procedure I, 27 (0.07 g, 0.258 mmol) and butyryl chloride (40 μL, 0.387 mmol) were reacted to afford 0.054 g (61%) of 31 as a beige solid. Mp = 287–288 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 8.28 (d, J = 8.8 Hz, 1H), 7.76 (d, J = 2.1 Hz, 1H), 7.72 (s, 1H), 7.55 (s, 1H), 7.49 (dd, J = 8.8, 2.1 Hz, 1H), 4.00 (s, 3H), 3.88 (s, 3H), 2.32 (t, J = 7.4 Hz, 2H), 1.67–1.57 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 171.64, 160.14, 155.26, 150.74, 149.38, 140.66, 129.86, 123.94, 115.20, 112.78, 112.42, 109.77, 106.26, 103.73, 56.38, 55.75, 38.41, 18.41, 13.65.
N-(8,9-dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)butyramide (35)
Following general procedure B, 31 (0.04 g, 0.117 mmol) was reacted to afford 0.031 g (84%) of 35 a pale-pink-brown solid. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.42 (br s, 1H), 10.18 (s, 1H), 10.13 (br s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 2.1 Hz, 1H), 7.52 (s, 1H), 7.50 (s, 1H), 7.47 (dd, J = 8.8, 2.1 Hz, 1H), 2.33 (t, J = 7.3 Hz, 2H), 1.68–1.58 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 171.57, 160.13, 153.41, 150.44, 146.74, 140.12, 128.50, 123.04, 115.35, 114.26, 112.97, 111.65, 107.29, 106.34, 38.41, 18.45, 13.64; HRMS (ESI) calculated for C17H16NO5 [M + H]+ 314.1023, found 314.1031.
N-(8,9-Dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)methanesulfonamide (36)
N-(8,9-Dimethoxy-6-oxo-6H-benzo[c]chromen-3-yl)methanesulfonamide (32)
To a suspension of 27 (0.0045 g, 0.166 mmol) in pyridine (1.6 mL) at 0 °C, mesyl chloride (15 μL, 0.18 mmol) was added. The reaction mixture was stirred from 0 °C to room temperature for 15 h. Then, a second portion of mesyl chloride (15 μL, 0.18 mmol) was added and the reaction mixture was stirred for 2 h. Then, the reaction mixture was diluted with EtOAc (50 mL), washed with a saturated aqueous solution of Cu2SO4 (1 × 10 mL), water (1 × 10 mL), an aqueous solution of HCl (1 M, 2 × 10 mL), and water (3 × 20 mL). The organic layer was concentrated under reduced pressure and the residue was dried by co-evaporation with toluene (×3) to afford 0.042 g (73%) of 32 as a pale-brown solid. Mp = 304–306 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.33 (d, J = 8.4 Hz, 1H), 7.73 (s, 1H), 7.56 (s, 1H), 7.21–7.18 (m, 2H), 4.02 (s, 3H), 3.89 (s, 3H), 3.12 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 159.92, 155.27, 150.99, 149.51, 139.86, 129.61, 124.65, 115.11, 113.31, 112.47, 109.78, 106.24, 103.82, 56.42, 55.75.
N-(8,9-Dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl)methanesulfonamide (36)
Following general procedure B, 32 (0.033 g, 0.095 mmol) was reacted and triturated with Et2O and CH2Cl2 to afford 0.014 g (46%) of 35 a pale-brown solid. Mp = 323–325 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.44 (br s, 1H), 10.17 (br s, 1H), 10.15 (br s, 1H), 8.01 (d, J = 8.6 Hz, 1H), 7.53 (app s, 2H), 7.18–7.14 (m, 2H), 3.09 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 159.9, 153.5, 150.7, 146.9, 139.3, 128.3, 123.8, 115.3, 114.3, 113.6, 111.7, 107.4, 106.4, 39.6; HRMS (ESI) calculated for C14H12NO6S [M + H]+ 322.0380, found 322.0374.
3-Fluoro-8,9-dihydroxy-6H-benzo[c]chromen-6-one (61)
Methyl 4′-fluoro-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylate (43)
Following general procedure D, 9 (0.50 g, 1.81 mmol) and 4-fluorobenzeneboronic acid (37) (0.51 g, 2.00 mmol) were reacted. The compound was extracted and used directly for the next step.
4′-Fluoro-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylic acid (49)
Following general procedure E, crude 43 was reacted to afford 0.25 (48% over 2 steps) of 49 as a white solid. Mp = 212–214 °C; 1H NMR (400 MHz, CD3OD) δ 7.48 (s, 1H), 7.32–7.26 (m, 2H), 7.11–7.04 (m, 2H), 6.84 (s, 1H), 3.90 (s, 3H), 3.88 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 163.55 (d, J = 243.9 Hz), 139.27 (d, J = 3.4 Hz), 131.53 (d, J = 8.1 Hz), 115.47 (d, J = 21.7 Hz); 19F NMR (659 MHz, DMSO-d6) δ -116.48.
3-Fluoro-8,9-dimethoxy-6H-benzo[c]chromen-6-one (55)
Following general procedure F, 49 (0.2 g, 0.72 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.1 g (50%) of 55 as a brown solid. Mp = 256–258 °C; 1H NMR (400 MHz, CDCl3) δ 7.93 (dd, J = 8.6, 5.8 Hz, 1H), 7.74 (s, 1H), 7.37 (s, 1H), 7.13–7.02 (m, 2H), 4.10 (s, 3H), 4.01 (s, 3H); 13C NMR (101 MHz, cdcl3) δ 163.08 (d, J = 250.4 Hz), 151.98 (d, J = 12.3 Hz), 123.76 (d, J = 9.7 Hz), 114.83 (d, J = 3.1 Hz), 113.80 (d, J = 1.3 Hz), 112.35 (d, J = 22.5 Hz), 105.21 (d, J = 25.2 Hz); 19F NMR (659 MHz, DMSO) δ -116.48.
3-Fluoro-8,9-dihydroxy-6H-benzo[c]chromen-6-one (61)
Following general procedure B, 55 (0.05 g, 0.36 mmol) was reacted and triturated with Et2O to afford 0.26 g (85%) of 61 as a white solid. Mp = 312–314 °C; 1H NMR (700 MHz, DMSO-d6) δ 10.51 (s, 1H), 10.26 (s, 1H), 8.13 (dd, J = 9.5, 6.3 Hz, 1H), 7.59 (s, 1H), 7.55 (s, 1H), 7.34 (dd, J = 9.7, 3.1 Hz, 1H), 7.23 (tt, J = 9.6, 4.8 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 162.37 (d, J = 246.7 Hz), 160.22, 154.02, 151.45 (d, J = 12.6 Hz), 147.70, 129.07–123.24 (m), 115.43, 114.76, 112.49 (d, J = 22.0 Hz), 112.19, 108.36, 104.81 (d, J = 25.5 Hz); 19F NMR (564 MHz, DMSO) δ -111.40; HRMS (ESI) calculated for C13H8FO4 [M + H]+ 247.0407, found 247.0409.
8,9-Dihydroxy-3-(trifluoromethyl)-6H-benzo[c]chromen-6-one (62)
Methyl 4,5-dimethoxy-4′-(trifluoromethyl)-[1,1′-biphenyl]-2-carboxylate (44)
Following general procedure D, 9 (0.50 g, 1.81 mmol) and 4-(trifluoromethyl)benzeneboronic acid (38) (0.38 g, 2.00 mmol) were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.55 (89%) of 44 as a beige solid. Mp = 94–97 °C; 1H-NMR (700 MHz, CDCl3) δ 7.64 (d, J = 8.0 Hz, 2H), 7.50 (s, 1H), 7.40 (d, J = 8.0 Hz, 2H), 6.75 (s, 1H), 3.98 (s, 3H), 3.93 (s, 3H), 3.64 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 167.6, 151.6, 148.3, 145.7 (q, J = 1.2 Hz), 136.3, 129.2 (q, J = 32.5 Hz) 129.0, 124.9 (q, J = 3.8 Hz), 124.4 (q, J = 272.1 Hz), 121.7, 113.5, 113.2, 56.3, 56.2, 52.0; 19F-NMR (659 MHz, CDCl3) δ –62.36.
4,5-Dimethoxy-4′-(trifluoromethyl)-[1,1′-biphenyl]-2-carboxylic acid (50)
Following general procedure E, 44 (0.5 g, 0.99 mmol) was reacted to afford 0.46 g (98%) of 50 as a pale-yellow solid. Mp = 190–193 °C; 1H-NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 2H), 7.58 (s, 1H), 7.41 (d, J = 8.0 Hz, 2H), 6.73 (s, 1H), 3.97 (s, 3H), 3.93 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 170.32, 152.26, 148.32, 145.40, 137.44, 129.18, 124.93, 124.41 (q, J = 272.3 Hz), 120.06, 113.89, 113.86, 56.35, 56.33.
8,9-Dimethoxy-3-(trifluoromethyl)-6H-benzo[c]chromen-6-one (56)
Following general procedure F, 50 (0.44 g, 1.35 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.36 g (84%) of 56 as a brown solid. Mp = 233–234 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.67 (d, J = 8.1 Hz, 1H), 7.95 (s, 1H), 7.82 (d, J = 1.7 Hz, 1H), 7.74 (dd, J = 8.1, 1.7 Hz, 1H), 7.66 (s, 1H), 4.06 (s, 3H), 3.94 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 159.4, 155.2, 150.8, 150.2, 129.4 (q, J = 32.7 Hz), 128.1, 125.0, 123.6 (q, J = 270.2 Hz), 121.6, 120.7 (q, J = 3.7 Hz), 114.4 (q, J = 3.7 Hz), 114.2, 109.9, 105.0, 56.6, 55.9; 19F-NMR (659 MHz, DMSO-d6) δ –60.88.
8,9-Dihydroxy-3-(trifluoromethyl)-6H-benzo[c]chromen-6-one (62)
Following general procedure B, 56 (0.34 g, 1.05 mmol) was reacted and triturated with Et2O to afford 0.26 g (84%) of 62 as a grey solid. Mp = 293–295 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.59 (br s, 1H), 10.44 (br s, 1H), 8.24 (d, J = 8.6 Hz, 1H), 7.69–7.68 (m, 2H), 7.62 (dd, J = 8.6, 1.7 Hz, 1H), 7.59 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 159.36, 153.44, 149.89, 148.4, 128.82 (q, J = 32.6 Hz), 126.64, 123.98, 123.71 (q, J = 272.2 Hz), 121.76, 120.82 (q, J = 3.8 Hz), 114.50, 114.22 (q, J = 3.8 Hz), 113.18, 108.61; 19F-NMR (659 MHz, DMSO-d6) δ –60.96; HRMS (ESI) calculated for C14H8F3O4 [M + H]+ 297.0369, found 297.0360.
8,9-Dihydroxy-3-methyl-6H-benzo[c]chromen-6-one (63)
Methyl 4,5-dimethoxy-4′-methyl-[1,1′-biphenyl]-2-carboxylate (45)
Following general procedure D, 9 (0.50 g, 1.81 mmol) and 4-tolylboronic acid (39) (0.3 g, 2.181 mmol). were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.44 (84%) of 45 as a beige solid. Mp = 87–91 °C; 1H-NMR (400 MHz, DMSO-d6) δ 7.30 (s, 1H), 7.20–7.15 (m, 4H), 6.88 (s, 1H), 3.84 (s, 3H), 3.82 (s, 3H), 3.56 (s, 3H), 2.34 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 167.92, 150.85, 147.33, 137.88, 136.02, 135.65, 128.53, 128.23, 121.86, 113.64, 112.62, 55.71, 55.68, 51.62, 20.70.
4,5-Dimethoxy-4′-methyl-[1,1′-biphenyl]-2-carboxylic acid (51)
Following general procedure E, 45 (0.39 g, 1.35 mmol) was reacted to afford 0.28 g (76%) of 51 as a white solid. Mp = 226–229 °C; 1H-NMR (400 MHz, DMSO-d6) δ 12.41 (br s, 1H), 7.31 (s, 1H), 7.20–7.15 (m, 4H), 6.82 (s, 1H), 3.82 (s, 3H), 3.82 (s, 3H), 2.33 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 168.83, 150.48, 147.18, 138.32, 135.82, 135.53, 128.42, 128.38, 123.07, 113.75, 112.81, 55.65, 55.63, 20.70.
8,9-Dimethoxy-3-methyl-6H-benzo[c]chromen-6-one (57)
Following general procedure F, 51 (0.25 g, 0.91 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.09 g (38%) of 60 as a yellow solid. Mp = 217–221 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.28 (d, J = 8.5 Hz, 1H), 7.80 (s, 1H), 7.59 (s, 1H), 7.23–7.22 (m, 2H), 4.03 (s, 3H), 3.90 (s, 3H), 2.41 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 160.08, 155.20, 150.37, 149.60, 140.07, 129.79, 125.44, 123.38, 117.02, 115.26, 112.92, 109.76, 103.98, 56.40, 55.74, 20.83.
8,9-Dihydroxy-3-methyl-6H-benzo[c]chromen-6-one (63)
Following general procedure B, 57 (0.083 g, 0.307 mmol) was reacted and triturated with Et2O to afford 0.07 g (97%) of 67 as a brown solid. Mp = 270–272 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.84 (br s, 2H), 7.91 (d, J = 8.5 Hz, 1H), 7.56 (s, 1H), 7.54 (s, 1H), 7.16–7.15 (m, 2H), 2.37 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 160.10, 153.35, 150.16, 147.00, 139.33, 128.42, 125.49, 122.43, 117.03, 115.42, 114.30, 112.13, 107.54, 20.81; HRMS (ESI) calculated for C14H11O4 [M + H]+ 243.0657, found 243.0659.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carbaldehyde (64)
Methyl 4′-formyl-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylate (46)
Following general procedure D, 9 (0.600 g, 2.18 mmol), 4-formylbenzenboronic acid (40) (0.36 g, 2.40 mmol), were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.56 g (85%) of 46 as a white solid. Mp = 142–144 °C. 1H-NMR (400 MHz, CDCl3): δ 10.01 (s, 1H), 7.86 (d, J = 8.2 Hz, 2H), 7.46 (s, 1H,), 7.41 (d, J = 8.2 Hz, 2H), 6.75 (s, 1H), 3.93 (s, 3H), 3.89 (s, 3H), 3.59 (s, 3H), 13C NMR (101 MHz, CDCl3) δ 191.91, 167.39, 151.41, 148.24, 148.22, 136.15, 134.93, 129.26, 121.58, 113.17, 113.05, 56.13, 56.10, 51.83.
4′-Formyl-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylic acid (52)
Following general procedure E, 46 (0.65 g, 2.16 mmol) was reacted to afford 0.55 g (89%) of 52 as a white solid. Mp = 249–251 °C; 1H NMR (400 MHz, CDCl3) δ 10.04 (s, 1H), 7.91–7.83 (m, 2H), 7.56 (s, 1H), 7.48–7.41 (m, 2H), 6.74 (s, 1H), 3.95 (s, 3H), 3.93 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 192.17, 171.83, 152.15, 148.21, 148.05, 137.42, 135.02, 129.40, 129.31, 120.03, 113.72, 113.51, 56.17.
8,9-Dimethoxy-6-oxo-6H-benzo[c]chromene-3-carbaldehyde (58)
Following general procedure F, 52 (0.55 g, 1.92 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.22 g (40%) of 58 as a white powder. Mp = 301–303 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.60 (d, J = 8.1 Hz, 1H), 8.02–7.75 (m, 3H), 7.62 (s, 1H), 4.04 (s, 3H), 3.92 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 192.54, 160.00, 155.62, 151.40, 150.91, 136.98, 128.75, 125.00, 124.73, 123.54, 118.66, 110.44, 105.64, 57.04, 56.38.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carbaldehyde (64)
Following general procedure B, 58 (0.07 g, 0.24 mmol) was reacted and triturated with Et2O to afford 0.030 g (47%) of 64 as yellow solid. Mp = 352–354 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.73–10.35 (m, 2H), 10.04 (s, 1H), 8.26 (d, J = 8.5 Hz, 1H), 7.83 (dq, J = 3.8, 1.6 Hz, 2H), 7.70 (s, 1H), 7.59 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 192.51, 159.99, 153.83, 150.66, 149.06, 136.52, 127.33, 124.95, 124.07, 123.80, 118.63, 114.99, 113.87, 109.35; HRMS (ESI) m/z: 255.0301 [M − H]−, calculated for C14H7O5 255.0299.
8,9-Dihydroxy-3-(methylsulfonyl)-6H-benzo[c]chromen-6-one (65)
Methyl 4,5-dimethoxy-4′-(methylsulfonyl)-[1,1′-biphenyl]-2-carboxylate (47)
Following general procedure D, 9 (0.500 g, 1.82 mmol), 4-(methanesulfonyl)phenylboronic acid (41) (400 mg, 2.00 mmol), were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.52 g (82%) of 47 as a white solid. Mp = 301–303 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.99–7.87 (m, 2H), 7.59–7.49 (m, 2H), 7.41 (s, 1H), 6.95 (s, 1H), 3.86 (s, 6H), 3.59 (s, 3H), 3.26 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.95, 151.21, 148.05, 146.24, 139.15, 134.52, 129.46, 126.52, 121.43, 113.83, 112.88, 55.85, 55.78, 51.81, 43.59.
4,5-Dimethoxy-4′-(methylsulfonyl)-[1,1′-biphenyl]-2-carboxylic acid (53)
Following general procedure E, 47 (0.47 g, 1.34 mmol) was reacted to afford 0.31 g (69%) of 53 as a beige crystal. Mp = 201–203 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.62 (s, 1H), 7.95–7.88 (m, 2H), 7.60–7.53 (m, 2H), 7.42 (s, 1H), 6.89 (s, 1H), 3.84 (s, 3H), 3.85 (s, 3H), 3.26 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 167.98, 150.84, 147.92, 146.77, 139.02, 134.38, 129.60, 126.40, 122.69, 113.87, 113.09, 55.81, 55.72, 43.56.
8,9-Dimethoxy-3-(methylsulfonyl)-6H-benzo[c]chromen-6-one (59)
Following general procedure I, 53 (0.2 g, 0.59 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.18 g (92%) of 59 as a white powder. Mp = 353–355 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 8.4 Hz, 1H), 7.97 (s, 1H), 7.92 (d, J = 1.8 Hz, 1H), 7.89 (dd, J = 8.3, 1.9 Hz, 1H), 7.67 (s, 1H), 4.07 (s, 3H), 3.95 (s, 3H), 3.34 (s, 3H); 13C NMR (201 MHz, DMSO-d6) δ 158.97, 155.22, 150.98, 149.83, 141.07, 127.76, 124.64, 122.20, 121.87, 115.60, 114.23, 110.14, 105.18, 56.43, 55.83, 43.20.
8,9-Dihydroxy-3-(methylsulfonyl)-6H-benzo[c]chromen-6-one (65)
Following general procedure B, 59 (0.07 g, 0.21 mmol) was reacted and triturated with Et2O to afford 0.044 g (69%) of 65 as beige solid. Mp = 319–321 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 2H), 8.33 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 1.9 Hz, 1H), 7.83 (dd, J = 8.3, 1.9 Hz, 1H), 7.71 (s, 1H), 7.60 (s, 1H), 3.31 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 159.32, 153.45, 149.74, 148.65, 140.49, 126.54, 124.00, 122.64, 122.35, 115.89, 114.51, 113.35, 108.92, 43.34; HRMS (ESI) m/z: 305.0125 [M − H]−, calculated for C14H9O6S 300.0127.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-sulfonamide (66)
Methyl 4,5-dimethoxy-4′-sulfamoyl-[1,1′-biphenyl]-2-carboxylate (48)
Following general procedure D, 9 (0.2 g, 0.73 mmol), (4-aminosulfonylphenyl)boronic acid (42) (0.16 g, 0.80 mmol), were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.15 g (58%) of 48 as a white solid. Mp = 198–200 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.83–7.77 (m, 2H), 7.46–7.41 (m, 2H), 7.37 (d, J = 2.0 Hz, 3H), 6.92 (s, 1H), 3.83 (s, 3H), 3.83 (s, 3H), 3.57 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.95, 151.21, 148.05, 146.24, 139.15, 134.52, 129.46, 126.52, 121.43, 113.83, 112.88, 55.85, 55.78, 51.81, 43.59.
4,5-Dimethoxy-4′-sulfamoyl-[1,1′-biphenyl]-2-carboxylic acid (54)
Following general procedure E, 48 (0.5 g, 1.42 mmol) was reacted to afford 0.48 g (quant.) of 54 as a white solid. Mp = 223–225 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 7.85–7.77 (m, 2H), 7.52–7.44 (m, 2H), 7.40 (d, J = 3.5 Hz, 3H), 6.88 (s, 1H), 3.84 (s, 3H), 3.84 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 168.17, 150.80, 147.80, 144.92, 142.37, 134.66, 129.15, 125.14, 122.73, 113.91, 113.07, 55.82, 55.80, 55.74.
8,9-Dimethoxy-6-oxo-6H-benzo[c]chromene-3-sulfonamide (60)
Following general procedure I, 54 (0.2 g, 0.59 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.12 g (60%) of 60 as a beige powder. Mp = 320–322 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.66–8.61 (m, 1H), 7.93 (s, 1H), 7.79 (dd, J = 8.3, 1.9 Hz, 1H), 7.76 (d, J = 1.7 Hz, 1H), 7.66 (s, 1H), 7.57 (s, 2H), 4.06 (s, 3H), 3.94 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 159.52, 155.24, 150.73, 149.88, 144.63, 128.27, 124.61, 121.17, 120.90, 114.33, 114.10, 109.97, 104.98, 56.61, 55.94.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-sulfonamide (66)
Following general procedure B, 60 (0.12 g, 0.35 mmol) was reacted and triturated with Et2O to afford 0.073 g (67%) of 66 as beige solid. Mp = 319–321 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 10.47 (s, 1H), 8.27 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 8.3, 1.9 Hz, 1H), 7.71 (d, J = 1.8 Hz, 1H), 7.68 (s, 1H), 7.59 (s, 1H), 7.54 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.50, 153.44, 149.61, 148.32, 144.05, 126.88, 123.70, 121.34, 121.07, 114.47, 114.30, 113.11, 108.67; HRMS (ESI) m/z: 308.0229 [M + H]+, calculated for C13H10NO6S 308.0231.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carboxylic acid (81)
2-Bromo-4,5-dimethoxybenzaldehyde (68)
The compound was obtained following the literature procedure [
25]. To a solution of 3,4-dimethoxybenzaldehyde (5.00 g, 30.09 mmol) in acetic acid (40.0 mL), a solution of bromine (4.65 mL, 90.27 mmol) in acetic acid (15.0 mL) was added slowly through an addition funnel at ice-cold conditions. The resulting reaction mixture was allowed to stir at room temperature overnight. Addition of ice-cold water (40.0 mL) to the reaction mixture led to a solid precipitate, which was filtered and thoroughly washed with cold water. The crude product was recrystallized from a methanol–water mixture (6:1) to yield 6.0 g (81%) of
68 as a colorless crystalline solid. Mp = 156–157 °C;
1H NMR (400 MHz, DMSO-d
6) δ 10.04 (s, 1H), 7.31 (s, 1H), 7.30 (s, 1H), 3.88 (s, 3H), 3.80 (s, 3H);
13C NMR (101 MHz, DMSO-d
6) δ 190.65, 154.94, 149.04, 126.16, 119.77, 116.41, 110.95, 56.94, 56.15.
Ethyl 2′-formyl-4′,5′-dimethoxy-[1,1′-biphenyl]-4-carboxylate (72)
Following general procedure D, 68 (1.00 g, 4.08 mmol), 4-ethoxycarbonylphenylboronic acid (69) (0.38 g, 4.48 mmol), were reacted and purified by flash chromatography to afford 0.95 g (74%) of 72 as a white solid. Mp = 96–98 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 1H), 8.08–7.95 (m, 2H), 7.65–7.55 (m, 2H), 7.43 (s, 1H), 7.03 (s, 1H), 4.34 (q, J = 7.1 Hz, 2H), 3.90 (s, 3H), 3.86 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 190.18, 165.92, 153.74, 149.26, 142.42, 139.56, 131.05, 129.64, 129.45, 126.53, 113.56, 109.26, 61.32, 56.52, 56.12, 14.63.
4′-(Ethoxycarbonyl)-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylic acid (75)
Following general procedure J, 72 (0.500 g, 1.59 mmol) was reacted to afford 0.40 g (76%) of 75 as a white solid. Mp = 151–153 °C; 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 8.09–8.01 (m, 2H), 7.57 (s, 1H), 7.42–7.32 (m, 2H), 6.74 (s, 1H), 4.40 (q, J = 7.1 Hz, 2H), 3.96 (s, 3H), 3.93 (s, 3H), 1.41 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 171.94, 166.58, 152.06, 148.04, 146.23, 137.77, 130.05, 129.55, 129.13, 128.69, 120.05, 113.68, 60.98, 56.16, 56.15, 14.37.
Ethyl 8,9-dimethoxy-6-oxo-6H-benzo[c]chromene-3-carboxylate (78)
The compound was obtained following the literature procedure [
19]. In a 20 mL vial
75 (0.38 g, 1.15 mmol), Pd(OAc)
2 (0.013 g, 0.05 mmol), N-acetylglycine (0.020 g, 0.17 mmol), KOAc (0.22 g, 2.30 mmol), and diacetoxiodo benzene (0.74 g, 2.30 mmol) were solubilized in t-BuOH (15.0 mL). The resulting mixture was stirred at 80 °C for 12 h. The solution was then rinsed with EtOAc (20.0 mL) and filtered through a Celite cake. Water (40.0 mL) was then added, and the aqueous layer was extracted with EtOAc (3 × 30 mL). The organic phase was dried (Na
2SO
4), filtered, and concentrated in vacuum to give 0.3 g of crude product. Crystallization from MeOH afforded 0.04 g (11%) of
78 as a white solid. Mp = 275–277 °C;
1H NMR (400 MHz, CDCl
3) δ 8.04–7.89 (m, 3H), 7.75 (s, 1H), 7.45 (s, 1H), 4.42 (q,
J = 7.1 Hz, 2H), 4.11 (s, 3H), 4.01 (s, 3H), 1.43 (t,
J = 7.1 Hz, 3H);
13C NMR (101 MHz, CDCl
3) δ 165.35, 160.53, 155.14, 150.94, 150.54, 131.31, 128.73, 125.06, 122.11, 121.82, 118.91, 115.19, 110.62, 103.19, 61.48, 56.40, 56.38, 14.28.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carboxylic acid (81)
Following general procedure B, 78 (0.030 g, 0.09 mmol) was reacted and triturated with Et2O to afford 0.015 g (60%) of 81 as dark-brown powder. Mp = 352–354 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.48 (br s, 3H), 8.16 (d, J = 8.4 Hz, 1H), 7.85 (dd, J = 8.3, 1.6 Hz, 1H), 7.77 (d, J = 1.6 Hz, 1H), 7.65 (s, 1H), 7.57 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 171.56, 156.76, 156.59, 152.65, 144.03, 143.40, 142.46, 130.93, 118.23, 109.29, 108.81, 100.74, 100.59; HRMS (ESI) m/z: 271.0252 [M − H]−, calculated for C14H7O6 271.0248.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carboxamide (82):
2′-Formyl-4′,5′-dimethoxy-[1,1′-biphenyl]-4-carboxamide (73)
Following general procedure D, 68 (500 mg, 2.04 mmol), (4-carbamoylphenyl)boronic acid (70) (0.4 g, 2.45 mmol), were reacted and purified by flash chromatography to afford 0.49 g (84%) of 73 as a white solid. Mp = 173–175 °C. 1H-NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.08 (br s, 1H), 8.00–7.97 (m, 2H), 7.55–7.53 (m, 2H), 7.46 (br s, 1H), 7.43 (s, 1H), 7.04 (s, 1H), 3.92 (s, 3H), 3.87 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 189.83, 167.41, 153.30, 148.66, 139.98, 139.64, 133.58, 130.13, 127.44, 126.06, 113.19, 108.65, 56.06, 55.67.
4′-Carbamoyl-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylic acid (76)
Following general procedure J, 73 (0.46 g, 1.62 mmol) was reacted to afford 0.44 g (91%) of 76 as a white solid. Mp = 241–242 °C. 1H-NMR (400 MHz, DMSO-d6) δ 12.49 (br s, 1H), 7.99 (br s, 1H), 7.88–7.85 (m, 2H), 7.38–7.35 (m, 4H), 6.88 (s, 1H), 3.84 (s, 3H), 3.84 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 168.45, 167.69, 150.65, 147.60, 144.23, 135.03, 132.52, 128.45, 126.99, 122.95, 113.74, 112.94, 55.74, 55.69.
8,9-Dimethoxy-6-oxo-6H-benzo[c]chromene-3-carboxamide (79)
Following general procedure G, 76 (0.2 g, 0.66 mmol) was reacted and triturated from MeOH and CH2Cl2 to afford 0.07 g (34%) of 79 as a yellow solid. Mp = 333–335 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.49 (d, J = 8.2 Hz, 1H), 8.17 (br s, 1H), 7.89–7.86 (m, 3H), 7.62 (s, 1H), 7.59 (br s, 1H), 4.05 (s, 3H), 3.92 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 166.43, 159.81, 155.18, 150.43, 150.03, 135.04, 128.78, 123.65, 123.31, 120.30, 115.99, 113.89, 109.88, 104.77, 56.54, 55.85.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carboxamide (82)
Following general procedure B, 79 (0.048 g, 0.16 mmol) was reacted and triturated with Et2O to afford 0.035 g (81%) of 82 as a white powder. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.51 (br s, 1H), 10.38 (br s, 1H), 8.15–8.13 (m, 1H), 8.12 (br s, 1H), 7.85–7.82 (m, 2H), 7.67 (s, 1H), 7.59 (s, 1H), 7.54 (br s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 166.53, 159.83, 153.37, 149.80, 147.98, 134.51, 127.42, 123.47, 122.72, 120.52, 115.99, 114.42, 112.99, 108.45; HRMS (ESI) calculated for C14H10NO5 [M + H]+ 272.0553, found 272.0546.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carbonitrile (83)
2′-Formyl-4′,5′-dimethoxy-[1,1′-biphenyl]-4-carbonitrile (74)
Following general procedure D, 68 (490 mg, 2.00 mmol), 4-cyanophenylboronic acid (71) (0.32 g, 2.20 mmol), were reacted and purified by flash chromatography to afford 0.43 g (81%) of 74 as a white solid. Mp = 218–220 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.70 (s, 1H), 7.97–7.95 (m, 2H), 7.69–7.67 (m, 2H), 7.46 (s, 1H), 7.05 (s, 1H), 3.92 (s, 3H), 3.88 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 189.64, 153.27, 148.96, 142.22, 138.32, 132.14, 131.18, 126.06, 118.74, 113.26, 110.60, 109.12, 56.12, 55.72.
4′-Cyano-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylic acid (77)
Following general procedure J, 74 (200 mg, 0.75 mmol) was reacted to afford 0.17 g (82%) of 77 as a white solid. Mp = 232–233 °C; 1H-NMR (400 MHz, DMSO-d6) δ 12.59 (br s, 1H), 7.85–7.83 (m, 2H), 7.51–7.48 (m, 2H), 7.41 (s, 1H), 6.88 (s, 1H), 3.84 (s, 3H), 3.84 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 167.97, 150.83, 147.95, 146.47, 134.35, 131.63, 129.75, 122.62, 119.00, 113.70, 113.07, 109.40, 55.81, 55.70.
8,9-Dimethoxy-6-oxo-6H-benzo[c]chromene-3-carbonitrile (80)
Following general procedure G, 77 (0.16 g, 0.56 mmol) was reacted and triturated from MeOH and CH2Cl2 to afford 0.03 g (19%) of 80 as a pale-yellow solid. Mp 337–339 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.64 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 1.6 Hz, 1H), 7.95 (s, 1H), 7.86 (dd, J = 8.4, 1.6 Hz, 1H), 7.65 (s, 1H), 4.05 (s, 3H), 3.94 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 159.19, 155.24, 151.07, 149.98, 127.94, 127.76, 124.97, 122.44, 121.11, 118.08, 114.41, 111.30, 110.01, 105.27, 56.67, 55.97.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromene-3-carbonitrile (83)
Following general procedure B, 80 (0.025 g, 0.09 mmol) was reacted and triturated with Et2O to afford 0.008 g (37%) of 83 as a pale-brown solid. Mp = 348–350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.25 (d, J = 8.3 Hz, 1H), 7.94 (d, J = 1.6 Hz, 1H), 7.75 (dd, J = 8.3, 1.6 Hz, 1H), 7.69 (s, 1H), 7.58 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 159.17, 153.70, 149.76, 148.82, 127.77, 126.51, 124.00, 122.68, 121.06, 118.17, 114.36, 113.18, 110.61, 108.85; HRMS (ESI) calculated for C14H6NO4 [M − H]– 252.0302, found 252.0310.
8,9-Dihydroxy-3-methoxy-6H-benzo[c]chromen-6-one (90)
Methyl 4-(benzyloxy)-2-bromo-5-hydroxybenzoate (84)
To a solution of 8 (1.20 g, 4.59 mmol) in CH2Cl2 (50 mL) BBr3 in CH2Cl2 (1.0 M, 27.57 mmol) was added dropwise at 0 °C. The solution was stirred at room temperature for 2 h. MeOH (60.0 mL) was added to quench the reaction and the resulting solvent was concentrated under reduced pressure. Thionyl chloride (10.0 mL) was added, and the solution was refluxed for 2 h. The solvent was then removed under reduced pressure and MeOH (30.0 mL) was added. The resulting solution was stirred at room temperature for 1 h (until full conversion, reaction monitored by TLC). The solvent was then removed under reduces pressure to afford 1.1 g (97%) of 84 as a pink solid. Mp = 149–151 °C; 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 7.17 (s, 1H), 3.90 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.88, 148.05, 142.49, 122.66, 121.07, 118.56, 113.56, 52.62.
Methyl 4,5-bis(benzyloxy)-2-bromobenzoate (85)
Following general procedure K, 84 (0.700 g, 2.83 mmol) was reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.96 g (79%) of 85 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.52 (s, 1H), 7.46–7.41 (m, 4H), 7.41–7.29 (m, 7H), 7.18 (s, 1H), 5.17 (s, 2H), 5.15 (s, 2H), 3.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.73, 152.03, 147.41, 136.37, 135.86, 128.65, 128.55, 128.21, 128.08, 127.39, 127.25, 123.30, 119.53, 117.56, 114.59, 71.42, 71.09, 52.25.
Methyl 4,5-bis(benzyloxy)-4′-methoxy-[1,1′-biphenyl]-2-carboxylate (87)
Following general procedure D, 85 (0.96 g, 2.24 mmol), 4-methoxyphenylboronic acid (86) (0.34 g, 2.24 mmol), were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.71 g (69%) of 87 as a white solid. Mp = 151–153 °C; 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 1.0 Hz, 1H), 7.50–7.45 (m, 2H), 7.46–7.41 (m, 2H), 7.41–7.29 (m, 6H), 7.19–7.12 (m, 2H), 6.94–6.88 (m, 2H), 6.88 (d, J = 0.9 Hz, 1H), 5.21 (s, 2H), 5.19 (s, 2H), 3.84 (s, 3H), 3.63 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.25, 158.72, 151.10, 147.27, 137.38, 136.85, 136.51, 133.79, 129.51, 128.54, 128.52, 127.98, 127.93, 127.39, 127.26, 122.42, 116.57, 116.44, 113.31, 71.40, 70.90, 55.24, 51.79.
4,5-Bis(benzyloxy)-4′-methoxy-[1,1′-biphenyl]-2-carboxylic acid (88)
Following general procedure E, 87 (0.71 g, 1.56 mmol) was reacted to afford 0.65 g (95%) of 88 as a white solid. Mp = 218–220 °C; 1H NMR (400 MHz, CDCl3) δ 7.62 (s, 1H), 7.52–7.46 (m, 2H), 7.46–7.41 (m, 2H), 7.42–7.28 (m, 6H), 7.22–7.14 (m, 2H), 6.93–6.86 (m, 2H), 6.86 (s, 1H), 5.21 (s, 2H), 5.20 (s, 2H), 3.84 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 172.18, 158.86, 151.77, 147.16, 138.49, 136.76, 136.37, 133.47, 129.73, 128.56, 128.53, 128.03, 127.95, 127.40, 127.25, 120.77, 117.05, 116.77, 113.40, 71.29, 70.84, 55.24.
8,9-Bis(benzyloxy)-3-methoxy-6H-benzo[c]chromen-6-one (89)
Following general procedure F, 88 (0.65 g, 1.47 mmol) was reacted to afford 0.21 g (33%) of 89 as a white powder. Mp = 208–210 °C; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 1.9 Hz, 1H), 7.70 (d, J = 8.9 Hz, 1H), 7.57–7.45 (m, 4H), 7.45–7.28 (m, 7H), 6.87 (dd, J = 8.8, 2.6 Hz, 1H), 6.82 (d, J = 2.5 Hz, 1H), 5.35 (s, 2H), 5.24 (s, 2H), 3.85 (d, J = 1.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 161.24, 160.84, 154.88, 152.25, 148.89, 136.27, 136.02, 130.50, 128.74, 128.58, 128.25, 128.05, 127.32, 127.10, 123.07, 113.23, 113.00, 112.28, 111.18, 104.72, 101.48, 71.02, 70.91, 55.63.
8,9-Dihydroxy-3-methoxy-6H-benzo[c]chromen-6-one (90)
Following general procedure H, 89 (0.21 g, 0.48 mmol) was reacted to afford 0.12 g (97%) of 90 as a white powder. Mp = 338–340 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.56–9.86 (m, 2H), 7.93 (d, J = 9.6 Hz, 1H), 7.49 (d, J = 7.9 Hz, 2H), 6.97–6.83 (m, 2H), 3.81 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.63, 160.52, 153.91, 151.86, 146.89, 129.20, 124.11, 114.65, 112.44, 111.58, 111.49, 107.61, 101.77, 56.10; HRMS (ESI) m/z: 257.0464 [M − H]−, calculated for C14H9O5 257.0455.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl acetate (94)
4,5-Bis(benzyloxy)-2-bromobenzoic acid (91)
Following general procedure E, 85 (3.30 g, 7.37 mmol) was reacted to afford 3.09 g (97%) of 91 as a white solid. Mp = 152–154 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 1.2 Hz, 1H), 7.50–7.26 (m, 10H), 7.21 (s, 1H), 5.19 (s, 2H), 5.16 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 169.92, 152.71, 147.33, 136.24, 135.72, 128.68, 128.59, 128.27, 128.11, 127.39, 127.26, 121.62, 119.68, 118.11, 115.71, 71.28, 71.08.
8,9-Bis(benzyloxy)-3-hydroxy-6H-benzo[c]chromen-6-one (92)
Following general procedure A, 91 (0.64 g, 1.55 mmol) was reacted to afford 0.32 g of 92 that was ca. 85% pure according to NMR and was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (OH, 1H), 7.82–7.25 (m, 15H), 5.43 (s, 2H), 5.26 (s, 2H)
8,9-Bis(benzyloxy)-6-oxo-6H-benzo[c]chromen-3-yl acetate (93)
To a solution of 92 (85% purity 150 mg = 128 mg, 0.301 mmol) in acetic anhydride (3 mL), sodium acetate (87 mg, 1.06 mmol) was added. The reaction mixture was stirred at 50 °C for 18 h. Then, the reaction mixture was cooled down to room temperature and water (5 mL) was added. The formed precipitate was collected by filtration, washed with water, dried, and triturated with MeOH to afford 0.094 g (67%) of 93 as a white solid. Mp = 200–205 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.43 (d, J = 8.7 Hz, 1H), 8.00 (s, 1H), 7.73 (s, 1H), 7.56–7.21 (m, 12H), 5.46 (s, 2H), 5.30 (s, 2H), 2.31 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 168.94, 159.70, 154.55, 151.18, 150.74, 149.07, 136.56, 136.30, 129.28, 128.56, 128.48, 128.14, 127.95, 127.82, 127.45, 118.47, 115.66, 113.11, 110.66, 106.07, 70.39, 70.03, 20.89.
8,9-Dihydroxy-6-oxo-6H-benzo[c]chromen-3-yl acetate (94)
Following general procedure H, 89 (0.08 g, 0.17 mmol) was reacted and purified by flash chromatography (CH2Cl2/MeOH) to afford 0.01 g (20%) of 94 as a white powder. Mp = 210–215 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 9.1 Hz, 1H), 7.58 (s, 1H), 7.55 (s, 1H), 7.21 (s, 1H), 7.14 (d, J = 9.1 Hz, 1H), 2.30 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 168.97, 159.79, 153.49, 150.61, 150.44, 147.33, 127.82, 123.55, 118.43, 115.88, 114.30, 112.02, 110.58, 107.91, 20.89; HRMS (ESI) calculated for C15H9O6 [M − H]– 285.0405, found 285.0416.
7,8-Dihydroxyisochromeno[3,4-e]indol-5(1H)-one (98)
1H-Indol-4-yl 4,5-bis(benzyloxy)-2-bromobenzoate (96)
A solution of 91 (1.24 g, 3.00 mmol) in SOCl2 (20.0 mL) was refluxed for 2 h. The solvent was removed under reduced pressure and diluted with dichloromethane (20.0 mL). The benzyl chloride solution was then added dropwise at 0 °C to a solution of 4-hydroxyindole (95) (0.40 g, 3.00 mmol) and triethylamine (0.91 g, 9.00 mmol) in dichloromethane (20.0 mL). The reaction was stirred for 2 h at room temperature and then quenched with water (40.0 mL). The organic layer was washed with water (30.0 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a yellow oil. Purification by flash chromatography (65/35 pentane/EtOAc) afforded 1.03 g (65%) of 96 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.52 (t, J = 2.3 Hz, 1H), 7.91 (s, 1H), 7.53–7.36 (m, 10H), 7.34 (s, 1H), 7.22–7.17 (m, 2H), 7.07–7.03 (m, 1H), 7.00 (dd, J = 3.3, 2.4 Hz, 1H), 6.47 (ddd, J = 3.3, 2.1, 0.5 Hz, 1H), 5.22 (s, 4H); 13C NMR (101 MHz, cdcl3) δ 163.99, 152.55, 147.52, 143.58, 137.85, 136.33, 135.83, 128.77, 128.69, 128.37, 128.22, 127.60, 127.44, 124.95, 122.74, 121.96, 121.21, 119.75, 118.06, 115.56, 111.80, 109.65, 99.20, 71.50, 71.19.
7,8-Bis(benzyloxy)isochromeno[3,4-e]indol-5(1H)-one (97)
The compound was obtained following the literature procedure [
26]. To a solution of
96 (1.00 g, 1.89 mmol) in dry DMA (10.0 mL), NaOAc (0.31 g, 3.78 mmol), Pd(OAc)
2 (0.042 g, 0.19 mmol), and tricyclohexylphosphine tetrafluoroborate (0.210 g, 0.57 mmol) were added. The mixture was degassed and then heated at 175 °C for 1 h in the microwave. The reaction mixture was cooled to room temperature and then diluted with diethyl ether and filtered through Celite to remove the catalyst. The solvent was removed under reduced pressure to give 1.5 g of a dark oil. Purification by flash chromatography (60/40 pentane/EtOAc) and recrystallization from MeOH afforded 0.24 g (28%) of
97 as a white solid. Mp = 197–199 °C;
1H NMR (400 MHz, DMSO-d
6) δ 11.55 (s, 1H), 7.99 (d,
J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.72 (s, 1H), 7.58–7.51 (m, 2H), 7.51–7.45 (m, 2H), 7.45–7.26 (m, 8H), 6.69 (ddd,
J = 3.0, 2.0, 0.9 Hz, 1H), 5.45 (s, 2H), 5.27 (s, 2H);
13C NMR (101 MHz, DMSO-d
6) δ 160.85, 155.06, 148.27, 144.66, 137.89, 137.20, 136.94, 132.45, 128.99, 128.90, 128.50, 128.33, 128.17, 127.89, 126.61, 117.14, 116.60, 112.72, 112.42, 109.42, 108.46, 105.96, 98.96, 70.67, 70.45.
7,8-Dihydroxyisochromeno[3,4-e]indol-5(1H)-one (98)
Following general procedure H, 97 (0.10 g, 0.22 mmol) was reacted to afford 0.05 g (84%) of 98 as a yellow solid. Mp > 350 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 10.09 (br s, 2H), 7.68 (d, J = 8.7 Hz, 1H), 7.54 (s, 1H), 7.52 (s, 1H), 7.40 (d, J = 3.1 Hz, 1H), 7.37 (dd, J = 8.6, 0.9 Hz, 1H), 6.65 (dd, J = 3.1, 0.8 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 160.95, 153.91, 146.35, 144.24, 137.50, 130.95, 126.39, 117.22, 115.82, 114.75, 111.54, 109.42, 108.62, 107.68, 98.82; HRMS (ESI) m/z: 266.0472 [M − H]−, calculated for C15H8NO4 266.0459.
8,9-Dihydroxy-6H-dibenzo[c,h]chromen-6-one (103)
Methyl 4,5-dimethoxy-2-(naphthalen-2-yl)benzoate (100)
Following general procedure D, 9 (0.55 g, 2.00 mmol), 2-naphtalenlboronic acid (99) (0.38 g, 2.20 mmol), were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.37 g (57%) of 100 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.90–7.75 (m, 4H), 7.53 (s, 1H), 7.51–7.39 (m, 3H), 6.91 (s, 1H), 3.97 (s, 3H), 3.90 (s, 3H), 3.58 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.15, 151.34, 147.87, 139.42, 137.36, 133.26, 132.39, 128.00, 127.72, 127.54, 127.09, 126.66, 126.18, 125.88, 122.00, 113.94, 113.03, 56.12, 56.05, 51.78.
4,5-Dimethoxy-2-(naphthalen-2-yl)benzoic acid (101)
Following general procedure E, 100 (0.37 g, 1.14 mmol) was reacted to afford 0.29 g (82%) of 101 as a white solid. Mp = 233–235 °C; 1H NMR (400 MHz, CDCl3) δ 7.92–7.81 (m, 2H), 7.81–7.73 (m, 2H), 7.57 (s, 1H), 7.54–7.45 (m, 2H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 6.85 (s, 1H), 3.95 (s, 3H), 3.93 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 172.31, 151.97, 147.74, 139.21, 138.70, 133.15, 132.43, 128.02, 127.67, 127.63, 127.10, 126.73, 126.10, 125.89, 120.31, 114.29, 113.65, 56.14, 56.12.
8,9-dimethoxy-6H-dibenzo[c,h]chromen-6-one (102)
Following general procedure F, 101 (0.29 g, 0.99 mmol) was reacted to afford 0.200 g (69%) of 102 as a white powder. Mp = 202–204 °C; 1H NMR (400 MHz, CDCl3) δ 8.50–8.35 (m, 1H), 7.78 (d, J = 8.9 Hz, 1H), 7.76–7.72 (m, 1H), 7.63 (s, 1H), 7.62–7.58 (m, 1H), 7.58–7.48 (m, 2H), 7.30 (s, 1H), 4.04 (s, 3H), 3.96 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 161.11, 155.06, 149.77, 146.43, 133.57, 130.41, 127.48, 127.40, 126.90, 124.22, 123.65, 121.93, 118.69, 114.06, 112.81, 110.20, 102.71, 56.24, 56.22.
8,9-Dihydroxy-6H-dibenzo[c,h]chromen-6-one (103)
Following general procedure B, 102 (0.120 g, 0.66 mmol) was reacted to afford 0.075 g (69%) of 103 as a dark brown solid. Mp > 350 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.50 (br s, 1H), 10.28 (br s, 1H), 8.36–8.30 (m, 1H), 8.11 (d, J = 8.9 Hz, 1H), 8.02–7.94 (m, 1H), 7.88–7.78 (m, 1H), 7.71–7.58 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 160.32, 153.99, 147.79, 145.80, 133.55, 129.24, 128.36, 127.72, 127.63, 124.62, 123.52, 121.45, 120.31, 114.82, 113.73, 113.00, 108.71; HRMS (ESI) m/z: 277.0515 [M − H]−, calculated for C17H9O4 277.0506.
3-Hydroxy-8,9-dimethoxy-6H-benzo[c]chromen-6-one (118)
Following general procedure A, 8 (3.13 g, 12.00 mmol) was reacted to afford 2.91 g (89%) of 118 as a white solid. Mp = 329–331 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.15 (s, 1H), 7.81–7.31 (m, 2H), 7.07–6.43 (m, 2H), 3.98 (s, 3H), 3.85 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.68, 159.43, 155.46, 152.12, 149.23, 130.74, 124.99, 113.16, 112.00, 109.87, 103.67, 103.13, 56.63, 56.00; HRMS (ESI) m/z: 271.0617 [M − H]−, calculated for C15H11O5 271.0612.
3,9-Dihydroxy-8-methoxy-6H-benzo[c]chromen-6-one (119)
5-Bromo-4-formyl-2-methoxyphenyl acetate (106)
To a solution of KBr (6.7 g, 52.10 mmol) and 4-formyl-2-methoxyphenyl acetate (104) (3.2 g, 16.50 mmol) in H2O (80 mL) was added dropwise Br2 (3.2 g, 20.02 mmol). The resulting mixture was stirred at room temperature for 12 h. The precipitate formed was collected by filtration to give 4.5 g (95%) of 106 as an orange powder. Mp = 137–139 °C; 1H NMR (400 MHz, CDCl3) δ 10.26 (d, J = 0.4 Hz, 1H), 7.51 (s, 1H), 7.35 (s, 1H), 3.88 (s, 3H), 2.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 190.81, 167.90, 151.24, 144.95, 131.61, 127.98, 117.87, 112.11, 56.27, 20.53.
4-Acetoxy-2-bromo-5-methoxybenzoic acid (112)
The compound was synthetized following the literature procedure [
27]. Compound
106 (1.00 g, 3.66 mmol) was dissolved in acetone (20.0 mL) and heated to reflux. A mixture of water (20.0 mL) and KMnO
4 (1.15 g, 7.32 mmol) was added at refluxing temperature and stirred for 90 min at 50 °C. The solvent was removed under reduced pressure. The undesired by-product MnO
2 was removed by filtration on a Celite bed and washed with 10% KOH solution (10 mL). The pH of the filtrate was adjusted to 2–2.5 using 1 N aq. HCl and extracted with EtOAc (3 × 30.0 mL). The organic phase was dried (Na
2SO
4), filtered, and concentrated under reduced pressure afforded 0.96 g (99%) of
112 as a white solid. The compound was used directly for the next step.
2-Bromo-4-hydroxy-5-methoxybenzoic acid (113)
The compound was synthetized following the literature procedure [
28]. A solution of
112 (0.96 g, 3.64 mmol) in NaOH aq. solution (5%, 20.0 mL) was heated to reflux for 1 h. The reaction was then cooled down to 0 °C, acidified with 1M HCl and extracted with EtOAc (3 × 30.0 mL). The organic phase was dried (Na
2SO
4), filtered, and concentrated under reduced pressure to afford 0.43 g (45%) of
113 as a white solid. Mp = 193–195 °C;
1H-NMR (400 MHz, DMSO-d
6) δ 12.91 (br s, 1H), 10.23 (s, 1H), 7.38 (s, 1H), 7.05 (s, 1H), 3.79 (s, 3H);
13C-NMR (101 MHz, DMSO-d
6) δ 166.34, 150.38, 146.66, 122.09, 120.42, 114.81, 112.57, 55.76.
3,9-Dihydroxy-8-methoxy-6H-benzo[c]chromen-6-one (119)
Following general procedure A, 113 (0.2 g, 0.81 mmol) was reacted to afford 0.1 g (40%) of 119 as a dark brown solid. Mp = 320–322 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 10.15 (s, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.53 (s, 1H), 7.47 (s, 1H), 6.79 (dd, J = 8.6, 2.4 Hz, 1H), 6.69 (d, J = 2.4 Hz, 1H), 3.88 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.74, 159.41, 154.44, 152.14, 148.57, 130.99, 124.50, 113.40, 111.07, 110.96, 109.89, 107.17, 103.22, 56.20; HRMS (ESI) m/z: 257.0456 [M − H]−, calculated for C14H9O5 257.0454.
3,8-Dihydroxy-9-methoxy-6H-benzo[c]chromen-6-one (120)
4-Bromo-5-formyl-2-methoxyphenyl acetate (107)
The compound was synthetized following the literature procedure [
29]. To a stirred solution of 2-bromo-4-hydroxy-5-methoxybenzaldehyde (
105) (1.00 g, 4.33 mmol) in CH
2Cl
2 (30.0 mL), acetic anhydride (0.82 mL, 8.66 mmol), DMAP (0.53 g, 4.33 mmol), and Et
3N (0.60 mL, 4.33 mmol) were added at room temperature. The reaction mixture was stirred until all the starting material was consumed as monitored by TLC. Water (25.0 mL) and CH
2Cl
2 (25 mL) were added, and the two phases were separated. The aqueous layer was extracted with CH
2Cl
2 (20 mL). The combined organic layers were washed with brine (10.0 mL), dried over Na
2SO
4, filtered, and concentrated under reduced pressure to give the crude product. Purification by flash chromatography (40/60 EtOAc/hexanes) afforded 1.05 g (89%) of
107 as a white solid. Mp = 97–99 °C;
1H NMR (400 MHz, CDCl
3) δ 10.18 (s, 1H), 7.62 (s, 1H), 7.17 (s, 1H), 3.92 (s, 3H), 2.32 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 189.94, 168.32, 156.34, 139.60, 126.84, 125.61, 123.72, 116.81, 56.57, 20.47.
5-Acetoxy-2-bromo-4-methoxybenzoic acid (114)
The compound was synthetized following the literature procedure [
27]. Compound
107 (1.00 g, 3.66 mmol) was dissolved in acetone (20.0 mL) and heated to reflux. A mixture of water (20.0 mL) and KMnO
4 (1.15 g, 7.32 mmol) was added at refluxing temperature and stirred for 90 min at 50 °C. The solvent was removed under reduced pressure. The undesired by-product MnO
2 was removed by filtration on a Celite bed and washed with 10% KOH solution (10 mL). The pH of the filtrate was adjusted to 2–2.5 using 1 N aq. HCl and extracted with EtOAc (3 × 30.0 mL). The organic phase was dried (Na
2SO
4), filtered, and concentrated under reduced pressure afforded 0.92 g (87%) of
114 as a white solid. Mp = 182–184 °C;
1H NMR (400 MHz, CDCl
3) δ 7.81 (s, 1H), 7.25 (s, 1H), 3.90 (s, 3H), 2.33 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 169.40, 168.40, 154.84, 138.43, 127.25, 121.70, 121.56, 118.60, 56.42, 20.49.
2-Bromo-5-hydroxy-4-methoxybenzoic acid (115)
The compound was synthetized following the literature procedure [
28]. A solution of
114 (0.92 g, 3.18 mmol) in NaOH aq. solution (5%, 20.0 mL) was heated to reflux for 1 h. The reaction was then cooled down to 0 °C, acidified with 1M HCl, and extracted with EtOAc (3 × 30.0 mL). The organic phase was dried (Na
2SO
4), filtered, and concentrated under reduced pressure to afford 0.71 g (90%) of
115 as a white solid. Mp = 203–205 °C;
1H NMR (400 MHz, DMSO-d
6) δ 12.72 (s, 1H), 9.64 (s, 1H), 7.28 (s, 1H), 7.15 (s, 1H), 3.82 (s, 3H);
13C NMR (101 MHz, DMSO-d
6) δ 166.82, 151.21, 145.98, 124.27, 118.20, 117.59, 110.77, 56.47.
3,8-Dihydroxy-9-methoxy-6H-benzo[c]chromen-6-one (120)
Following general procedure A, 115 (0.64 g, 2.59 mmol) was reacted to afford 0.31 g (46%) of 120 as a dark-brown solid. Mp = 341–343 °C; 1H NMR (400 MHz, D2O, Na salt) δ 6.63 (s, 1H), 6.49 (d, J = 8.1 Hz, 1H), 6.45 (s, 1H), 5.81 (d, J = 2.4 Hz, 1H), 5.68 (dd, J = 8.1, 2.5 Hz, 1H), 3.57 (s, 3H); 13C NMR (101 MHz, d2o) δ 180.59, 165.83, 164.41, 152.94, 149.83, 133.05, 131.04, 125.17, 119.97, 116.67, 115.52, 108.98, 105.53, 55.83; HRMS (ESI) m/z: 257.0456 [M − H]−, calculated for C14H9O5 257.0455.
9-Amino-3,8-dihydroxy-6H-benzo[c]chromen-6-one (124)
2-Bromo-5-methoxy-4-nitrobenzoic acid (116)
To a suspension of 3-methoxy-4-nitrobenzoic acid (108) (2.00 g, 10.15 mmol) in concentrated H2SO4 (80 mL), NBS (1.896 g, 10.65 mmol) was added in 3 portions (every 15 min) at room temperature. The reaction mixture was stirred at room temperature for 2 days. Then, the reaction mixture was poured into ice/water (ca. 200 mL) and the yellow precipitate filtered off and dried by co-evaporation with toluene to afford 1.98 g (71%) of 116 as a yellow solid. Mp 195–197 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 7.63 (s, 1H), 3.96 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.35, 150.87, 140.34, 139.26, 129.08, 115.63, 108.73, 57.27.
3-Hydroxy-8-methoxy-9-nitro-6H-benzo[c]chromen-6-one (121)
Following general procedure A, 116 (1.00 g, 3.62 mmol) was reacted and triturated with MeOH to afford 0.67 g (64%) of 121 as a yellow solid. Mp = 285–287 °C; 1H NMR (400 MHz, acetone-d6) δ 9.30 (s, 1H), 8.61 (s, 1H), 8.15 (d, J = 8.7 Hz, 1H), 7.99 (s, 1H), 6.94 (dd, J = 8.7, 2.4 Hz, 1H), 6.83 (d, J = 2.4 Hz, 1H), 4.13 (s, 3H); 13C-NMR (101 MHz, acetone-d6) δ 160.81, 160.14, 153.24, 151.09, 129.98, 125.51, 123.85, 118.90, 114.54, 114.28, 110.92, 110.08, 104.18, 57.62.
9-Amino-3-hydroxy-8-methoxy-6H-benzo[c]chromen-6-one (123)
To a suspension of 121 (250 mg, 0.870 mmol) in EtOH (5 mL), SnCl2 (825 mg, 4.35 mmol) was added. The reaction mixture was stirred at 70 °C for 30 h. Then, the reaction mixture was poured into ice/water, and neutralized by addition of a saturated aqueous solution of NaHCO3. The mixture was extracted with EtOAc (6 × 100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography (CH2Cl2:MeOH 100:0 to 98:2) afforded 0.08 g (36%) of 123 as a white solid. Mp = 330–332 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.10 (br s, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.38 (s, 1H), 7.19 (s, 1H), 6.80 (dd, J = 8.8, 2.4 Hz, 1H), 6.68 (d, J = 2.4 Hz, 1H), 6.15 (br s, 2H), 3.89 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 160.50, 158.69, 151.79, 146.13, 145.73, 130.72, 123.51, 112.74, 109.72, 108.55, 106.47, 102.76, 102.20, 55.53.
9-Amino-3,8-dihydroxy-6H-benzo[c]chromen-6-one (124)
Following general procedure B, 123 (0.035 g, 0.136 mmol) was reacted and triturated with Et2O, to afford 0.021 g (64%) of 124 as a brown solid. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.02 (br s, 1H), 10.00 (br s, 1H), 7.71 (d, J = 8.8 Hz, 1H), 7.34 (s, 1H), 7.15 (s, 1H), 6.77 (dd, J = 8.8, 2.4 Hz, 1H), 6.65 (d, J = 2.4 Hz, 1H), 5.92 (br s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ 160.43, 158.29, 151.54, 145.32, 144.08, 129.51, 123.21, 112.60, 112.02, 110.05, 106.81, 102.73, 102.55; HRMS (ESI) calculated for C13H11NO4 [M + H]+ 244.0604, found 244.0614.
N-(3,8-Dihydroxy-6-oxo-6H-benzo[c]chromen-9-yl)acetamide (127)
To a suspension of 124 (58 mg, 0.226 mmol) in 1,4-dioxane (4 mL), a saturated aqueous solution of NaHCO3 (2 mL) and acetic anhydride (5 mL) were added. The reaction mixture was stirred at 100 °C for 1 h. The reaction mixture was cooled down to 0 °C, water was added, and a white solid precipitated, which was collected by filtration and washed with water. The crude (triacetylated compound) was suspended in CH2Cl2 (0.85 mL) and BBr3 (1 M in CH2Cl2, 0.85 mL, 0.85 mmol) was added. The reaction mixture was stirred at room temperature for 48 h. Then, the reaction was quenched with MeOH and the solvent removed under reduced pressure. Purification by flash column chromatography (CH2Cl2:MeOH 10:0 to 9:1) afforded 0.022 g (41%) of 127 as a white solid. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.15 (s, 1H), 9.55 (s, 1H), 8.89 (s, 1H), 7.77 (d, J = 8.8 Hz, 1H), 7.59 (s, 1H), 6.84 (dd, J = 8.8, 2.4 Hz, 1H), 6.72 (d, J = 2.4 Hz, 1H), 2.21 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 169.76, 160.16, 158.68, 151.35, 146.72, 134.48, 127.76, 123.36, 114.02, 113.13, 113.07, 111.70, 109.79, 102.95, 24.30; HRMS (ESI) calculated for C15H12NO5 [M + H]+ 286.0710, found 286.0700.
8-Amino-3,9-dihydroxy-6H-benzo[c]chromen-6-one (126)
Methyl 2-bromo-4,5-dinitrobenzoate (110)
To a solution of 2-bromo-4-nitrobenzoic acid methyl ester (109) (5.00 g, 19.23 mmol) in sulfuric acid (15.0 mL), nitric acid (8.0 mL) was added. The reaction was stirred at 50 °C for 1 h and then poured into ice-cold water. The solid obtained was recrystallized from MeOH to afford 4.95 g (84%) of 110 as a yellow solid. Mp = 98–100 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.60 (s, 1H), 3.15 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.90, 143.59, 140.18, 137.29, 130.99, 128.07, 127.27, 54.02.
Methyl 2-bromo-4-methoxy-5-nitrobenzoate (111)
To a solution of 110 (2.00 g, 6.55 mmol) in MeOH (30.0 mL) at 0 °C, a solution of KOH (0.72 g, 13.11 mmol) in MeOH (10.0 mL) was added. The reaction was stirred at room temperature for 1 h. The solvent was removed under reduced pressure, and the solid obtained was taken up with water and filtered. Recrystallization from MeOH afforded 1.62 g (85%) of 111 as a white solid. Mp = 137–139 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.37 (s, 1H), 7.74 (s, 1H), 4.02 (s, 3H), 3.84 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.28, 154.68, 137.96, 128.50, 127.94, 123.48, 120.79, 58.26, 53.22.
2-Bromo-4-methoxy-5-nitrobenzoic acid (117)
Following general procedure E, 111 (1.5 g, 5.17 mmol) was reacted to afford 1.05 g (74%) of 117 as a white solid. Mp = 175–177 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.60 (s, 1H), 8.34 (s, 1H), 7.70 (s, 1H), 4.01 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.38, 154.32, 137.96, 128.33, 127.86, 124.74, 120.70, 58.13.
3-Hydroxy-9-methoxy-8-nitro-6H-benzo[c]chromen-6-one (122)
Following general procedure A, 117 (1.05 g, 3.80 mmol) was reacted to afford 0.100 g (9%) of 122 as a yellow solid. Mp = 355–357 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 8.57 (s, 1H), 8.34 (d, J = 8.9 Hz, 1H), 7.92 (s, 1H), 6.86 (dd, J = 8.8, 2.4 Hz, 1H), 6.74 (d, J = 2.4 Hz, 1H), 4.14 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.04, 159.57, 157.37, 153.75, 141.19, 138.71, 128.24, 127.14, 113.85, 111.87, 108.76, 106.44, 103.48, 58.26, 40.59, 40.43, 40.22.
8-Amino-3-hydroxy-9-methoxy-6H-benzo[c]chromen-6-one (125)
Following general procedure H, 25 122 (0.08 g, 0.28 mmol) was reacted to afford to afford 0.070 g (98%) of 125 as a yellow solid. Mp = 340–342 °C; 1H NMR (400 MHz, DMF-d7) δ 10.46 (s, 1H), 8.30 (d, J = 8.7 Hz, 1H), 7.80 (s, 1H), 7.69 (s, 1H), 7.03 (dd, J = 8.7, 2.4 Hz, 1H), 6.95 (d, J = 2.3 Hz, 1H), 5.59 (s, 2H), 4.27 (s, 3H); 13C NMR (101 MHz, DMF-d7) δ 161.13, 158.79, 153.49, 151.79, 139.18, 126.81, 123.82, 113.21, 112.86, 111.44, 111.25, 103.02, 102.10, 56.10.
8-Amino-3,9-dihydroxy-6H-benzo[c]chromen-6-one (126)
Following general procedure B, 125 (0.070 g, 0.27 mmol) was reacted to afford 0.06 g (91%) of 126 as a white solid. Mp > 350 °C; 1H NMR (400 MHz, DMF-d7) δ 8.40 (s, 1H), 8.09 (d, J = 8.7 Hz, 1H), 8.00 (s, 1H), 7.10 (dd, J = 8.7, 2.4 Hz, 1H), 6.99 (d, J = 2.4 Hz, 1H); 13C-NMR (101 MHz, DMF-d7): δ 162.3, 162.2, 158.2, 154.6, 136.6, 127.1, 126.3, 124.7, 115.2, 113.4, 111.4, 108.6, 105.0; HRMS (ESI) m/z: 242.0461 [M − H]−, calculated for C13H8NO4 242.0459.
N-(3,9-Dihydroxy-6-oxo-6H-benzo[c]chromen-8-yl)acetamide (128)
To a solution of 126 (50 mg, 0.206 mmol) in 1,4-dioxane (0.5 mL), a saturated aqueous solution of NaHCO3 (0.5 mL) and acetic anhydride (0.2 mL, 2.06 mmol) were added. The reaction mixture was stirred at room temperature for 1 h. Then, the solvent was removed under reduced pressure and dried by co-evaporation with toluene. The crude (triacetylated compound) was suspended in CH2Cl2 (0.85 mL) and BBr3 (1 M in CH2Cl2, 0.85 mL, 0.85 mmol) was added. The reaction mixture was stirred at room temperature for 48 h. Then, the reaction was quenched with MeOH and the solvent removed under reduced pressure. Purification by flash column chromatography (CH2Cl2:MeOH 10:0 to 9:1) afforded 0.031 g (53%) of 128 as a pale-brown/beige solid. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.27 (br s, 1H), 9.47 (s, 1H), 8.76 (s, 1H), 7.82 (d, J = 8.7 Hz, 1H), 7.49 (s, 1H), 6.83 (dd, J = 8.7, 2.4 Hz, 1H), 6.71 (d, J = 2.4 Hz, 1H), 2.15 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 169.2, 160.3, 159.4, 154.4, 152.0, 132.2, 127.5, 124.0, 121.8, 113.1, 110.4, 109.2, 105.9, 102.9, 23.9. HRMS (ESI) calculated for C15H12NO5 [M + H]+ 286.0710, found 286.0699.
8,9-Diamino-3-hydroxy-6H-benzo[c]chromen-6-one (135)
2-Bromo-4,5-dinitrobenzoic acid (129)
To a solution of 2-bromo-4-nitrobenzoic acid (5.00 g, 20.3 mmol) in H2SO4 (15.0 mL), HNO3 (8.0 mL) was added. The reaction mixture was stirred at 50 °C for 100 min and then poured into ice/water. The solid obtained was filtered, washed with water, and dried by co-evaporation with toluene (×3) to give 4.5 g (76%) of 129 as a pale-yellow solid. Mp = 170–172 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.55 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 164.65, 142.64, 140.00, 138.71, 130.52, 126.99, 126.16.
Methyl 4-(benzylamino)-2-bromo-5-nitrobenzoate (130)
To a solution of 129 (2.180 g, 7.49 mmol) in DMF (60 mL), benzyl amine (2.5 mL, 23.2 mmol) was added. The reaction mixture was stirred at room temperature for 16 h. Then, the solvent was removed under reduced pressure, and the resulting solid was triturated with Et2O and a mixture of Et2O:CH2Cl2 to afford a yellow solid. This compound was dissolved in MeOH (100 mL), and H2SO4 (1.5 mL) was added. The reaction mixture was stirred at reflux for 8 h. Then, the reaction mixture was cooled down to room temperature, a saturated aqueous solution of NaHCO3 (20 mL) was added, and the solvent was removed under reduced pressure. A saturated aqueous solution of NaHCO3 (100 mL) and EtOAc (100 mL) was added to the residue and the phases were separated, the aqueous layer was extracted with EtOAc (3 × 80 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography (pentane:EtOAc 100:0 to 0:100) afforded 2.15 g (46%) of 130 as a yellow solid. Mp = 187–189 °C; 1H-NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.52 (br s, 1H), 7.43–7.33 (m, 5H), 7.18 (s, 1H), 4.55 (d, J = 5.5 Hz, 2H), 3.90 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 164.33, 146.28, 135.95, 131.56, 131.19, 129.35, 128.43, 127.43, 119.86, 117.84, 52.54, 47.56.
Methyl 5-(benzylamino)-4′-(benzyloxy)-4-nitro-[1,1′-biphenyl]-2-carboxylate (132)
Following general procedure D, 130 (0.25 g, 0.68 mmol) and 4-(benzyloxy)phenylboronic acid (131) (0.23 g, 1.03 mmol) were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.32 g (90%) of 132 as a yellow solid. Mp = 193–195 °C; 1H-NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 8.63 (t, J = 5.5 Hz, 1H), 7.47–7.32 (m, 10H), 7.16–7.13 (m, 2H), 7.01–6.97 (m, 2H), 6.74 (s, 1H), 5.10 (s, 2H), 4.57 (d, J = 5.5 Hz, 2H), 3.70 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 166.59, 159.04, 150.61, 146.14, 136.88, 136.57, 132.69, 130.93, 129.42, 129.19, 128.76, 128.20, 128.16, 127.67, 127.42, 117.72, 116.39, 116.12, 114.47, 70.16, 52.09, 47.42.
5-(Benzylamino)-4′-(benzyloxy)-4-nitro-[1,1′-biphenyl]-2-carboxylic acid (133)
Following general procedure E, 132 (0.3 g, 0.64 mmol) was reacted to afford 0.15 g (52%) of 133 as a yellow solid. Mp = 216–218 °C; 1H-NMR (400 MHz, DMSO-d6) δ 12.59 (br s, 1H), 8.97 (t, J = 6.0 Hz, 1H), 8.57 (s, 1H), 7.48–7.45 (m, 2H), 7.42–7.38 (m, 2H), 7.37–7.32 (m, 5H), 7.29–7.25 (m, 1H), 7.11–7.09 (m, 2H), 7.01–6.98 (m, 2H), 6.72 (s, 1H), 5.11 (s, 2H), 4.72 (d, J = 6.0 Hz, 2H); 13C-NMR (101 MHz, DMSO-d6) δ 167.09, 158.42, 148.82, 145.62, 138.07, 136.96, 132.15, 129.62, 129.43, 129.30, 128.65, 128.47, 127.92, 127.78, 127.21, 126.99, 117.93, 116.68, 114.15, 69.28, 45.77.
9-(Benzylamino)-3-(benzyloxy)-8-nitro-6H-benzo[c]chromen-6-one (134)
Following general procedure G, 133 (0.14 g, 0.31 mmol) was reacted and purified by flash chromatography (CH2Cl2/MeOH) to afford 0.06 g (43%) of 134 as a yellow solid. Mp = 284–286 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.10 (t, J = 6.1 Hz, 1H), 8.84 (s, 1H), 8.13 (d, J = 8.6 Hz, 1H), 7.51–7.47 (m, 5H), 7.43–7.34 (m, 5H), 7.28–7.24 (m, 1H), 7.08–7.05 (m, 2H), 5.23 (s, 2H), 4.87 (d, J = 6.1 Hz, 2H); 13C-NMR (201 MHz, DMSO-d6) δ 161.65, 159.23, 153.29, 147.62, 139.53, 137.99, 136.25, 131.91, 130.50, 128.63, 128.51, 128.10, 127.92, 127.31, 127.27, 125.99, 113.19, 109.55, 107.36, 105.07, 102.58, 69.90, 45.79.
8,9-Diamino-3-hydroxy-6H-benzo[c]chromen-6-one (135)
Following general procedure H, 134 (0.03 g, 0.07 mmol) was reacted to afford 0.009 g (53%) of 135 as a pale brown solid. M.p. > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.23 (s, 1H), 7.09 (s, 1H), 6.75 (dd, J = 8.7, 2.4 Hz, 1H), 6.63 (d, J = 2.4 Hz, 1H), 5.82 (br s, 2H), 5.09 (br s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ 160.68, 157.67, 151.15, 143.45, 135.40, 127.04, 122.68, 112.47, 111.75, 110.50, 107.97, 102.71, 102.69; HRMS (ESI) calculated for C13H11N2O3 [M + H]+ 243.0764, found 243.0762.
N,N′-(3-Hydroxy-6-oxo-6H-benzo[c]chromene-8,9-diyl)diacetamide (140)
Methyl 4,5-diamino-2-bromobenzoate (136)
To a solution of 110 (1.81 g, 5.94 mmol) in acetic acid (120 mL), iron (3.98 g, 71.28 mmol) was added. The reaction mixture was stirred at 75 °C for 3 h. Then, the reaction was cooled down to room temperature and filtered to remove the iron residues (wash with water and EtOAc, ca. 100 mL of each). Then, the solvent was removed under reduced pressure, water (200 mL) and EtOAc (200 mL) were added, the layers were separated, and the aqueous layer was extracted with EtOAc (3 × 200 mL), the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography (CH2Cl2:MeOH 99:1 to 98:2) afforded 0.85 g (58%) of 136 as a pale-brown solid. Mp = 111–112 °C; 1H-NMR (400 MHz, CDCl3) δ 7.33 (s, 1H), 6.93 (s, 1H), 3.86 (s, 3H), 3.79 (br s, 2H), 3.33 (br s, 2H); 13C-NMR (101 MHz, CDCl3) δ 166.22, 140.44, 132.49, 120.95, 120.77, 120.47, 113.82, 52.07.
Methyl 4,5-diacetamido-2-bromobenzoate (137)
To a suspension of 136 (1.77 g, 7.22 mmol) in 1,4-dioxane (50 mL), a saturated aqueous solution of NaHCO3 (50 mL) and acetyl chloride (2.6 mL, 36.1 mmol) were added. The reaction mixture was stirred at room temperature for 24 h. Then, more portions of NaHCO3 (2.0 g, 23.8 mmol) and acetyl chloride (2.6 mL, 36.1 mmol) were added 6 times over the following 6 days to achieve full conversion. The reaction mixture was poured into ice/water (200 mL) and the solid was filtered off, washed with water, and dried to give 1.13 g (48%) of 137 as a pale-orange solid. Mp = 251–252 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.60 (bs, 2H), 8.16 (s, 1H), 8.05 (s, 1H), 3.83 (s, 3H), 2.12 (s, 3H), 2.09 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 169.16, 169.03, 165.08, 134.74, 128.45, 128.04, 127.56, 126.11, 115.64, 52.45, 23.97, 23.72.
Methyl 4,5-diacetamido-2′,4′-dimethoxy-[1,1′-biphenyl]-2-carboxylate (139)
Following general procedure D, 137 (0.8 g, 2.43 mmol) 2,4-dimethoxybenzeneboronic acid (138) (0.88 g, 4.86 mmol) were reacted and purified by flash chromatography (CH2Cl2/MeOH) to afford 0.68 g (73%) of 139 as a pale-brown solid. Mp = 110–112 °C; 1H-NMR (400 MHz, CDCl3) δ 8.76 (s, 1H), 8.72 (s, 1H), 7.81 (s, 1H), 7.18 (s, 1H), 7.07 (d, J = 8.3 Hz, 1H), 6.53 (dd, J = 8.3, 2.3 Hz, 1H), 6.44 (d, J = 2.3 Hz, 1H), 3.84 (s, 3H), 3.71 (s, 3H), 3.65 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 170.86, 170.49, 167.55, 160.84, 156.99, 136.95, 133.22, 130.39, 129.02, 128.53, 127.80, 127.07, 122.15, 104.39, 98.30, 55.50, 55.20, 51.93, 23.65, 23.59.
N,N′-(3-Hydroxy-6-oxo-6H-benzo[c]chromene-8,9-diyl)diacetamide (140)
Following general procedure B, 139 (0.6 g, 1.55 mmol) was reacted and triturated with MeOH and CH2Cl2 to afford 0.43 g (84%) of 140 as a pale-gray solid. Mp 340–342 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 9.67 (s, 1H), 9.58 (s, 1H), 8.54 (s, 1H), 8.38 (s, 1H), 7.89 (d, J = 8.7 Hz, 1H), 6.85 (dd, J = 8.7, 2.4 Hz, 1H), 6.74 (d, J = 2.4 Hz, 1H), 2.18 (s, 3H), 2.13 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 169.32, 169.12, 160.03, 159.61, 152.01, 137.32, 131.73, 129.23, 125.42, 124.21, 115.24, 114.66, 113.25, 109.12, 103.02, 24.16, 23.82; HRMS (ESI) calculated for C17H15N2O5 [M + H]+ 327.0975, found 327.0967.
3-Hydroxy-9-methylbenzo[3,4]isochromeno[6,7-d]imidazol-6(10H)-one (141)
A solution of 140 (0.04 mg, 0.011 mmol) in concentrated HCl (3 mL) was stirred at 105 °C in a sealed microwave vial for 4 h. The reaction mixture was cooled down to room temperature, diluted with ice/water, and the precipitate was filtered off to afford 0.015 g (50%) of 141 as a gray solid. Mp > 350 °C; 1H-NMR (400 MHz, CD3OD) δ 8.51 (d, J = 0.7 Hz, 1H), 8.31 (s, 1H), 8.09 (d, J = 8.8 Hz, 1H), 6.87 (dd, J = 8.8, 2.4 Hz, 1H), 6.72 (d, J = 2.4 Hz, 1H), 2.88 (s, 3H); 13C-NMR (101 MHz, CD3OD) δ 162.66, 161.68, 157.25, 153.27, 138.42, 134.48, 132.77, 125.74, 118.78, 117.08, 114.59, 110.78, 106.67, 104.23, 13.09; HRMS (ESI) calculated for C15H11N2O3 [M + H]+ 267.0764, found 267.0769.
3-Hydroxy-6H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-c]chromen-6-one (144)
3-Hydroxy-6H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-c]chromen-6-one (142)
A mixture of 84 (0.50 g, 2.02 mmol), diiodomethane (0.81 g, 3.03 mmol) and anhydrous K2CO3 (1.40 g, 10.12 mmol) in dry DMF (15 mL) was heated at reflux for 2 h. After cooling, the solvent was evaporated under reduced pressure to give the crude compound. Purification by flash chromatography (pentane/EtOAc) afforded 0.32 g (61%) of 142 as a white solid. Mp = 85–87 °C; 1H NMR (400 MHz, CDCl3) δ 7.33 (s, 1H), 7.09 (s, 1H), 6.05 (s, 2H), 3.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.71, 150.95, 147.11, 129.53, 124.50, 114.38, 110.99, 102.48, 52.34.
6-bromobenzo[d][1,3]dioxole-5-carboxylic acid (143)
Following general procedure E, 142 (0.32 g, 1.23 mmol) was reacted to afford 0.21 g (69%) of 143 as a white solid. Mp = 202–204 °C; 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.14 (s, 1H), 6.08 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 166.71, 150.42, 147.01, 126.00, 113,66 113.07, 112.91, 110.20, 102.77.
3-Hydroxy-6H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-c]chromen-6-one (144)
Following general procedure A, 143 (0.21 g, 0.87 mmol) was reacted to afford 0.16 g (73%) of 144 as a white solid. Mp = 331–333 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.80 (s, 1H), 7.49 (s, 1H), 6.79 (dd, J = 8.7, 2.4 Hz, 1H), 6.71 (d, J = 2.4 Hz, 1H), 6.22 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 160.48, 159.77, 154.52, 152.01, 147.96, 133.28, 125.19, 113.58, 113.45, 110.15, 107.35, 103.09, 103.06, 101.31; HRMS (ESI) m/z: 255.0308 [M − H]−, calculated for C14H7O5 255.0299.
3-Hydroxy-8,10-dihydrobenzo[3,4]isochromeno[6,7-d]imidazole-6,9-dione (150)
Methyl 6-bromo-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxylate (145)
To a solution of 136 (0.23 g, 0.939 mmol) in anhydrous THF (3 mL), CDI (0.23 g, 1.41 mmol) was added. The reaction mixture was stirred under an atmosphere of nitrogen at room temperature for 16 h. Then, the solvent was removed under reduced pressure. Flash column chromatography (CH2Cl2:MeOH 97:3 to 90:10) afforded the desired product together with imidazole, and after trituration with MeOH to give 0.2 g (78%) of 145 obtained as a pale-pink solid. Mp = 287–289 °C; 1H-NMR (400 MHz, DMSO-d6) δ 11.05 (br s, 2H), 7.36 (s, 1H), 7.20 (s, 1H), 3.81 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 165.78, 155.19, 133.71, 129.00, 122.76, 113.27, 112.27, 110.79, 52.25.
6-Bromo-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxylic acid (149)
Following general procedure E, 145 (0.17 g, 0.65 mmol) was reacted to afford 0.16 g (98%) of 149 as a pale-pink solid. Mp = 360–361 °C; 1H-NMR (400 MHz, DMSO-d6) δ 12.97 (br s, 1H), 11.03 (br s, 1H), 10.94 (br s, 1H), 7.36 (s, 1H), 7.17 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 166.85, 155.21, 133.38, 128.94, 123.89, 113.22, 112.27, 110.86.
3-Hydroxy-8,10-dihydrobenzo[3,4]isochromeno[6,7-d]imidazole-6,9-dione (150)
Following general procedure A, 149 (0.13 g, 0.51 mmol) was reacted and recrystallized from MeOH to afford 0.04 g (33%) of 150 as a brown solid. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 11.38 (br s, 1H), 11.09 (br s, 1H), 10.18 (br s, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.63 (s, 1H), 7.61 (s, 1H), 6.81 (dd, J = 8.8, 2.4 Hz, 1H), 6.72 (d, J = 2.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 160.88, 158.86, 155.53, 151.16, 136.75, 130.16, 130.03, 124.22, 112.96, 112.15, 110.00, 107.68, 102.76, 99.85; HRMS (ESI) calculated for C14H7N2O4 [M − H]– 267.0411, found 267.0413.
3-Hydroxybenzo[3,4]isochromeno[6,7-d]imidazol-6(10H)-one (153)
Methyl 6-bromo-1H-benzo[d]imidazole-5-carboxylate (146)
This compound was synthesized following a reported method for the synthesis of benzimidazoles [
30]. To a solution of
136 (0.8 mg, 3.26 mmol) in DMF (10 mL), PhSiH
3 (1.61 mL, 13.06 mmol) was added. The reaction mixture was stirred at 120 °C for 15 h. Then, the reaction was cooled down to room temperature and water (100 mL) was added (a white solid precipitates). The mixture was extracted with EtOAc (3 × 80 mL), the combined organic layers were dried over Na
2SO
4, filtered, and concentrated in vacuo. Purification by flash column chromatography (CH
2Cl
2:MeOH 100:0 to 95:5) afforded 0.27 g (32%) of
146 as a yellow solid. Mp 95–97 °C.
1H-NMR (400 MHz, DMSO-d
6) δ 8.41 (s, 1H), 8.05 (s, 1H), 7.94 (s, 1H), 3.86 (s, 3H);
13C-NMR (101 MHz, DMSO-d
6) δ 166.65, 52.42; only peaks of ester were visible (CO and OMe), but not the heterocycle.
Methyl 6-(2,4-dimethoxyphenyl)-1H-benzo[d]imidazole-5-carboxylate (151)
Following general procedure D, 146 (0.1 g, 0.39 mmol) and 138 (0.14 g, 0.78 mmol) were reacted and purified by flash chromatography (CH2Cl2/MeOH) to afford 0.07 g (56%) of 151 as a yellow solid. Mp = 110–112 °C. 1H-NMR (400 MHz, DMSO-d6) δ 12.68 (br s, 1H), 8.35 (s, 1H), 7.97 (s, 1H), 7.39 (s, 1H), 7.16 (d, J = 8.2 Hz, 1H), 6.59 (dd, J = 8.2, 2.4 Hz, 1H), 6.55 (d, J = 2.4 Hz, 1H), 3.81 (s, 3H), 3.61 (s, 3H), 3.60 (s, 3H); 13C-NMR (201 MHz, DMSO-d6) δ 168.24, 159.80, 156.86, 144.09, 130.30, 125.77, 123.52, 104.62, 98.01, 55.18, 55.01, 51.41 (5 carbons of the benzimidazole ring do not appear, probably because of tautomerism).
3-Hydroxybenzo[3,4]isochromeno[6,7-d]imidazol-6(10H)-one (153)
Following general procedure B, 151 (0.05 g, 0.16 mmol) was reacted and triturated with Et2O to afford 0.011 g (37%) of 153 as a pale-yellow solid. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.30 (br s, 1H), 9.14 (br s, 1H), 8.53 (s, 1H), 8.49 (s, 1H), 8.28 (d, J = 8.9 Hz, 1H), 6.87 (dd, J = 8.9, 2.4 Hz, 1H), 6.77 (d, J = 2.4 Hz, 1H); 13C-NMR (201 MHz, DMSO-d6) δ 160.61, 159.77, 151.35, 145.55, 137.71, 132.35, 131.77, 125.11, 117.05, 116.66, 113.29, 109.36, 106.75, 102.98; HRMS (ESI) calculated for C14H9N2O3 [M + H]+ 253.0608, found 253.0608.
3-Hydroxybenzo[3,4]isochromeno[6,7-d][1,2,3]triazol-6(10H)-one (154)
Methyl 6-bromo-1H-benzo[d][1,2,3]triazole-5-carboxylate (147)
To a solution of 136 (0.72 g, 2.96 mmol) in MeOH (25 mL) at 0 °C, NaNO2 (0.22 mg, 3.25 mmol) and an aqueous solution of HCl (1 M, 22 mL) were added. The reaction mixture was stirred from 0 °C to room temperature for 15 h. Then, MeOH was removed under reduced pressure, a saturated aqueous solution of NaHCO3 (50 mL) was added and the mixture was extracted with EtOAc (3 × 60 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure, to afford 0.73 g (96%) of 147 as a brown-orange solid. Mp = 174–175 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.39 (d, J = 0.6 Hz, 1H), 8.33 (d, J = 0.6 Hz, 1H), 3.90 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 166.27, 139.22, 138.65, 129.29, 119.18, 118.74, 116.06, 52.80.
Methyl 1-acetyl-6-bromo-1H-benzo[d][1,2,3]triazole-5-carboxylate + Methyl 1-acetyl-5-bromo-1H-benzo[d][1,2,3]triazole-6-carboxylate (148)
To a suspension of 147 (0.6 mg, 2.34 mmol) in dry CH2Cl2 (20 mL) at 0 °C, Et3N (0.52 mL, 3.75 mmol) and AcCl (0.23 mL, 3.28 mmol) were added. The reaction mixture was stirred at room temperature for 3 h. Then, an aqueous solution of HCl (1 M, 10 mL) was added, the mixture was diluted with water (20 mL) and CH2Cl2 (40 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 40 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give 0.7 g (99%) of 148 as a pale-brown solid. The product was obtained as a mixture of regioisomers (major: minor 6:4). 1H-NMR (400 MHz, CDCl3) δ 8.66 (d, J = 0.6 Hz, 1H major), 8.64 (d, J = 0.6 Hz, 1H minor), 8.55 (d, J = 0.6 Hz, 1H major), 8.44 (d, J = 0.6 Hz, 1H minor), 4.01 (s, 6H, major and minor overlap), 3.02 (s, 3H, minor), 3.01 (s, 3H, major); 13C-NMR (101 MHz, CDCl3) δ 169.22, 169.19, 166.16, 165.80, 147.62, 144.80, 134.63, 132.68, 130.61, 129.70, 125.35, 123.74, 123.16, 119.94, 117.65, 116.74, 53.23, 53.12, 23.27, 23.22 (both major and minor isomers reported).
Methyl 6-(2,4-dimethoxyphenyl)-1H-benzo[d][1,2,3]triazole-5-carboxylate (152)
Following general procedure D, 148 (0.5 g, 1.68 mmol) and 138 (0.61 g, 3.35 mmol) were reacted and purified by flash chromatography (CH2Cl2/MeOH) to afford 0.27 g (51%) of 152 as a white solid. Mp = 98–100 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.29 (br s, 1H), 7.70 (br s, 1H), 7.26 (d, J = 8.3 Hz, 1H), 6.63 (dd, J = 8.3, 2.4 Hz, 1H), 6.57 (d, J = 2.4 Hz, 1H), 3.82 (s, 3H), 3.65 (s, 3H), 3.63 (s, 3H).;13C-NMR (201 MHz, DMSO-d6) δ 167.62, 160.39, 156.76, 130.51, 122.17, 104.93, 98.02, 55.26, 55.05, 51.79 (the 6 carbons of the benzotriazole ring do not appear, probably because of tautomerism).
3-Hydroxybenzo[3,4]isochromeno[6,7-d][1,2,3]triazol-6(10H)-one (154)
Following general procedure B, 152 (0.04 g, 0.121 mmol) was reacted and triturated with Et2O to afford 0.011 g (37%) of 154 as a pale-yellow solid. Mp > 350 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.35 (br s, 1H), 8.86 (br s, 1H), 8.64 (br s, 1H), 8.34 (d, J = 8.8 Hz, 1H), 6.86 (dd, J = 8.8, 2.4 Hz, 1H), 6.76 (d, J = 2.4 Hz, 1H); 13C-NMR (201 MHz, DMSO-d6) δ 160.80, 159.84, 151.39, 125.44, 113.24, 109.51, 102.99 (the 6 carbons of the benzotriazole ring do not appear, probably because of tautomerism); HRMS (ESI) calculated for C13H8N3O3 [M + H]+ 254.0560, found 254.0563.
2,3,8-Trihydroxy-6H-benzo[c]chromen-6-one (161)
Methyl 2-bromo-5-methoxybenzoate (156)
Following general procedure C, 2-bromo-5-methoxynenzoic acid (155) (1.50 g, 6.49 mmol) was reacted to afford 1.57 g (99%) of 156 as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.8 Hz, 1H), 7.31 (d, J = 3.1 Hz, 1H), 6.89 (dd, J = 8.8, 3.1 Hz, 1H), 3.93 (d, J = 0.3 Hz, 3H), 3.81 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.47, 158.51, 135.02, 132.68, 119.04, 116.20, 111.93, 55.64, 52.51.
Methyl 3′,4,4′-trimethoxy-[1,1′-biphenyl]-2-carboxylate (158)
Following general procedure B, 156 (1.00 g, 4.08 mmol) and 3,4-methoxyphenylboronic acid (157) (0.74 g, 4.08 mmol), were reacted to afford 1.04 g (84%) of 158 as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.33–7.24 (m, 2H), 7.05 (dd, J = 8.5, 2.8 Hz, 1H), 6.92–6.78 (m, 3H), 3.91 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.66 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.33, 158.40, 148.46, 148.10, 134.41, 133.68, 131.90, 131.76, 120.60, 117.38, 114.12, 111.79, 110.79, 55.85, 55.56, 52.11.
3′,4,4′-Trimethoxy-[1,1′-biphenyl]-2-carboxylic acid (159)
Following general procedure E, 158 (1.04 g, 3.44 mmol) was reacted to afford 0.91 g (92%) of 159 as a white solid. Mp = 166–168 °C; 1H NMR (400 MHz, CDCl3) δ 11.67 (s, 1H), 7.43 (d, J = 2.7 Hz, 1H), 7.28 (d, J = 8.6 Hz, 1H), 7.07 (dd, J = 8.5, 2.8 Hz, 1H), 6.85 (dd, J = 3.5, 1.5 Hz, 3H), 3.88 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 173.77, 158.36, 148.36, 148.23, 135.42, 133.42, 132.34, 130.21, 120.89, 118.35, 114.92, 112.18, 110.86, 55.82, 55.81, 55.55.
2,3,8-Trimethoxy-6H-benzo[c]chromen-6-one (160)
Following general procedure F, 159 (0.91 g, 3.15 mmol) was reacted to afford 0.45 g (50%) of 160 as a white powder. Mp = 201–203 °C; 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.8 Hz, 1H), 7.76 (d, J = 2.7 Hz, 1H), 7.36 (dd, J = 8.8, 2.8 Hz, 1H), 7.31 (s, 1H), 6.84 (d, J = 1.2 Hz, 1H), 3.99 (s, 3H), 3.93 (s, 3H), 3.92 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 161.65, 159.14, 150.53, 146.47, 145.28, 128.56, 124.35, 122.72, 121.23, 111.02, 110.14, 103.51, 100.74, 56.42, 56.20, 55.74.
2,3,8-Trihydroxy-6H-benzo[c]chromen-6-one (161)
Following general procedure B, 160 (0.30 g, 1.05 mmol) was reacted to afford 0.24 g (94%) of 161 as yellow solid. Mp > 350 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 9.78 (s, 1H), 9.15 (s, 1H), 7.92 (d, J = 8.9 Hz, 1H), 7.49 (d, J = 2.7 Hz, 1H), 7.41 (s, 1H), 7.29 (dd, J = 8.7, 2.7 Hz, 1H), 6.73 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.23, 157.25, 147.64, 143.87, 143.57, 127.41, 124.49, 123.90, 120.81, 114.02, 109.70, 108.03, 103.89; HRMS (ESI) m/z: 243.0306 [M − H]−, calculated for C13H7O5 243.0299.
3,8,9-Trihydroxyphenanthridin-6(5H)-one (165)
2-Bromo-4,5-dimethoxybenzamide (162)
A solution of 8 (4.00 g, 15.3 mmol) in SOCl2 (35 mL) was stirred at reflux for 4 h. The reaction mixture was cooled down to room temperature and evaporated under reduced pressure to afford the corresponding acyl chloride as a pale-yellow solid. This solid was dissolved in THF (80 mL) and added dropwise to a solution of NH3 in THF (0.4 M, 115 mL) at 0 °C, and the reaction mixture was stirred from 0 °C to room temperature for 20 h. Then, the solvent was removed under reduced pressure. Purification by flash column chromatography (CH2Cl2:MeOH 98.5:1.5) afforded 3.03 g (76%) of 162 as a white solid. Mp = 177–179 °C; 1H-NMR (400 MHz, CDCl3) δ 7.35 (s, 1H), 7.01 (s, 1H), 6.50 (br s, 1H), 6.18 (br s, 1H), 3.90 (s, 3H), 3.90 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 168.55, 151.32, 148.51, 127.72, 116.05, 113.53, 110.39, 110.13, 56.43, 56.28.
4,4′,5-Trimethoxy-[1,1′-biphenyl]-2-carboxamide (163)
Following general procedure B, 162 (1.00 g, 3.85 mmol) and 86 (0.64 g, 4.23 mmol), were reacted and purified by flash chromatography (CH2Cl2/MeOH) to afford 0.61 g (55%) of 163 as a white solid. Mp = 167–168 °C; 1H-NMR (400 MHz, CDCl3) δ 7.45 (s, 1H), 7.36–7.32 (m, 2H), 6.98–6.95 (m, 2H), 6.74 (s, 1H), 5.50 (br s, 1H), 5.20 (br s, 1H), 3.96 (s, 3H), 3.91 (s, 3H), 3.85 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 170.45, 159.61, 150.63, 148.22, 133.51, 132.68, 130.34, 125.73, 114.37, 113.23, 112.56, 56.26, 56.20, 55.50.
3,8,9-Trimethoxyphenanthridin-6(5H)-one (164)
To a solution of 163 (0.3 g, 1.04 mmol) in o-xylene (30 mL), CuI (0.07 g, 0.366 mmol), PPh3 (0.19 g, 0.731 mmol), and KOtBu (0.82 mg, 7.31 mmol) were added in 3 portions over 3 days, the mixture was stirred at 120 °C for 4 days in total. Then, water (200 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. Recrystallization from MeOH afforded 0.05 g (18%) of 164 as a pale-yellow solid. Mp = 275–277 °C. 1H-NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H), 8.28 (d, J = 8.7 Hz, 1H), 7.76 (s, 1H), 7.65 (s, 1H), 6.87–6.83 (m, 2H), 4.00 (s, 3H), 3.88 (s, 3H), 3.81 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 160.73, 159.58, 153.33, 148.56, 137.47, 129.43, 124.61, 117.85, 111.28, 109.87, 107.77, 103.56, 99.24, 56.04, 55.48, 55.25.
3,8,9-Trihydroxyphenanthridin-6(5H)-one (165)
Following general procedure B, 164 (0.024 g, 0.0084 mmol) was reacted to afford and purified by reversed phase (C18) flash column chromatography (H2O:CH3CN 10:0 to 7:3) to afford 0.02 g (88%) of 165 as a red solid. Mp > 350 °C; 1H-NMR (400 MHz, CD3OD) δ 7.92 (d, J = 8.7 Hz, 1H), 7.67 (s, 1H), 7.56 (s, 1H), 6.77–6.73 (m, 2H); 13C-NMR (176 MHz, CD3OD) δ 13C NMR (176 MHz, MeOD) δ 164.17, 159.21, 152.96, 146.86, 137.96, 131.48, 124.77, 118.09, 113.04, 112.85, 112.84, 107.43, 102.47; HRMS (ESI) m/z: 244.0610 [M + H]+, calculated for C13H9NO4 244.0612.
3,8,9-Trihydroxy-5-methylphenanthridin-6(5H)-one (167)
3,8,9-Trimethoxy-5-methylphenanthridin-6(5H)-one (166)
To a suspension of 164 (0.016 g, 0.056 mmol) and Cs2CO3 (0.091 g, 0.28 mmol) in THF (6 mL) under an atmosphere of nitrogen, methyl iodide (11 µL, 0.17 mmol) was added. The mixture was stirred at reflux (sealed microwave vial, pre-heated heating block at 86 °C) for 5 h. Then, the solvent was removed under reduced pressure. Water (30 mL) and CH2Cl2 (30 mL) were added, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was triturated with Et2O to afford 0.014 (83%) 166 as a yellow solid. Mp = 179–180 °C; 1H-NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.6 Hz, 1H), 7.87 (s, 1H), 7.45 (s, 1H), 6.88 (dd, J = 8.7, 2.4 Hz, 1H), 6.85 (d, J = 2.4 Hz, 1H), 4.07 (s, 3H), 4.02 (s, 3H), 3.93 (s, 3H), 3.77 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 161.66, 160.26, 153.44, 149.14, 139.10, 128.72, 124.05, 118.32, 112.94, 109.17, 108.93, 102.14, 100.28, 56.31, 56.20, 55.68, 30.12.
3,8,9-Trihydroxy-5-methylphenanthridin-6(5H)-one (167)
Following general procedure B, 166 (0.01 g, 0.033 mmol) was reacted and triturated with MeOH to afford 0.003 g (35%) of 167 as yellow solid. 1H-NMR (400 MHz, CD3OD) δ 8.00 (d, J = 8.8 Hz, 1H), 7.69 (s, 1H), 7.55 (s, 1H), 6.91 (d, J = 2.3 Hz, 1H), 6.81 (dd, J = 8.8, 2.3 Hz, 1H), 3.72 (s, 3H); 13C-NMR (176 MHz, CD3OD) δ 163.53, 159.48, 152.77, 146.98, 139.62, 130.30, 125.21, 117.87, 113.61, 113.39, 112.32, 107.09, 102.38, 30.38; HRMS (ESI) m/z: 258.0766 [M + H]+, calculated for C14H12NO4 258.0765.
Phenanthrene-2,3,7-triol (178)
4,4′,5-Trimethoxy-[1,1′-biphenyl]-2-carbaldehyde (170)
Following general procedure D, 68 (2.0 g, 8.16 mmol) and 86 (1.275 g, 8.39 mmol) were reacted and purified by flash chromatography (pentane/EtOAc) to afford 1.81 g (81%) of 170 as a white solid. Mp = 106–107 °C; 1H NMR (400 MHz, CDCl3) δ 9.83 (s, 1H), 7.52 (s, 1H), 7.32–7.28 (m, 2H), 7.02–6.97 (m, 2H), 6.83 (s, 1H), 3.98 (s, 3H), 3.97 (s, 3H), 3.87 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 191.36, 159.67, 153.48, 148.62, 141.33, 131.42, 129.93, 127.02, 113.90, 112.69, 108.67, 56.28, 56.19, 55.49.
(E/Z)-4,4′,5-Trimethoxy-2-(2-methoxyvinyl)-1,1′-biphenyl (172)
To a solution of methoxymethyltriphenylphosphonium chloride (2.57 g, 7.5 mmol) in THF (25 mL), a solution of KOtBu (842 mg, 7.5 mmol) in THF (15 mL) was added at 0 °C. After stirring for 30 min, a solution of 170 (0.91 mg, 5.0 mmol) in THF (10 mL) was added and stirred at room temperature for 2 h. The reaction mixture was quenched with water (40 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layer was dried over MgSO4, and the organic solvent was removed under reduced pressure. The crude was purified by flash chromatography (pentane/EtOAc 90/10) to give 0.5 g (90%) of 172 as a colorless oil (mixture of E/Z isomer). 1H NMR (400 MHz, CDCl3) δ 7.32–7.25 (m, 2H), 6.97–6.92 (m, 2H), 6.88 (d, J = 5.5 Hz, 1H), 6.76 (d, J = 9.6 Hz, 1H), 5.76 (d, J = 12.9 Hz, 1H), 3.93 (d, J = 1.6 Hz, 3H), 3.87 (d, J = 1.9 Hz, 3H), 3.84 (d, J = 2.9 Hz, 3H), 3.75 (s, 1H), 3.55 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 158.48, 158.45, 148.14, 147.79, 147.54, 147.37, 146.84, 146.40, 133.96, 133.63, 133.00, 132.15, 130.95, 130.80, 126.47, 125.97, 113.47, 113.40, 112.93, 112.21, 108.15, 104.51, 103.81, 60.54, 56.43, 55.98, 55.97, 55.86, 55.84, 55.27.
2,3,7-Trimethoxyphenanthrene (175)
To a solution of 172 (0.41 g, 1.3 mmol) in CH2Cl2 (10 mL) CF3SO3H (0.1 mL) was added under argon at 0 °C, and the solution was stirred overnight at room temperature. The organic layers were evaporated to dryness. The residue was purified by crystallization from MeOH to give 0.25 g (68%) of 175 as a grey solid. Mp = 157–158 °C; 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 8.9 Hz, 1H), 7.91 (s, 1H), 7.67–7.54 (m, 2H), 7.29–7.23 (m, 2H), 4.10 (s, 3H), 4.03 (s, 3H), 3.96 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 157.52, 149.44, 148.59, 132.61, 126.54, 125.95, 125.08, 124.68, 124.22, 123.73, 117.03, 108.32, 108.30, 102.77, 55.96, 55.89, 55.38.
Phenanthrene-2,3,7-triol (178)
Following general procedure B, 175 (0.08 g, 0.30 mmol) was reacted and triturated with MeOH to afford 0.05 g (77%) of 178 as a grey solid. Mp = 266–268 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.84–9.06 (m, 3H), 8.23 (d, J = 8.9 Hz, 1H), 7.83 (s, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.35 (d, J = 8.8 Hz, 1H), 7.13 (s, 1H), 7.11–7.04 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 155.33, 147.17, 145.76, 132.57, 126.58, 125.23, 124.85, 123.97, 123.42, 123.08, 117.25, 112.43, 111.40, 106.87; HRMS (ESI) calculated for C14H9O3 [M − H]− 225.0557, found 225.0571.
Phenanthridine-3,8,9-triol (179)
(E)-4,4′,5-trimethoxy-[1,1′-biphenyl]-2-carbaldehyde O-acetyl oxime (173)
To a solution of 170 (1.69 g, 6.20 mmol) in EtOH (16 mL), pyridine (1.41 mL, 17.38 mmol) and hydroxylamine hydrochloride (0.65 g, 9.31 mmol) were added. The resulting mixture was stirred at 60 °C for 75 min. Then, the reaction mixture was cooled to room temperature, and water (40 mL) and EtOAc (40 mL) were added. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with aqueous HCl (1 M, 1 × 10 mL) and brine (1 × 10 mL), and dried over Na2SO4, filtered, and concentrated under reduced pressure. Then, the resulting crude was dissolved in pyridine (16 mL), and DMAP (ca 8 mg, 64 μmol) and acetic anhydride (1.17 mL, 12.41 mmol) were added. The reaction mixture was stirred at room temperature for 1.5 h. Then, water (40 mL) and EtOAc (40 mL) were added. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with aqueous HCl (1 M, 1 × 40 mL) and brine (1 × 40 mL), and dried over Na2SO4, filtered, and concentrated under reduced pressure to give 1.9 g (95%) of 173 as a pale-yellow solid. Mp = 132–133 °C; 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.56 (s, 1H), 7.22–7.18 (m, 2H), 6.99–6.95 (m, 2H), 6.79 (s, 1H), 3.96 (s, 3H), 3.91 (s, 3H), 3.86 (s, 3H), 2.15 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.79, 159.35, 155.41, 151.58, 148.42, 137.83, 131.16, 130.99, 119.98, 113.90, 112.56, 108.50, 56.25, 56.01, 55.40, 19.66.
3,8,9-Trimethoxyphenanthridine (176)
This compound was synthesized following a reported method for the synthesis of phenanthridines [
22]. To a solution of
173 (1.68 g, 5.11 mmol) in acetic acid (35 mL), Fe(acac)
3 (0.36 g, 1.02 mmol) was added. The reaction mixture was stirred under an atmosphere of nitrogen at 80 °C for 2 h. The reaction mixture was then allowed to cool down to room temperature, neutralized with a saturated aqueous solution of NaHCO
3, and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over Na
2SO
4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography (CH
2Cl
2/MeOH 95/5) afforded 0.42 g (31%) of the desired product
176 as a yellow solid. Mp = 167–168 °C;
1H NMR (400 MHz, CDCl
3) δ 9.11 (s, 1H), 8.33 (d,
J = 9.0 Hz, 1H), 7.78 (s, 1H), 7.55 (d,
J = 2.7 Hz, 1H), 7.33 (s, 1H), 7.28 (dd,
J = 9.0, 2.6 Hz, 1H), 4.13 (s, 3H), 4.06 (s, 3H), 3.98 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 159.57, 153.22, 152.24, 149.39, 145.75, 128.72, 123.03, 121.04, 118.15, 118.06, 109.82, 107.85, 101.43, 56.30, 56.23, 55.67.
Phenanthridine-3,8,9-triol (179)
To a solution of 176 (0.1 g, 0.371 mmol) in acetic acid (1.1 mL), HBr (48% aqueous, 3.3 mL) was added. The reaction mixture was stirred at 120 °C for 6 days. Then, the solvent was removed under reduced pressure. Et2O was added and the precipitate formed was filtered off. Purification by flash chromatography (CH2Cl2/MeOH) afforded 0.01 g (11%) of 179 as an orange solid. Mp = 203–205 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.91 (s, 1H), 8.23 (d, J = 8.9 Hz, 1H), 7.78 (s, 1H), 7.33 (s, 1H), 7.26 (s, 1H), 7.12 (d, J = 8.7 Hz, 1H); 13C NMR (176 MHz, DMSO-d6) δ 163.50, 161.57, 157.33, 154.45, 150.76, 123.78, 120.01, 117.70, 116.92, 112.06, 105.56, 41.80, 41.68; HRMS (ESI) calculated for C13H10NO3 [M + H]+ 228.0655, found 228.0658.
Phenanthridine-2,3,8-triol (180)
Bromo-5-methoxybenzaldehyde (169)
To a solution of KBr (1.19 g, 10.0 mmol) in water (100 mL), m-anisaldehyde (168) (1.22 mL, 10.0 mmol) was added. Bromine (0.52 mL, 10.0 mmol) was added dropwise, and the reaction mixture was stirred at room temperature for 4 h. Then, the precipitate was filtered off, washed with water, and dried by co-evaporation with toluene (3 × 10 mL) to afford 1.91 g (89%) of 169 as a pale-orange solid. Mp = 63–65 °C; 1H-NMR (400 MHz, CDCl3) δ 10.31 (s, 1H), 7.52 (d, J = 8.8 Hz, 1H), 7.41 (d, J = 3.2 Hz, 1H), 7.03 (dd, J = 8.8, 3.2 Hz, 1H), 3.84 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 191.93, 159.40, 134.70, 134.10, 123.28, 118.12, 112.80, 55.88.
3′,4,4′-Trimethoxy-[1,1′-biphenyl]-2-carbaldehyde (171)
Following general procedure D, 169 (0.43 g, 2.00 mmol) and 157 (0,40 g, 2.20 mmol) were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.45 (83%) of 171 as a white solid. Mp = 103–104 °C; 1H-NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 7.49 (d, J = 2.8 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H), 7.19 (dd, J = 8.5, 2.8 Hz, 1H), 6.96–6.93 (m, 1H), 6.88–6.85 (m, 2H), 3.94 (s, 3H), 3.90 (s, 3H), 3.90 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 192.61, 159.09, 149.05, 148.93, 139.10, 134.72, 132.14, 130.25, 123.07, 121.54, 113.34, 111.08, 109.86, 56.14, 56.13, 55.76.
(E)-3′,4,4′-trimethoxy-[1,1′-biphenyl]-2-carbaldehyde O-acetyl oxime (174)
To a solution of 171 (0.42 g, 1.54 mmol) in EtOH (4 mL), pyridine (0.35 mL, 4.32 mmol) and hydroxylamine hydrochloride (0.16 g, 2.31 mmol) were added. The resulting mixture was stirred at 60 °C for 75 min. Then, the reaction mixture was cooled to room temperature, and water (10 mL) and EtOAc (10 mL) were added. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with aqueous HCl (1 M, 1 × 10 mL) and brine (1 × 10 mL), and dried over Na2SO4, filtered, and concentrated under reduced pressure. Then, the resulting crude (444 mg, white solid) was dissolved in pyridine (4 mL), and DMAP (ca 2 mg, 16 μmol) and acetic anhydride (0.30 mL, 3.08 mmol) were added. The reaction mixture was stirred at room temperature for 1.5 h. Then, water (10 mL) and EtOAc (10 mL) were added. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with aqueous HCl (1 M, 1 × 10 mL) and brine (1 × 10 mL), and dried over Na2SO4, filtered, and concentrated under reduced pressure to give 0.5 g (99%) of. 174 as a pale-yellow solid. Mp = 107–109 °C; 1H-NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 7.57 (d, J = 2.7 Hz, 1H), 7.30 (d, J = 8.6 Hz, 1H), 7.07 (dd, J = 8.6, 2.7 Hz, 1H), 6.94–6.92 (m, 1H), 6.81–6.78 (m, 2H), 3.93 (s, 3H), 3.89 (s, 3H), 3.88 (s, 3H), 2.18 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ 168.83, 158.94, 155.90, 148.85, 148.77, 136.48, 131.52, 131.49, 128.79, 122.51, 119.01, 113.16, 111.13, 110.33, 56.12, 56.11, 55.82, 19.74.
2,3,8-Trimethoxyphenanthridine (177)
This compound was synthesized following a reported method for the synthesis of phenanthridines [
22]. To a solution of
174 (0.47 g, 1.43 mmol) in acetic acid (9 mL), Fe(acac)
3 (0.1 g, 0.286 mmol) was added. The reaction mixture was stirred under an atmosphere of nitrogen at 80 °C for 2 h. The reaction mixture was then allowed to cool down to room temperature, neutralized with a saturated aqueous solution of NaHCO
3, and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over Na
2SO
4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography (CH
2Cl
2/MeOH 95/5) afforded 0.11 g (49%) of the desired product
177 as a pale-orange sticky oil (undesired regioisomer was also recovered).
1H-NMR (400 MHz, CDCl
3) δ 9.07 (s, 1H), 8.32 (d,
J = 9.1 Hz, 1H), 7.71 (s, 1H), 7.53 (s, 1H), 7.41 (dd,
J = 9.1, 2.6 Hz, 1H), 7.30 (d,
J = 2.6 Hz, 1H), 4.08 (s, 3H), 4.04 (s, 3H), 3.96 (s, 3H),
13C-NMR (101 MHz, CDCl
3) δ 158.14, 150.52, 150.28, 149.74, 139.70, 127.03, 126.84, 123.16, 122.12, 118.69, 109.96, 107.60, 101.35, 56.19, 56.15, 55.63.
Phenanthridine-2,3,8-triol (180)
To a solution of 177 (0.1 g, 0.371 mmol) in acetic acid (1.1 mL), HBr (48% aqueous, 3.3 mL) was added. The reaction mixture was stirred at 120 °C for 6 days. Then, the solvent was removed under reduced pressure. Et2O was added and the precipitate formed was filtered off. Purification by preparative HPLC (H2O (0.1% formic acid): CH3CN (0.1% formic acid) 9:1 to 6:4 over 25 min) afforded 0.037 G (37%) of 180 formate salt as a dark-yellow-orange solid. Mp = 210–211 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.97–9.72 (m, 3H), 8.91 (s, 1H), 8.29 (d, J = 8.8 Hz, 1H), 8.14 (s, 1H, formic acid), 7.79 (s, 1H), 7.35–7.30 (m, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 163.11(formic acid), 155.58, 149.19, 146.98, 146.90, 138.38, 126.62, 124.72, 123.18, 121.54, 117.82, 113.08, 110.51, 105.26; HRMS (ESI) calculated for C13H10NO3 [M + H]+ 228.0655, found 228.0660.
6,6-dimethyl-6H-benzo[c]chromene-3,8,9-triol (183)
3,8,9-tris(benzyloxy)-6H-benzo[c]chromen-6-one (181)
Following general procedure K, 1 (0.50 g, 2.04 mmol) was reacted to afford 0.79 g of 181 (75%) as a brown solid. Mp = 173–175 °C; 1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.60–7.28 (m, 15H), 6.95 (dd, J = 8.8, 2.5 Hz, 1H), 6.90 (d, J = 2.5 Hz, 1H), 5.35 (s, 2H), 5.25 (s, 2H), 5.12 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 161.25, 159.90, 154.89, 152.17, 148.94, 136.25, 136.09, 135.99, 130.47, 128.75, 128.70, 128.58, 128.26, 128.07, 127.51, 127.33, 127.10, 123.12, 113.28, 113.01, 112.97, 111.43, 104.77, 102.61, 71.02, 70.92, 70.39.
3,8,9-Tris(benzyloxy)-6,6-dimethyl-6H-benzo[c]chromene (182)
The compound was synthetized following the literature procedure [
31]. To a solution of methyl magnesium bromide in diethyl ether (3.0 M, 2.43 mL, 7.29 mmol) under nitrogen at 0 °C, a suspension of
181 (0.75 g, 1.46 mmol) in anhydrous THF (30.0 mL) was added. The resulting mixture was refluxed for 12 h and then cooled to room temperature. The solution was poured into concentrated HCl conc. (3.0 mL) and stirred for 5 min. Water (30.0 mL) was then added and the solution was extracted with EtOAc (3 × 30.0 mL). The organic phase was dried (Na
2SO
4), filtered, and concentrated under reduced pressure to give 0.7 g of crude product. Purification by flash chromatography (90/10 pemtane/EtOAc) afforded 0.31 g (40%) of
182 as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 7.61–7.46 (m, 6H), 7.46–7.29 (m, 8H), 7.26 (d,
J = 2.0 Hz, 1H), 6.83 (s, 1H), 6.69 (dd,
J = 8.5, 2.5 Hz, 1H), 6.64 (d,
J = 2.5 Hz, 1H), 5.25 (s, 2H), 5.21 (s, 2H), 5.09 (s, 2H), 1.60 (s, 6H);
13C NMR (101 MHz, CDCl
3) δ 159.69, 153.54, 149.04, 148.12, 137.29, 137.26, 136.92, 131.85, 128.61, 128.60, 128.53, 128.02, 127.95, 127.63, 127.59, 127.46, 123.21, 122.86, 115.71, 111.66, 108.92, 108.69, 103.88, 77.73, 72.13, 71.64, 70.09, 27.65.
6,6-dimethyl-6H-benzo[c]chromene-3,8,9-triol (183)
Following general procedure H, 182 (0.16 g, 0.30 mmol) was reacted to afford 0.05 g (66%) of 183 as a yellow oil. 1H NMR (400 MHz, CD3OD) δ 7.38 (d, J = 8.5 Hz, 1H), 7.05 (s, 1H), 6.67 (s, 1H), 6.42 (dd, J = 8.4, 2.5 Hz, 1H), 6.30 (d, J = 2.4 Hz, 1H), 1.51 (s, 6H); 13C NMR (101 MHz, CD3OD) δ 157.43, 153-07, 144.50, 144.13, 130.26, 122.54, 121.03, 114.91, 110.13, 106.53, 107.95, 103.96, 77.10, 26.62; HRMS (ESI) m/z: 257.0826 [M − H]−, calculated for C15H13O4 257.0819
2,3,7-Trihydroxy-9H-fluoren-9-one (192)
Methyl 4,4′,5-trimethoxy-[1,1′-biphenyl]-2-carboxylate (186)
Following general procedure D, 9 (0.55 g, 2.00 mmol) and 86 (0.33 g, 2.20 mmol), were reacted and purified by flash chromatography (pentane/EtOAc) to afford 0.54 g (89%) of 186 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.41 (s, 1H), 7.22 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 6.78 (s, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.85 (s, 3H), 3.64 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.31, 158.72, 151.13, 147.48, 137.02, 133.99, 129.54, 121.86, 113.63, 113.32, 112.81, 56.11, 56.03, 55.25, 51.80.
2,3,7-Trimethoxy-9H-fluoren-9-one (191)
The compound was synthetized following the literature procedure [
32]. To a solution of
186 (0.500 g, 1.73 mmol) in CHCl
3 (20.0 mL) trifluoromethanesulfonic acid (1.30 g, 8.67 mmol) was added. The reaction was refluxed for 2 h and then quenched with a solution of saturated NaHCO
3 (20.0 mL). The organic phase was separate, dried (Na
2SO
4), filtered, and concentrated under reduced pressure to give the crude compound. Crystallization from MeOH afforded 0.44 g (94%) of
191 as red crystals. Mp = 178–180 °C;
1H NMR (400 MHz, CDCl
3) δ 7.18 (d,
J = 8.1 Hz, 1H), 7.10 (s, 1H), 7.07 (d,
J = 2.5 Hz, 1H), 6.87–6.81 (m, 2H), 3.96 (s, 3H), 3.88 (s, 3H), 3.80 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 192.88, 160.23, 154.68, 148.75, 140.15, 136.55, 136.19, 126.74, 119.98, 118.83, 109.70, 107.30, 102.90, 56.28, 56.18, 55.65.
2,3,7-Trihydroxy-9H-fluoren-9-one (192)
Following general procedure B, 191 (0.12 g, 0.44 mmol) was reacted to afford 0.16 g (95%) of 192 as brown powder. Mp = 340–342 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.28–9.52 (m, 2H), 9.35 (br s, 1H), 7.25 (d, J = 7.8 Hz, 1H), 6.89 (s, 1H), 6.86 (s, 1H), 6.79 (d, J = 2.2 Hz, 1H), 6.78–6.71 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 192.57, 158.12, 152.71, 145.11, 139.20, 136.71, 134.94, 125.47, 121.08, 120.06, 111.86, 111.13, 108.22; HRMS (ESI) m/z: 227.0359 [M − H]−, calculated for C13H7O4 227.0350.
2,3,6-trihydroxy-9H-fluoren-9-one (194)
Methyl 3′,4′,5-trimethoxy-[1,1′-biphenyl]-2-carboxylate (187)
Following general procedure D, methyl 2-bromo-4-methoxybenzoate (184) (0.60 g, 2.44 mmol) and 157 (0.49 g, 2.69 mmol) were reacted and purified by flash chromatography to afford 0.62 g (84%) of 187 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.6 Hz, 1H), 6.93–6.80 (m, 5H), 3.92 (s, 3H), 3.88 (s, 3H), 3.87 (s, 3H), 3.65 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.52, 161.63, 148.36, 144.82, 134.24, 132.18, 122.85, 120.51, 116.28, 112.24, 111.74, 110.63, 55.89, 55.85, 55.47, 51.77.
2,3,6-Trimethoxy-9H-fluoren-9-one (193)
The compound was synthetized following the literature procedure. [
32] To a solution of
187 (0.200 g, 0.69 mmol) in CHCl
3 (20.0 mL), trifluoromethanesulfonic acid (0.52 g, 3.47 mmol) was added. The reaction was refluxed for 2 h and then quenched with a solution of saturated NaHCO
3 (20.0 mL). The organic phase was separated, dried (Na
2SO
4), filtered, and concentrated under reduced pressure to give the crude compound. Crystallization from MeOH afforded 0.12 g (64%) of
193 as orange crystals. Mp = 173–175 °C;
1H NMR (400 MHz, CDCl
3) δ 7.48 (dd,
J = 8.2, 1.5 Hz, 1H), 7.14 (s, 1H), 6.93 (d,
J = 1.9 Hz, 1H), 6.90–6.78 (m, 1H), 6.61 (dd,
J = 8.2, 2.1 Hz, 1H), 3.99 (s, 3H), 3.91 (s, 3H), 3.87 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 191.95, 165.14, 154.03, 149.83, 146.49, 138.03, 128.13, 127.58, 125.59, 111.22, 106.82, 106.68, 103.36, 56.33, 56.22, 55.70.
2,3,6-trihydroxy-9H-fluoren-9-one (194)
Following general procedure B, 193 (0.12 g, 0.44 mmol) was reacted to afford 0.75 g (74%) of 194 as a brown powder. Mp> 350 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.36 (br s, 1H), 9.86 (br s, 1H), 9.55 (br s, 1H), 7.25 (d, J = 8.0 Hz, 1H), 6.97 (s, 1H), 6.87 (s, 1H), 6.84 (d, J = 2.1 Hz, 1H), 6.49 (dd, J = 8.0, 2.1 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 191.36, 164.12, 151.68, 147.28, 146.45, 136.65, 127.00, 126.07, 125.63, 113.56, 111.29, 108.99, 107.98; HRMS (ESI) m/z: 227.0355 [M − H]−, calculated for C13H7O4 227.0350.
2,3,6,7-tetrahydroxy-9H-fluoren-9-one (196)
Methyl 3′,4,4′,5-tetramethoxy-[1,1′-biphenyl]-2-carboxylate (188)
Following general procedure D, 93 (0.60 g, 2.18 mmol) and 157 (0.50, 2.74 mmol), were reacted and purified by flash chromatography to afford 0.62 g (86%) of 188 as a yellow solid. Mp = 123–125 °C; 1H NMR (400 MHz, CDCl3) δ 7.39 (s, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.85 (dd, J = 8.2, 2.0 Hz, 1H), 6.81 (d, J = 1.5 Hz, 2H), 3.96 (s, 3H), 3.93 (s, 3H), 3.92 (s, 3H), 3.88 (s, 3H), 3.64 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.62, 151.21, 148.47, 148.32, 147.75, 137.04, 134.49, 122.28, 120.77, 113.67, 112.84, 112.11, 110.77, 56.29, 56.22, 56.06, 56.02, 52.04.
2,3,6,7-Tetramethoxy-9H-fluoren-9-one (195)
The compound was synthetized following the literature procedure. [
32] To a solution of
188 (0.3 g, 0.90 mmol) in CHCl
3 (20.0 mL) trifluoromethanesulfonic acid (0.68 g, 4.51 mmol) was added. The reaction was refluxed for 2 h and then quenched with a solution of saturated NaHCO
3 (20.0 mL). The organic phase was separate, dried (Na
2SO
4), filtered, and concentrated under reduced pressure to give the crude compound. Crystallization from MeOH afforded 0.15 g (54%) of
195 red crystals. Mp = 201–203 °C;
1H NMR (400 MHz, CDCl
3) δ 7.12 (s, 2H), 6.87 (s, 2H), 4.00 (s, 6H), 3.90 (s, 6H);
13C NMR (101 MHz, CDCl
3) δ 192.61, 154.03, 148.85, 138.81, 127.22, 107.34, 102.95, 56.30.
2,3,6,7-tetrahydroxy-9H-fluoren-9-one (195)
Following general procedure B, 194 (0.15 g, 0.56 mmol) was reacted to afford 0.082 g (70%) of 195 as dark-brown powder. Mp = 346–348 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 2H), 9.26 (s, 2H), 6.79 (s, 2H), 6.79 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 191.99, 151.57, 144.95, 137.79, 126.40, 111.55, 108.16; HRMS (ESI) m/z: 243.0311 [M − H]−, calculated for C13H7O5 243.0299.
7-Hydroxy-2,3-dimethoxy-9H-fluoren-9-one (197)
Methyl 4′-hydroxy-4,5-dimethoxy-[1,1′-biphenyl]-2-carboxylate (189)
Following general procedure D, 9 (0.55 g, 2.00 mmol), 4-hydroxyphenylboronic acid (185) (0.30 g, 2.20 mmol), were reacted and purified by flash chromatography to afford 0.46 g (80%) of 189 as a white solid. Mp = 189–191 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 7.22 (s, 1H), 7.05 (d, J = 8.5 Hz, 2H), 6.83 (s, 1H), 6.74 (d, J = 8.6 Hz, 2H), 3.81 (s, 3H), 3.78 (s, 3H), 3.54 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 168.69, 156.98, 151.20, 147.45, 136.11, 131.79, 129.85, 122.25, 115.25, 113.94, 113.01, 56.13, 56.08, 52.04.
7-Hydroxy-2,3-dimethoxy-9H-fluoren-9-one (197)
To a solution of 189 (0.29 g, 1.00 mmol) in CHCl3 (10.0 mL) was added trifluoromethanesulfonic acid (0.75 g, 5.00 mmol). The reaction was refluxed for 12 h and then quenched with a solution of saturated NaHCO3 (20.0 mL). The organic phase was separates, dried (Na2SO4), filtered, and concentrated under reduced pressure to give the crude compound. Crystallization from MeOH afforded 0.09 g (35%) of 197 as a red solid. Mp = 257–259 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 7.41 (d, J = 7.9 Hz, 1H), 7.26 (s, 1H), 7.05 (s, 1H), 6.83 (d, J = 2.3 Hz, 1H), 6.80 (dd, J = 7.9, 2.4 Hz, 1H), 3.88 (s, 3H), 3.76 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 192.71, 158.51, 155.34, 148.71, 140.62, 136.37, 134.68, 125.83, 121.70, 120.19, 111.38, 107.74, 104.60, 56.55, 56.23; HRMS (ESI) m/z: 255.0663 [M − H]−, calculated for C15H11O4 255.0651.
7-Hydroxy-9H-fluoreno[2,3-d][1,3]dioxol-9-one (198)
Methyl 6-(4-hydroxyphenyl)benzo[d][1,3]dioxole-5-carboxylate (190)
Following general procedure D, 142 (0.26 g, 1.00 mmol), 4-hydroxyphenylboronic acid (185) (0.15 g, 1.10 mmol), were reacted and purified by flash chromatography to afford 0.46 g (80%) of 189 as a white solid. Mp = 135–137 °C; 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 1H), 7.08 (d, J = 8.5 Hz, 2H), 6.79–6.70 (m, 3H), 6.33–6.22 (m, 1H), 6.03 (s, 2H), 3.68 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.90, 155.32, 150.13, 146.44, 139.06, 133.24, 129.50, 123.28, 115.13, 111.03, 109.81, 101.88, 52.20.
7-Hydroxy-9H-fluoreno[2,3-d][1,3]dioxol-9-one (198)
The compound was synthetized following the literature procedure. [
32] To a solution of
190 (0.20 g, 0.73 mmol) in CHCl
3 (10.0 mL) was added trifluoromethanesulfonic acid (0.52 g, 3.47 mmol). The reaction was refluxed for 12 h and then quenched with a solution of saturated NaHCO
3 (20.0 mL). The organic phase was separated, dried (Na
2SO
4), filtered, and concentrated under reduced pressure to give the crude compound. Crystallization from MeOH afforded 0.04 g (23%) of
198 as a red solid. Mp = 298–300 °C;
1H NMR (400 MHz, DMSO-d
6) δ 9.89 (s, 1H), 7.38 (d,
J = 7.9 Hz, 1H), 7.24 (s, 1H), 7.01 (s, 1H), 6.87–6.73 (m, 2H), 6.09 (s, 2H);
13C NMR (101 MHz, DMSO-d
6) δ 192.01, 158.68, 153.89, 147.50, 142.97, 136.22, 134.23, 127.66, 121.87, 120.35, 111.39, 104.96, 102.75, 102.19; HRMS (ESI) m/z: 239.0350 [M − H]
−, calculated for C
14H
7O
4 239.0359.
9H-Fluorene-2,3,7-triol (202)
2,3,7-tris(benzyloxy)-9H-fluoren-9-one (199)
Following general procedure K, 1 (0.30 g, 1.31 mmol) was reacted to afford 0.63 g (96%) of 199 as a red solid. Mp = 141–143 °C; 1H NMR (400 MHz, CDCl3) δ 7.50–7.28 (m, 14H), 7.23 (s, 1H), 7.21–7.14 (m, 2H), 6.98–6.90 (m, 2H), 5.26 (s, 2H), 5.16 (s, 2H), 5.08 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 192.69, 159.41, 154.69, 148.48, 140.32, 136.64, 136.62, 136.50, 136.39, 136.33, 128.65, 128.63, 128.52, 128.12, 127.96, 127.46, 127.30, 127.12, 120.20, 120.08, 110.92, 110.60, 105.78, 71.37, 71.15, 70.40.
2,3,7-Tris(benzyloxy)-9H-fluoren-9-ol (200)
To a solution of 199 (0.65 g, 1.30 mmol) in THF (20.0 mL) and MeOH (20.0 mL) at 0 °C, NaBH4 (0.10 g, 2.60 mmol) was added. The mixture was stirred at room temperature for 2 h. The reaction was then quenched with water (20.0 mL). The mixture was extracted with CH2Cl2 (3 × 20.0 mL), dried over Na2SO4, and the solvent removed under reduced pressure to afford 0.51 g (78%) of 200 as a white solid. Mp = 145–147 °C; 1H NMR (400 MHz, CDCl3) δ 7.59–7.26 (m, 16H), 7.21–7.13 (m, 2H), 7.07 (s, 1H), 6.93 (dd, J = 8.3, 2.4 Hz, 1H), 5.37–5.26 (m, 1H), 5.14 (d, J = 3.8 Hz, 2H), 5.11 (s, 2H), 5.03 (s, 2H), 2.26 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 158.38, 149.95, 148.34, 147.85, 138.53, 137.18, 136.91, 133.50, 133.04, 128.59, 128.51, 128.51, 128.48, 127.99, 127.85, 127.82, 127.49, 127.39, 127.34, 119.74, 115.59, 112.07, 111.72, 106.32, 74.89, 71.54, 71.48, 70.25, 30.37.
2,3,7-tris(benzyloxy)-9-methoxy-9H-fluorene (201)
The compound was synthetized following the literature procedure [
33]. To a solution of
200 (0.30 g, 0.60 mmol) in MeOH (30.0 mL) was added H
2SO
4 (0.5 mL). The mixture was refluxed for 1 h and then concentrated under reduced pressure to afford 0.28 g (91%) of
201 as a white solid. Mp = 131–133 °C;
1H NMR (400 MHz, CDCl
3) δ 7.58–7.42 (m, 7H), 7.43–7.29 (m, 11H), 7.20 (d,
J = 2.4 Hz, 1H), 7.18 (d,
J = 0.7 Hz, 1H), 7.17 (s, 1H), 6.97 (dd,
J = 8.2, 2.4 Hz, 1H), 5.44 (s, 1H), 5.23 (s, 2H), 5.20 (s, 2H), 5.11 (s, 2H), 2.96 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 158.28, 150.31, 148.19, 144.55, 137.23, 137.19, 136.90, 135.02, 134.73, 134.18, 128.58, 128.51, 128.44, 127.98, 127.85, 127.80, 127.53, 127.47, 127.35, 119.72, 115.55, 112.78, 112.12, 106.24, 71.68, 71.59, 70.34, 51.73.
9H-Fluorene-2,3,7-triol (202)
Following general procedure H, 201 (0.270 g, 0.52 mmol) was reacted to afford 0.075 g (67%) of 202 as a brown solid. Mp = 282–284 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.69 (s, 2H), 7.35 (d, J = 8.2 Hz, 1H), 7.01 (s, 1H), 6.85 (q, J = 1.0 Hz, 2H), 6.67 (dd, J = 8.2, 2.3 Hz, 1H), 3.57 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 155.98, 145.10, 144.89, 144.22, 133.79, 133.51, 133.42, 119.47, 113.96, 112.66, 112.49, 106.44, 36.19; HRMS (ESI) m/z: 213.0557 [M − H]−, calculated for C13H9O3 213.0561
(3,4-Dihydroxyphenyl)(3-hydroxyphenyl)methanone (209)
(3,4-Dimethoxyphenyl)(3-methoxyphenyl)methanone (206)
The compound was synthetized following the literature procedure [
34]. A solution of 3-methoxybenzoic acid (
203) (0.82 g, 5.42 mmol) in SOCl
2 (10.0 mL) was refluxed for 2 h. The solvent was removed under reduced pressure, washed with dichloromethane, and concentrated again, twice. The substituted benzoyl chloride was isolated as a yellow oil in quantitative yield. This was transferred into a round-bottomed flask and diluted with dichloromethane (20.0 mL). Aluminum trichloride (0.53 g, 3.98 mmol) was then added and the mixture was cooled to 0 °C. 1,2-Dimethoxybenzene (
205) (0.50 g, 3.62 mmol) was added dropwise, and the mixture was refluxed for 2 h. The temperature was cooled to room temperature and the organic layer was washed with ice water (30.0 mL), 2 M HCl (20.0 mL), and saturated aqueous sodium hydrogen carbonate (20.0 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a yellow oil. Purification by flash chromatography (90/10 pentane/EtOAc) afforded 0.62 g (63%) of
206 as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 7.49 (d,
J = 2.0 Hz, 1H), 7.41–7.34 (m, 2H), 7.30 (ddd,
J = 5.2, 2.6, 1.3 Hz, 2H), 7.11 (ddd,
J = 8.1, 2.6, 1.2 Hz, 1H), 6.89 (d,
J = 8.4 Hz, 1H), 3.96 (s, 3H), 3.94 (s, 3H), 3.85 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 195.30, 159.44, 153.01, 148.95, 139.57, 130.16, 129.08, 125.48, 122.34, 118.19, 114.21, 112.08, 109.70, 56.08, 56.04, 55.44.
(3,4-Dihydroxyphenyl)(3-hydroxyphenyl)methanone (209)
Following general procedure B, 206 (0.27 g, 1.00 mmol) was reacted to afford 0.21 g (91%) of 209 as yellow powder. Mp = 193–195 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.84 (br s, 1H), 9.73 (br s, 1H), 9.39 (br s, 1H), 7.29 (t, J = 7.8 Hz, 1H), 7.21 (d, J = 2.1 Hz, 1H), 7.09 (dd, J = 8.2, 2.1 Hz, 1H), 7.06–6.92 (m, 3H), 6.83 (d, J = 8.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 194.71, 157.53, 150.97, 145.50, 140.04, 129.78, 128.74, 123.73, 120.33, 119.09, 117.32, 116.03, 115.49; HRMS (ESI) m/z: 243.0514 [M − H]−, calculated for C13H11O4 229.0506.
(3,4-Dihydroxyphenyl)(4-hydroxyphenyl)methanone (210)
(3,4-Dimethoxyphenyl)(4-methoxyphenyl)methanone (207)
The compound was synthetized following the literature procedure [
34]. A solution of 4-methoxybenzoic acid (
204) (0.82 g, 5.42 mmol) in SOCl
2 (10.0 mL) was refluxed for 2 h. The solvent was removed under reduced pressure, washed with dichloromethane, and concentrated again, twice. The substituted benzoyl chloride was isolated as a yellow oil in quantitative yield. This was transferred into a round-bottomed flask and diluted with dichloromethane (20.0 mL). Aluminum trichloride (0.53 g, 3.98 mmol) was then added and the mixture was cooled to 0 °C.
205 (0.50 g, 3.62 mmol) was added dropwise and the mixture was refluxed for 2 h. The temperature was cooled to room temperature and the organic layer was washed with ice water (30.0 mL), 2 M HCl (20.0 mL), and saturated aqueous sodium hydrogen carbonate (20.0 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a yellow oil. Crystallization from MeOH afforded 0.61 g (62%) of 207 as a white powder. Mp = 87–89 °C;
1H NMR (400 MHz, CDCl
3) δ 7.84–7.74 (m, 2H), 7.43 (d,
J = 2.0 Hz, 1H), 7.36 (dd,
J = 8.3, 1.9 Hz, 1H), 7.01–6.92 (m, 2H), 6.90 (d,
J = 8.3 Hz, 1H), 3.96 (s, 3H), 3.94 (s, 3H), 3.89 (s, 3H);
13C NMR (101 MHz, CDCl
3) δ 194.46, 162.82, 152.55, 148.87, 132.21, 130.79, 130.70, 124.80, 113.43, 112.20, 109.69, 56.06, 56.02, 55.46.
(3,4-Dihydroxyphenyl)(4-hydroxyphenyl)methanone (210)
Following general procedure B, 207 (0.27 g, 1.00 mmol) was reacted to afford 0.20 g (88%) of 210 as yellow powder. Mp = 202–204 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.63–7.49 (m, 2H), 7.16 (d, J = 2.1 Hz, 1H), 7.04 (dd, J = 8.1, 2.0 Hz, 1H), 6.89–6.82 (m, 2H), 6.81 (d, J = 8.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 193.54, 161.52, 150.24, 145.40, 132.38, 129.52, 129.39, 123.19, 117.26, 115.35; HRMS (ESI) m/z: 243.0512 [M − H]−, calculated for C13H11O4 229.0506.
Bis(3,4-dihydroxyphenyl)methanone (211)
Bis(3,4-dimethoxyphenyl)methanone (208)
The compound was synthetized following the literature procedure. [
34] A solution of
7 (1.00 g, 5.49 mmol) in SOCl
2 (10.0 mL) was refluxed for 2h. It was concentrated in vacuo, washed with dichloromethane, and concentrated again, twice. The substituted benzoyl chloride was isolated as a yellow oil in quantitative yield. This was transferred into a round-bottomed flask and diluted with dichloromethane (40.0 mL). Aluminum trichloride (0.80 g, 6.03 mmol) was then added and the mixture was cooled to 0 °C. Compound
205 (0.76 g, 5.48 mmol) was added dropwise and the mixture was refluxed for 2 h. The temperature was cooled to room temperature and the organic layer was washed with ice water (30 mL), 2 M HCl (20 mL) and saturated aqueous sodium hydrogen carbonate (20.0 mL), dried over anhydrous sodium sulfate, and concentrated in vacuo to give a yellow oil. Purification by flash chromatography (90/10 pentane/EtOAc) afforded 1.05 g (63%) of
208 as a white solid. Mp = 149–151 °C;
1H NMR (400 MHz, cdcl
3) δ 7.42 (d,
J = 2.1 Hz, 2H), 7.37 (ddd,
J = 8.3, 2.0, 0.6 Hz, 2H), 6.89 (d,
J = 8.3 Hz, 2H), 3.95 (s, 6H), 3.93 (s, 6H).
Bis(3,4-dihydroxyphenyl)methanone (211)
Following general procedure B, 208 (0.20 g, 0.66 mmol) was reacted to afford 0.14 g (89%) of 211 as yellow powder. Mp = 244–246 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 2H), 9.31 (s, 2H), 7.14 (d, J = 2.1 Hz, 2H), 7.04 (dd, J = 8.2, 2.1 Hz, 2H), 6.81 (d, J = 8.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 193.61, 150.08, 145.29, 129.70, 123.06, 117.29, 115.30; HRMS (ESI) m/z: 245.0462 [M − H]−, calculated for C13H12O5 245.0455.


 ,
, 



















{kind=link}
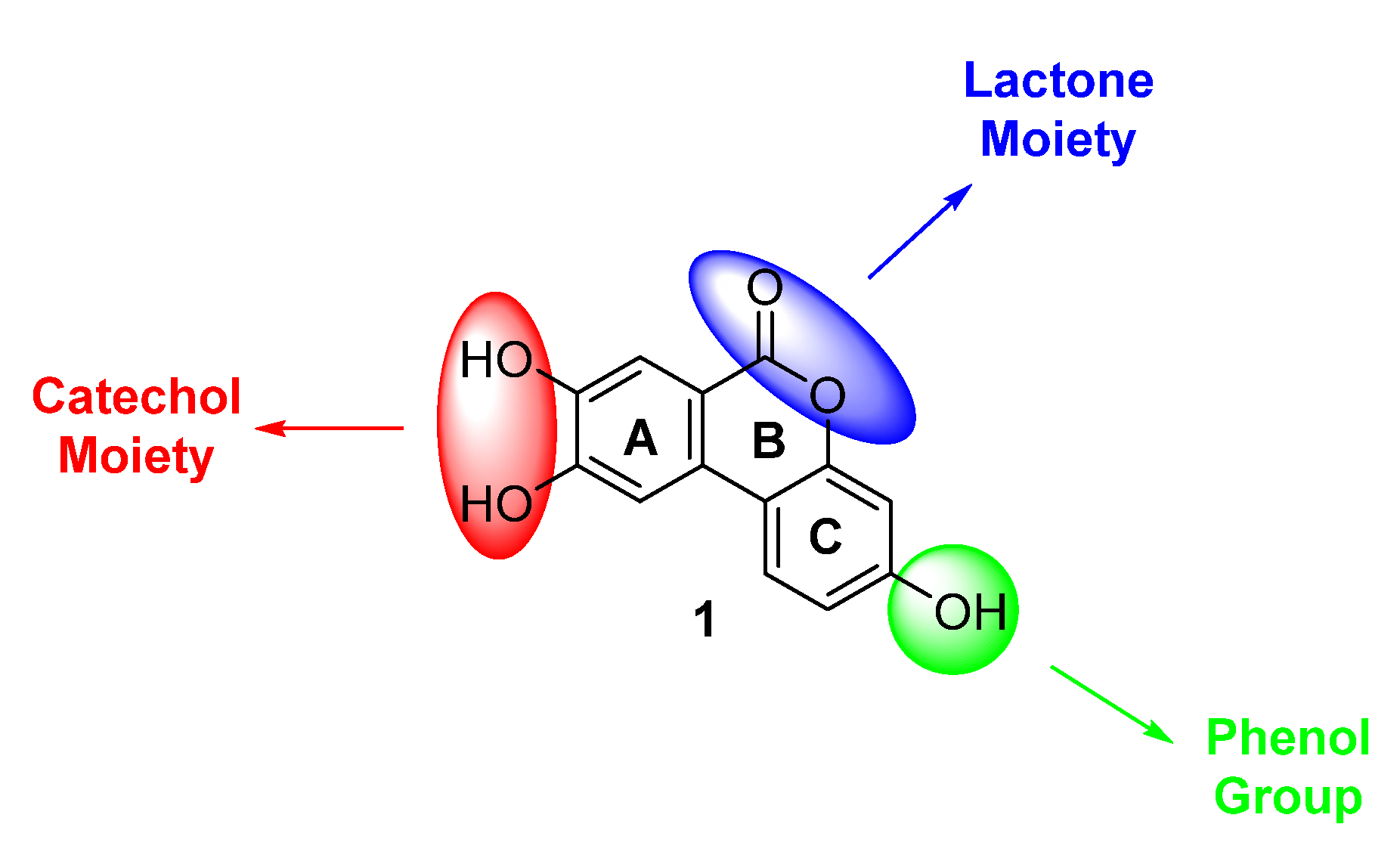{kind=link}
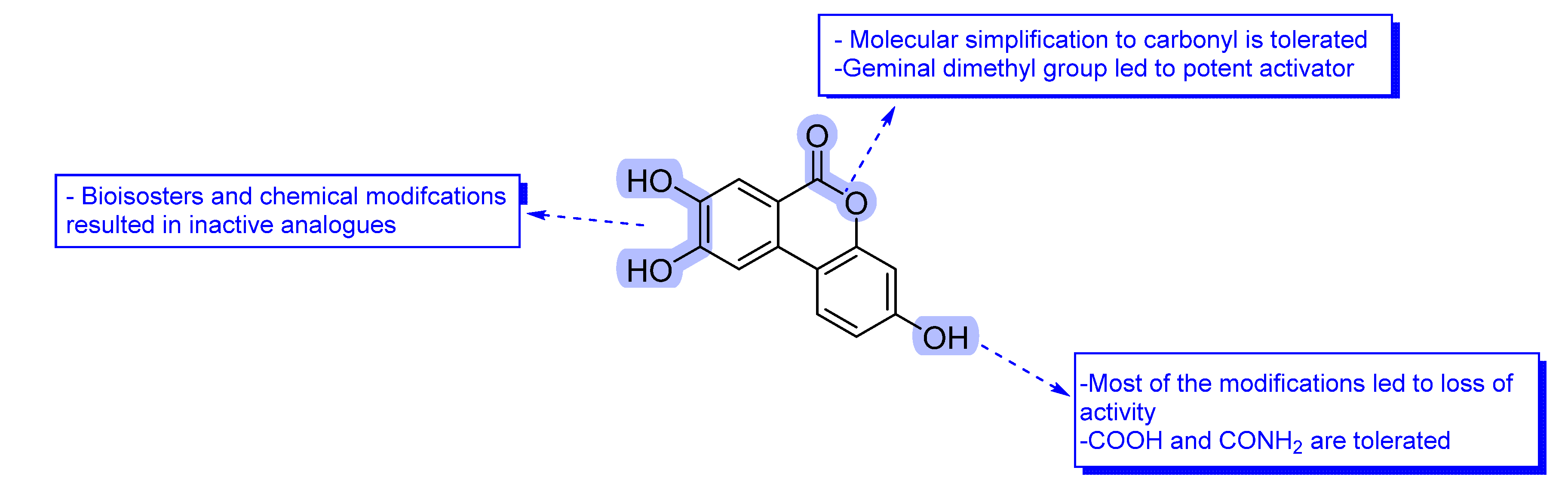{kind=link}
{kind=link}
{kind=link}
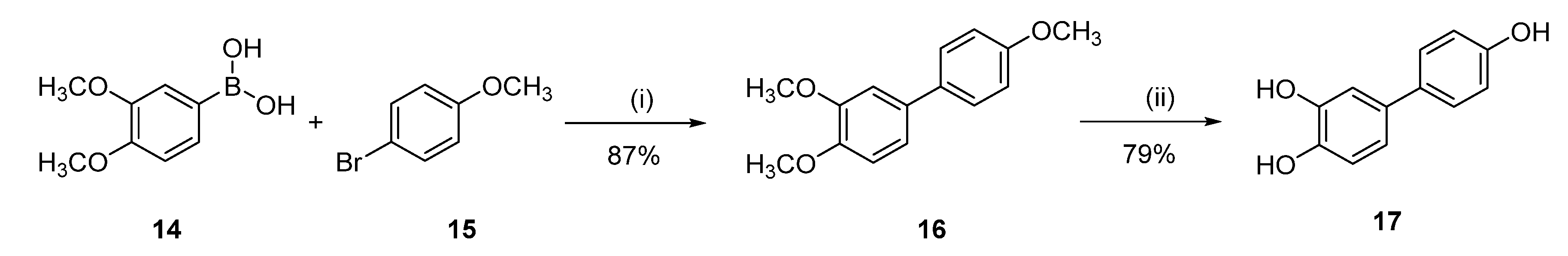{kind=link}
{kind=link}
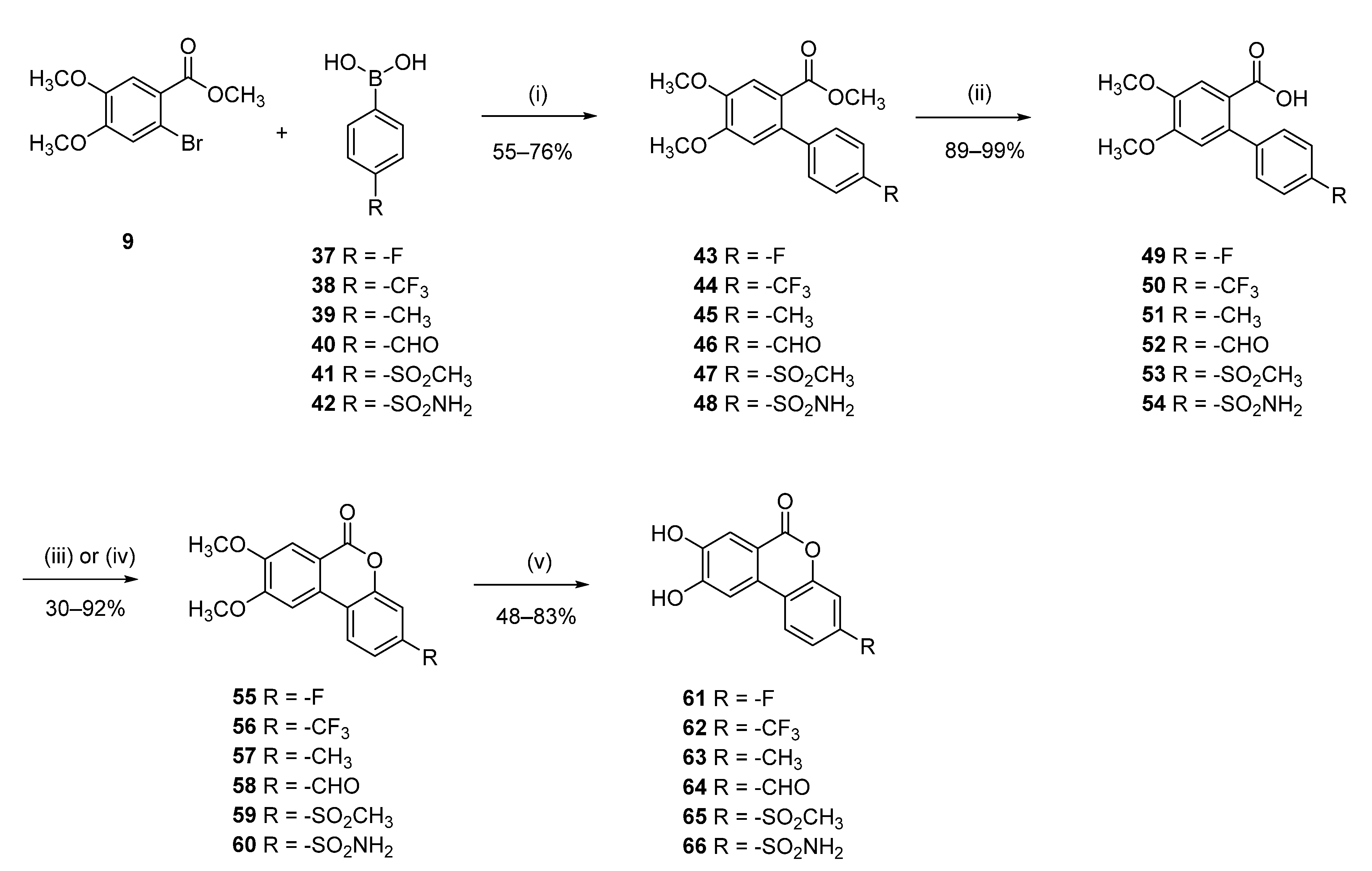{kind=link}
{kind=link}
{kind=link}
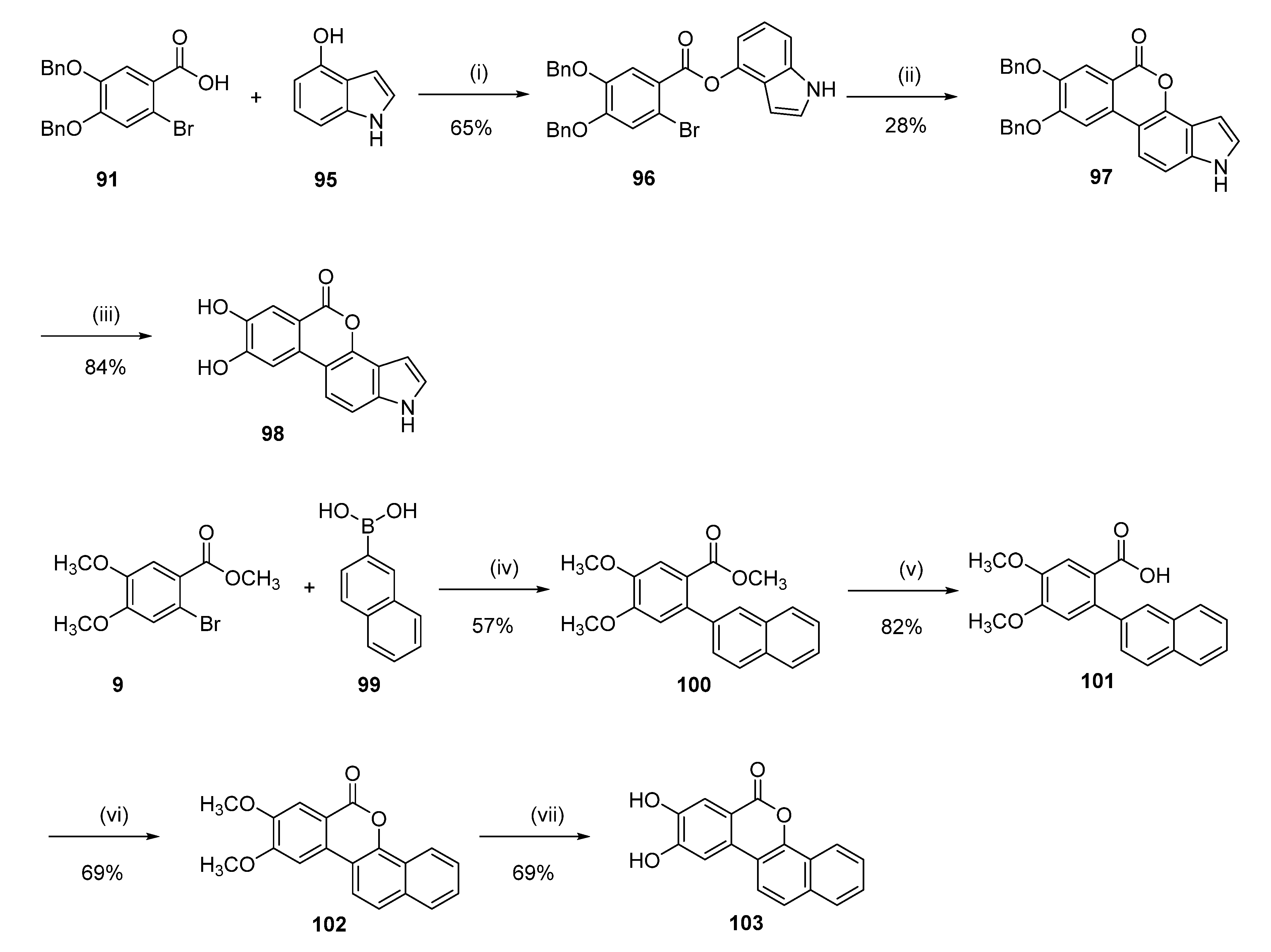{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
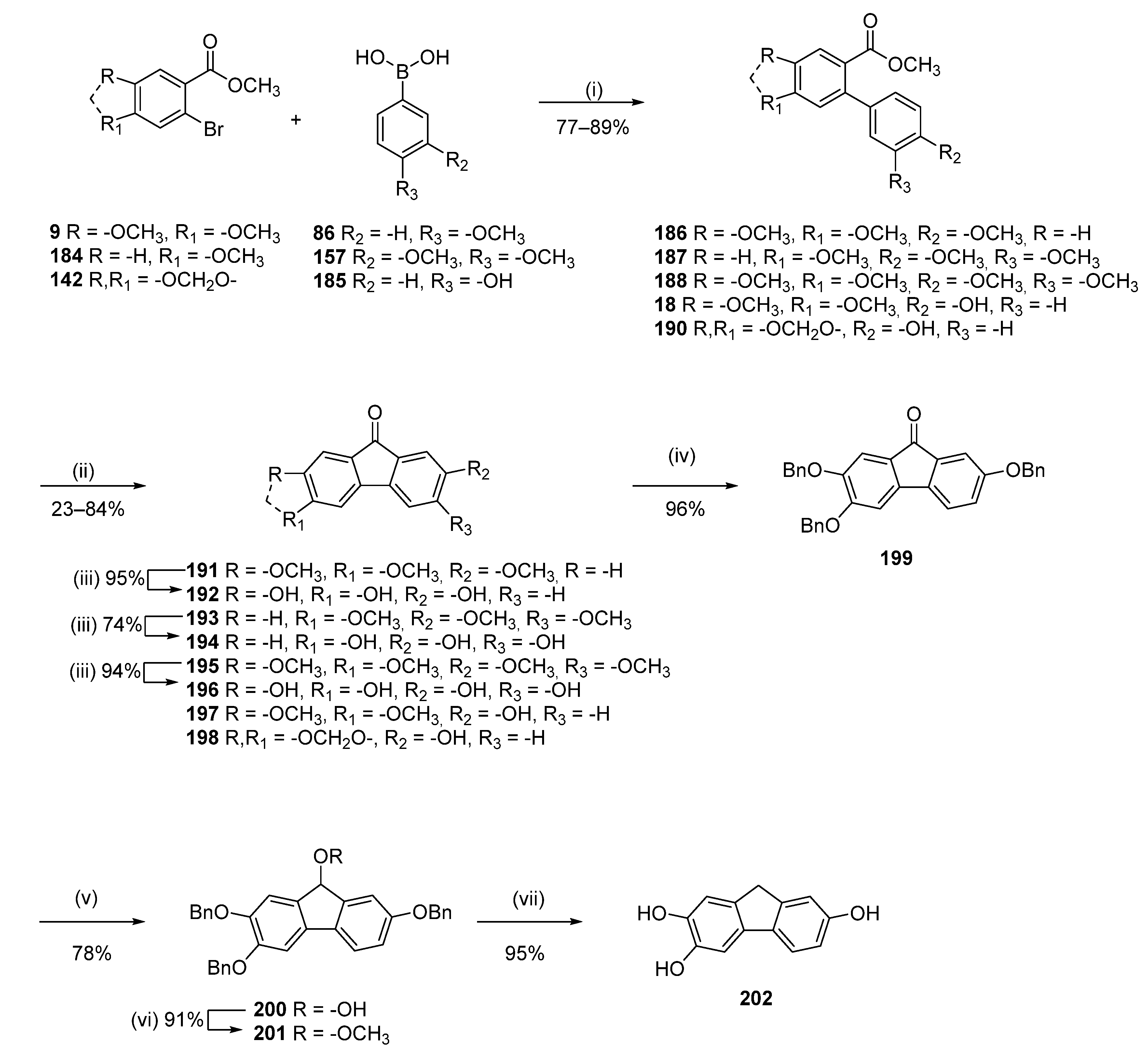{kind=link}
{kind=link}






























