



At the Crossroads of the cGAS-cGAMP-STING Pathway and the DNA Damage Response: Implications for Cancer Progression and Treatment

 , , and
, , and 
Abstract
:1. Introduction
- (1)
- TLR9 (Toll-like receptor 9) is found almost exclusively in the endosomal membrane of B cells and plasmacytoid DCs (dendritic cells). It detects external DNA and is particularly sensitive to unmethylated or hypomethylated CpG islands [23];
- (2)
- IFI16 and AIM2 belong to the PYHIN family [24]. IFI16 detects both DNA and RNA. It predominantly resides in the nucleus and cytosol and primarily detects DNA strands around 70 base pairs in length. Upon activation, IFI16 stimulates the inflammasome. It is expressed in many cell types and is particularly abundant in leukocytes. AIM2, located in the cytosol, is an active defender against viruses and bacteria. When activated, it facilitates the formation of the inflammasome, which can trigger the activation of caspases. This activation leads to the release of cytokines IL-1β and IL-18 and induces pyroptosis, especially in response to DNA damage caused by acute ionizing radiation. AIM2 exhibits high specificity for T and B cells, as well as spermatids;
- (3)
- (4)
- ZBP1 is highly enriched in immune cells (plasma cells, NK cells) and is a sensor for dsDNA and dsRNA helices in the left-handed Z conformation, namely, Z-DNA and Z-RNA [27] ZBP1 plays a key role in mediating various forms of cell death. Its activation leads to RIPK3 stimulation and subsequent MLKL phosphorylation, causing disruption of the nuclear envelope and release of cellular DNA into the cytosol. ZBP1 stabilizes Z-form mtDNA and forms a cytosolic complex with cGAS;
- (5)
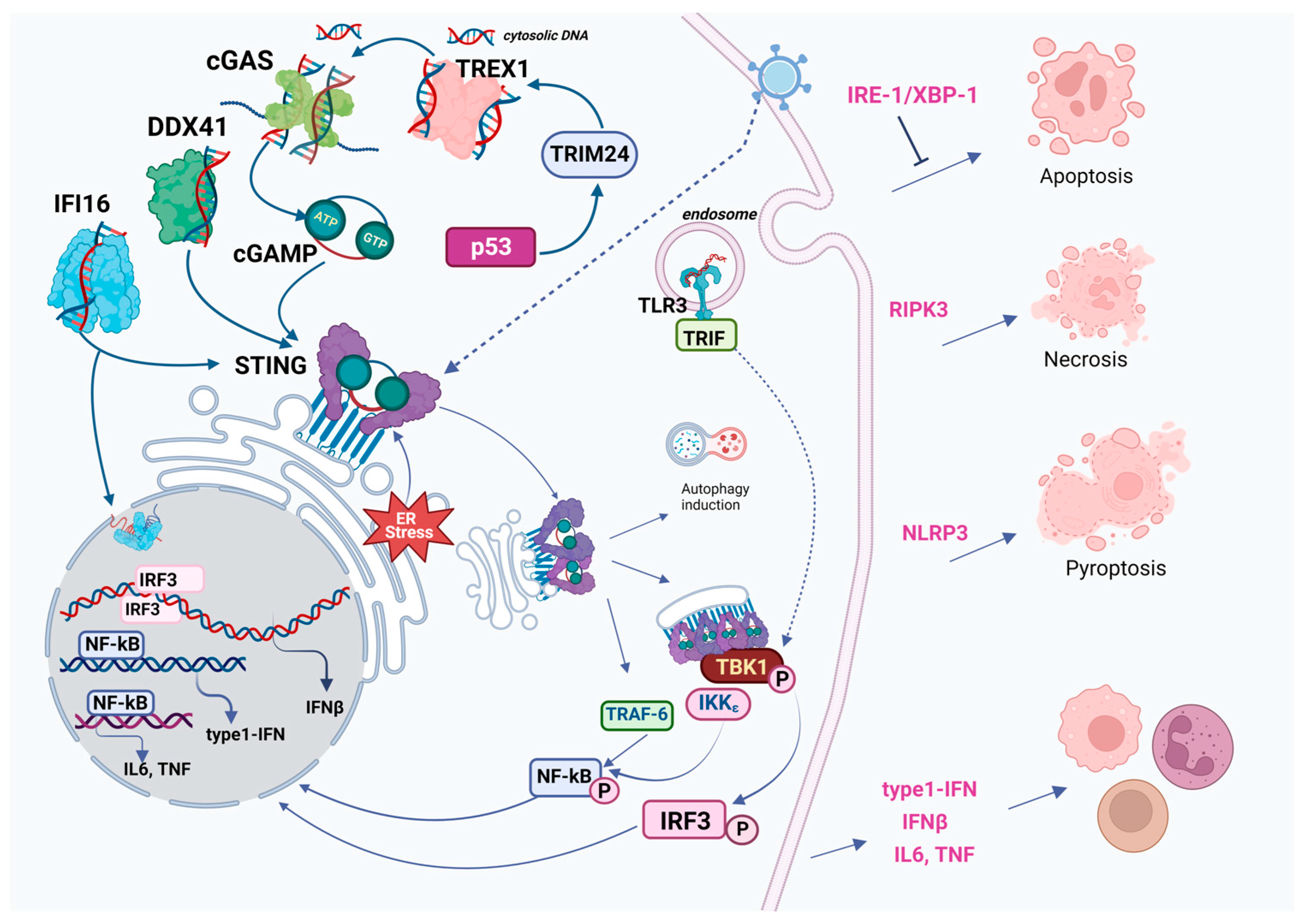
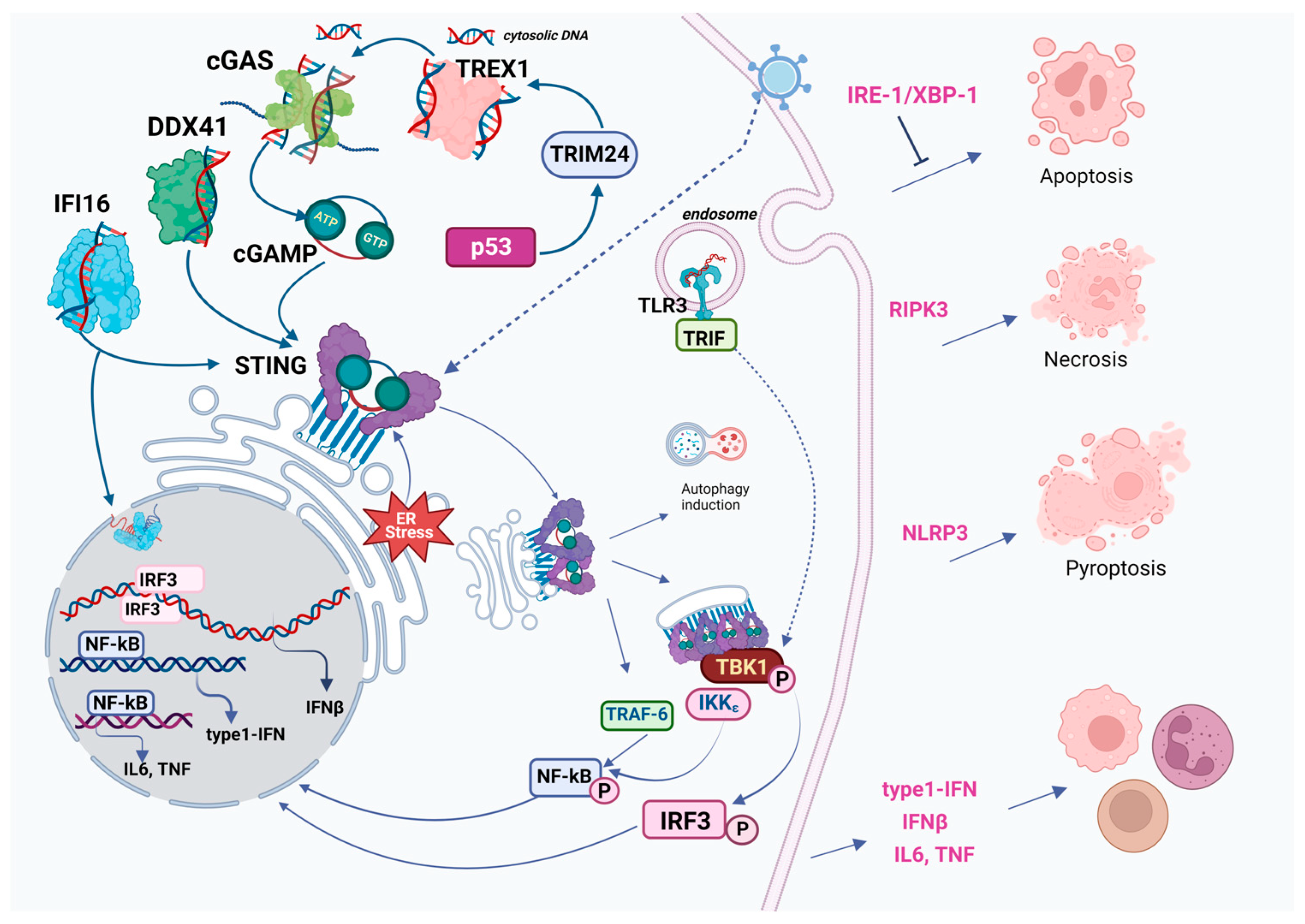
- Among the cytosolic DNA sensors, however, cGAS appears to be one of the most widespread and important at most stages of infection (Figure 1).
2. Overview of the Classical cGAS-cGAMP-STING-TBK1-IRF3 Pathway: Structure of Key Components and Their Molecular Evolution
2.1. cGAS (Cyclic GMP-AMP Synthase)
2.2. cGAMP (Cyclic GMP-AMP)
2.3. STING (Stimulator of Interferon Genes)
2.4. TBK1 (TANK-Binding Kinase 1)
2.5. IRF3, NF-κB and Downstream Signaling
3. cGAS-STING in Major DNA Damage Response Pathways
3.1. Efficient Oncosuppressive Function of cGAS-STING in DNA Damage Response
3.2. Senescence as the Route out of Cancer and the Role of the STING Pathway
3.3. DDR and Its Links to Carcinogenesis and Tumor Formation
3.4. STING and BRCA1,2
3.5. STING and Oncogenic Infections
4. Cross-Talk of cGAS-STING with Major Cell Death Pathways
4.1. Necroptosis and cGAS-STING
4.2. Pyroptosis and cGAS-STING
4.3. Ferroptosis and cGAS-STING
4.4. Autophagy and cGAS-STING
5. Roles of the cGAS-STING Pathway in Response to Radio- and Chemotherapies
5.1. Radiotherapy Elicits DNA Damage and Activates cGAS-STING
5.2. Chemotherapy and cGAS-STING
5.3. PARPi and cGAS-STING
5.4. Immune Checkpoint Blockade and cGAS-STING Pathway
6. Duality of the cGAS-STING Pathway Effects in Cancer
6.1. cGAS-STING Pathway Inhibition Promotes Carcinogenesis
6.2. Tumorigenic Effects of the Excessive cGAS-STING Activity
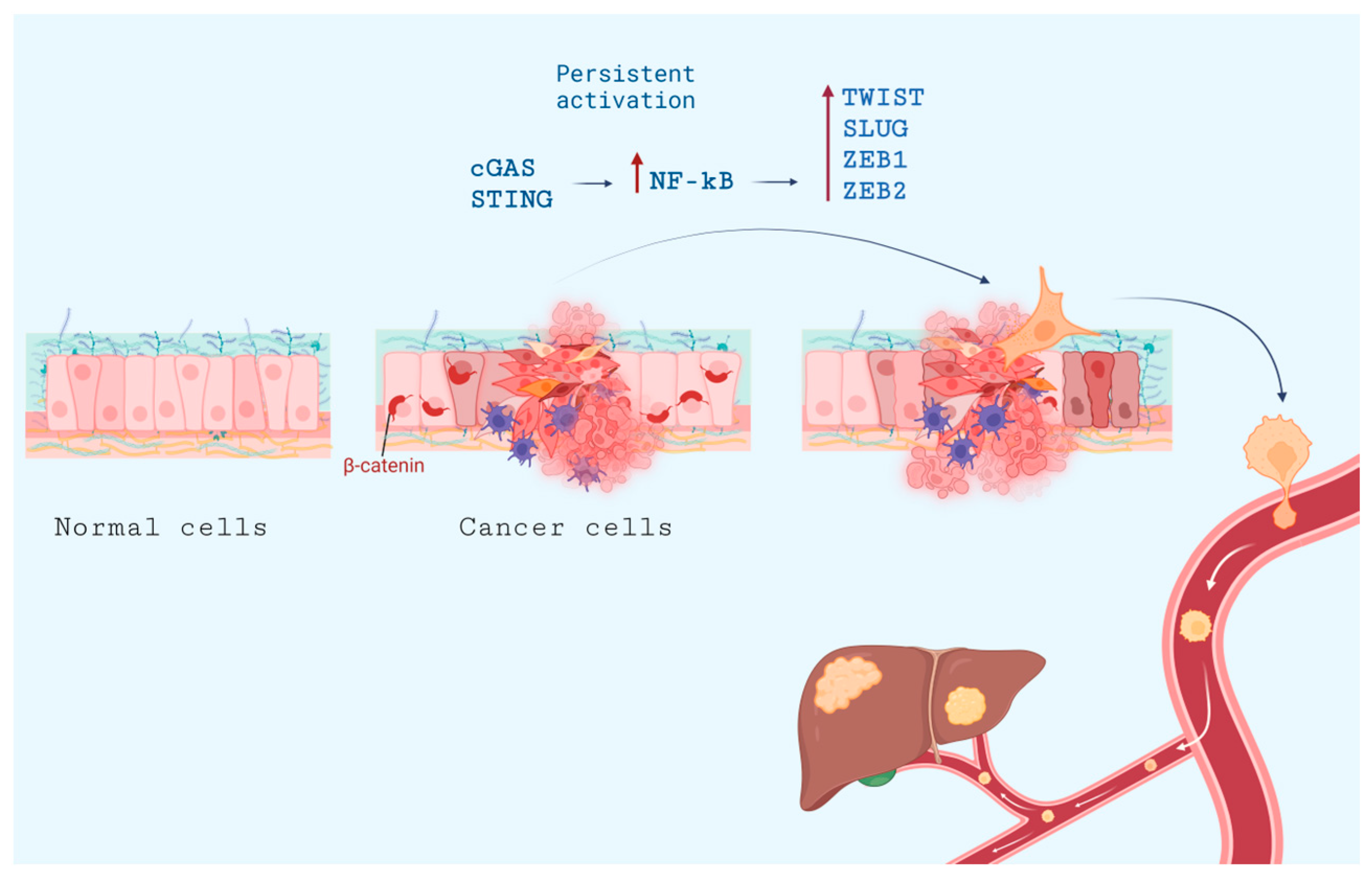
7. Metastasis and cGAS-STING Pathway
8. Pharmaceutical Achievements in the Regulation of the cGAS-STING Pathway
8.1. Administration Schedules
8.2. STING Antagonists
8.3. Nanosystems for Efficient Delivery of cGAS-STING Modulators
8.4. STING as a Drug
9. Agonists of cGAS- STING Pathway in Distinctive Cancer Types and Perspectives of Personalization
9.1. Gliomas
9.2. Pancreatic Ductal Adenocarcinoma (PDAC)
9.3. Melanoma
9.4. Oncohematology
9.5. Personalization on the Basis of Genomic Profiling
9.6. Modulating cGAS-STING According to the Tumor Peculiarities
10. Perspectives for Efficient Synergies between cGAS-STING Regulation and Other Anti-Cancer Therapeutic Modalities
10.1. Interactions between Different Anti-Cancer Drugs
10.2. Cell-Based Therapies
10.3. Immunotherapies Such as Oncolytic Viruses (OV)
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING Is a Direct Innate Immune Sensor of Cyclic Di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS Produces a 2’-5’-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2’,5’)pA(3’,5’)p] Is the Metazoan Second Messenger Produced by DNA-Activated Cyclic GMP-AMP Synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, S.; Wu, Y.; Li, Q.; Zhang, T.; Min, K.; Feng, D.; Liu, M.; Wei, J.; Zhu, L.; et al. A Bibliometric Analysis of the Innate Immune DNA Sensing cGAS-STING Pathway from 2013 to 2021. Front. Immunol. 2022, 13, 916383. [Google Scholar] [CrossRef]
- Yoon, S.H.; Waters, C.M. The Ever-Expanding World of Bacterial Cyclic Oligonucleotide Second Messengers. Curr. Opin. Microbiol. 2021, 60, 96–103. [Google Scholar] [CrossRef]
- Wein, T.; Sorek, R. Bacterial Origins of Human Cell-Autonomous Innate Immune Mechanisms. Nat. Rev. Immunol. 2022, 22, 629–638. [Google Scholar] [CrossRef]
- Bose, D. cGAS/STING Pathway in Cancer: Jekyll and Hyde Story of Cancer Immune Response. Int. J. Mol. Sci. 2017, 18, 2456. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Song, Y.; Chu, Q.; Wu, K. Activating cGAS-STING Pathway for the Optimal Effect of Cancer Immunotherapy. J. Hematol. Oncol. 2019, 12, 35. [Google Scholar] [CrossRef]
- Hoong, B.Y.D.; Gan, Y.H.; Liu, H.; Chen, E.S. cGAS-STING Pathway in Oncogenesis and Cancer Therapeutics. Oncotarget 2020, 11, 2930–2955. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.-C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA Damage Response in Immuno-Oncology: Developments and Opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.N.; Victorelli, S.G.; Salmonowicz, H.; Dasgupta, N.; Liu, T.; Passos, J.F.; Adams, P.D. Cytoplasmic DNA: Sources, Sensing, and Role in Aging and Disease. Cell 2021, 184, 5506–5526. [Google Scholar] [CrossRef] [PubMed]
- Vashi, N.; Bakhoum, S.F. The Evolution of STING Signaling and Its Involvement in Cancer. Trends Biochem. Sci. 2021, 46, 446–460. [Google Scholar] [CrossRef]
- Du, J.-M.; Qian, M.-J.; Yuan, T.; Chen, R.-H.; He, Q.-J.; Yang, B.; Ling, Q.; Zhu, H. cGAS and Cancer Therapy: A Double-Edged Sword. Acta Pharmacol. Sin. 2022, 43, 2202–2211. [Google Scholar] [CrossRef]
- Huang, R.; Ning, Q.; Zhao, J.; Zhao, X.; Zeng, L.; Yi, Y.; Tang, S. Targeting STING for Cancer Immunotherapy: From Mechanisms to Translation. Int. Immunopharmacol. 2022, 113, 109304. [Google Scholar] [CrossRef]
- Li, J.; Bakhoum, S.F. The Pleiotropic Roles of cGAS-STING Signaling in the Tumor Microenvironment. J. Mol. Cell Biol. 2022, 14, mjac019. [Google Scholar] [CrossRef]
- Samson, N.; Ablasser, A. The cGAS-STING Pathway and Cancer. Nat. Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef]
- Xu, Y.; Nowsheen, S.; Deng, M. DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy. Cancers 2023, 15, 1619. [Google Scholar] [CrossRef]
- Yum, S.; Li, M.; Frankel, A.E.; Chen, Z.J. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 323–344. [Google Scholar] [CrossRef]
- Huérfano, S.; Šroller, V.; Bruštíková, K.; Horníková, L.; Forstová, J. The Interplay between Viruses and Host DNA Sensors. Viruses 2022, 14, 666. [Google Scholar] [CrossRef] [PubMed]
- Kou, M.; Wang, L. Surface Toll-like Receptor 9 on Immune Cells and Its Immunomodulatory Effect. Front. Immunol. 2023, 14, 1259989. [Google Scholar] [CrossRef] [PubMed]
- Bosso, M.; Kirchhoff, F. Emerging Role of PYHIN Proteins as Antiviral Restriction Factors. Viruses 2020, 12, 1464. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, Y.; Wang, Y.; Niu, Q.; Gao, Q.; Fu, Q.; Ma, J.; Wang, H.; Yan, Y.; Ding, C.; et al. Chicken DNA Virus Sensor DDX41 Activates IFN-β Signaling Pathway Dependent on STING. Dev. Comp. Immunol. 2017, 76, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The Helicase DDX41 Senses Intracellular DNA Mediated by the Adaptor STING in Dendritic Cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) Is a Cytosolic DNA Sensor and an Activator of Innate Immune Response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural Mechanism of Cytosolic DNA Sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef]
- Cai, H.; Imler, J.-L. cGAS-STING: Insight on the Evolution of a Primordial Antiviral Signaling Cassette. Fac. Rev. 2021, 10, 54. [Google Scholar] [CrossRef]
- Kranzusch, P.J. cGAS and CD-NTase Enzymes: Structure, Mechanism, and Evolution. Curr. Opin. Struct. Biol. 2019, 59, 178–187. [Google Scholar] [CrossRef]
- Kuchta, K.; Knizewski, L.; Wyrwicz, L.S.; Rychlewski, L.; Ginalski, K. Comprehensive Classification of Nucleotidyltransferase Fold Proteins: Identification of Novel Families and Their Representatives in Human. Nucleic Acids Res. 2009, 37, 7701–7714. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Lee, A.S.-Y.; Berger, J.M.; Doudna, J.A. Structure of Human cGAS Reveals a Conserved Family of Second-Messenger Enzymes in Innate Immunity. Cell Rep. 2013, 3, 1362–1368. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS Senses Long and HMGB/TFAM-Bound U-Turn DNA by Forming Protein-DNA Ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Herzner, A.-M.; Hagmann, C.A.; Goldeck, M.; Wolter, S.; Kübler, K.; Wittmann, S.; Gramberg, T.; Andreeva, L.; Hopfner, K.-P.; Mertens, C.; et al. Sequence-Specific Activation of the DNA Sensor cGAS by Y-Form DNA Structures as Found in Primary HIV-1 cDNA. Nat. Immunol. 2015, 16, 1025–1033. [Google Scholar] [CrossRef]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Höning, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.P.; Hornung, V. Cytosolic RNA:DNA Hybrids Activate the cGAS-STING Axis. EMBO J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.P.; Song, C.; Bocek, M.J.; Choi, J.-H.; Kousorous, J.; Sathirachinda, A.; Lin, C.; Brickner, J.R.; Bai, G.; Lans, H.; et al. R-Loop-Derived Cytoplasmic RNA-DNA Hybrids Activate an Immune Response. Nature 2023, 613, 187–194. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 Acts at Stalled Replication Forks to Prevent Interferon Induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Ye, B.; Du, Y.; Li, C.; Xiong, Z.; Qu, Y.; Fan, Z. A Circular RNA Protects Dormant Hematopoietic Stem Cells from DNA Sensor cGAS-Mediated Exhaustion. Immunity 2018, 48, 688–701.e7. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-Viral Specificity of IFN-Induced Genes Reveals New Roles for cGAS in Innate Immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Chen, S.; Rong, M.; Lv, Y.; Zhu, D.; Xiang, Y. Regulation of cGAS Activity by RNA-Modulated Phase Separation. EMBO Rep. 2023, 24, e51800. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, J.; Zheng, J.; Fu, J.; Wang, W.; Duan, L.; Qian, X.; Yang, Y. Phase Separation in cGAS-STING Signaling: Cytosolic DNA Sensing and Regulatory Functions. Chembiochem 2023, 24, e202300147. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Juszkiewicz, S.; Blears, D.; Bajpe, P.K.; Han, Z.; Faull, P.; Mitter, R.; Stewart, A.; Snijders, A.P.; Hegde, R.S.; et al. Translation Stress and Collided Ribosomes Are Co-Activators of cGAS. Mol. Cell 2021, 81, 2808–2822.e10. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.; Valadao, A.-L.; Vlachakis, D.; Polak, K.; Vila, I.K.; Taffoni, C.; Prabakaran, T.; Marriott, A.S.; Kaczmarek, R.; Houel, A.; et al. Lysyl-tRNA Synthetase Produces Diadenosine Tetraphosphate to Curb STING-Dependent Inflammation. Sci. Adv. 2020, 6, eaax3333. [Google Scholar] [CrossRef]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311.e11. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-Induced Liquid Phase Condensation of cGAS Activates Innate Immune Signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e11. [Google Scholar] [CrossRef]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.P.F.; Puig Lombardi, E.; Herve, S.; De Silva, N.S.; Rookhuizen, D.C.; Zueva, E.; Goudot, C.; et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep. 2019, 26, 2377–2393.e13. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Cambier, S.; Gray, E.E.; Stetson, D.B. Tight Nuclear Tethering of cGAS Is Essential for Preventing Autoreactivity. Elife 2019, 8, e47491. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Lama, L.; Adura, C.; Tomita, D.; Glickman, J.F.; Tuschl, T.; Patel, D.J. Human cGAS Catalytic Domain Has an Additional DNA-Binding Interface That Enhances Enzymatic Activity and Liquid-Phase Condensation. Proc. Natl. Acad. Sci. USA 2019, 116, 11946–11955. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.-J.; Xing, J.-Q.; Zhao, M.; Huang, Y.-J.; Chen, S.; et al. G3BP1 Promotes DNA Binding and Activation of cGAS. Nat. Immunol. 2019, 20, 18–28. [Google Scholar] [CrossRef]
- Hornung, V.; Hartmann, R.; Ablasser, A.; Hopfner, K.-P. OAS Proteins and cGAS: Unifying Concepts in Sensing and Responding to Cytosolic Nucleic Acids. Nat. Rev. Immunol. 2014, 14, 521–528. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type-I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Slavik, K.M.; Kranzusch, P.J. CBASS to cGAS-STING: The Origins and Mechanisms of Nucleotide Second Messenger Immune Signaling. Annu. Rev. Virol. 2023, 10, 423–453. [Google Scholar] [CrossRef] [PubMed]
- Morehouse, B.R.; Govande, A.A.; Millman, A.; Keszei, A.F.A.; Lowey, B.; Ofir, G.; Shao, S.; Sorek, R.; Kranzusch, P.J. STING Cyclic Dinucleotide Sensing Originated in Bacteria. Nature 2020, 586, 429–433. [Google Scholar] [CrossRef]
- Shu, C.; Yi, G.; Watts, T.; Kao, C.C.; Li, P. Structure of STING Bound to Cyclic Di-GMP Reveals the Mechanism of Cyclic Dinucleotide Recognition by the Immune System. Nat. Struct. Mol. Biol. 2012, 19, 722–724. [Google Scholar] [CrossRef]
- Sauer, J.-D.; Sotelo-Troha, K.; von Moltke, J.; Monroe, K.M.; Rae, C.S.; Brubaker, S.W.; Hyodo, M.; Hayakawa, Y.; Woodward, J.J.; Portnoy, D.A.; et al. The N-Ethyl-N-Nitrosourea-Induced Goldenticket Mouse Mutant Reveals an Essential Function of Sting in the in Vivo Interferon Response to Listeria Monocytogenes and Cyclic Dinucleotides. Infect. Immun. 2011, 79, 688–694. [Google Scholar] [CrossRef]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide That Activates Human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef]
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-Function Analysis of STING Activation by c[G(2’,5’)pA(3’,5’)p] and Targeting by Antiviral DMXAA. Cell 2013, 154, 748–762. [Google Scholar] [CrossRef]
- Shi, H.; Wu, J.; Chen, Z.J.; Chen, C. Molecular Basis for the Specific Recognition of the Metazoan Cyclic GMP-AMP by the Innate Immune Adaptor Protein STING. Proc. Natl. Acad. Sci. USA 2015, 112, 8947–8952. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.-C.; Zhang, X. Cryo-EM Structures of STING Reveal Its Mechanism of Activation by Cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Shang, G.; Li, J.; Lu, Y.; Bai, X.-C.; Zhang, X. Activation of STING by Targeting a Pocket in the Transmembrane Domain. Nature 2022, 604, 557–562. [Google Scholar] [CrossRef]
- Tang, E.D.; Wang, C.-Y. Single Amino Acid Change in STING Leads to Constitutive Active Signaling. PLoS ONE 2015, 10, e0120090. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A Conserved PLPLRT/SD Motif of STING Mediates the Recruitment and Activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef]
- Ergun, S.L.; Fernandez, D.; Weiss, T.M.; Li, L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 2019, 178, 290–301.e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Carlson, R.J.; Pires, I.S.; Gentili, M.; Feng, E.; Hellier, Q.; Schwartz, M.A.; Blainey, P.C.; Irvine, D.J.; Hacohen, N. Human STING Is a Proton Channel. Science 2023, 381, 508–514. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, L.; Shen, J.; Zhai, Y.; Jiang, Q.; Yi, M.; Deng, X.; Ruan, Z.; Fang, R.; Chen, Z.; et al. The STING Phase-Separator Suppresses Innate Immune Signalling. Nat. Cell Biol. 2021, 23, 330–340. [Google Scholar] [CrossRef]
- Corbet, G.A.; Burke, J.M.; Parker, R. Nucleic Acid-Protein Condensates in Innate Immune Signaling. EMBO J. 2023, 42, e111870. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with Covalent Small-Molecule Inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is an Endogenous High-Affinity Ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef]
- Wheeler, O.P.G.; Unterholzner, L. DNA Sensing in Cancer: Pro-Tumour and Anti-Tumour Functions of cGAS-STING Signalling. Essays Biochem. 2023, 67, 905–918. [Google Scholar] [CrossRef]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal 2012, 5, ra20. [Google Scholar] [CrossRef]
- Jin, L.; Xu, L.-G.; Yang, I.V.; Davidson, E.J.; Schwartz, D.A.; Wurfel, M.M.; Cambier, J.C. Identification and Characterization of a Loss-of-Function Human MPYS Variant. Genes. Immun. 2011, 12, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-Linked Ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.-M.; et al. NEMO-IKKβ Are Essential for IRF3 and NF-κB Activation in the cGAS-STING Pathway. J. Immunol. 2017, 199, 3222–3233. [Google Scholar] [CrossRef] [PubMed]
- Chamma, H.; Guha, S.; Laguette, N.; Vila, I.K. Protocol to Induce and Assess cGAS-STING Pathway Activation in Vitro. STAR Protoc. 2022, 3, 101384. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Fang, Y.; Lin, Z.; Yang, L.; Zheng, L.; Hu, H.; Yu, T.; Huang, B.; Chen, S.; Wang, H.; et al. Inhibition of cGAS-Mediated Interferon Response Facilitates Transgene Expression. iScience 2020, 23, 101026. [Google Scholar] [CrossRef]
- An, J.; Zhang, C.-P.; Qiu, H.-Y.; Zhang, H.-X.; Chen, Q.-B.; Zhang, Y.-M.; Lei, X.-L.; Zhang, C.-X.; Yin, H.; Zhang, Y. Enhancement of the Viability of T Cells Electroporated with DNA via Osmotic Dampening of the DNA-Sensing cGAS-STING Pathway. Nat. Biomed. Eng. 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS Cooperate in the Activation of STING during DNA Sensing in Human Keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-Canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e5. [Google Scholar] [CrossRef] [PubMed]
- Ka, N.-L.; Lim, G.Y.; Hwang, S.; Kim, S.-S.; Lee, M.-O. IFI16 Inhibits DNA Repair That Potentiates Type-I Interferon-Induced Antitumor Effects in Triple Negative Breast Cancer. Cell Rep. 2021, 37, 110138. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Raulet, D.H. The DNA Damage Response Arouses the Immune System. Cancer Res. 2006, 66, 3959–3962. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. C-Di-AMP Secreted by Intracellular Listeria Monocytogenes Activates a Host Type I Interferon Response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Liu, X.-Y.; Du, X.-X.; Jiang, Z.-F.; Su, X.-D. The Structural Basis for the Sensing and Binding of Cyclic Di-GMP by STING. Nat. Struct. Mol. Biol. 2012, 19, 728–730. [Google Scholar] [CrossRef]
- Chen, F.; Wu, R.; Liu, J.; Kang, R.; Li, J.; Tang, D. The STING1-MYD88 Complex Drives ACOD1/IRG1 Expression and Function in Lethal Innate Immunity. iScience 2022, 25, 104561. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Demaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef]
- Hu, S.; Fang, Y.; Chen, X.; Cheng, T.; Zhao, M.; Du, M.; Li, T.; Li, M.; Zeng, Z.; Wei, Y.; et al. cGAS Restricts Colon Cancer Development by Protecting Intestinal Barrier Integrity. Proc. Natl. Acad. Sci. USA 2021, 118, e2105747118. [Google Scholar] [CrossRef]
- Thomsen, M.K.; Skouboe, M.K.; Boularan, C.; Vernejoul, F.; Lioux, T.; Leknes, S.L.; Berthelsen, M.F.; Riedel, M.; Cai, H.; Joseph, J.V.; et al. The cGAS-STING Pathway Is a Therapeutic Target in a Preclinical Model of Hepatocellular Carcinoma. Oncogene 2020, 39, 1652–1664. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef]
- Kim, Y.; Cho, N.-Y.; Jin, L.; Jin, H.Y.; Kang, G.H. Prognostic Significance of STING Expression in Solid Tumor: A Systematic Review and Meta-Analysis. Front. Oncol. 2023, 13, 1244962. [Google Scholar] [CrossRef]
- Sellaththurai, S.; Jung, S.; Kim, M.-J.; Nadarajapillai, K.; Ganeshalingam, S.; Jeong, J.B.; Lee, J. CRISPR/Cas9-Induced Knockout of Sting Increases Susceptibility of Zebrafish to Bacterial Infection. Biomolecules 2023, 13, 324. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through cGAS Promotes Senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS Is Essential for Cellular Senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-A.; Shen, Y.-L.; Hsia, H.-Y.; Tiang, Y.-P.; Sung, T.-L.; Chen, L.-Y. Extrachromosomal Telomere Repeat DNA Is Linked to ALT Development via cGAS-STING DNA Sensing Pathway. Nat. Struct. Mol. Biol. 2017, 24, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING Drives Ageing-Related Inflammation and Neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
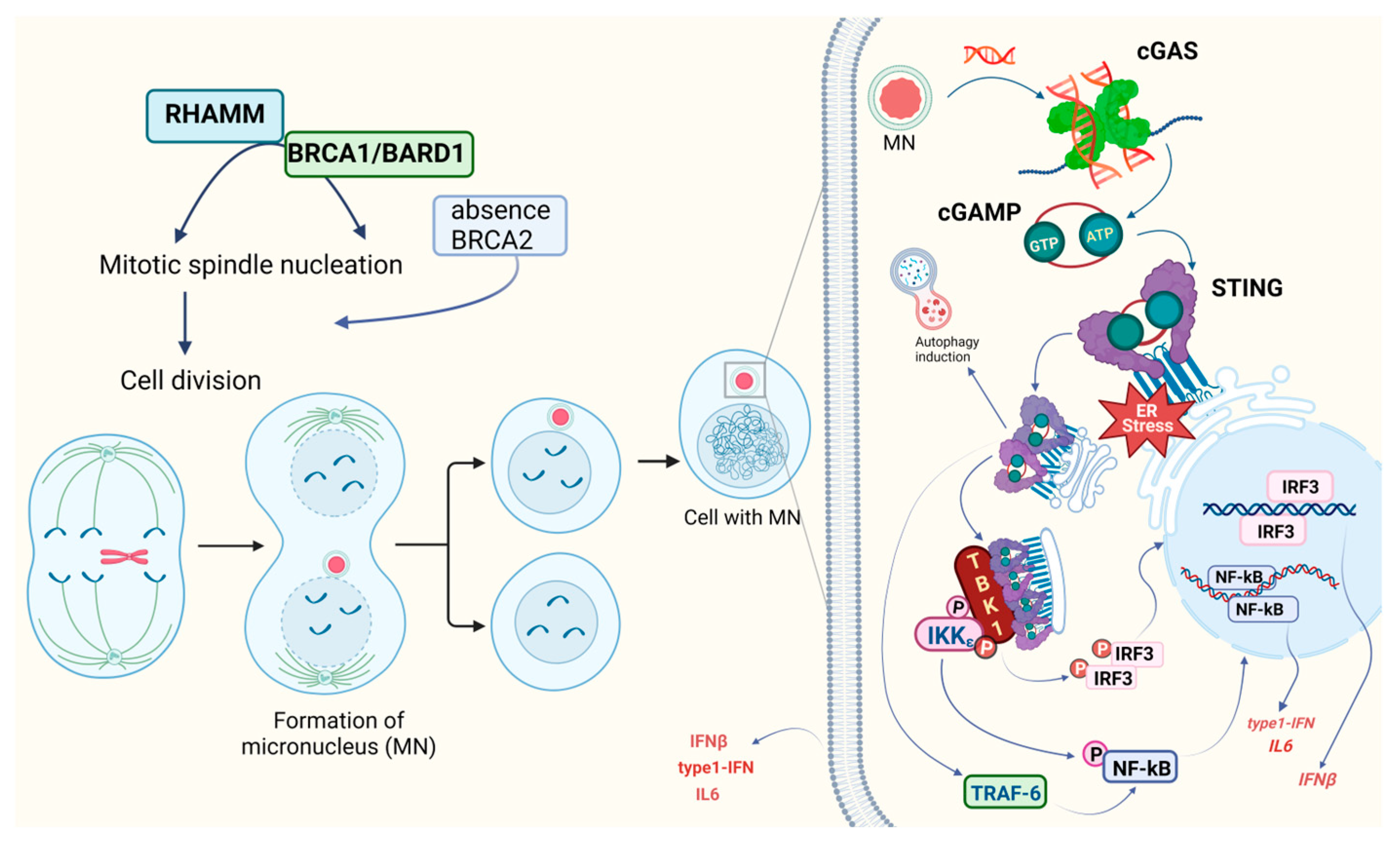
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular Mechanisms of Micronucleus, Nucleoplasmic Bridge and Nuclear Bud Formation in Mammalian and Human Cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Haarer, E.L.; Theodore, C.J.; Guo, S.; Frier, R.B.; Campellone, K.G. Genomic Instability Caused by Arp2/3 Complex Inactivation Results in Micronucleus Biogenesis and Cellular Senescence. PLoS Genet. 2023, 19, e1010045. [Google Scholar] [CrossRef]
- Haimovici, A.; Höfer, C.; Badr, M.T.; Bavafaye Haghighi, E.; Amer, T.; Boerries, M.; Bronsert, P.; Glavynskyi, I.; Fanfone, D.; Ichim, G.; et al. Spontaneous Activity of the Mitochondrial Apoptosis Pathway Drives Chromosomal Defects, the Appearance of Micronuclei and Cancer Metastasis through the Caspase-Activated DNAse. Cell Death Dis. 2022, 13, 315. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.M.; Nicholson-Puthenveedu, S.; Tageldein, M.M.; Khasnis, S.; Arrowsmith, C.H.; Harding, S.M. Antecedent Chromatin Organization Determines cGAS Recruitment to Ruptured Micronuclei. Nat. Commun. 2023, 14, 556. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.; Cho, M.-G.; Kim, E.-Y.; Kwon, D.; Kang, S.-J.; Lee, J.-H. The cGAS/STING/TBK1/IRF3 Innate Immunity Pathway Maintains Chromosomal Stability through Regulation of P21 Levels. Exp. Mol. Med. 2020, 52, 643–657. [Google Scholar] [CrossRef]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.-D.; Carroll, T.; Funabiki, H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 2019, 178, 302–315.e23. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Li, J.; Montrose, D.C.; Martinez, L.A. P53 Engages the cGAS/STING Cytosolic DNA Sensing Pathway for Tumor Suppression. Mol. Cell 2023, 83, 266–280.e6. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant P53 Suppresses Innate Immune Signaling to Promote Tumorigenesis. Cancer Cell 2021, 39, 494–508.e5. [Google Scholar] [CrossRef]
- Budke, B.; Zhong, A.; Sullivan, K.; Park, C.; Gittin, D.I.; Kountz, T.S.; Connell, P.P. Noncanonical NF-κB Factor P100/P52 Regulates Homologous Recombination and Modulates Sensitivity to DNA-Damaging Therapy. Nucleic Acids Res. 2022, 50, 6251–6263. [Google Scholar] [CrossRef]
- Chen, H.; Chen, H.; Zhang, J.; Wang, Y.; Simoneau, A.; Yang, H.; Levine, A.S.; Zou, L.; Chen, Z.; Lan, L. cGAS Suppresses Genomic Instability as a Decelerator of Replication Forks. Sci. Adv. 2020, 6, eabb8941. [Google Scholar] [CrossRef] [PubMed]
- Mateo, F.; He, Z.; Mei, L.; de Garibay, G.R.; Herranz, C.; García, N.; Lorentzian, A.; Baiges, A.; Blommaert, E.; Gómez, A.; et al. Modification of BRCA1-Associated Breast Cancer Risk by HMMR Overexpression. Nat. Commun. 2022, 13, 1895. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Kang, Z.; Foo, T.K.; Shen, Z.; Xia, B. Disrupted BRCA1-PALB2 Interaction Induces Tumor Immunosuppression and T-Lymphocyte Infiltration in HCC through cGAS-STING Pathway. Hepatology 2023, 77, 33–47. [Google Scholar] [CrossRef]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 Deficiency Instigates cGAS-Mediated Inflammatory Signaling and Confers Sensitivity to Tumor Necrosis Factor-Alpha-Mediated Cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar] [CrossRef]
- Banerjee, D.; Langberg, K.; Abbas, S.; Odermatt, E.; Yerramothu, P.; Volaric, M.; Reidenbach, M.A.; Krentz, K.J.; Rubinstein, C.D.; Brautigan, D.L.; et al. A Non-Canonical, Interferon-Independent Signaling Activity of cGAMP Triggers DNA Damage Response Signaling. Nat. Commun. 2021, 12, 6207. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Peng, P.; Tang, Z.; Zhao, J.; Wu, W.; Li, H.; Shao, M.; Li, L.; Yang, C.; Duan, F.; et al. Decreased Expression of STING Predicts Poor Prognosis in Patients with Gastric Cancer. Sci. Rep. 2017, 7, 39858. [Google Scholar] [CrossRef]
- Gong, W.; Liu, P.; Zhao, F.; Liu, J.; Hong, Z.; Ren, H.; Gu, G.; Wang, G.; Wu, X.; Zheng, T.; et al. STING-Mediated Syk Signaling Attenuates Tumorigenesis of Colitis-associated Colorectal Cancer Through Enhancing Intestinal Epithelium Pyroptosis. Inflamm. Bowel Dis. 2022, 28, 572–585. [Google Scholar] [CrossRef]
- Lou, M.; Huang, D.; Zhou, Z.; Shi, X.; Wu, M.; Rui, Y.; Su, J.; Zheng, W.; Yu, X.-F. DNA Virus Oncoprotein HPV18 E7 Selectively Antagonizes cGAS-STING-Triggered Innate Immune Activation. J. Med. Virol. 2023, 95, e28310. [Google Scholar] [CrossRef]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA Sensing Pathway by Gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Yan, L.; Liu, N.; Xu, M.; Cai, H. IFI16 Promotes Cervical Cancer Progression by Upregulating PD-L1 in Immunomicroenvironment through STING-TBK1-NF-kB Pathway. Biomed. Pharmacother. 2020, 123, 109790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Kang, R.; Tang, D. The STING1 Network Regulates Autophagy and Cell Death. Signal Transduct. Target. Ther. 2021, 6, 208. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.e18. [Google Scholar] [CrossRef]
- Tang, C.-H.A.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.-C.A. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016, 76, 2137–2152. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, A.; Xia, N.; Chen, N.; Meurens, F.; Zhu, J. How the Innate Immune DNA Sensing cGAS-STING Pathway Is Involved in Apoptosis. Int. J. Mol. Sci. 2023, 24, 3029. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Weindel, C.G.; Smirnova, I.; Tang, A.Y.; Ilyukha, V.; Sorokin, M.; Buzdin, A.; Fitzgerald, K.A.; et al. Constitutive Interferon Signaling Maintains Critical Threshold of MLKL Expression to License Necroptosis. Cell Death Differ. 2019, 26, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Liu, Q.; Li, X.; Yang, Y.; Wu, L.; Wu, X.; Zhao, Y.; Ren, J. RIPK3-MLKL Necroptotic Signalling Amplifies STING Pathway and Exacerbates Lethal Sepsis. Clin. Transl. Med. 2023, 13, e1334. [Google Scholar] [CrossRef] [PubMed]
- Brault, M.; Olsen, T.M.; Martinez, J.; Stetson, D.B.; Oberst, A. Intracellular Nucleic Acid Sensing Triggers Necroptosis through Synergistic Type I IFN and TNF Signaling. J. Immunol. 2018, 200, 2748–2756. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy Induction via STING Trafficking Is a Primordial Function of the cGAS Pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef]
- Li, C.; Liu, J.; Hou, W.; Kang, R.; Tang, D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front. Cell Dev. Biol. 2021, 9, 698679. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox Homeostasis Maintained by GPX4 Facilitates STING Activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Jin, L.; Yu, B.; Wang, H.; Shi, L.; Yang, J.; Wu, L.; Gao, C.; Pan, H.; Han, F.; Lin, W.; et al. STING Promotes Ferroptosis through NCOA4-Dependent Ferritinophagy in Acute Kidney Injury. Free Radic. Biol. Med. 2023, 208, 348–360. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Baird, J.R.; Friedman, D.; Cottam, B.; Dubensky, T.W.; Kanne, D.B.; Bambina, S.; Bahjat, K.; Crittenden, M.R.; Gough, M.J. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016, 76, 50–61. [Google Scholar] [CrossRef]
- Hou, Y.; Liang, H.; Rao, E.; Zheng, W.; Huang, X.; Deng, L.; Zhang, Y.; Yu, X.; Xu, M.; Mauceri, H.; et al. Non-Canonical NF-κB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 2018, 49, 490–503.e4. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-Dependent MDSC Mobilization Drives Extrinsic Radiation Resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, M.; Cao, D.; Yang, C.; Jin, J.; Wu, L.; Hong, X.; Li, W.; Lu, L.; Li, J.; et al. ZBP1-MLKL Necroptotic Signaling Potentiates Radiation-Induced Antitumor Immunity via Intratumoral STING Pathway Activation. Sci. Adv. 2021, 7, eabf6290. [Google Scholar] [CrossRef]
- Ahn, J.; Xia, T.; Konno, H.; Konno, K.; Ruiz, P.; Barber, G.N. Inflammation-Driven Carcinogenesis Is Mediated through STING. Nat. Commun. 2014, 5, 5166. [Google Scholar] [CrossRef]
- Li, T.; Cheng, H.; Yuan, H.; Xu, Q.; Shu, C.; Zhang, Y.; Xu, P.; Tan, J.; Rui, Y.; Li, P.; et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci. Rep. 2016, 6, 19049. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Wu, Y.; Zhao, G.; Chen, X.; Xu, Z.; Sun, J.; Tian, J.; Cheng, Z.; Shi, Y.; Jin, B. Activation of cGAS-STING Signal to Inhibit the Proliferation of Bladder Cancer: The Immune Effect of Cisplatin. Cells 2022, 11, 3011. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Wang, Q.; Bergholz, J.S.; Ding, L.; Lin, Z.; Kabraji, S.K.; Hughes, M.E.; He, X.; Xie, S.; Jiang, T.; Wang, W.; et al. STING Agonism Reprograms Tumor-Associated Macrophages and Overcomes Resistance to PARP Inhibition in BRCA1-Deficient Models of Breast Cancer. Nat. Commun. 2022, 13, 3022. [Google Scholar] [CrossRef]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 Abrogation Triggers Innate Immune Responses Potentiated by Treatment with PARP Inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP Inhibition Enhances Tumor Cell-Intrinsic Immunity in ERCC1-Deficient Non-Small Cell Lung Cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, Q.; Martincuks, A.; Kearns, M.J.; Jiang, T.; Lin, Z.; Cheng, X.; Qian, C.; Xie, S.; Kim, H.-J.; et al. STING Agonism Overcomes STAT3-Mediated Immunosuppression and Adaptive Resistance to PARP Inhibition in Ovarian Cancer. J. Immunother. Cancer 2023, 11, e005627. [Google Scholar] [CrossRef] [PubMed]
- Kornepati, A.V.R.; Rogers, C.M.; Sung, P.; Curiel, T.J. The Complementarity of DDR, Nucleic Acids and Anti-Tumour Immunity. Nature 2023, 619, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, B.; Yang, F.; Su, Y.; Yang, D.; Yao, Y.; Wang, S.; Wu, Y.; Tao, L.; Xu, T. Emerging Role of the cGAS-STING Signaling Pathway in Autoimmune Diseases: Biologic Function, Mechanisms and Clinical Prospection. Autoimmun. Rev. 2022, 21, 103155. [Google Scholar] [CrossRef]
- Falahat, R.; Berglund, A.; Putney, R.M.; Perez-Villarroel, P.; Aoyama, S.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. Epigenetic Reprogramming of Tumor Cell-Intrinsic STING Function Sculpts Antigenicity and T Cell Recognition of Melanoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2013598118. [Google Scholar] [CrossRef]
- Bruand, M.; Barras, D.; Mina, M.; Ghisoni, E.; Morotti, M.; Lanitis, E.; Fahr, N.; Desbuisson, M.; Grimm, A.; Zhang, H.; et al. Cell-Autonomous Inflammation of BRCA1-Deficient Ovarian Cancers Drives Both Tumor-Intrinsic Immunoreactivity and Immune Resistance via STING. Cell Rep. 2021, 36, 109412. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. cGAS-STING Drives the IL-6-Dependent Survival of Chromosomally Instable Cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef]
- Al-Asmari, S.S.; Rajapakse, A.; Ullah, T.R.; Pépin, G.; Croft, L.V.; Gantier, M.P. Pharmacological Targeting of STING-Dependent IL-6 Production in Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 709618. [Google Scholar] [CrossRef] [PubMed]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarró, L.; Couturier, D.-L.; Liu, L.; Schneider, M.; et al. A Pan-Cancer Compendium of Chromosomal Instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tijhuis, A.E.; Foijer, F. The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells 2019, 8, 1228. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, L.; Qiao, Z.; Mai, S.; Zhu, J.; Zhang, F.; Chen, S.; Li, L.; Shen, F.; Qin, Y.; et al. STING-Mediated Degradation of IFI16 Negatively Regulates Apoptosis by Inhibiting P53 Phosphorylation at Serine 392. J. Biol. Chem. 2021, 297, 100930. [Google Scholar] [CrossRef]
- Cheng, H.; Xu, Q.; Lu, X.; Yuan, H.; Li, T.; Zhang, Y.; Tan, X. Activation of STING by cGAMP Regulates MDSCs to Suppress Tumor Metastasis via Reversing Epithelial-Mesenchymal Transition. Front. Oncol. 2020, 10, 896. [Google Scholar] [CrossRef]
- Hu, J.; Sánchez-Rivera, F.J.; Wang, Z.; Johnson, G.N.; Ho, Y.-J.; Ganesh, K.; Umeda, S.; Gan, S.; Mujal, A.M.; Delconte, R.B.; et al. STING Inhibits the Reactivation of Dormant Metastasis in Lung Adenocarcinoma. Nature 2023, 616, 806–813. [Google Scholar] [CrossRef]
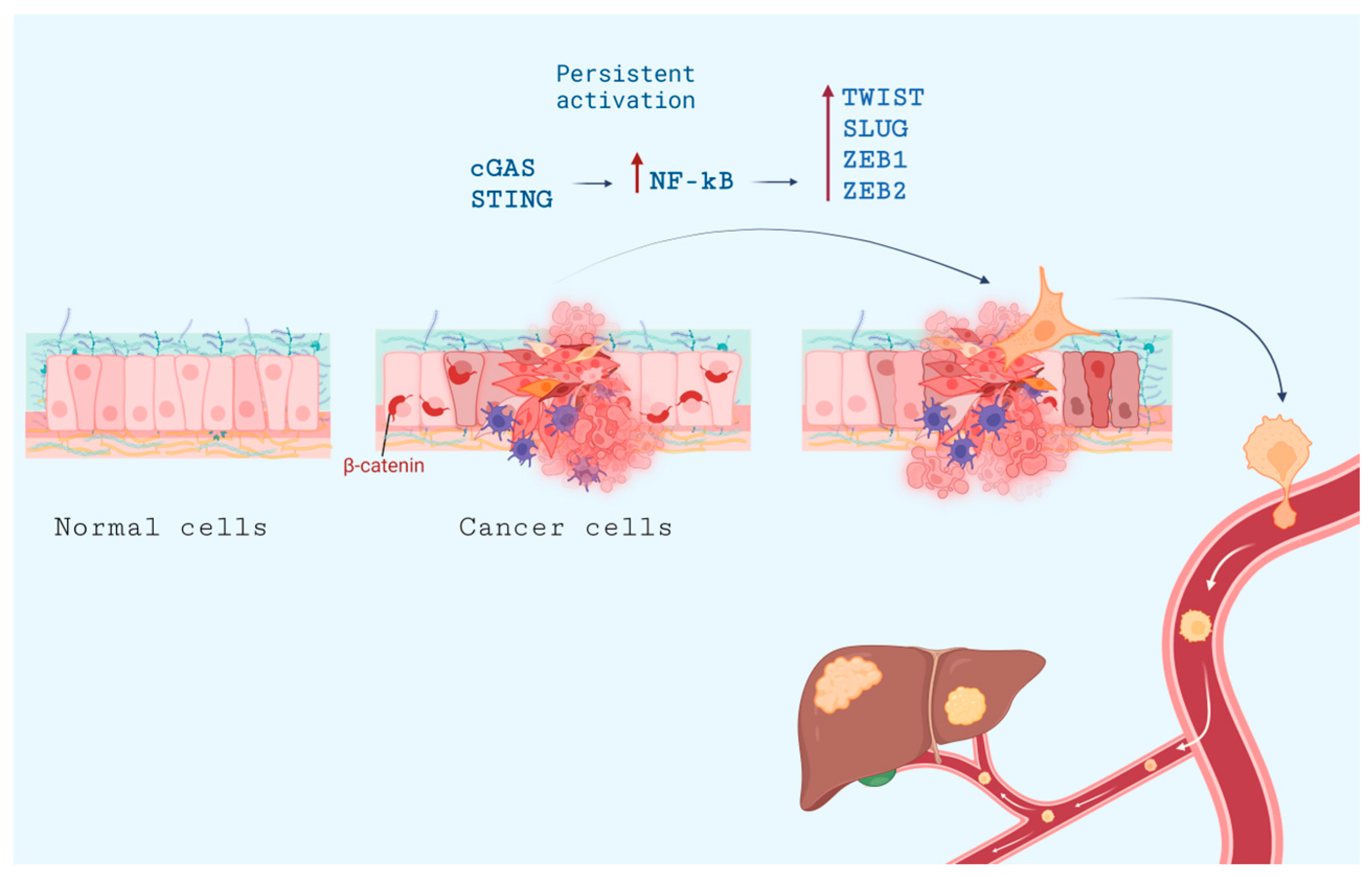
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Oh, A.; Pardo, M.; Rodriguez, A.; Yu, C.; Nguyen, L.; Liang, O.; Chorzalska, A.; Dubielecka, P.M. NF-κB Signaling in Neoplastic Transition from Epithelial to Mesenchymal Phenotype. Cell Commun. Signal 2023, 21, 291. [Google Scholar] [CrossRef]
- Mirzaei, S.; Saghari, S.; Bassiri, F.; Raesi, R.; Zarrabi, A.; Hushmandi, K.; Sethi, G.; Tergaonkar, V. NF-κB as a Regulator of Cancer Metastasis and Therapy Response: A Focus on Epithelial-Mesenchymal Transition. J. Cell Physiol. 2022, 237, 2770–2795. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S.F.W. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, A.; Baruah, P.S.; Smith, J.C.; Wang, Z.; Sayles, N.M.; Andrews, P.; Kendall, J.; Leu, J.; Chunduri, N.K.; Levy, D.; et al. Single-Chromosomal Gains Can Function as Metastasis Suppressors and Promoters in Colon Cancer. Dev. Cell 2020, 52, 413–428.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hubisz, M.J.; Earlie, E.M.; Duran, M.A.; Hong, C.; Varela, A.A.; Lettera, E.; Deyell, M.; Tavora, B.; Havel, J.J.; et al. Non-Cell-Autonomous Cancer Progression from Chromosomal Instability. Nature 2023, 620, 1080–1088. [Google Scholar] [CrossRef]
- Shen, R.; Liu, D.; Wang, X.; Guo, Z.; Sun, H.; Song, Y.; Wang, D. DNA Damage and Activation of cGAS/STING Pathway Induce Tumor Microenvironment Remodeling. Front. Cell Dev. Biol. 2021, 9, 828657. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.; Chen, X.; Corrales, L.; Kline, D.E.; Dubensky, T.W.; Duttagupta, P.; Kortylewski, M.; Kline, J. STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep. 2016, 15, 2357–2366. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef]
- Daei Farshchi Adli, A.; Jahanban-Esfahlan, R.; Seidi, K.; Farajzadeh, D.; Behzadi, R.; Zarghami, N. Co-Administration of Vadimezan and Recombinant Coagulase-NGR Inhibits Growth of Melanoma Tumor in Mice. Adv. Pharm. Bull. 2021, 11, 385–392. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING Activation of Tumor Endothelial Cells Initiates Spontaneous and Therapeutic Antitumor Immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Ohkuri, T.; Kosaka, A.; Ishibashi, K.; Kumai, T.; Hirata, Y.; Ohara, K.; Nagato, T.; Oikawa, K.; Aoki, N.; Harabuchi, Y.; et al. Intratumoral Administration of cGAMP Transiently Accumulates Potent Macrophages for Anti-Tumor Immunity at a Mouse Tumor Site. Cancer Immunol. Immunother. 2017, 66, 705–716. [Google Scholar] [CrossRef]
- Qin, Z.; Liu, H.; Sheng, Q.; Dan, J.; Wu, X.; Li, H.; Wang, L.; Zhang, S.; Yuan, C.; Yuan, H.; et al. Mutant P53 Leads to Low-Grade IFN-I-Induced Inflammation and Impairs cGAS-STING Signalling in Mice. Eur. J. Immunol. 2023, 53, e2250211. [Google Scholar] [CrossRef] [PubMed]
- Semenov, O.; Daks, A.; Fedorova, O.; Shuvalov, O.; Barlev, N.A. Opposing Roles of Wild-Type and Mutant P53 in the Process of Epithelial to Mesenchymal Transition. Front. Mol. Biosci. 2022, 9, 928399. [Google Scholar] [CrossRef]
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 Affects Genotoxic Stress Response via the Mdm2 Axis. Oncotarget 2015, 6, 25843–25855. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Galluzzi, L. P53 Regulates the Mitochondrial Immune Checkpoint. Trends Immunol. 2023, 44, 245–247. [Google Scholar] [CrossRef]
- Bulatov, E.; Sayarova, R.; Mingaleeva, R.; Miftakhova, R.; Gomzikova, M.; Ignatyev, Y.; Petukhov, A.; Davidovich, P.; Rizvanov, A.; Barlev, N.A. Isatin-Schiff Base-Copper (II) Complex Induces Cell Death in P53-Positive Tumors. Cell Death Discov. 2018, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, P.; Aksenova, V.; Petrova, V.; Tentler, D.; Orlova, D.; Smirnov, S.; Gurzhiy, V.; Okorokov, A.L.; Garabadzhiu, A.; Melino, G.; et al. Discovery of Novel Isatin-Based P53 Inducers. ACS Med. Chem. Lett. 2015, 6, 856–860. [Google Scholar] [CrossRef]
- Krześniak, M.; Zajkowicz, A.; Gdowicz-Kłosok, A.; Głowala-Kosińska, M.; Łasut-Szyszka, B.; Rusin, M. Synergistic Activation of P53 by Actinomycin D and Nutlin-3a Is Associated with the Upregulation of Crucial Regulators and Effectors of Innate Immunity. Cell Signal 2020, 69, 109552. [Google Scholar] [CrossRef]
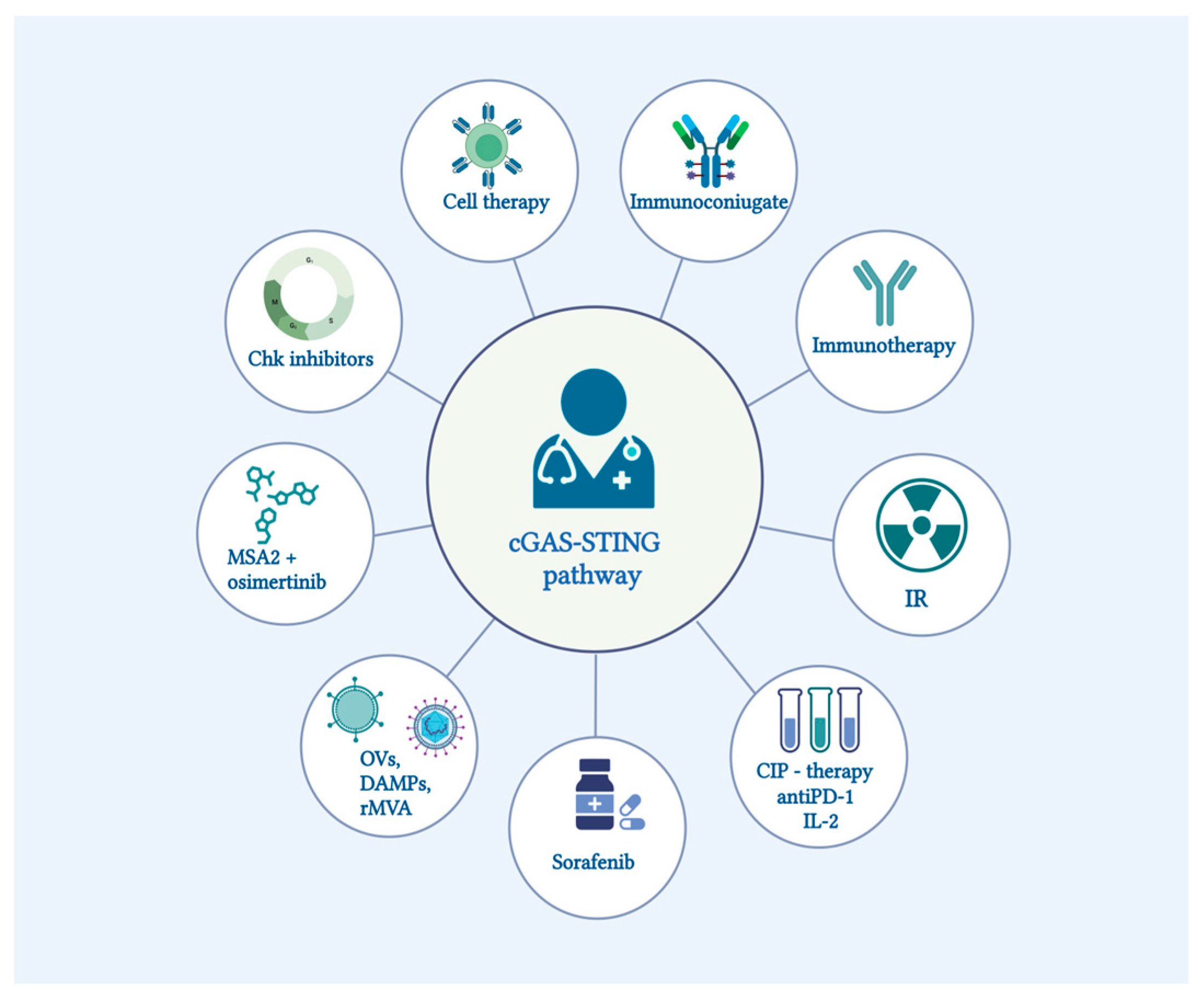
- Pan, X.; Zhang, W.; Guo, H.; Wang, L.; Wu, H.; Ding, L.; Yang, B. Strategies Involving STING Pathway Activation for Cancer Immunotherapy: Mechanism and Agonists. Biochem. Pharmacol. 2023, 213, 115596. [Google Scholar] [CrossRef]
- Zhao, K.; Huang, J.; Zhao, Y.; Wang, S.; Xu, J.; Yin, K. Targeting STING in Cancer: Challenges and Emerging Opportunities. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188983. [Google Scholar] [CrossRef] [PubMed]
- Vasiyani, H.; Wadhwa, B.; Singh, R. Regulation of cGAS-STING Signalling in Cancer: Approach for Combination Therapy. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188896. [Google Scholar] [CrossRef]
- Coderch, C.; Arranz-Herrero, J.; Nistal-Villan, E.; de Pascual-Teresa, B.; Rius-Rocabert, S. The Many Ways to Deal with STING. Int. J. Mol. Sci. 2023, 24, 9032. [Google Scholar] [CrossRef] [PubMed]
- Motedayen Aval, L.; Pease, J.E.; Sharma, R.; Pinato, D.J. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J. Clin. Med. 2020, 9, 3323. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhang, R.; Cen, S.; Zhou, J. STING Modulators: Predictive Significance in Drug Discovery. Eur. J. Med. Chem. 2019, 182, 111591. [Google Scholar] [CrossRef]
- Ding, C.; Song, Z.; Shen, A.; Chen, T.; Zhang, A. Small Molecules Targeting the Innate Immune cGAS‒STING‒TBK1 Signaling Pathway. Acta Pharm. Sin. B 2020, 10, 2272–2298. [Google Scholar] [CrossRef]
- Lara, P.N.; Douillard, J.-Y.; Nakagawa, K.; von Pawel, J.; McKeage, M.J.; Albert, I.; Losonczy, G.; Reck, M.; Heo, D.-S.; Fan, X.; et al. Randomized Phase III Placebo-Controlled Trial of Carboplatin and Paclitaxel with or without the Vascular Disrupting Agent Vadimezan (ASA404) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2011, 29, 2965–2971. [Google Scholar] [CrossRef]
- Aggarwal, C.; Somaiah, N.; Simon, G. Antiangiogenic Agents in the Management of Non-Small Cell Lung Cancer: Where Do We Stand Now and Where Are We Headed? Cancer Biol. Ther. 2012, 13, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.A.; Tan, N.Y.; Thiam, C.H.; Khatoo, M.; MacAry, P.A.; Angeli, V.; Gasser, S.; Zhang, Y.L. cGAS-STING Cytosolic DNA Sensing Pathway Is Suppressed by JAK2-STAT3 in Tumor Cells. Sci. Rep. 2021, 11, 7243. [Google Scholar] [CrossRef]
- Pan, B.-S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Trotter, B.W.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An Orally Available Non-Nucleotide STING Agonist with Antitumor Activity. Science 2020, 369, eaba6098. [Google Scholar] [CrossRef]
- Galenkamp, K.M.O.; Commisso, C. The Golgi as a “Proton Sink” in Cancer. Front. Cell Dev. Biol. 2021, 9, 664295. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sweis, R.F.; Kasper, S.; Hamid, O.; Bhatia, S.; Dummer, R.; Stradella, A.; Long, G.V.; Spreafico, A.; Shimizu, T.; et al. Combination of the STING Agonist MIW815 (ADU-S100) and PD-1 Inhibitor Spartalizumab in Advanced/Metastatic Solid Tumors or Lymphomas: An Open-Label, Multicenter, Phase Ib Study. Clin. Cancer Res. 2023, 29, 110–121. [Google Scholar] [CrossRef]
- Gogoi, H.; Mansouri, S.; Jin, L. The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines 2020, 8, 453. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sweis, R.F.; Hodi, F.S.; Messersmith, W.A.; Andtbacka, R.H.I.; Ingham, M.; Lewis, N.; Chen, X.; Pelletier, M.; Chen, X.; et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin. Cancer Res. 2022, 28, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.K.; Kim, M.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Beyond DNA Sensing: Expanding the Role of cGAS/STING in Immunity and Diseases. Arch. Pharm. Res. 2023, 46, 500–534. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Altman, M.D.; Lesburg, C.A.; Perera, S.A.; Piesvaux, J.A.; Schroeder, G.K.; Wyss, D.F.; Cemerski, S.; Chen, Y.; DiNunzio, E.; et al. Discovery of MK-1454: A Potent Cyclic Dinucleotide Stimulator of Interferon Genes Agonist for the Treatment of Cancer. J. Med. Chem. 2022, 65, 5675–5689. [Google Scholar] [CrossRef]
- Ivanov, G.S.; Tribulovich, V.G.; Pestov, N.B.; David, T.I.; Amoah, A.-S.; Korneenko, T.V.; Barlev, N.A. Artificial Genetic Polymers against Human Pathologies. Biol. Direct 2022, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Liu, Z.; Andresen, B.M.; Marzijarani, N.S.; Moore, J.C.; Marshall, N.M.; Borra-Garske, M.; Obligacion, J.V.; Fier, P.S.; Peng, F.; et al. A Kinase-cGAS Cascade to Synthesize a Therapeutic STING Activator. Nature 2022, 603, 439–444. [Google Scholar] [CrossRef]
- Diamond, J.R.; Henry, J.T.; Falchook, G.S.; Olszanski, A.J.; Singh, H.; Leonard, E.J.; Gregory, R.C.; Appleman, V.A.; Gibbs, J.; Harbison, C.; et al. Phase 1a/1b Study Design of the Novel STING Agonist, Immune-Stimulating Antibody-Conjugate (ISAC) TAK-500, with or without Pembrolizumab in Patients with Advanced Solid Tumors. JCO 2022, 40, TPS2690. [Google Scholar] [CrossRef]
- Luke, J.J.; Piha-Paul, S.A.; Medina, T.; Verschraegen, C.F.; Varterasian, M.; Brennan, A.M.; Riese, R.J.; Sokolovska, A.; Strauss, J.; Hava, D.L.; et al. Phase I Study of SYNB1891, an Engineered E. Coli Nissle Strain Expressing STING Agonist, with and without Atezolizumab in Advanced Malignancies. Clin. Cancer Res. 2023, 29, 2435–2444. [Google Scholar] [CrossRef]
- Zhou, L.; Yi, W.; Zhang, Z.; Shan, X.; Zhao, Z.; Sun, X.; Wang, J.; Wang, H.; Jiang, H.; Zheng, M.; et al. STING Agonist-Boosted mRNA Immunization via Intelligent Design of Nanovaccines for Enhancing Cancer Immunotherapy. Natl. Sci. Rev. 2023, 10, nwad214. [Google Scholar] [CrossRef]
- Mitrofanova, A.; Fontanella, A.; Tolerico, M.; Mallela, S.; Molina David, J.; Zuo, Y.; Boulina, M.; Kim, J.-J.; Santos, J.; Ge, M.; et al. Activation of Stimulator of IFN Genes (STING) Causes Proteinuria and Contributes to Glomerular Diseases. J. Am. Soc. Nephrol. 2022, 33, 2153–2173. [Google Scholar] [CrossRef]
- Hooy, R.M.; Massaccesi, G.; Rousseau, K.E.; Chattergoon, M.A.; Sohn, J. Allosteric Coupling between Mn2+ and dsDNA Controls the Catalytic Efficiency and Fidelity of cGAS. Nucleic Acids Res. 2020, 48, 4435–4447. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guan, Y.; Lv, M.; Zhang, R.; Guo, Z.; Wei, X.; Du, X.; Yang, J.; Li, T.; Wan, Y.; et al. Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity 2018, 48, 675–687.e7. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, Y.; Li, J.; Park, K.S.; Han, K.; Zhou, X.; Xu, Y.; Nam, J.; Xu, J.; Shi, X.; et al. Amplifying STING Activation by Cyclic Dinucleotide-Manganese Particles for Local and Systemic Cancer Metalloimmunotherapy. Nat. Nanotechnol. 2021, 16, 1260–1270. [Google Scholar] [CrossRef]
- Liu, J.; Yang, L.; Cao, X.; Chen, M.; Li, J.; Wang, X.; Wu, S.; Zhang, Z. PEGylated Mn Containing MOF Nanoparticles for Potential Immunotherapy of Pancreatic Cancer via Manganese Induced Activation of Anti-Tumor Immunity. Colloid Interface Sci. Commun. 2021, 42, 100409. [Google Scholar] [CrossRef]
- Hemphill, W.O.; Simpson, S.R.; Liu, M.; Salsbury, F.R.; Hollis, T.; Grayson, J.M.; Perrino, F.W. TREX1 as a Novel Immunotherapeutic Target. Front. Immunol. 2021, 12, 660184. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA Sensing by the cGAS-STING Pathway in Health and Disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Ghonim, M.A.; Ibba, S.V.; Tarhuni, A.F.; Errami, Y.; Luu, H.H.; Dean, M.J.; El-Bahrawy, A.H.; Wyczechowska, D.; Benslimane, I.A.; Del Valle, L.; et al. Targeting PARP-1 with Metronomic Therapy Modulates MDSC Suppressive Function and Enhances Anti-PD-1 Immunotherapy in Colon Cancer. J. Immunother. Cancer 2021, 9, e001643. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Zhu, Z.; Johnson, R.L.; Zhang, Z.; Herring, L.E.; Jiang, G.; Damania, B.; James, L.I.; Liu, P. Development of VHL-Recruiting STING PROTACs That Suppress Innate Immunity. Cell Mol. Life Sci. 2023, 80, 149. [Google Scholar] [CrossRef]
- Vinogradova, E.V.; Zhang, X.; Remillard, D.; Lazar, D.C.; Suciu, R.M.; Wang, Y.; Bianco, G.; Yamashita, Y.; Crowley, V.M.; Schafroth, M.A.; et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182, 1009–1026.e29. [Google Scholar] [CrossRef]
- Zhou, J.; Ventura, C.J.; Fang, R.H.; Zhang, L. Nanodelivery of STING Agonists against Cancer and Infectious Diseases. Mol. Aspects Med. 2022, 83, 101007. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Yi, J.; Cao, Y.; Qin, Z.; Zhong, Z.; Yang, W. Nanoparticle-Mediated STING Activation for Cancer Immunotherapy. Adv. Healthc. Mater. 2023, 12, e2300260. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Guo, B.; Qiu, X.; Xia, Y.; Qu, Y.; Cheng, L.; Meng, F.; Zhong, Z. Polymersome-Mediated Cytosolic Delivery of Cyclic Dinucleotide STING Agonist Enhances Tumor Immunotherapy. Bioact. Mater. 2022, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang-Bishop, L.; Kimmel, B.R.; Ngwa, V.M.; Madden, M.Z.; Baljon, J.J.; Florian, D.C.; Hanna, A.; Pastora, L.E.; Sheehy, T.L.; Kwiatkowski, A.J.; et al. STING-Activating Nanoparticles Normalize the Vascular-Immune Interface to Potentiate Cancer Immunotherapy. Sci. Immunol. 2023, 8, eadd1153. [Google Scholar] [CrossRef]
- Xu, L.; Deng, H.; Wu, L.; Wang, D.; Shi, L.; Qian, Q.; Huang, X.; Zhu, L.; Gao, X.; Yang, J.; et al. Supramolecular Cyclic Dinucleotide Nanoparticles for STING-Mediated Cancer Immunotherapy. ACS Nano 2023, 17, 10090–10103. [Google Scholar] [CrossRef]
- Ji, N.; Wang, M.; Tan, C. Liposomal Delivery of MIW815 (ADU-S100) for Potentiated STING Activation. Pharmaceutics 2023, 15, 638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cui, X.; Huang, Y.; Xu, X.; Feng, C.; Li, J. Anticancer Effect of STING Agonist-Encapsulated Liposomes on Breast Cancer. Molecules 2023, 28, 3740. [Google Scholar] [CrossRef]
- Jang, S.C.; Economides, K.D.; Moniz, R.J.; Sia, C.L.; Lewis, N.; McCoy, C.; Zi, T.; Zhang, K.; Harrison, R.A.; Lim, J.; et al. ExoSTING, an Extracellular Vesicle Loaded with STING Agonists, Promotes Tumor Immune Surveillance. Commun. Biol. 2021, 4, 497. [Google Scholar] [CrossRef]
- Xu, X.; Fan, H.; Yang, Y.; Yao, S.; Yu, W.; Guo, Z.; Tan, W. Virus-Like Particle-Induced cGAS-STING Activation and AIM2 Inflammasome-Mediated Pyroptosis for Robust Cancer Immunotherapy. Angew. Chem. Int. Ed. Engl. 2023, 62, e202303010. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Z.; Huang, H.; Shen, G.; Ren, Y.; Mao, X.; Wang, L.; Li, Z.; Wang, W.; Li, G.; et al. Cancer Immunotherapy Based on Cell Membrane-Coated Nanocomposites Augmenting cGAS/STING Activation by Efferocytosis Blockade. Small 2023, 19, e2302758. [Google Scholar] [CrossRef]
- Vornholz, L.; Isay, S.E.; Kurgyis, Z.; Strobl, D.C.; Loll, P.; Mosa, M.H.; Luecken, M.D.; Sterr, M.; Lickert, H.; Winter, C.; et al. Synthetic Enforcement of STING Signaling in Cancer Cells Appropriates the Immune Microenvironment for Checkpoint Inhibitor Therapy. Sci. Adv. 2023, 9, eadd8564. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hong, C.; Yan, E.Z.; Fletcher, S.J.; Zhu, G.; Yang, M.; Li, Y.; Sun, X.; Irvine, D.J.; Li, J.; et al. Self-Assembled cGAMP-STINGΔTM Signaling Complex as a Bioinspired Platform for cGAMP Delivery. Sci. Adv. 2020, 6, eaba7589. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Knelson, E.H.; Jimenez-Macias, J.L.; Nowicki, M.O.; Han, S.; Panagioti, E.; Lizotte, P.H.; Adu-Berchie, K.; Stafford, A.; Dimitrakakis, N.; et al. STING Activation Promotes Robust Immune Response and NK Cell-Mediated Tumor Regression in Glioblastoma Models. Proc. Natl. Acad. Sci. USA 2022, 119, e2111003119. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jin, J.; Chen, Y.; Wu, G.; Zhu, H.; Wang, Q.; Wang, J.; Li, S.; Grigore, F.-N.; Ma, J.; et al. GBP3 Promotes Glioblastoma Resistance to Temozolomide by Enhancing DNA Damage Repair. Oncogene 2022, 41, 3876–3885. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Banerjee, K.; Mujeeb, A.A.; Hartlage, C.S.; Núñez, F.M.; Núñez, F.J.; Alghamri, M.S.; Kadiyala, P.; Carney, S.; Barissi, M.N.; et al. H3.3-G34 Mutations Impair DNA Repair and Promote cGAS/STING-Mediated Immune Responses in Pediatric High-Grade Glioma Models. J. Clin. Investig. 2022, 132, e154229. [Google Scholar] [CrossRef]
- Rubin, S.J.S.; Sojwal, R.S.; Gubatan, J.; Rogalla, S. The Tumor Immune Microenvironment in Pancreatic Ductal Adenocarcinoma: Neither Hot nor Cold. Cancers 2022, 14, 4236. [Google Scholar] [CrossRef]
- Kabashima, A.; Matsuo, Y.; Ito, S.; Akiyama, Y.; Ishii, T.; Shimada, S.; Masamune, A.; Tanabe, M.; Tanaka, S. cGAS-STING Signaling Encourages Immune Cell Overcoming of Fibroblast Barricades in Pancreatic Cancer. Sci. Rep. 2022, 12, 10466. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Zhu, Y.; Zhang, Q.; Guan, H.; Liu, S.; Chen, S.; Mei, C.; Chen, C.; Liao, Z.; et al. A Non-Canonical cGAS-STING-PERK Pathway Facilitates the Translational Program Critical for Senescence and Organ Fibrosis. Nat. Cell Biol. 2022, 24, 766–782. [Google Scholar] [CrossRef]
- Jacoberger-Foissac, C.; Cousineau, I.; Bareche, Y.; Allard, D.; Chrobak, P.; Allard, B.; Pommey, S.; Messaoudi, N.; McNicoll, Y.; Soucy, G.; et al. CD73 Inhibits cGAS-STING and Cooperates with CD39 to Promote Pancreatic Cancer. Cancer Immunol. Res. 2023, 11, 56–71. [Google Scholar] [CrossRef]
- Hu, G.; Chen, Y.; Yang, X.; Wang, Y.; He, J.; Wang, T.; Fan, Q.; Deng, L.; Tu, J.; Tan, H.; et al. Mitotic SENP3 Activation Couples with cGAS Signaling in Tumor Cells to Stimulate Anti-Tumor Immunity. Cell Death Dis. 2022, 13, 640. [Google Scholar] [CrossRef]
- Francica, B.J.; Ghasemzadeh, A.; Desbien, A.L.; Theodros, D.; Sivick, K.E.; Reiner, G.L.; Hix Glickman, L.; Marciscano, A.E.; Sharabi, A.B.; Leong, M.L.; et al. TNFα and Radioresistant Stromal Cells Are Essential for Therapeutic Efficacy of Cyclic Dinucleotide STING Agonists in Nonimmunogenic Tumors. Cancer Immunol. Res. 2018, 6, 422–433. [Google Scholar] [CrossRef]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS Is Essential for the Antitumor Effect of Immune Checkpoint Blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Shinriki, S.; Matsui, H. Unique Role of DDX41, a DEAD-Box Type RNA Helicase, in Hematopoiesis and Leukemogenesis. Front. Oncol. 2022, 12, 992340. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Brendel, V.P.; Shu, C.; Li, P.; Palanathan, S.; Cheng Kao, C. Single Nucleotide Polymorphisms of Human STING Can Affect Innate Immune Response to Cyclic Dinucleotides. PLoS ONE 2013, 8, e77846. [Google Scholar] [CrossRef]
- Ruiz-Moreno, J.S.; Hamann, L.; Shah, J.A.; Verbon, A.; Mockenhaupt, F.P.; Puzianowska-Kuznicka, M.; Naujoks, J.; Sander, L.E.; Witzenrath, M.; Cambier, J.C.; et al. The Common HAQ STING Variant Impairs cGAS-Dependent Antibacterial Responses and Is Associated with Susceptibility to Legionnaires’ Disease in Humans. PLoS Pathog. 2018, 14, e1006829. [Google Scholar] [CrossRef]
- Tang, Z.; Zhao, J.; Li, Y.; Tomer, S.; Selvaraju, M.; Tien, N.; Sun, D.; Johnson, D.K.; Zhen, A.; Li, P.; et al. Structural and Biological Evaluations of a Non-Nucleoside STING Agonist Specific for Human STING A230 Variants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-Tumor Immunity. Cell Rep. 2018, 25, 3074–3085.e5. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 Axis Promotes cGAS/STING Signaling in ARID1A-Deficient Tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.-Y. ATM Inhibition Enhances Cancer Immunotherapy by Promoting mtDNA Leakage and cGAS/STING Activation. J. Clin. Investig. 2021, 131, e139333. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Veleeparambil, M.; Kessler, P.M.; Willard, B.; Chattopadhyay, S.; Sen, G.C. EGFR-Mediated Tyrosine Phosphorylation of STING Determines Its Trafficking Route and Cellular Innate Immunity Functions. EMBO J. 2020, 39, e104106. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Q.; Zhang, F.; Meng, F.; Liu, S.; Zhou, R.; Wu, Q.; Li, X.; Shen, L.; Huang, J.; et al. HER2 Recruits AKT1 to Disrupt STING Signalling and Suppress Antiviral Defence and Antitumour Immunity. Nat. Cell Biol. 2019, 21, 1027–1040. [Google Scholar] [CrossRef]
- Lt, O.; Wc, L.; S, M.; G, O.; Z, N.; Y, B.; M, Y.; Pl, L.; Jy, G.; P, W.; et al. IFI16-Dependent STING Signaling Is a Crucial Regulator of Anti-HER2 Immune Response in HER2+ Breast Cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2201376119. [Google Scholar] [CrossRef]
- Milling, L.E.; Garafola, D.; Agarwal, Y.; Wu, S.; Thomas, A.; Donahue, N.; Adams, J.; Thai, N.; Suh, H.; Irvine, D.J. Neoadjuvant STING Activation, Extended Half-Life IL2, and Checkpoint Blockade Promote Metastasis Clearance via Sustained NK-Cell Activation. Cancer Immunol. Res. 2022, 10, 26–39. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, Q.; Jiang, T.; Wang, W.; Zhao, J.J. Targeting Tumor-Associated Macrophages with STING Agonism Improves the Antitumor Efficacy of Osimertinib in a Mouse Model of EGFR-Mutant Lung Cancer. Front. Immunol. 2023, 14, 1077203. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liang, W.; Li, K.; Liao, X.; Chen, J.; Qiu, X.; Liu, K.; Qiu, D.; Qin, Y. Sorafenib Suppresses the Activation of Type I Interferon Pathway Induced by RLR-MAVS and cGAS-STING Signaling. Biochem. Biophys. Res. Commun. 2022, 623, 181–188. [Google Scholar] [CrossRef]
- Wayne, J.; Brooks, T.; Landras, A.; Massey, A.J. Targeting DNA Damage Response Pathways to Activate the STING Innate Immune Signaling Pathway in Human Cancer Cells. FEBS J. 2021, 288, 4507–4540. [Google Scholar] [CrossRef]
- Brooks, T.; Wayne, J.; Massey, A.J. Checkpoint Kinase 1 (Chk1) Inhibition Fails to Activate the Stimulator of Interferon Genes (STING) Innate Immune Signalling in a Human Coculture Cancer System. Mol. Biomed. 2021, 2, 19. [Google Scholar] [CrossRef]
- Gao, J.; Zheng, M.; Wu, X.; Zhang, H.; Su, H.; Dang, Y.; Ma, M.; Wang, F.; Xu, J.; Chen, L.; et al. CDK Inhibitor Palbociclib Targets STING to Alleviate Autoinflammation. EMBO Rep. 2022, 23, e53932. [Google Scholar] [CrossRef]
- Lee, J.H.; Kanwar, B.; Khattak, A.; Balentine, J.; Nguyen, N.H.; Kast, R.E.; Lee, C.J.; Bourbeau, J.; Altschuler, E.L.; Sergi, C.M.; et al. COVID-19 Molecular Pathophysiology: Acetylation of Repurposing Drugs. Int. J. Mol. Sci. 2022, 23, 13260. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; VanPortfliet, J.J.; Chen, Y.-F.; Bryant, J.D.; Li, Y.; Fails, D.; Torres-Odio, S.; Ragan, K.B.; Deng, J.; Mohan, A.; et al. Cooperative Sensing of Mitochondrial DNA by ZBP1 and cGAS Promotes Cardiotoxicity. Cell 2023, 186, 3013–3032.e22. [Google Scholar] [CrossRef]
- Smith, T.T.; Moffett, H.F.; Stephan, S.B.; Opel, C.F.; Dumigan, A.G.; Jiang, X.; Pillarisetty, V.G.; Pillai, S.P.S.; Wittrup, K.D.; Stephan, M.T. Biopolymers Codelivering Engineered T Cells and STING Agonists Can Eliminate Heterogeneous Tumors. J. Clin. Investig. 2017, 127, 2176–2191. [Google Scholar] [CrossRef]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING Agonist Promotes CAR T Cell Trafficking and Persistence in Breast Cancer. J. Exp. Med. 2021, 218, e20200844. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef]
- de Queiroz, N.M.G.P.; Xia, T.; Konno, H.; Barber, G.N. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol. Cancer Res. 2019, 17, 974–986. [Google Scholar] [CrossRef]
- Whelan, J.T.; Singaravelu, R.; Wang, F.; Pelin, A.; Tamming, L.A.; Pugliese, G.; Martin, N.T.; Crupi, M.J.F.; Petryk, J.; Austin, B.; et al. CRISPR-Mediated Rapid Arming of Poxvirus Vectors Enables Facile Generation of the Novel Immunotherapeutic STINGPOX. Front. Immunol. 2022, 13, 1050250. [Google Scholar] [CrossRef]
- Kolyasnikova, N.M.; Pestov, N.B.; Sanchez-Pimentel, J.P.; Barlev, N.A.; Ishmukhametov, A.A. Anti-Cancer Virotherapy in Russia: Lessons from the Past, Current Challenges and Prospects for the Future. Curr. Pharm. Biotechnol. 2023, 24, 266–278. [Google Scholar]
- Yang, N.; Wang, Y.; Liu, S.; Tariq, S.B.; Luna, J.M.; Mazo, G.; Tan, A.; Zhang, T.; Wang, J.; Yan, W.; et al. OX40L-Expressing Recombinant Modified Vaccinia Virus Ankara Induces Potent Antitumor Immunity via Reprogramming Tregs. J. Exp. Med. 2023, 220, e20221166. [Google Scholar] [CrossRef]
- Dai, P.; Wang, W.; Yang, N.; Serna-Tamayo, C.; Ricca, J.M.; Zamarin, D.; Shuman, S.; Merghoub, T.; Wolchok, J.D.; Deng, L. Intratumoral Delivery of Inactivated Modified Vaccinia Virus Ankara (iMVA) Induces Systemic Antitumor Immunity via STING and Batf3-Dependent Dendritic Cells. Sci. Immunol. 2017, 2, eaal1713. [Google Scholar] [CrossRef]
- Arwert, E.N.; Milford, E.L.; Rullan, A.; Derzsi, S.; Hooper, S.; Kato, T.; Mansfield, D.; Melcher, A.; Harrington, K.J.; Sahai, E. STING and IRF3 in Stromal Fibroblasts Enable Sensing of Genomic Stress in Cancer Cells to Undermine Oncolytic Viral Therapy. Nat. Cell Biol. 2020, 22, 758–766. [Google Scholar] [CrossRef]
- Kumar Bhardwaj, V.; Purohit, R.; Kumar, S. Himalayan Bioactive Molecules as Potential Entry Inhibitors for the Human Immunodeficiency Virus. Food Chem. 2021, 347, 128932. [Google Scholar] [CrossRef]
- Pang, E.S.; Daraj, G.; Balka, K.R.; De Nardo, D.; Macri, C.; Hochrein, H.; Masterman, K.-A.; Tan, P.S.; Shoppee, A.; Magill, Z.; et al. Discordance in STING-Induced Activation and Cell Death Between Mouse and Human Dendritic Cell Populations. Front. Immunol. 2022, 13, 794776. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Modality | Cancer Type | Compound | Status | Co-Treatment | Phase | Study ID |
|---|---|---|---|---|---|---|
| cGAS activation | var. | MnCl2 | A | PD-1 ab | I | NCT03991559 |
| var. | A | radiotherapy | I/II | NCT04873440 | ||
| PanC | A | nab-PX, GC, PD-1 ab | I/II | NCT03989310 | ||
| OC | A | nab-PX, Pt, PD-1 ab | I/II | NCT03989336 | ||
| BTC | A | nab-PX, GC, PD-1 ab | I/II | NCT04004234 | ||
| STING activation | var. | ONM-501 | A | PD-1 ab | I | NCT06022029 |
| var. | BI 1387446 | A | PD-1 ab | I | NCT04147234 | |
| data | BMS-986301 | A | PD-1 ab, ±CTLA-4 ab | I | NCT03956680 | |
| HNC | ADU-S100 | E | PD-1 ab | II | NCT03937141 | |
| var. | ADU-S100 | E | CTLA-4 ab | I | NCT02675439 | |
| var. | ADU-S100 | E | PD-1 ab | I | NCT03172936 | |
| var. | E7766 | C | N/A | I | NCT04144140 | |
| NMIBC | E7766 | E | N/A | I | NCT04109092 | |
| var. | CDK-002/ExoSTING | C | N/A | I,II | NCT04592484 | |
| var. | IMSA101 | C | ICI | I,II | NCT04020185 | |
| var. | IMSA101 | A | radiotherapy, PD-1 ab | II | NCT05846659 | |
| NSCLC, RCC | IMSA101 | A | radiotherapy, PD-1 ab | II | NCT05846646 | |
| var. | MK-1454 | C | PD-1 ab | I | NCT03010176 | |
| HNC | MK-1454 | C | PD-1 ab | II | NCT04220866 | |
| melanoma etc | SB11285 | A | PD-1 ab | I | NCT04096638 | |
| var. | GSK3745417 | A | PD-1 ab | I | NCT03843359 | |
| AML, HR-MDS | GSK3745417 | A | N/A | I | NCT05424380 | |
| var. | KL340399 | A | N/A | I | NCT05549804 | |
| var. | SNX281 | A | PD-1 ab | I | NCT04609579 | |
| var. | TAK-676 | A | PD-1 ab, Pt, 5-FU | I,II | NCT04420884 | |
| var. | TAK-676 | A | radiotherapy, PD-1 ab | I | NCT04879849 | |
| HNC | TAK-676 | C | Pt, 5-FU, PX | I | NCT06062602 | |
| sarcoma, MCC | CRD3874-SI | A | N/A | I | NCT06021626 | |
| STING activation by live bacteria | var. | SYNB1891 | C | PD-L1 | I | NCT04167137 |
| Antibody-directed STING activation | var. | TAK-500 | A | PD-1 ab | I | NCT05070247 |
| HER2+ cancers | XMT-2056 | E | N/A | I | NCT05514717 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korneenko, T.V.; Pestov, N.B.; Nevzorov, I.A.; Daks, A.A.; Trachuk, K.N.; Solopova, O.N.; Barlev, N.A. At the Crossroads of the cGAS-cGAMP-STING Pathway and the DNA Damage Response: Implications for Cancer Progression and Treatment. Pharmaceuticals 2023, 16, 1675. https://doi.org/10.3390/ph16121675
Korneenko TV, Pestov NB, Nevzorov IA, Daks AA, Trachuk KN, Solopova ON, Barlev NA. At the Crossroads of the cGAS-cGAMP-STING Pathway and the DNA Damage Response: Implications for Cancer Progression and Treatment. Pharmaceuticals. 2023; 16(12):1675. https://doi.org/10.3390/ph16121675
Chicago/Turabian StyleKorneenko, Tatyana V., Nikolay B. Pestov, Ivan A. Nevzorov, Alexandra A. Daks, Kirill N. Trachuk, Olga N. Solopova, and Nickolai A. Barlev. 2023. "At the Crossroads of the cGAS-cGAMP-STING Pathway and the DNA Damage Response: Implications for Cancer Progression and Treatment" Pharmaceuticals 16, no. 12: 1675. https://doi.org/10.3390/ph16121675
APA StyleKorneenko, T. V., Pestov, N. B., Nevzorov, I. A., Daks, A. A., Trachuk, K. N., Solopova, O. N., & Barlev, N. A. (2023). At the Crossroads of the cGAS-cGAMP-STING Pathway and the DNA Damage Response: Implications for Cancer Progression and Treatment. Pharmaceuticals, 16(12), 1675. https://doi.org/10.3390/ph16121675