
Development of Dapagliflozin Solid Lipid Nanoparticles as a Novel Carrier for Oral Delivery: Statistical Design, Optimization, In-Vitro and In-Vivo Characterization, and Evaluation

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Design of Dapagliflozin-SLNs
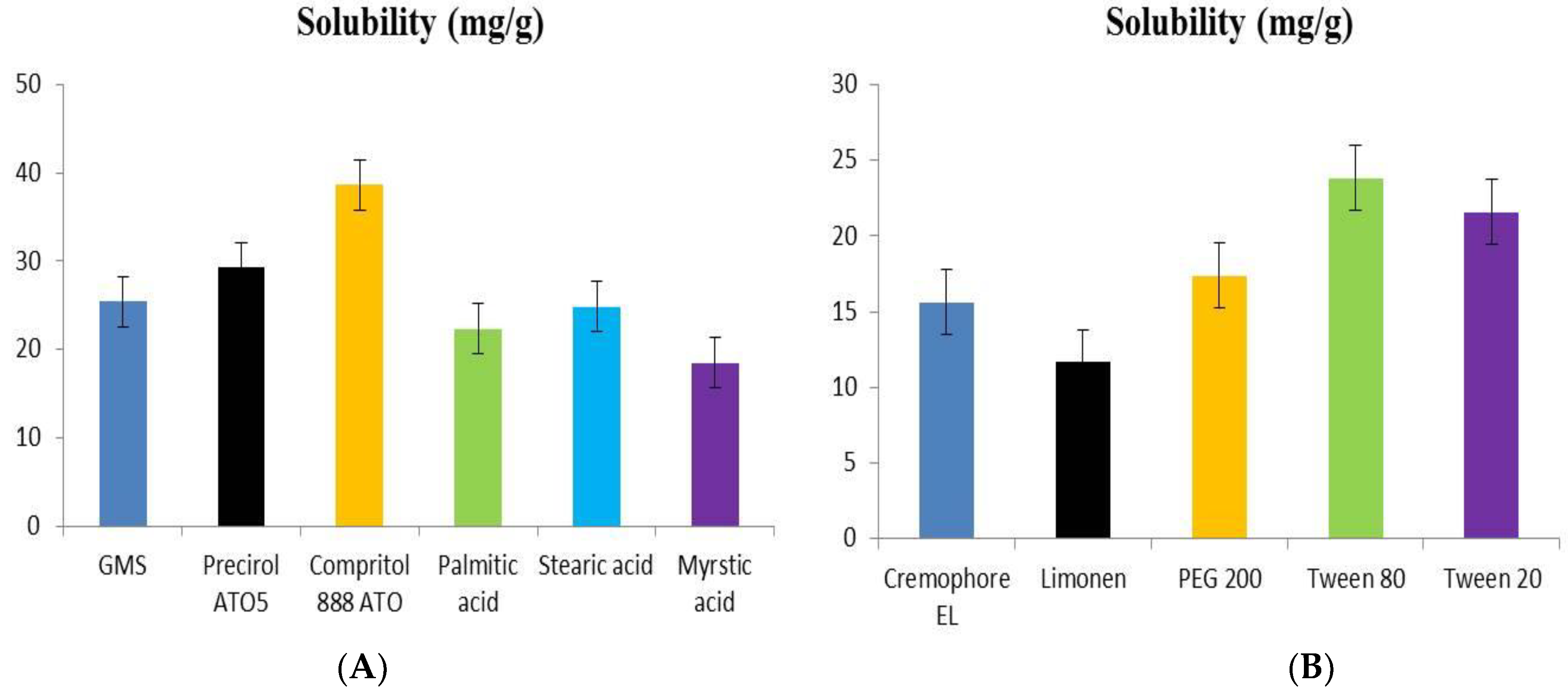
2.1.1. Solubility of Dapagliflozin in Lipids
2.1.2. Selection of Surfactant
2.1.3. Selection of Sonication Time and Amplitude
2.2. Preparation and Characterization of Dapagliflozin-SLNs
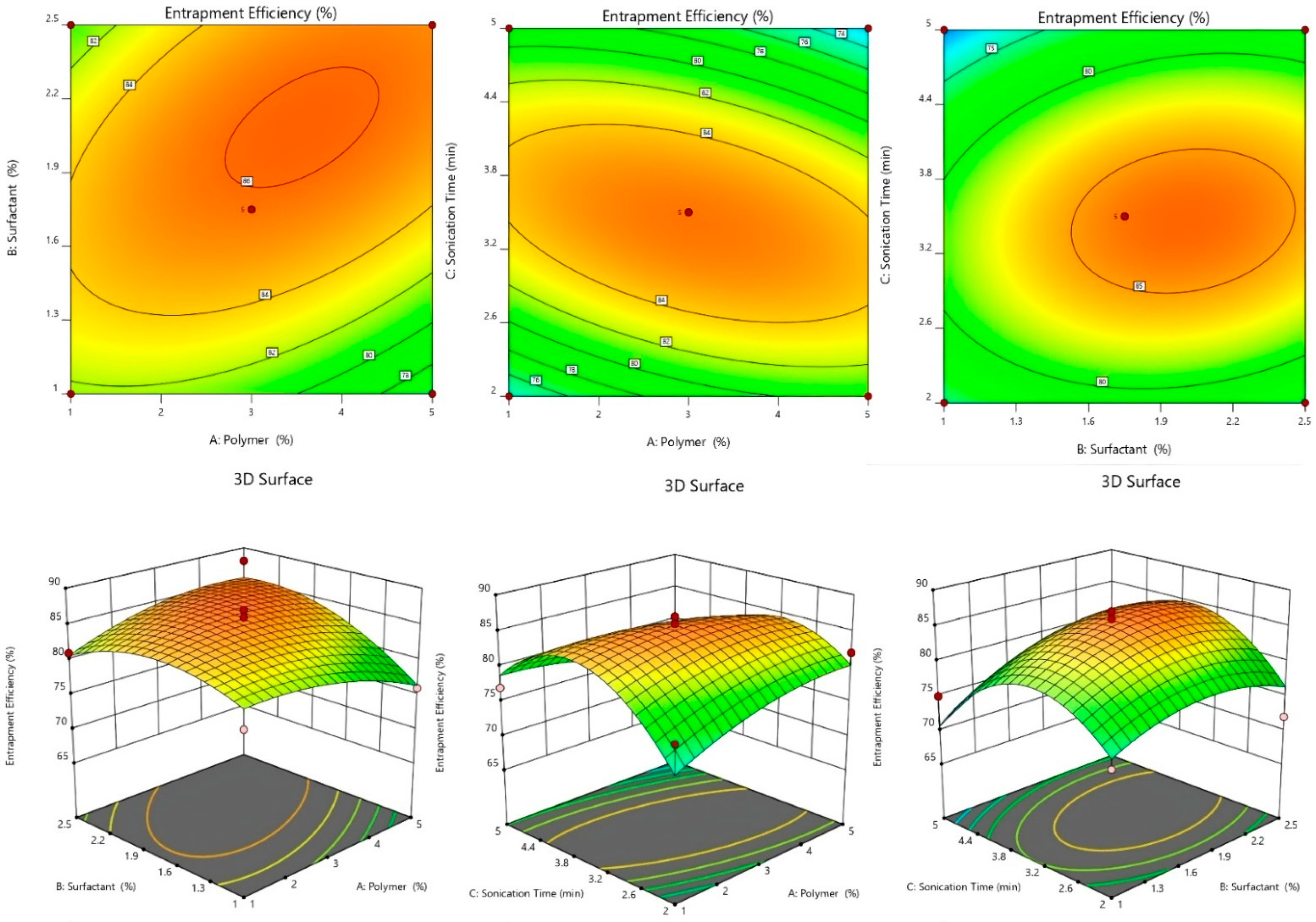
2.2.1. Effect of Independent Variables on %EE
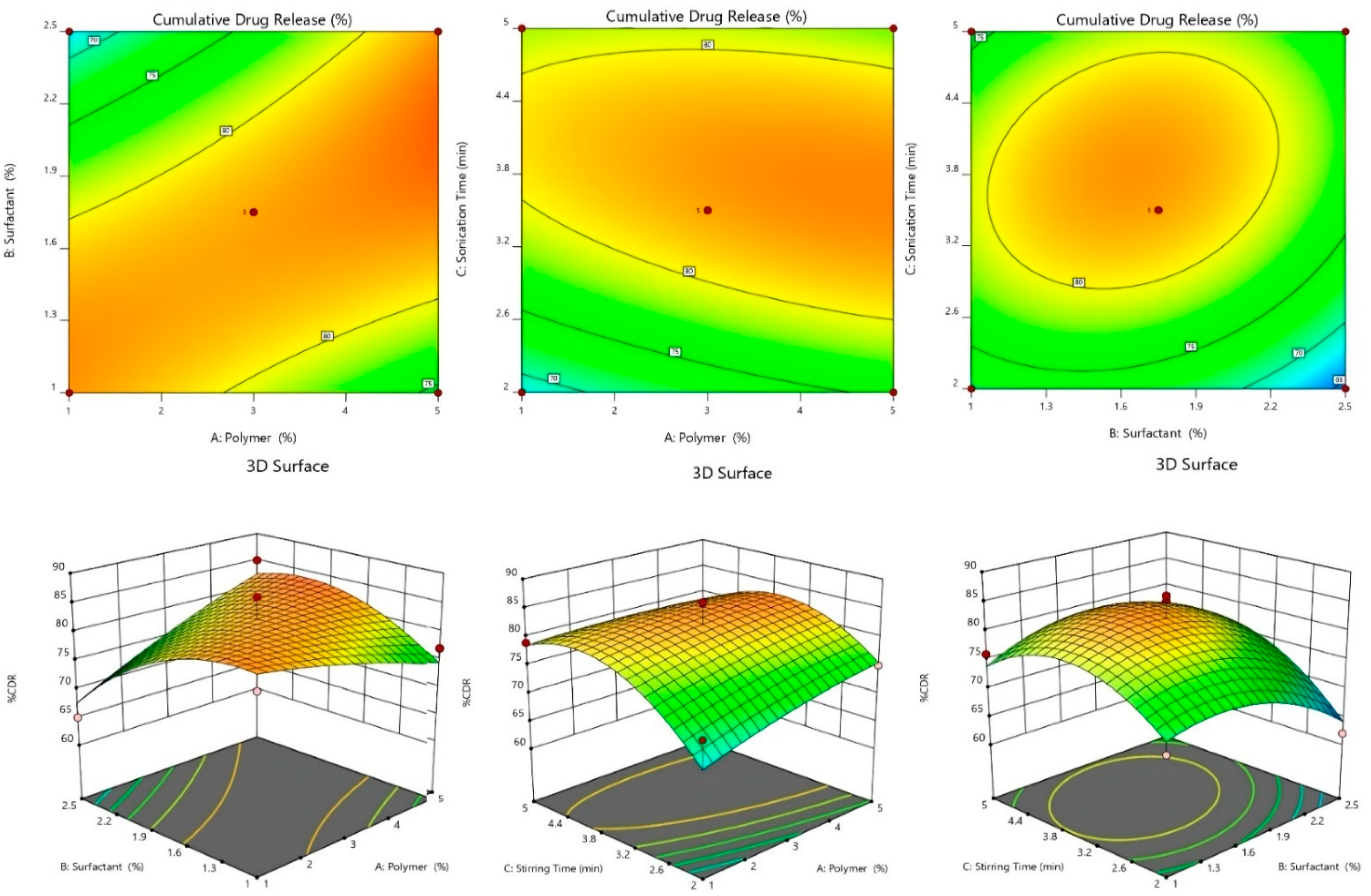
2.2.2. Effect of Independent Variables on %CDR
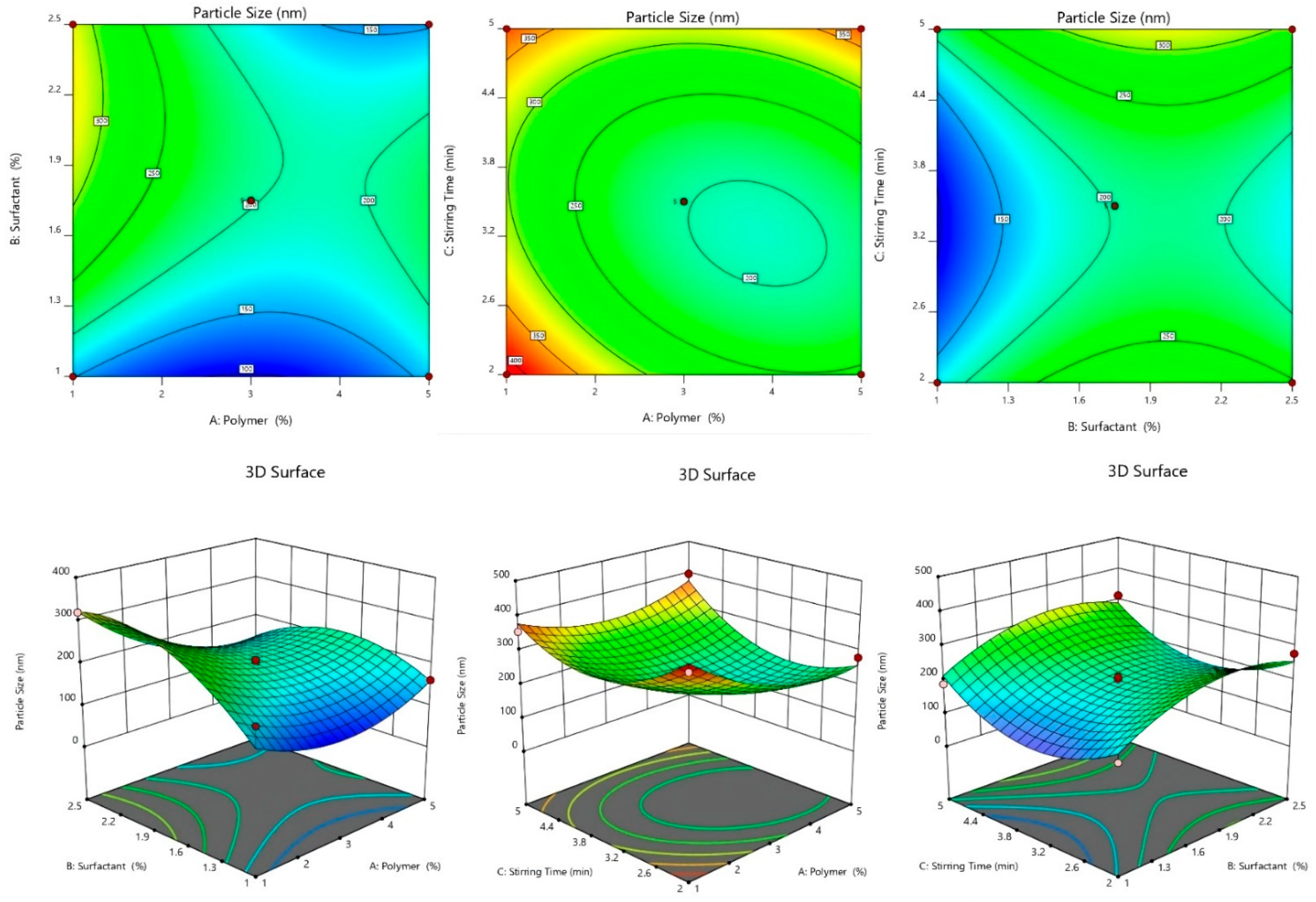
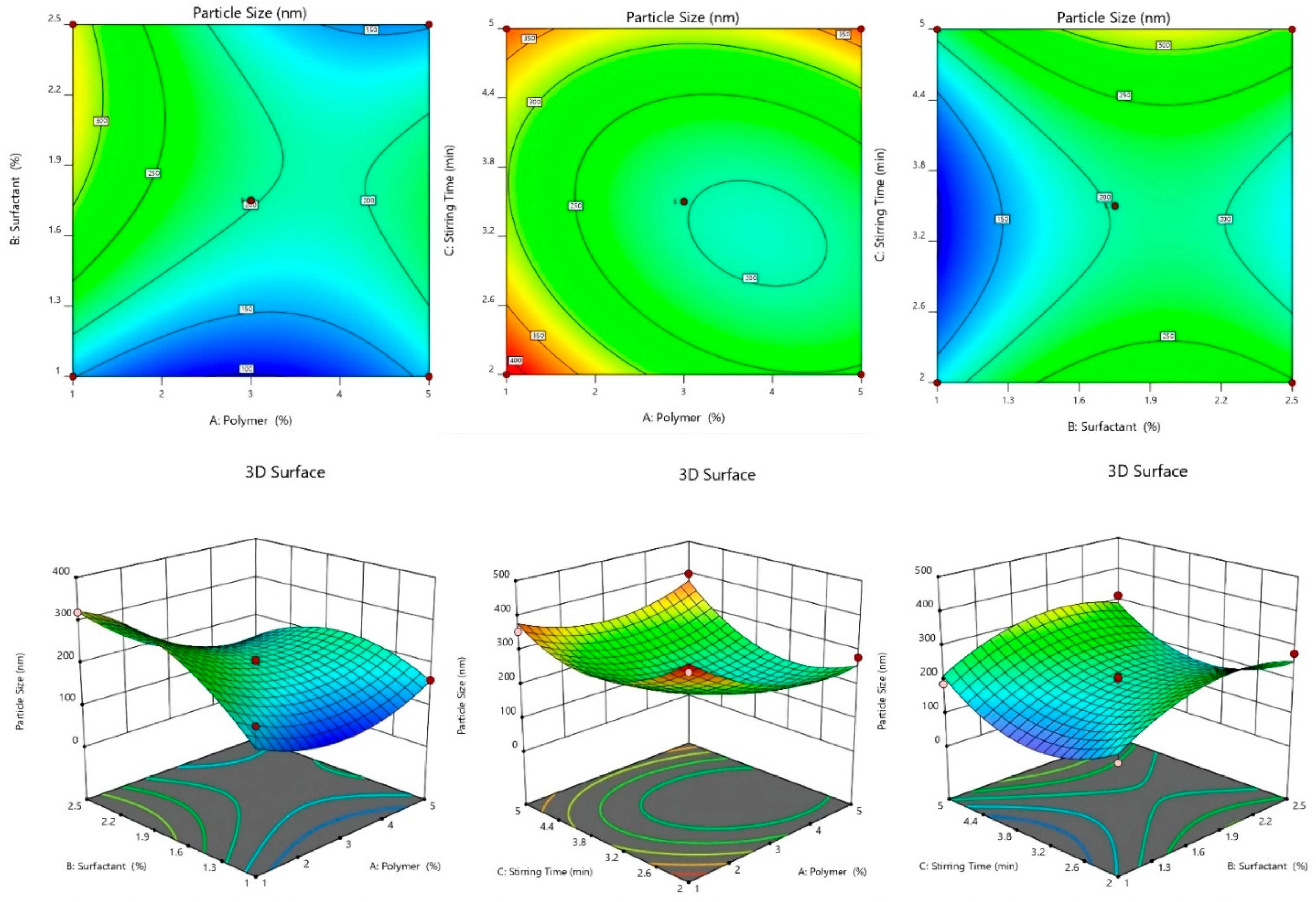
2.2.3. Effect of Independent Variables on Particle Size
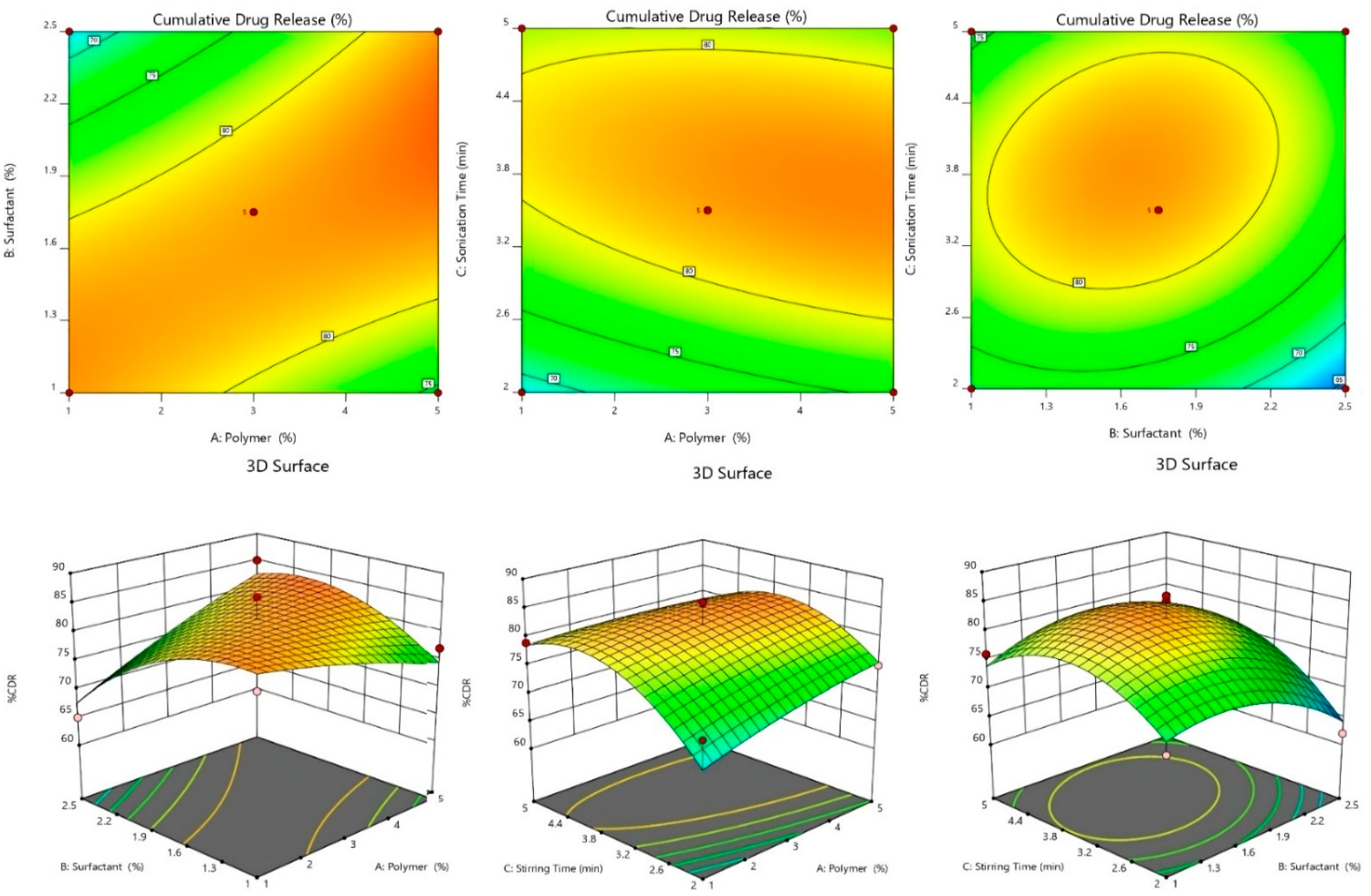
2.2.4. 3D-Response Surface and Contour Plot
2.2.5. Optimization of SLNs Formulation
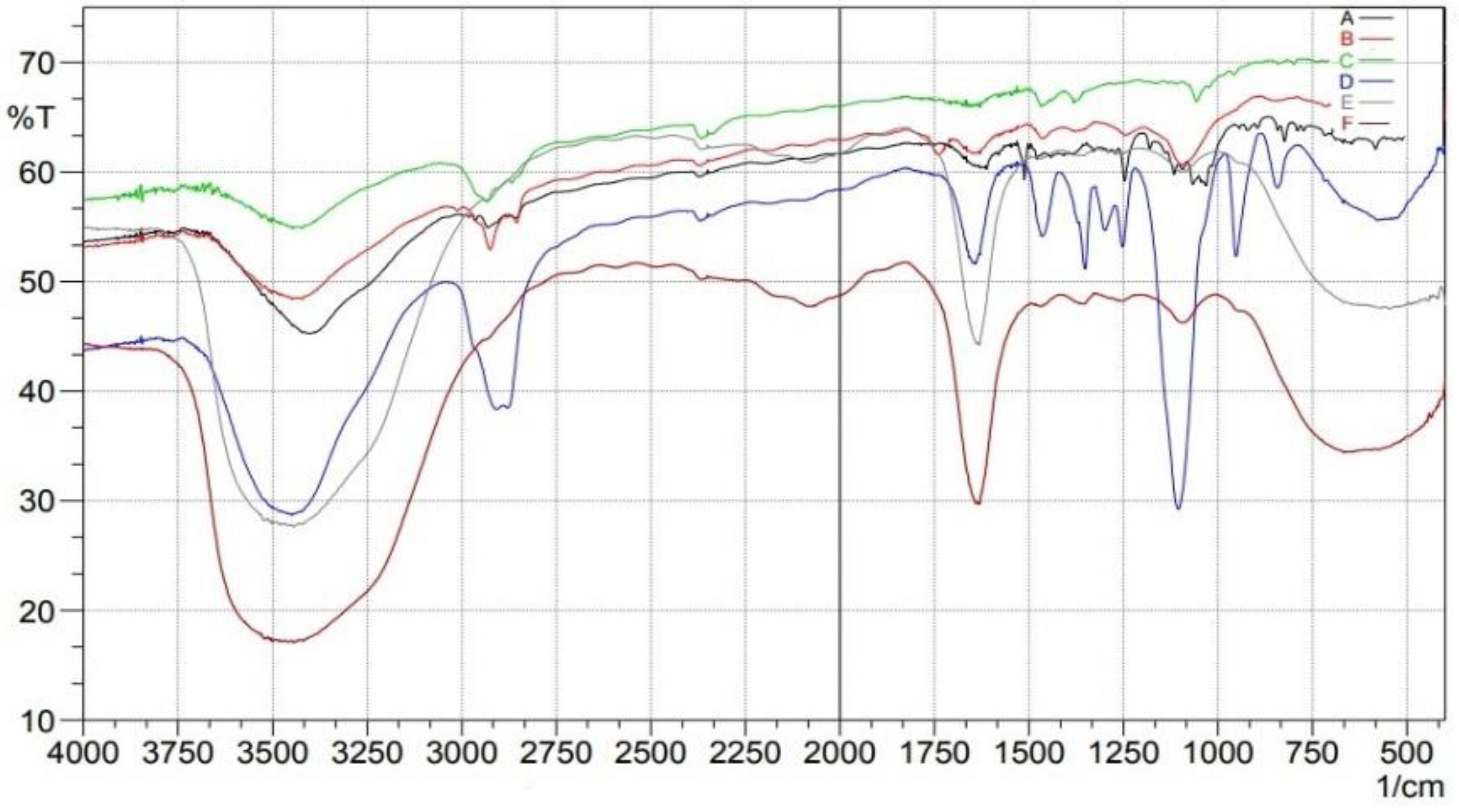
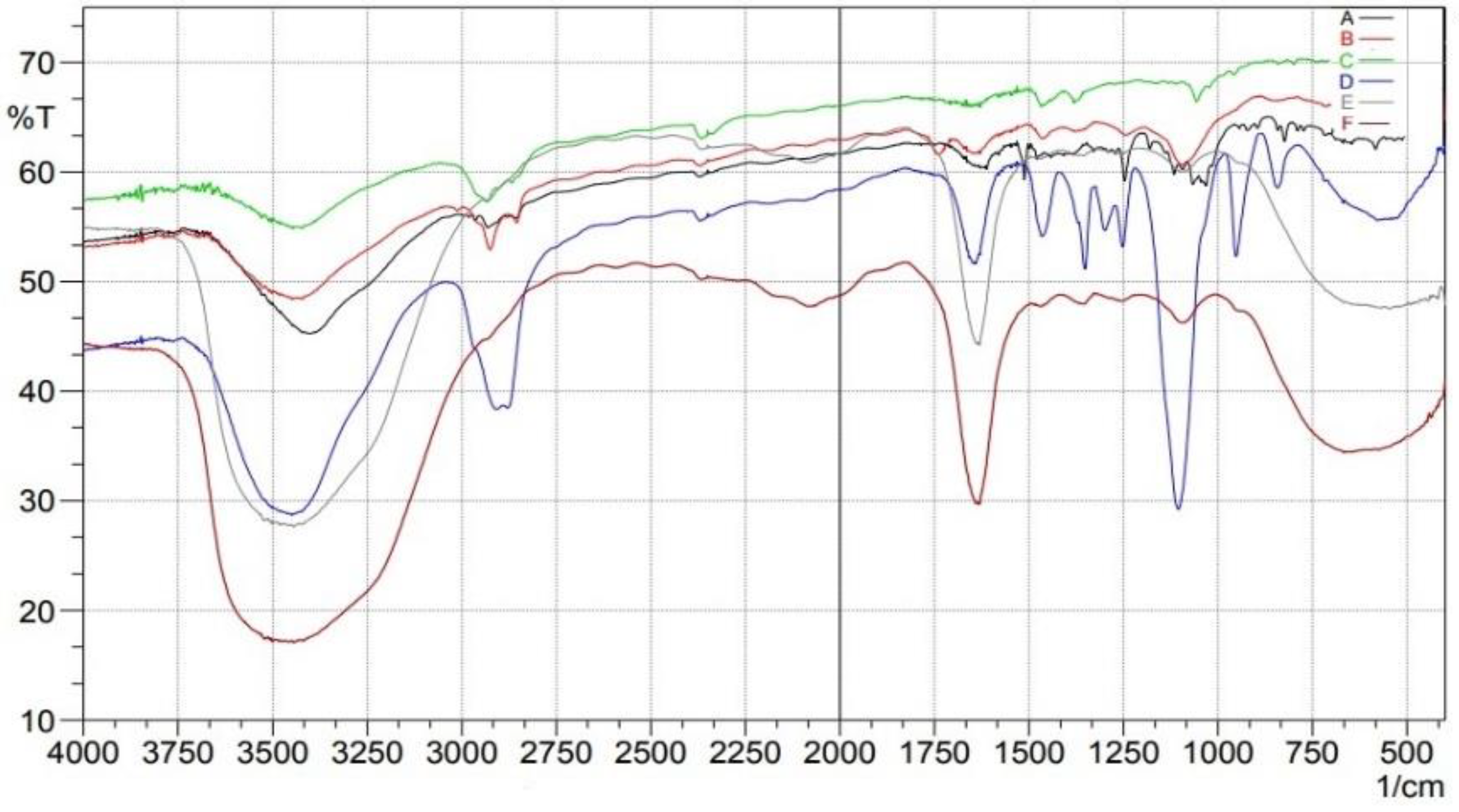
2.3. Fourier Transform Infrared (FTIR)
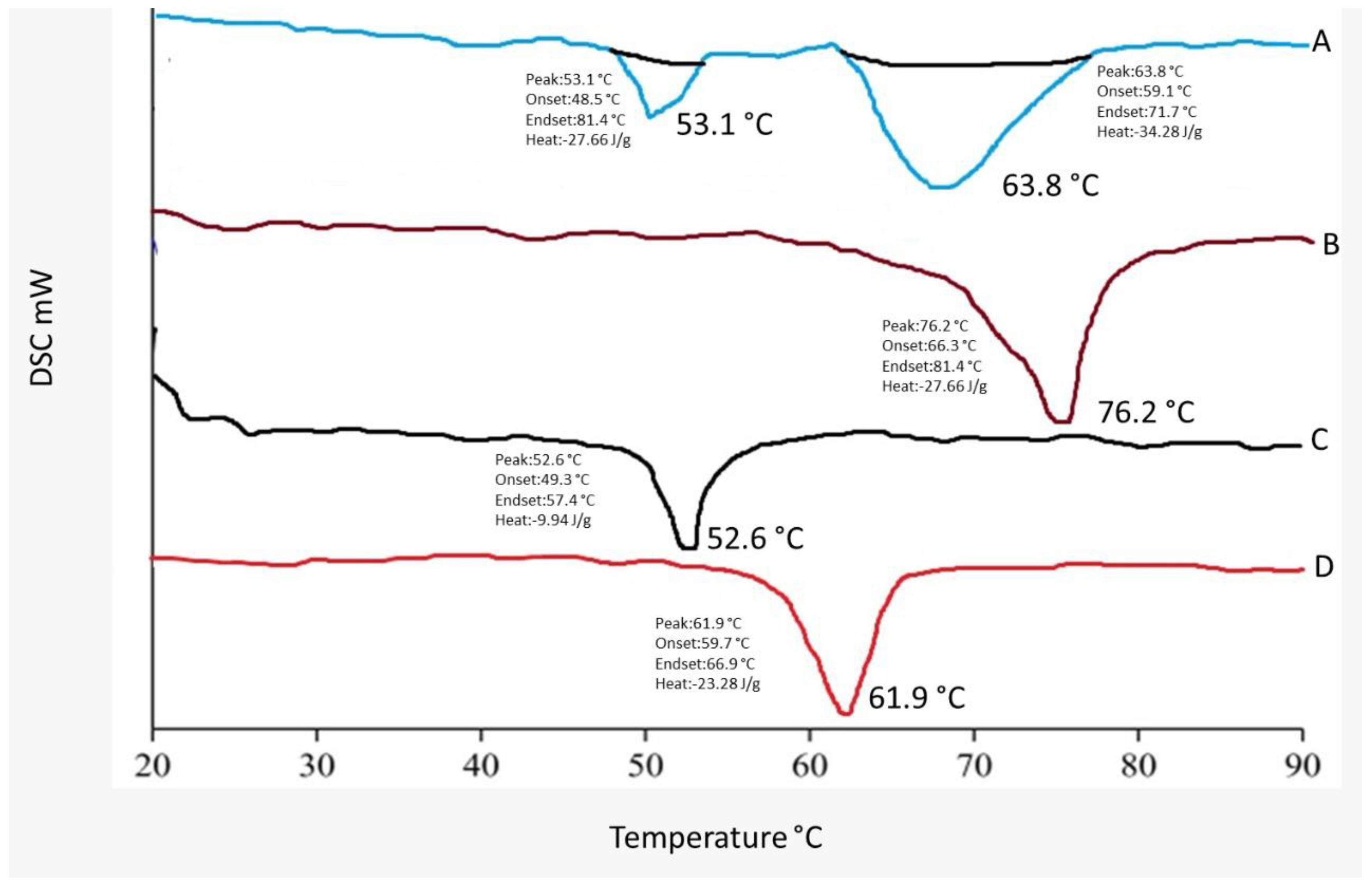
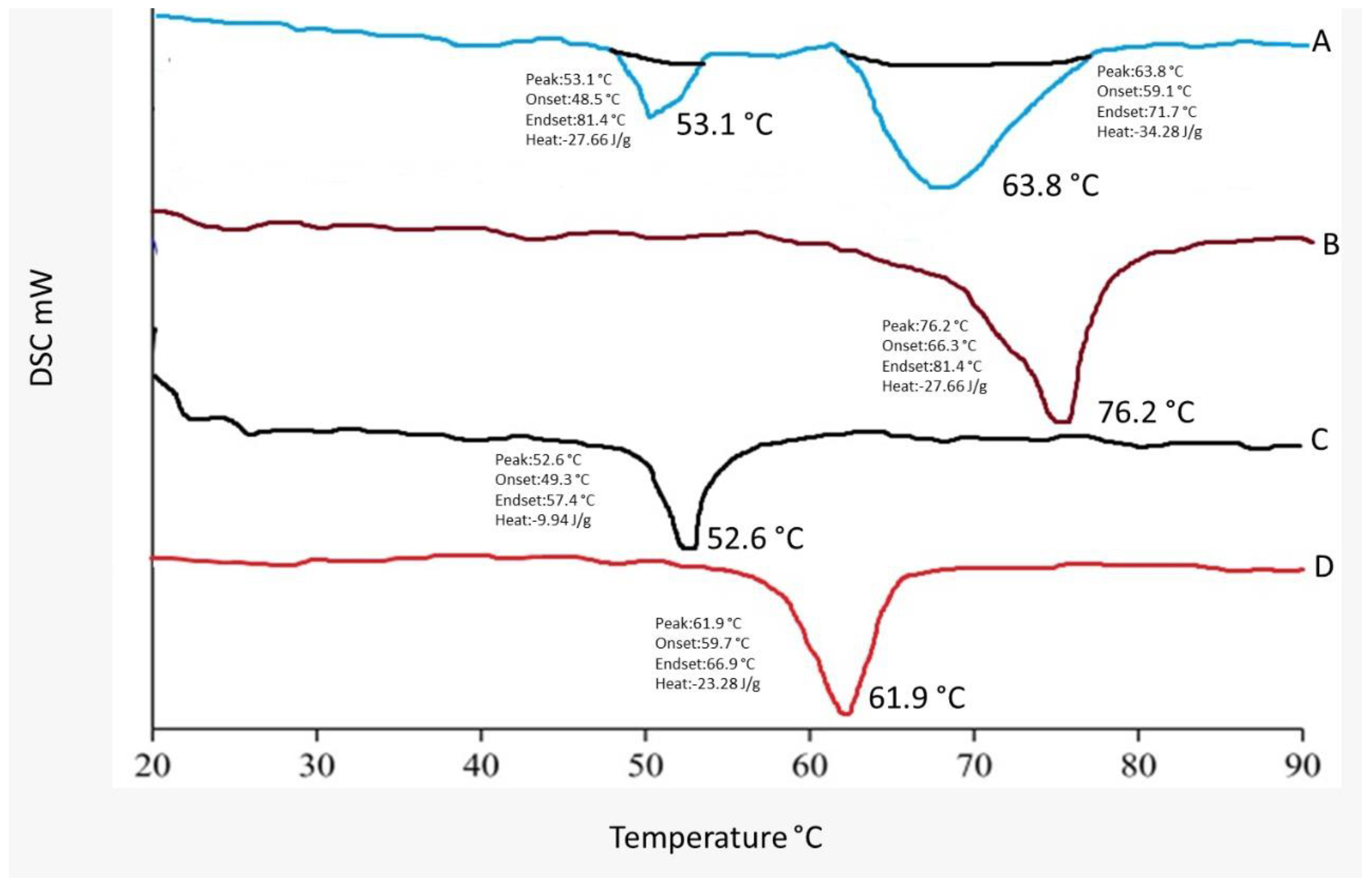
2.4. DSC Studies
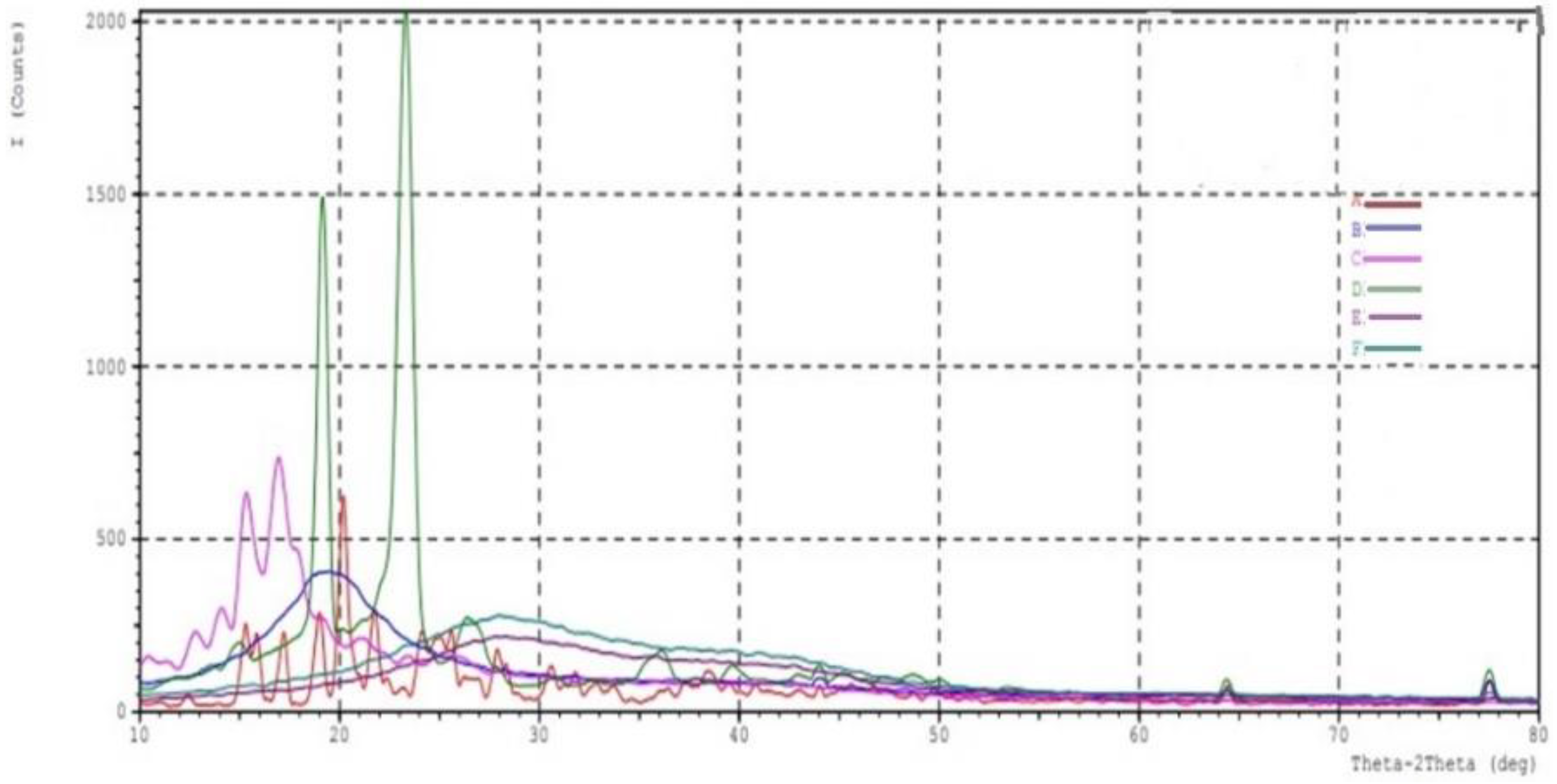
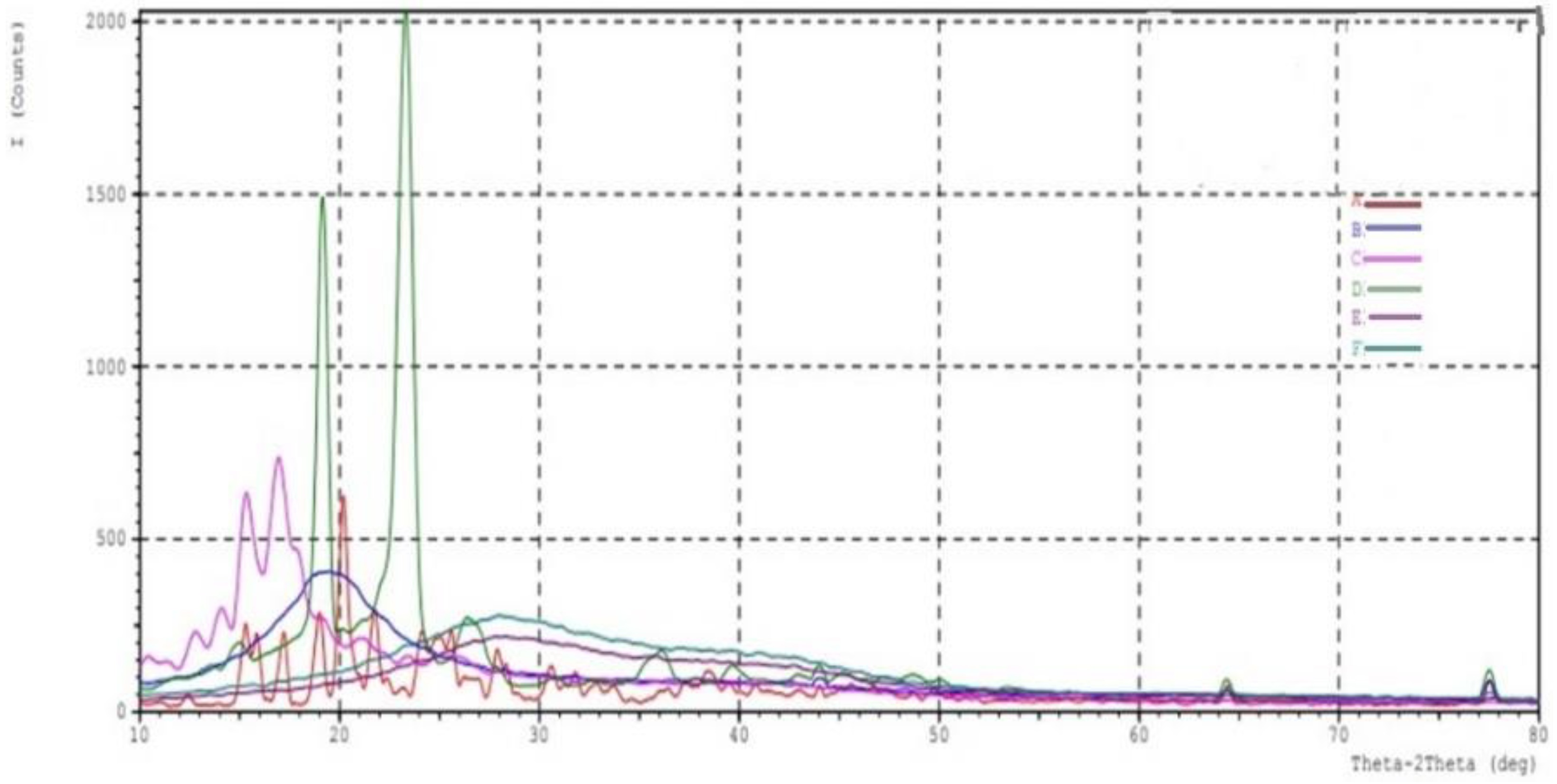
2.5. XRD Crystallography
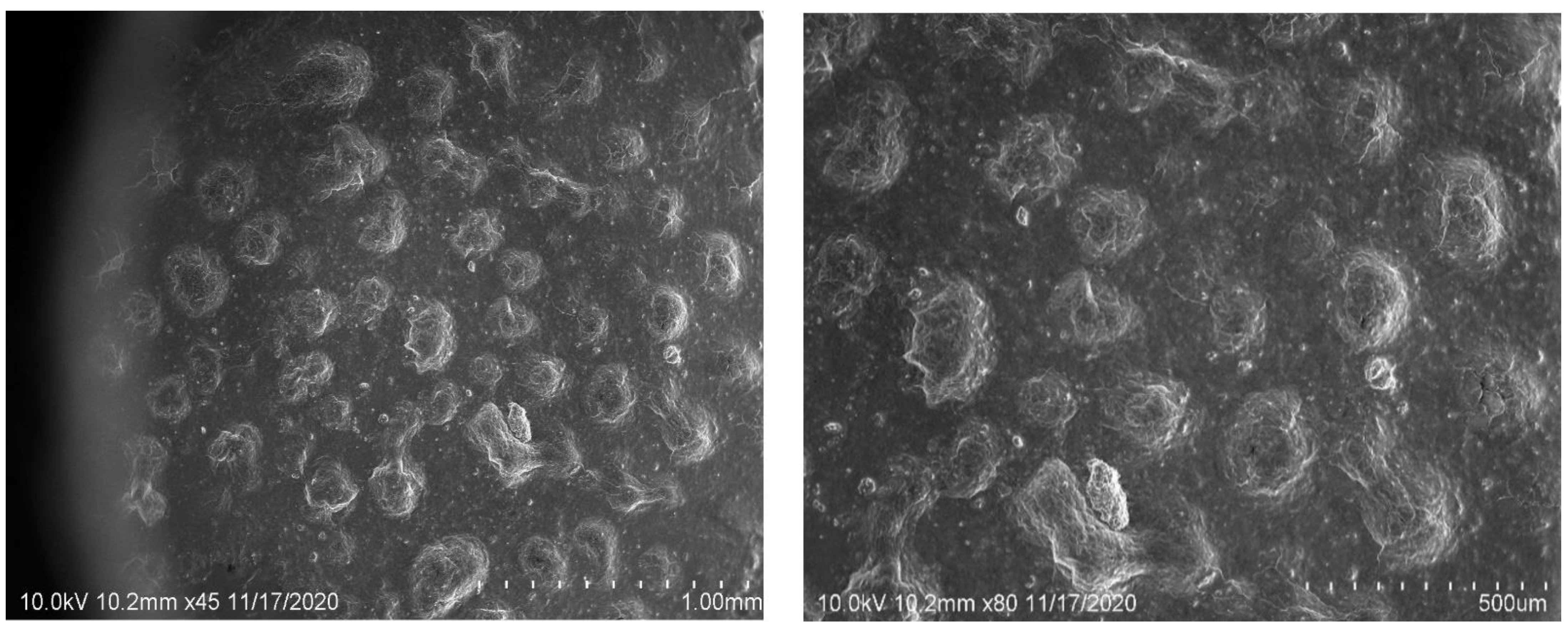
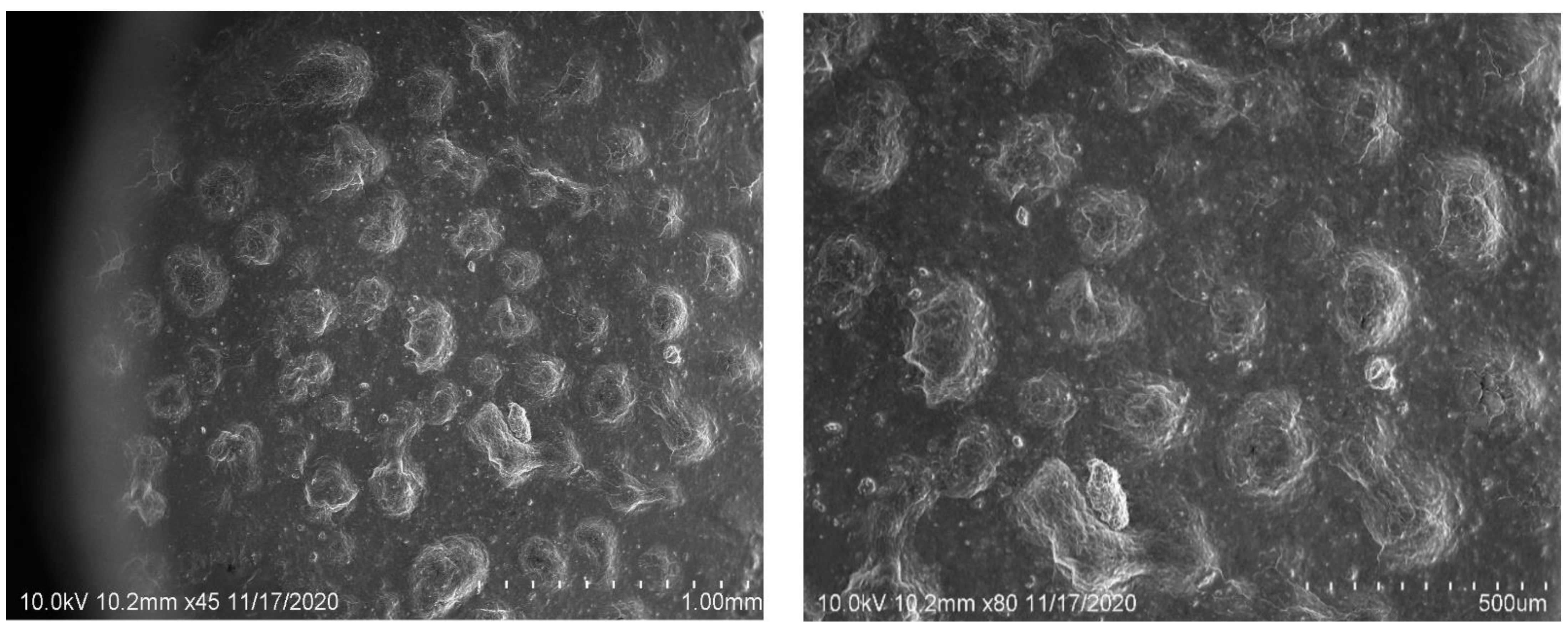
2.6. SEM Image Studies
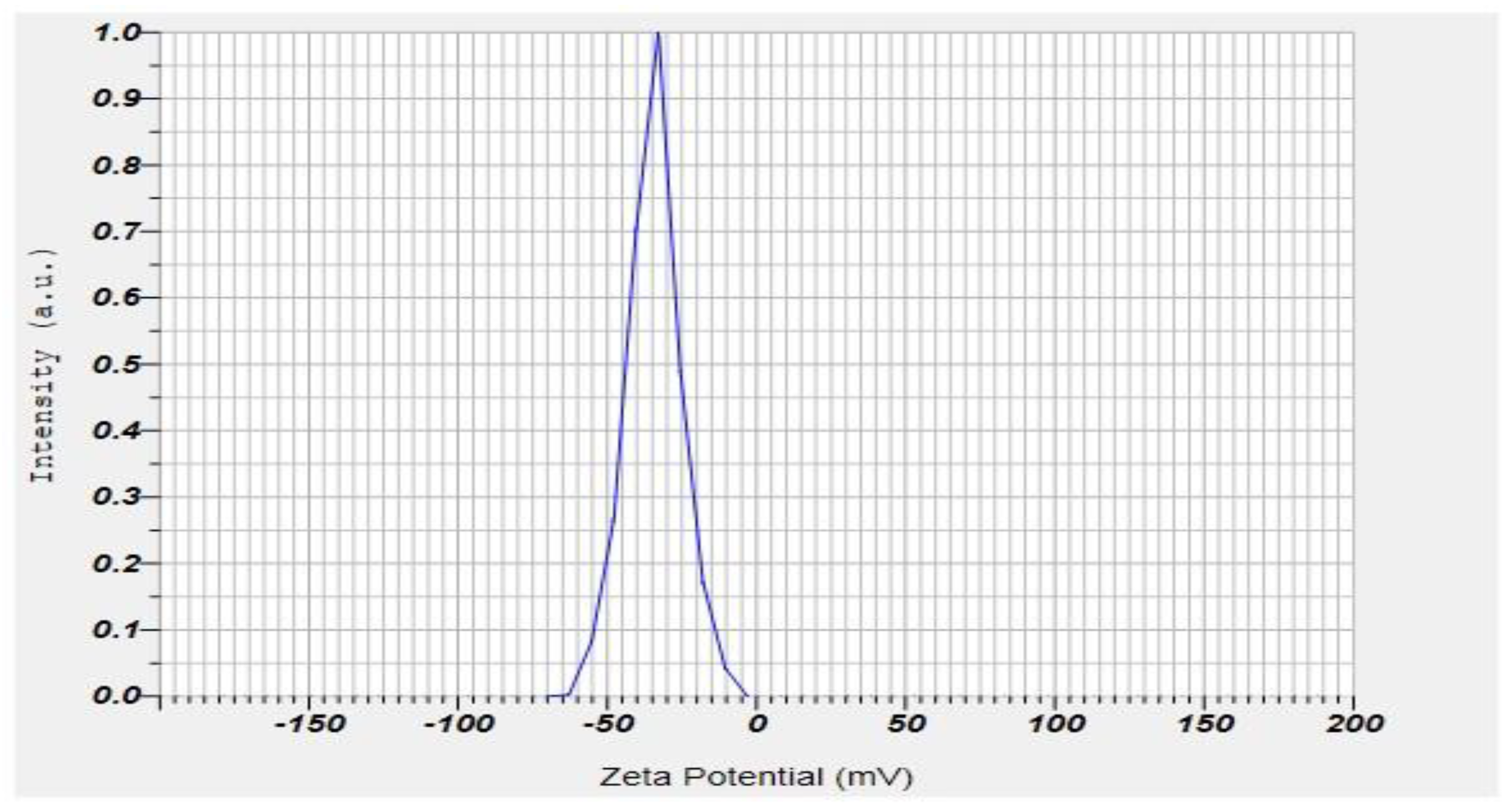
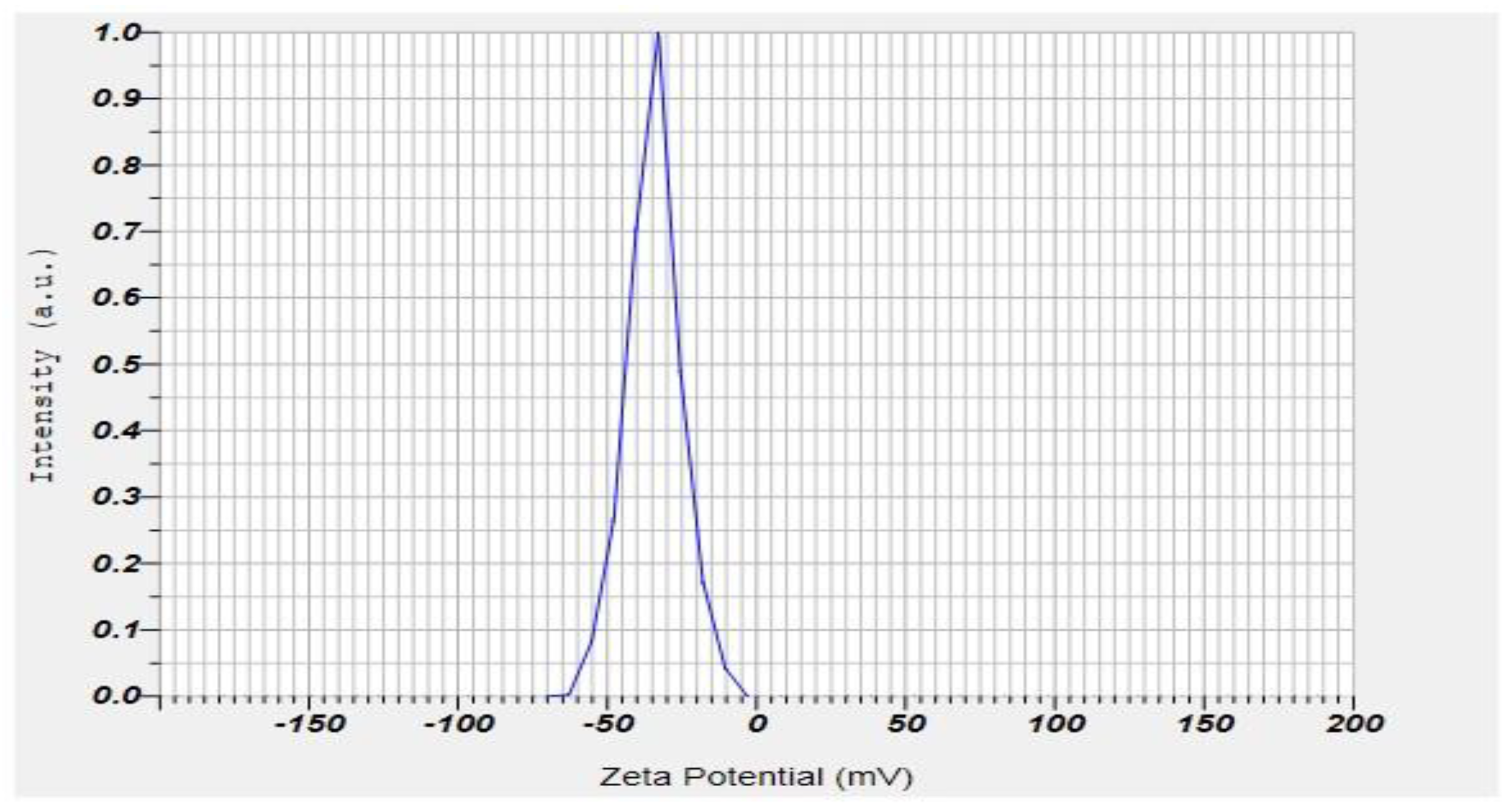
2.7. Zeta Potential and PDI of the Formulation
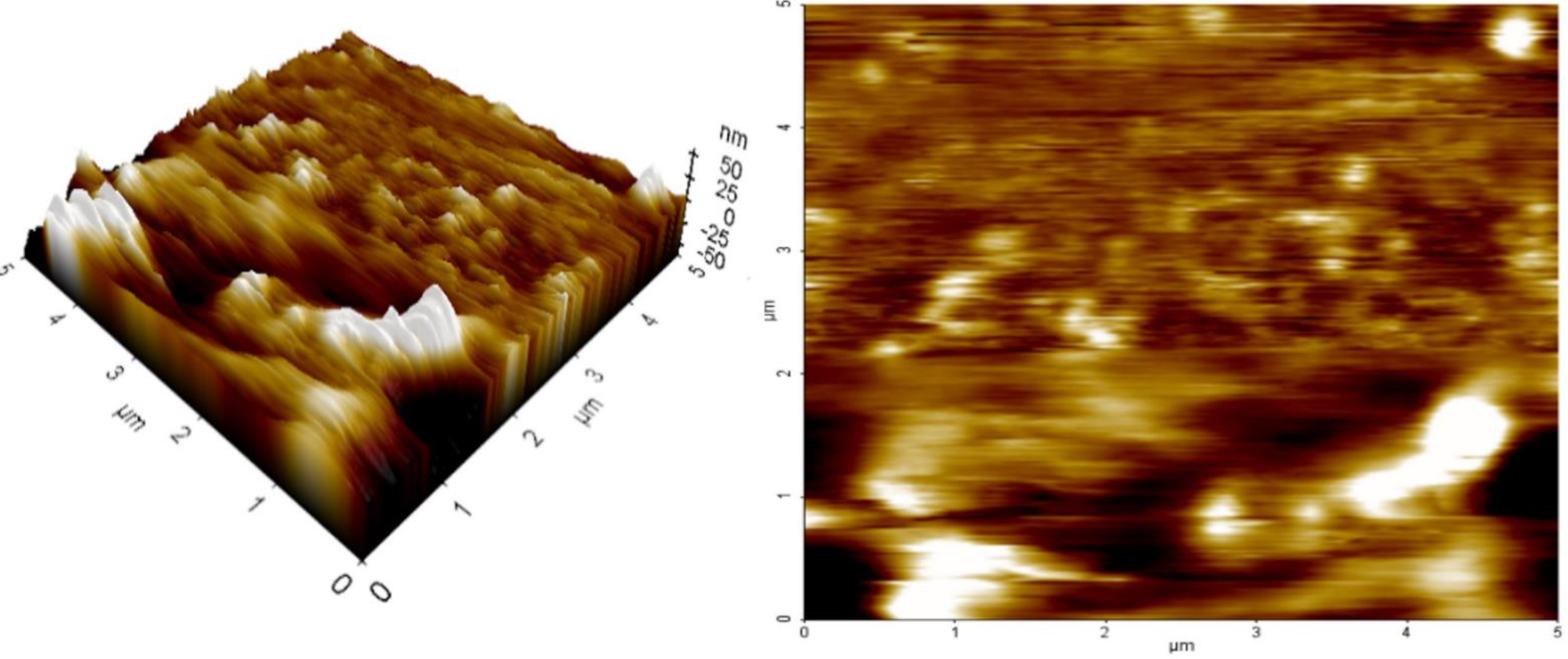
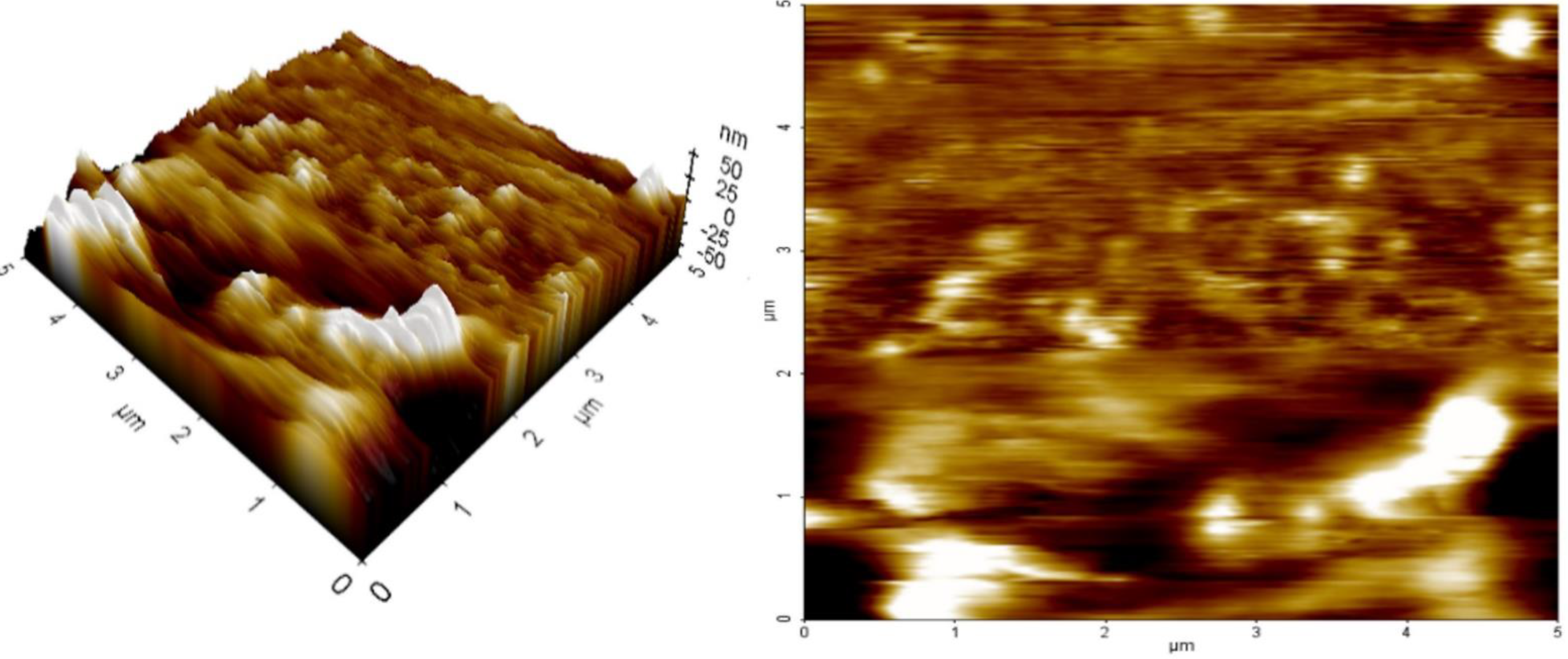
2.8. AFM
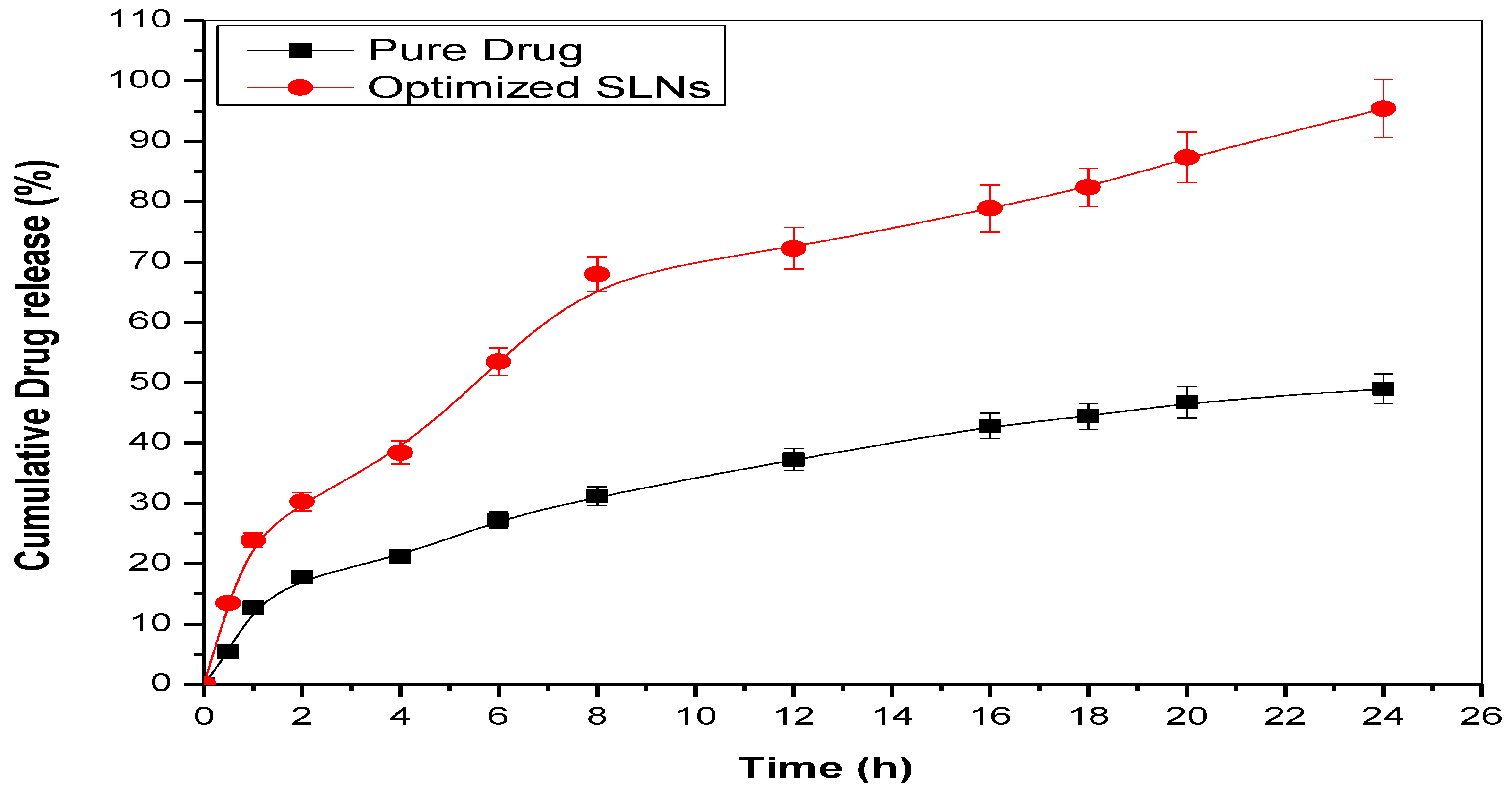
2.9. Studies of In Vitro Drug Release
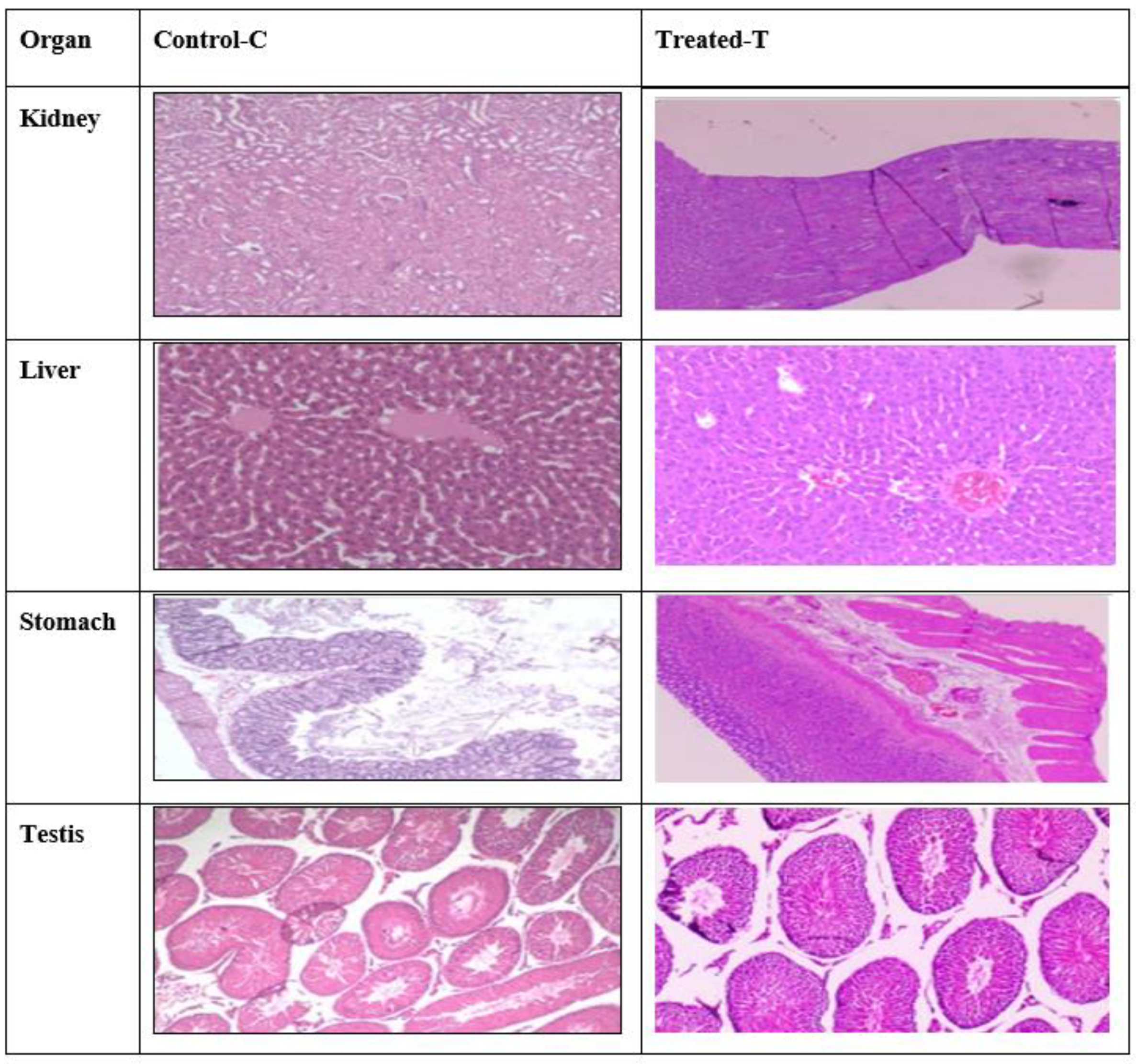
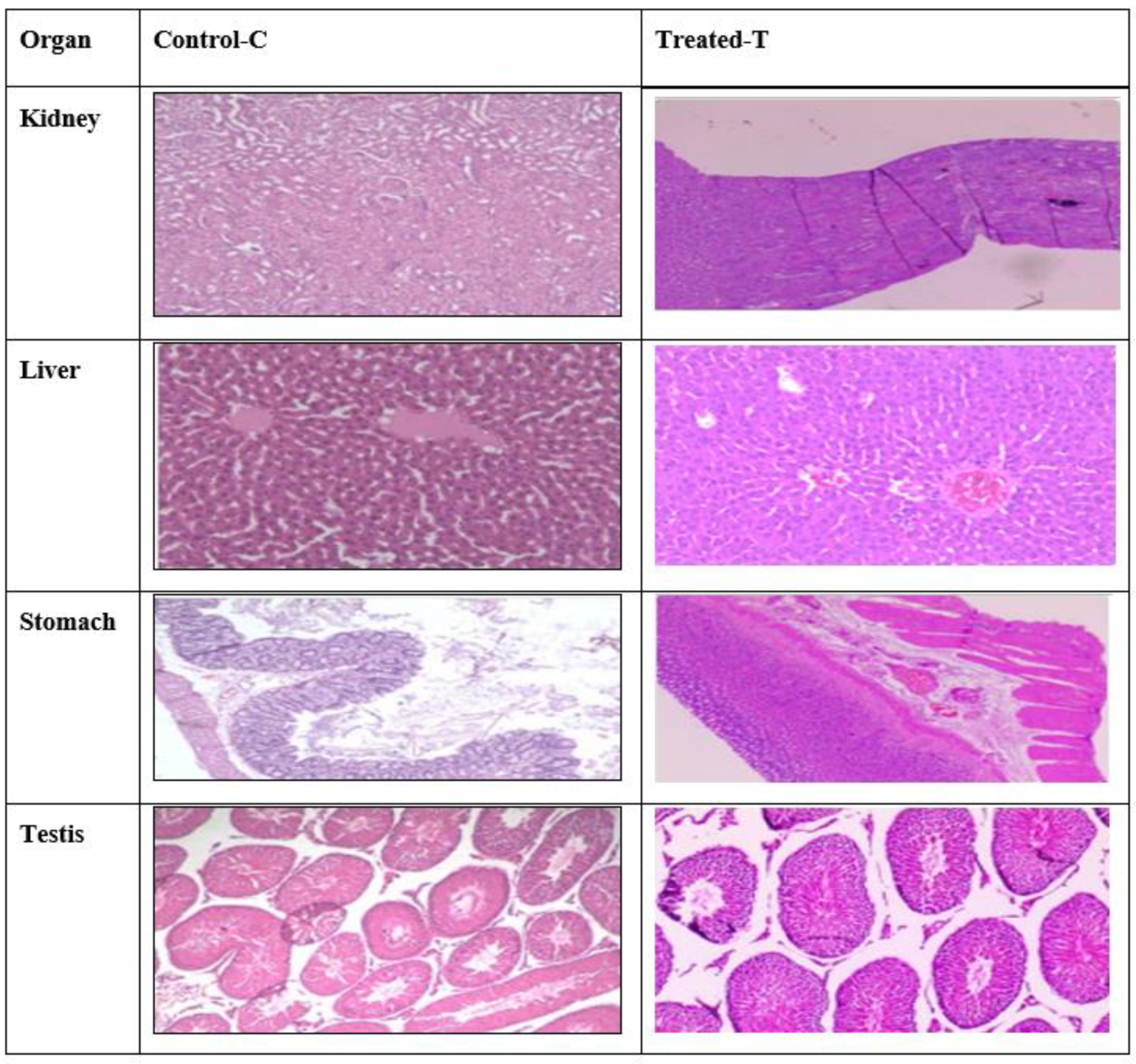
2.10. Histopathology Studies
2.11. In Vivo Study
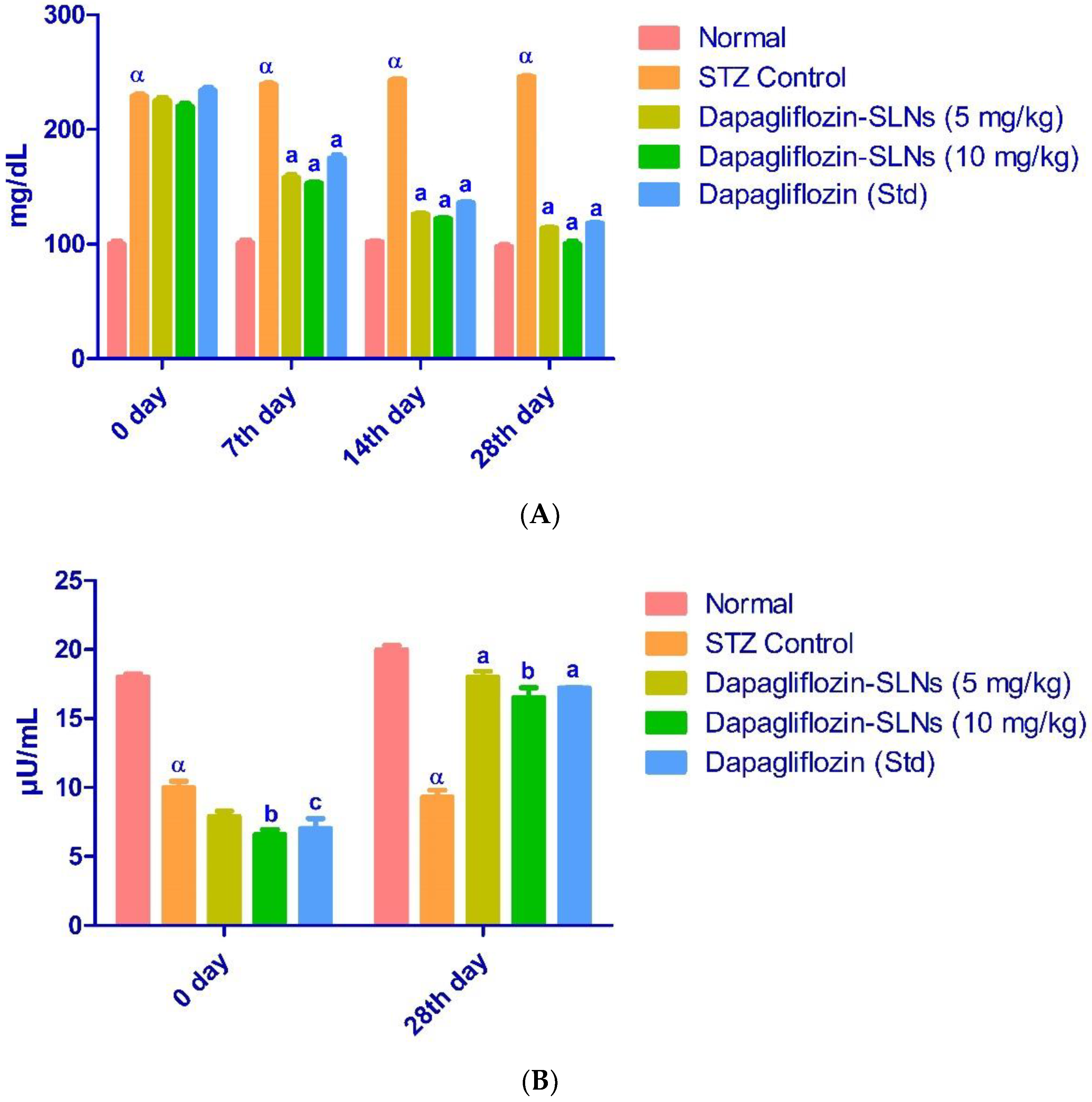
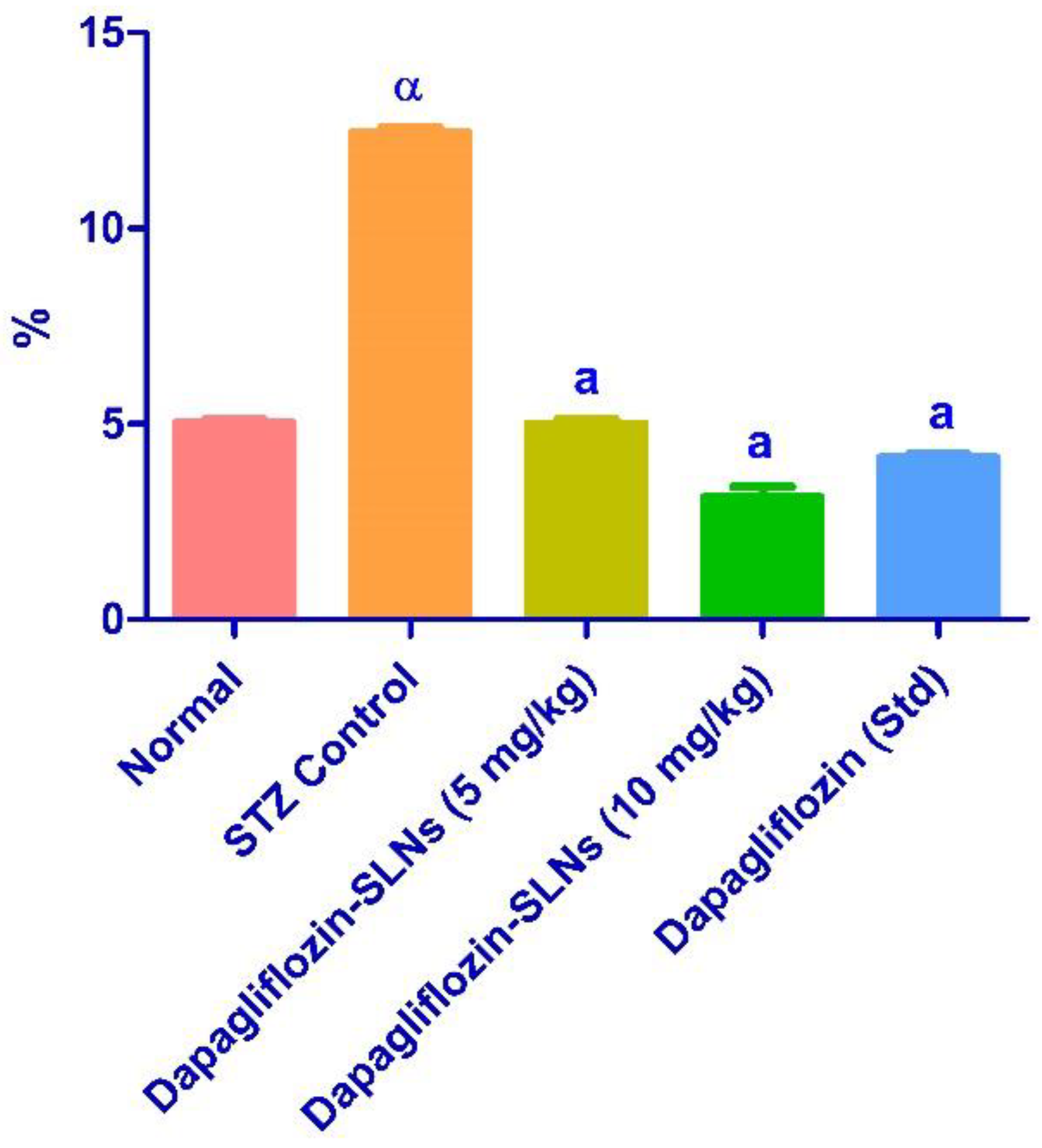
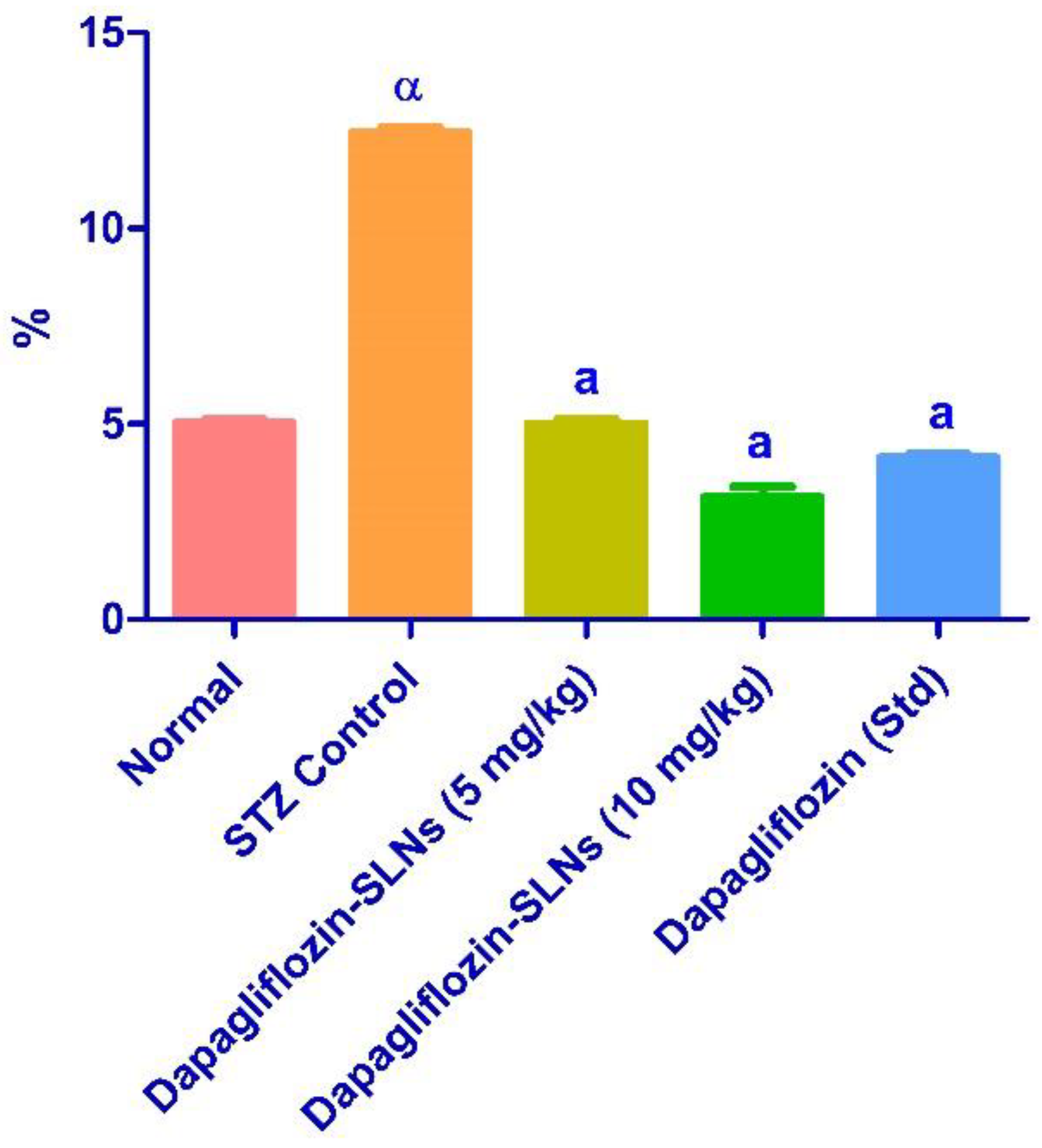
2.12. Effect of Dapagliflozin-SLNs on Insulin, HbA1c, and Blood Glucose Levels in STZ-Induced Diabetic Rats
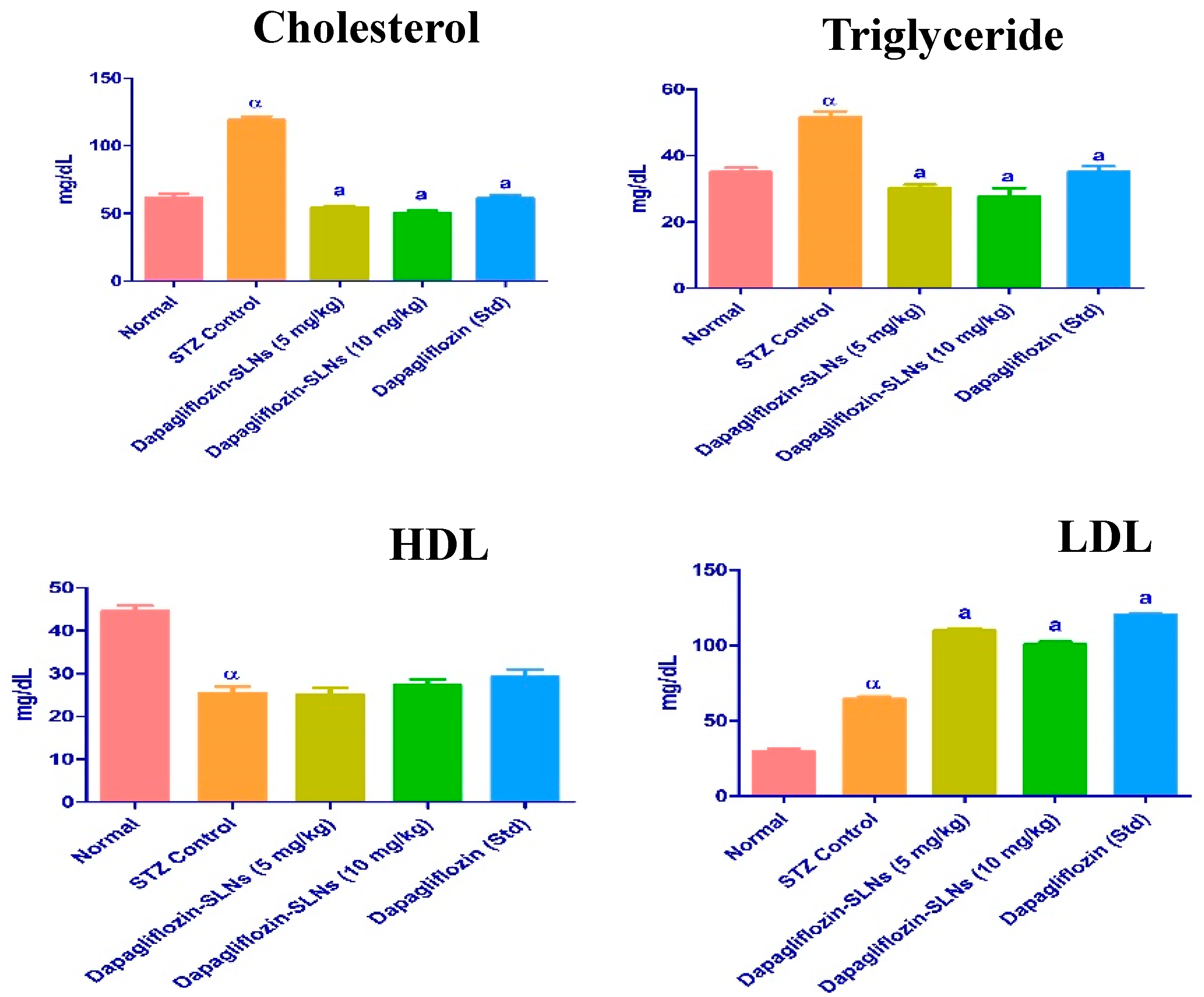
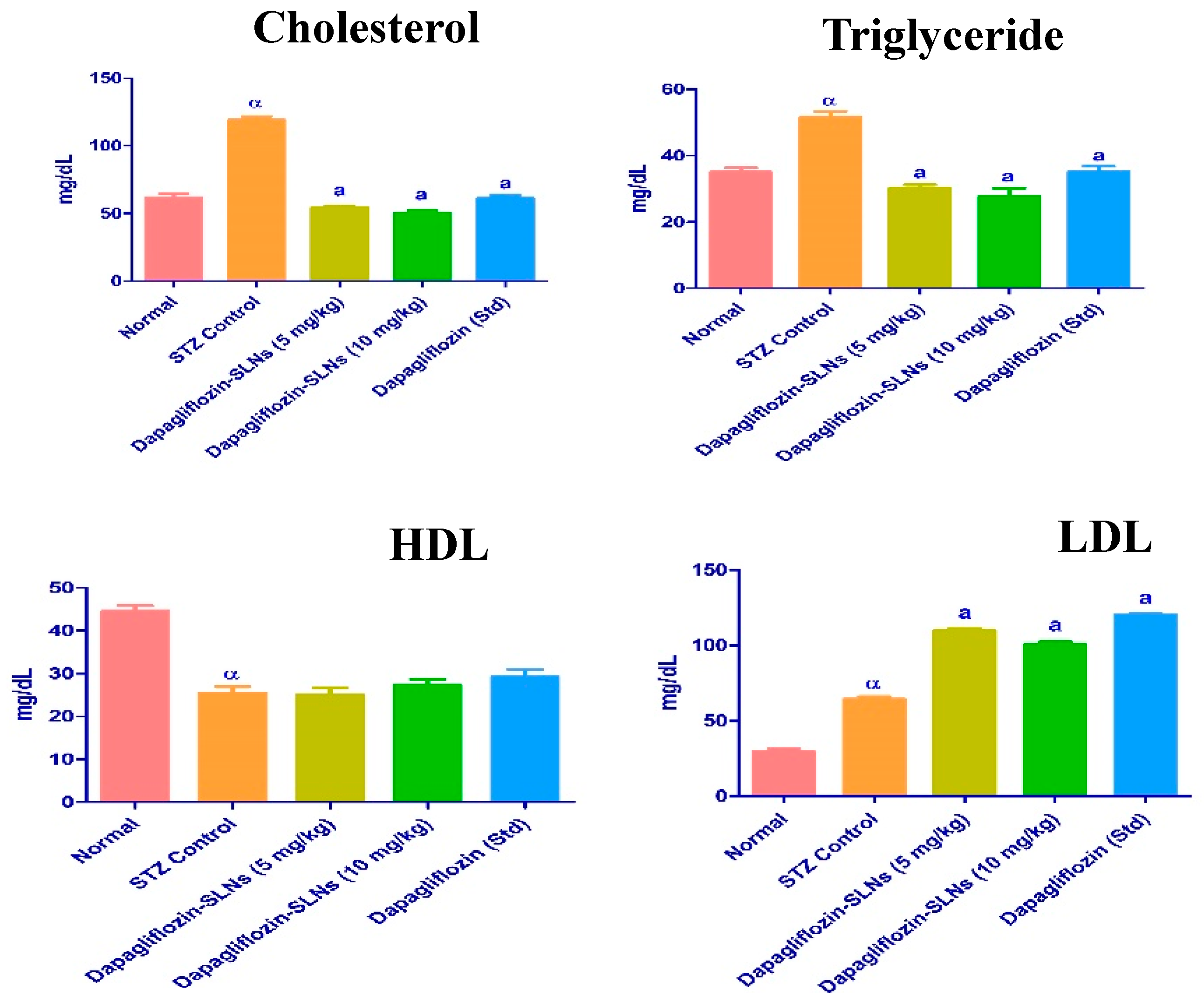
2.13. Lipid Profiles
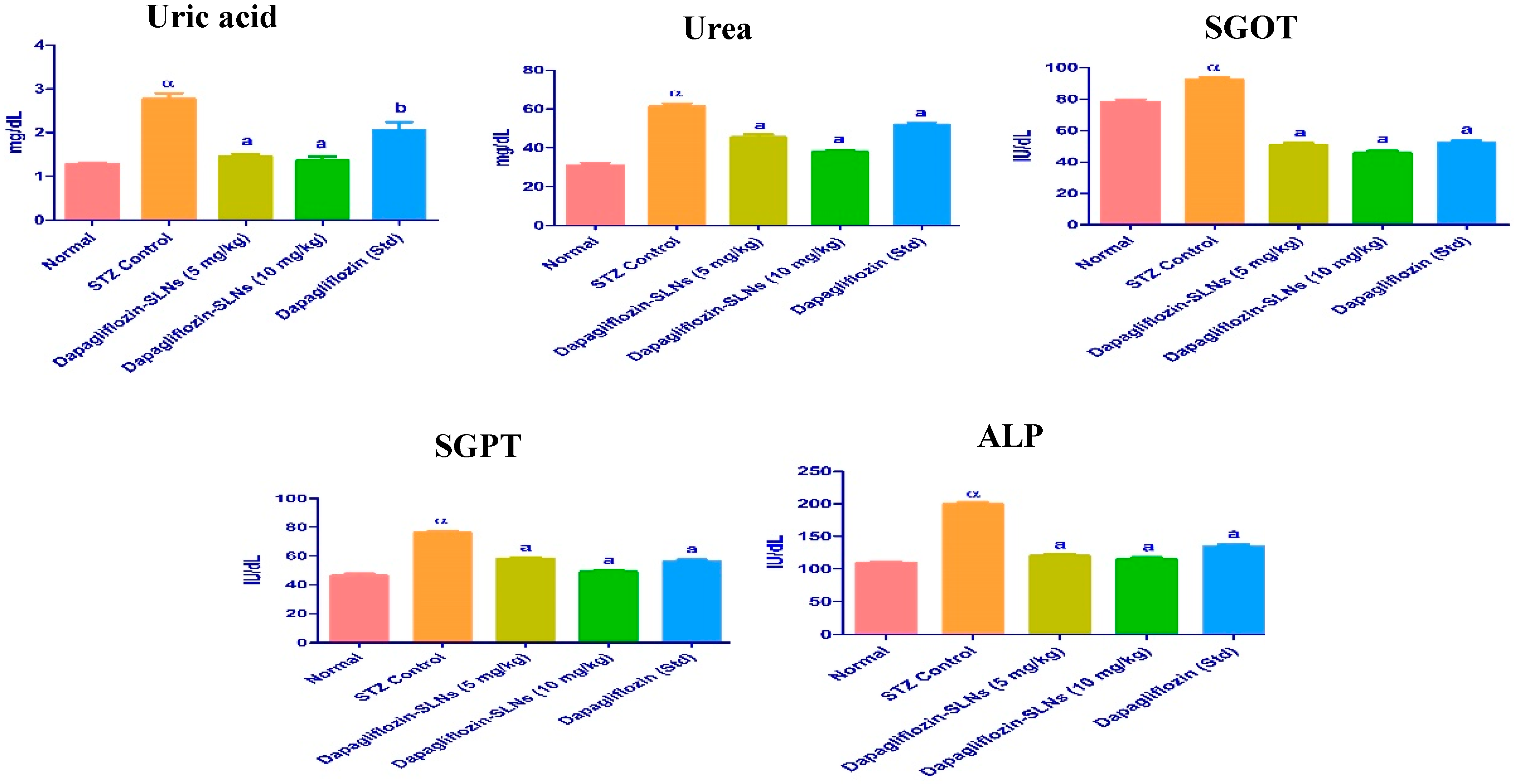
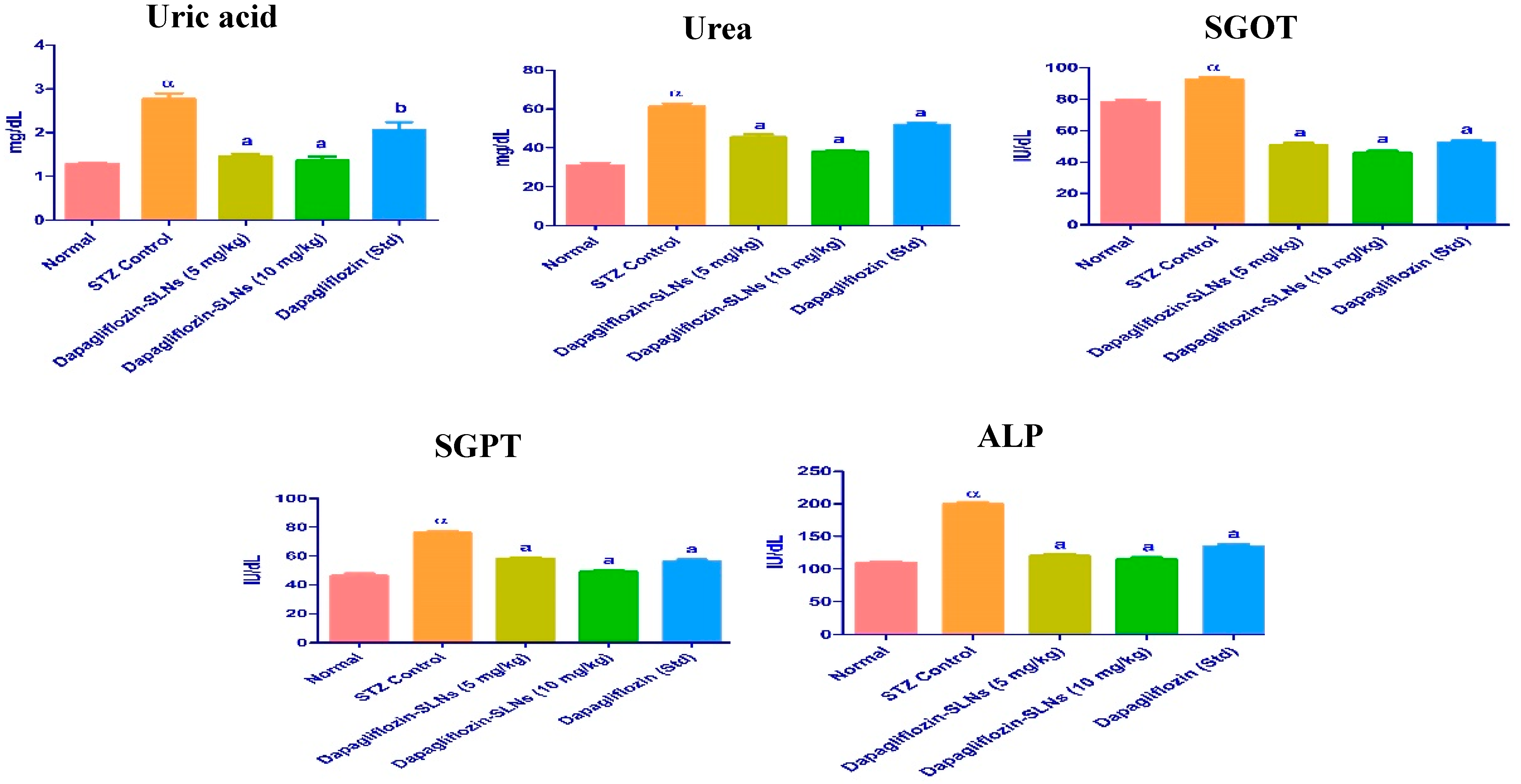
2.14. Biochemical Enzymes
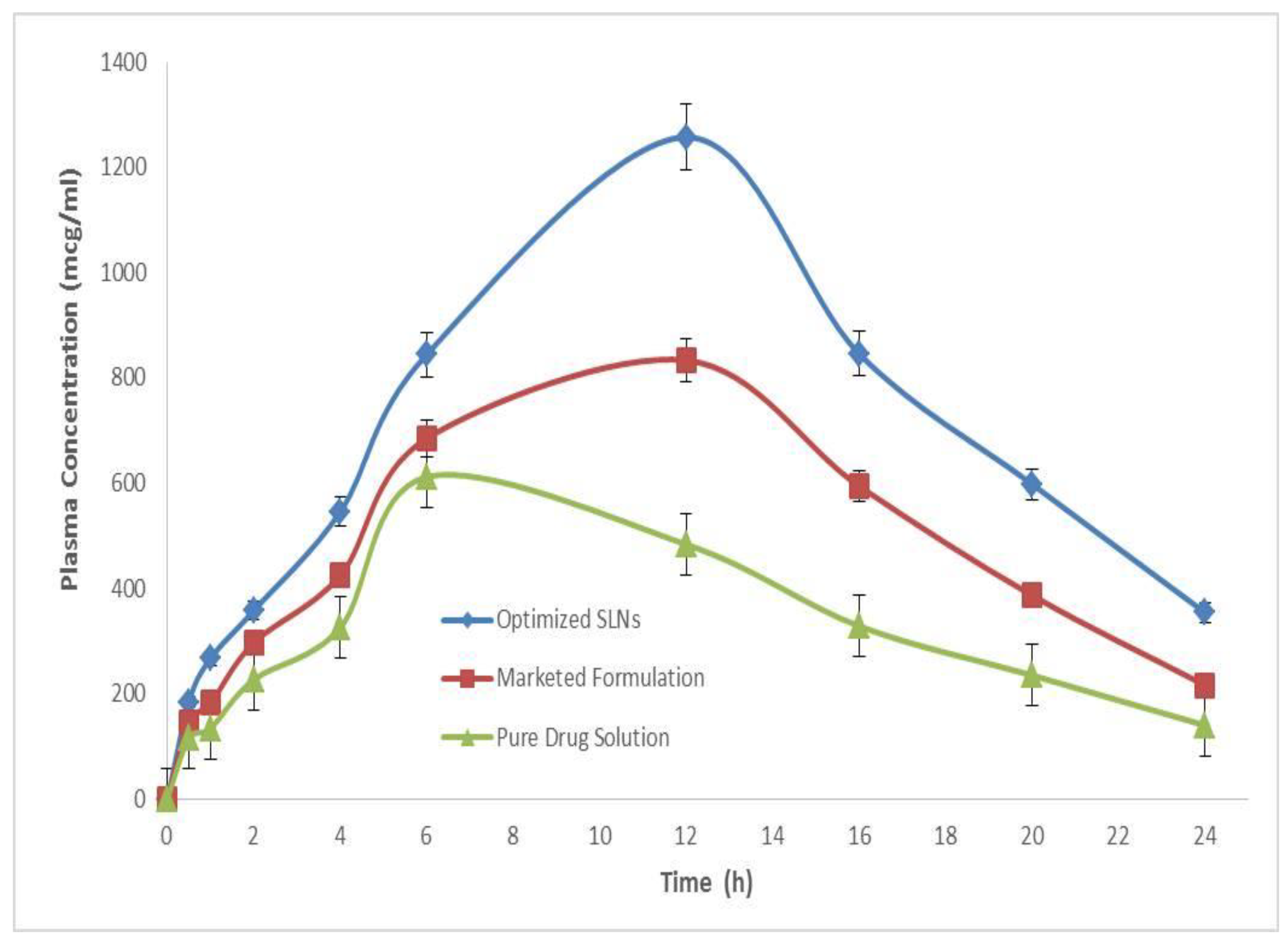
2.15. Oral Bioavailability Studies
2.16. Stability Studies
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Measurement of Dapagliflozin Solubility in Lipids
4.2.1. Selection of Appropriate Surfactant
4.2.2. Selection of Sonication Time and Amplitude
4.3. RP-HPLC Conditions for Analysis of Dapagliflozin
4.4. Preparation of SLNS
4.5. Experimental Design and Statistical Analysis
4.6. Measurement of Particle Size, PDI, and Zeta Potential of SLNs
4.7. Drug Loading
4.8. %EE
4.9. Solid-State Characterizations
4.9.1. FTIR
4.9.2. DSC
4.9.3. XRD
4.9.4. SEM
4.9.5. AFM
4.10. In Vitro Drug Release Studies and In Vivo Anti-Diabetic Studies
4.10.1. High-Fat Diet
4.10.2. Acute Toxicity Studies (Fixed-Dose Procedure)
4.10.3. Induction of Diabetes
4.10.4. Biochemical Estimations
4.10.5. Statistical Analysis
4.11. In Vivo Oral Bioavailability Studies
4.11.1. Animals
4.11.2. Pharmacokinetic Studies and Experimental Design
4.12. In Vitro UP-HPLC Analysis of Plasma Samples for Pharmacokinetic Assessment
4.13. Stability Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muzzalupo, R.; Tavano, L.; Cassano, R.; Trombino, S.; Ferrarelli, T.; Picci, N. A new approach for the evaluation of niosomes as effective transdermal drug delivery systems. Eur. J. Pharm. Biopharm. 2011, 79, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef]
- Canivell, S.; Mata-Cases, M.; Real, J.; Franch-Nadal, J.; Vlacho, B.; Khunti, K.; Gratacòs, M.; Mauricio, D. Glycaemic control after treatment intensification in patients with type 2 diabetes uncontrolled on two or more non-insulin antidiabetic drugs in a real-world setting. Diabetes Obes. Metab. 2019, 21, 1373–1380. [Google Scholar] [CrossRef]
- Spinks, C.B.; Zidan, A.S.; Khan, M.A.; Habib, M.J.; Faustino, P.J. Pharmaceutical characterization of novel tenofovir liposomal formulations for enhanced oral drug delivery: In vitro pharmaceutics and Caco-2 permeability investigations. Clin. Pharmacol. Adv. Appl. 2017, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Alsulays, B.B.; Anwer, M.K.; Soliman, G.A.; Alshehri, S.M.; Khafagy, E.-S. Impact of penetratin stereochemistry on the oral bioavailability of insulin-loaded solid lipid nanoparticles. Int. J. Nanomed. 2019, 14, 9127. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; Imam, S.S.; Aqil, M.; Ahad, A.; Sultana, Y.; Ameeduzzafar; Ali, A. Carvedilol nano lipid carriers: Formulation, characterization and in-vivo evaluation. Drug Deliv. 2016, 23, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Paudel, A.; Imam, S.S.; Fazil, M.; Khan, S.; Hafeez, A.; Ahmad, F.J.; Ali, A. Formulation and optimization of candesartan cilexetil nano lipid carrier: In vitro and in vivo evaluation. Curr. Drug Deliv. 2017, 14, 1005–1015. [Google Scholar] [CrossRef]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, P.; Ramalingam, P.; Karthivashan, G.; Ko, Y.T.; Choi, D.-K. Recent developments in solid lipid nanoparticle and surface-modified solid lipid nanoparticle delivery systems for oral delivery of phyto-bioactive compounds in various chronic diseases. Int. J. Nanomed. 2018, 13, 1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, B.; Peira, E.; Dianzani, C.; Gallarate, M.; Battaglia, L.; Gigliotti, C.L.; Boggio, E.; Dianzani, U.; Dosio, F. Development and characterization of solid lipid nanoparticles loaded with a highly active doxorubicin derivative. Nanomaterials 2018, 8, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevaskis, N.L.; Kaminskas, L.M.; Porter, C.J.H. From sewer to saviour—targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Cross, S.E.; Roberts, M.S. Physical enhancement of transdermal drug application: Is delivery technology keeping up with pharmaceutical development? Curr. Drug Deliv. 2004, 1, 81–92. [Google Scholar] [CrossRef]
- Jain, N.K. Advances in Controlled and Novel Drug Delivery; CBS Publishers & Distributors: New Delhi, India, 2008. [Google Scholar]
- Barry, B.W. Novel mechanisms and devices to enable successful transdermal drug delivery. Eur. J. Pharm. Sci. 2001, 14, 101–114. [Google Scholar] [CrossRef]
- Kandavilli, S.; Nair, V.; Panchagnula, R. Polymers in transdermal drug delivery systems. Pharm. Technol. 2002, 26, 62–81. [Google Scholar]
- Sharma, S. Topical preparations are used for the localized effects at the site of their application by virtue of drug penetration into the underlying layers of skin or mucous membranes. Pharm. Rev. 2008, 6, 1–10. [Google Scholar]
- Carbone, C.; Tomasello, B.; Ruozi, B.; Renis, M.; Puglisi, G. Preparation and optimization of PIT solid lipid nanoparticles via statistical factorial design. Eur. J. Med. Chem. 2012, 49, 110–117. [Google Scholar] [CrossRef]
- Ansari, M.D.; Ahmed, S.; Imam, S.S.; Khan, I.; Singhal, S.; Sharma, M.; Sultana, Y. CCD based development and characterization of nano-transethosome to augment the antidepressant effect of agomelatine on Swiss albino mice. J. Drug Deliv. Sci. Technol. 2019, 54, 101234. [Google Scholar] [CrossRef]
- Abedullahh, M.H. Box-behnken design for development and optimization of acetazolamide microspheres. India 2014, 5, 1228–1239. [Google Scholar]
- Abdallah, M.H.; Sabry, S.A.; Hasan, A.A. Enhancing Transdermal Delivery of Glimepiride Via Entrapment in Proniosomal Gel. J. Young Pharm. 2016, 8, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Hosny, K.M.; Khalid, M.; Alkhalidi, H.M. Quality by design approach to screen the formulation and process variables influencing the characteristics of carvedilol solid lipid nanoparticles. J. Drug Deliv. Sci. Technol. 2018, 45, 168–176. [Google Scholar] [CrossRef]
- Nepal, P.R.; Han, H.-K.; Choi, H.-K. Preparation and in vitro--in vivo evaluation of Witepsol®H35 based self-nanoemulsifying drug delivery systems (SNEDDS) of coenzyme Q10. Eur. J. Pharm. Sci. 2010, 39, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Nagarsenker, M.S. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int. J. Pharm. 2007, 329, 166–172. [Google Scholar] [CrossRef]
- Sharma, J.B.; Bhatt, S.; Saini, V.; Kumar, M. Pharmacokinetics and pharmacodynamics of curcumin-loaded solid lipid nanoparticles in the Management of Streptozotocin-Induced Diabetes Mellitus: Application of central composite design. Assay Drug Dev. Technol. 2021, 19, 262–279. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Yang, Y.; Shi, K.; Wu, J.; Zhao, W.; Mo, J. Preparation and characterization of loperamide-loaded dynasan 114 solid lipid nanoparticles for increased oral absorption in the treatment of diarrhea. Front. Pharmacol. 2016, 7, 332. [Google Scholar] [CrossRef] [Green Version]
- Manoochehri, S.; Darvishi, B.; Kamalinia, G.; Amini, M.; Fallah, M.; Ostad, S.N.; Atyabi, F.; Dinarvand, R. Surface modification of PLGA nanoparticles via human serum albumin conjugation for controlled delivery of docetaxel. DARU J. Pharm. Sci. 2013, 21, 58. [Google Scholar] [CrossRef] [Green Version]
- Lalani, J.; Patil, S.; Kolate, A.; Lalani, R.; Misra, A. Protein-functionalized PLGA nanoparticles of lamotrigine for neuropathic pain management. AAPS Pharmscitech 2015, 16, 413–427. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Ng, W.K.; Tan, R.B.H. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): Development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur. J. Pharm. Sci. 2012, 47, 139–151. [Google Scholar] [CrossRef]
- Ozturk, A.; Yenilmez, E.; Arslan, R.; Şenel, B.; Yazan, Y. Dexketoprofen trometamol loaded solid lipid nanoparticles (SLNs): Formulation, in vitro and in vivo evaluation. J. Res. Pharm. 2020, 24, 82–99. [Google Scholar]
- Shah, M.; Pathak, K. Development and statistical optimization of solid lipid nanoparticles of simvastatin by using 23 full-factorial design. AAPS Pharmscitech 2010, 11, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumana Chompootaweep, M.D. The pharmacokinetics of pioglitazone in Thai healthy subjects. J. Med. Assoc. Thail. 2006, 89, 2116–2122. [Google Scholar]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288. [Google Scholar] [PubMed]
- Kovacevic, A.; Savic, S.; Vuleta, G.; Mueller, R.H.; Keck, C.M. Polyhydroxy surfactants for the formulation of lipid nanoparticles (SLN and NLC): Effects on size, physical stability and particle matrix structure. Int. J. Pharm. 2011, 406, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, C.; Ahad, A.; Aqil, M.; Imam, S.S.; Sultana, Y. Formulation and optimization of nanostructured lipid carriers to enhance oral bioavailability of telmisartan using Box--Behnken design. J. Drug Deliv. Sci. Technol. 2018, 44, 431–439. [Google Scholar] [CrossRef]
- Venkateswarlu, V.; Manjunath, K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J. Control. Release 2004, 95, 627–638. [Google Scholar] [CrossRef]
- Kumar, V.V.; Chandrasekar, D.; Ramakrishna, S.; Kishan, V.; Rao, Y.M.; Diwan, P.V. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: Influence of wax and glyceride lipids on plasma pharmacokinetics. Int. J. Pharm. 2007, 335, 167–175. [Google Scholar] [CrossRef]
- Martin-Garcia, B.; Pimentel-Moral, S.; Gómez-Caravaca, A.M.; Arraez-Roman, D.; Segura-Carretero, A. Box-Behnken experimental design for a green extraction method of phenolic compounds from olive leaves. Ind. Crops Prod. 2020, 154, 112741. [Google Scholar] [CrossRef]
- Kushwaha, A.K.; Vuddanda, P.R.; Karunanidhi, P.; Singh, S.K.; Singh, S. Development and evaluation of solid lipid nanoparticles of raloxifene hydrochloride for enhanced bioavailability. Biomed Res. Int. 2013, 2013, 584549. [Google Scholar] [CrossRef] [Green Version]
- Mohsin, K.; Alamri, R.; Ahmad, A.; Raish, M.; Alanazi, F.K.; Hussain, M.D. Development of self-nanoemulsifying drug delivery systems for the enhancement of solubility and oral bioavailability of fenofibrate, a poorly water-soluble drug. Int. J. Nanomed. 2016, 11, 2829. [Google Scholar]
- Agarwal, S.; Murthy, R.S.R.; Harikumar, S.L.; Garg, R. Quality by design approach for development and characterisation of solid lipid nanoparticles of quetiapine fumarate. Curr. Comput. Aided. Drug Des. 2020, 16, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Gade, S.; Patel, K.K.; Gupta, C.; Anjum, M.M.; Deepika, D.; Agrawal, A.K.; Singh, S. An ex vivo evaluation of moxifloxacin nanostructured lipid carrier enriched in situ gel for transcorneal permeation on goat cornea. J. Pharm. Sci. 2019, 108, 2905–2916. [Google Scholar] [CrossRef]
- Pandey, S.; Patel, P.; Gupta, A. Novel solid lipid nanocarrier of glibenclamide: A factorial design approach with response surface methodology. Curr. Pharm. Des. 2018, 24, 1811–1820. [Google Scholar] [CrossRef]
- Gonçalves, L.M.D.; Maestrelli, F.; Mannelli, L.D.C.; Ghelardini, C.; Almeida, A.J.; Mura, P. Development of solid lipid nanoparticles as carriers for improving oral bioavailability of glibenclamide. Eur. J. Pharm. Biopharm. 2016, 102, 41–50. [Google Scholar] [CrossRef]
- Sefidgar, S.M.; Ahmadi-Hamedani, M.; Javan, A.J.; Sani, R.N.; Vayghan, A.J. Effect of crocin on biochemical parameters, oxidative/antioxidative profiles, sperm characteristics and testicular histopathology in streptozotocin-induced diabetic rats. Avicenna J. Phytomed. 2019, 9, 347. [Google Scholar]
- Nabi, S.A.; Kasetti, R.B.; Sirasanagandla, S.; Tilak, T.K.; Kumar, M.V.J.; Rao, C.A. Antidiabetic and antihyperlipidemic activity of Piper longum root aqueous extract in STZ induced diabetic rats. BMC Complement. Altern. Med. 2013, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.; Sharma, S.; Khuller, G.K. Oral solid lipid nanoparticle-based antitubercular chemotherapy. Tuberculosis 2005, 85, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Heiati, H.; Tawashi, R.; Phillips, N.C. Drug retention and stability of solid lipid nanoparticles containing azidothymidine palmitate after autoclaving, storage and lyophilization. J. Microencapsul. 1998, 15, 173–184. [Google Scholar] [CrossRef]
- Kazi, M.; Al-Swairi, M.; Ahmad, A.; Raish, M.; Alanazi, F.K.; Badran, M.M.; Khan, A.A.; Alanazi, A.M.; Hussain, M.D. Evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for poorly water-soluble talinolol: Preparation, in vitro and in vivo assessment. Front. Pharmacol. 2019, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- Kazi, M.; Shahba, A.A.; Alrashoud, S.; Alwadei, M.; Sherif, A.Y.; Alanazi, F.K. Bioactive self-nanoemulsifying drug delivery systems (Bio-SNEDDS) for combined oral delivery of curcumin and piperine. Molecules 2020, 25, 1703. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, G.; Zhang, T.; Ma, Z.; Wu, B. Effects of PEGylated lipid nanoparticles on the oral absorption of one BCS II drug: A mechanistic investigation. Int. J. Nanomed. 2014, 9, 5503. [Google Scholar]
- Agency, E.M. ICH Topic Q 1 A (R2) Stability Testing of New Drug Substances and Products. 2003. Available online: https://www.ema.europa.eu/en/ich-q1a-r2-stability-testing-new-drug-substances-drug-products (accessed on 17 January 2022).
- Ahmad, J.; Mir, S.R.; Kohli, K.; Amin, S. Effect of oil and co-surfactant on the formation of Solutol HS 15 based colloidal drug carrier by Box--Behnken statistical design. Colloids Surf. A Physicochem. Eng. Asp. 2014, 453, 68–77. [Google Scholar] [CrossRef]
- Ahmad, J.; Kohli, K.; Mir, S.R.; Amin, S. Formulation of self-nanoemulsifying drug delivery system for telmisartan with improved dissolution and oral bioavailability. J. Dispers. Sci. Technol. 2011, 32, 958–968. [Google Scholar] [CrossRef]
- Behbahani, E.S.; Ghaedi, M.; Abbaspour, M.; Rostamizadeh, K. Optimization and characterization of ultrasound assisted preparation of curcumin-loaded solid lipid nanoparticles: Application of central composite design, thermal analysis and X-ray diffraction techniques. Ultrason. Sonochem. 2017, 38, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Amasya, G.; Aksu, B.; Badilli, U.; Onay-Besikci, A.; Tarimci, N. QbD guided early pharmaceutical development study: Production of lipid nanoparticles by high pressure homogenization for skin cancer treatment. Int. J. Pharm. 2019, 563, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Bhatnagar, A.; Kumar, N.; Ali, A. Others Chitosan nanoparticles amplify the ocular hypotensive effect of cateolol in rabbits. Int. J. Biol. Macromol. 2014, 65, 479–491. [Google Scholar]
- Imam, S.S.; Bukhari, S.N.A.; Ahmad, J.; Ali, A. Others Formulation and optimization of levofloxacin loaded chitosan nanoparticle for ocular delivery: In-vitro characterization, ocular tolerance and antibacterial activity. Int. J. Biol. Macromol. 2018, 108, 650–659. [Google Scholar]
- Agarwal, V.; Siddiqui, A.; Ali, H.; Nazzal, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEDDS). Int. J. Pharm. 2009, 366, 44–52. [Google Scholar] [CrossRef]
- Atef, E.; Belmonte, A.A. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS). Eur. J. Pharm. Sci. 2008, 35, 257–263. [Google Scholar] [CrossRef]
- Shaveta, S.; Singh, J.; Afzal, M.; Kaur, R.; Imam, S.S.; Alruwaili, N.K.; Alharbi, K.S.; Alotaibi, N.H.; Alshammari, M.S.; Kazmi, I.; et al. Development of solid lipid nanoparticle as carrier of pioglitazone for amplification of oral efficacy: Formulation design optimization, in-vitro characterization and in-vivo biological evaluation. J. Drug Deliv. Sci. Technol. 2020, 57, 101674. [Google Scholar] [CrossRef]
- Ekambaram, P.; Sathali, A.A.H. Formulation and evaluation of solid lipid nanoparticles of ramipril. J. Young Pharm. 2011, 3, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Pandit, J.; Sultana, Y.; Mishra, A.K.; Hazari, P.P.; Aqil, M. Optimization by design of etoposide loaded solid lipid nanoparticles for ocular delivery: Characterization, pharmacokinetic and deposition study. Mater. Sci. Eng. C 2019, 100, 959–970. [Google Scholar] [CrossRef]
- Chadha, R.; Bhandari, S. Drug--excipient compatibility screening—role of thermoanalytical and spectroscopic techniques. J. Pharm. Biomed. Anal. 2014, 87, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Tamjidi, F.; Shahedi, M.; Varshosaz, J.; Nasirpour, A. Design and characterization of astaxanthin-loaded nanostructured lipid carriers. Innov. Food Sci. Emerg. Technol. 2014, 26, 366–374. [Google Scholar] [CrossRef]
- Montenegro, L.; Sinico, C.; Castangia, I.; Carbone, C.; Puglisi, G. Idebenone-loaded solid lipid nanoparticles for drug delivery to the skin: In vitro evaluation. Int. J. Pharm. 2012, 434, 169–174. [Google Scholar] [CrossRef]
- Colas, J.-C.; Shi, W.; Rao, V.S.N.M.; Omri, A.; Mozafari, M.R.; Singh, H. Microscopical investigations of nisin-loaded nanoliposomes prepared by Mozafari method and their bacterial targeting. Micron 2007, 38, 841–847. [Google Scholar] [CrossRef]
- Aljaeid, B.M.; Hosny, K.M. Miconazole-loaded solid lipid nanoparticles: Formulation and evaluation of a novel formula with high bioavailability and antifungal activity. Int. J. Nanomed. 2016, 11, 441. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Wang, Y.; Wang, K.; Ji, B.; Zhou, F. Stability of a type 2 diabetes rat model induced by high-fat diet feeding with low-dose streptozotocin injection. J. Zhejiang Univ. B 2018, 19, 559–569. [Google Scholar] [CrossRef]
- Trinder, P. Determination of glucose in blood using glucose oxidase with an alternative oxygen acceptor. Ann. Clin. Biochem. 1969, 6, 24–27. [Google Scholar] [CrossRef]
- Pappa, H.; Farru, R.; Vilanova, P.O.; Palacios, M.; Pizzorno, M.T. A new HPLC method to determine Donepezil hydrochloride in tablets. J. Pharm. Biomed. Anal. 2002, 27, 177–182. [Google Scholar] [CrossRef]
- Abdelbary, G.A.; Tadros, M.I. Brain targeting of olanzapine via intranasal delivery of core--shell difunctional block copolymer mixed nanomicellar carriers: In vitro characterization, ex vivo estimation of nasal toxicity and in vivo biodistribution studies. Int. J. Pharm. 2013, 452, 300–310. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Dependent Variables | |||||||
|---|---|---|---|---|---|---|---|---|
| F. Code | X1 (% w/v) | X2 (% w/v) | X3 (min) | Y1 (%EE) | Y2 (CDR %) | Y3 (PS) (nm) | Zeta | PDI |
| F1 | +1 | 0 | −1 | 92.06 ± 1.2 | 75.13 ± 2.8 | 280.23 ± 8.9 | −27.8 ± 1.01 | 0.45 ± 0.05 |
| F2 | 0 | 0 | 0 | 86.08 ± 2.3 | 86.06 ± 2.4 | 190.13 ± 4.6 | −26.4 ± 0.62 | 0.56 ± 0.01 |
| F3 | 0 | −1 | +1 | 75.31 ± 0.5 | 88.08 ± 0.7 | 189.08 ± 4.2 | −22.5 ± 0.72 | 0.42 ± 0.06 |
| F4 | −1 | −1 | 0 | 79.10 ± 0.7 | 89.28 ± 0.2 | 150.37 ± 4.6 | −35.8 ± 0.22 | 0.37 ± 0.01 |
| F5 | +1 | 0 | +1 | 68.26 ± 0.2 | 83.34 ± 1.1 | 398.49 ± 2.1 | −33.4 ± 0.62 | 0.41 ± 0.02 |
| F6 | +1 | +1 | 0 | 82.34 ± 1.8 | 76.29 ± 1.8 | 199.05 ± 2.8 | −31.9 ± 1.64 | 0.80 ± 0.07 |
| F7 | −1 | +1 | 0 | 84.61 ± 0.4 | 65.43 ± 2.7 | 320.11 ± 8.4 | −38.7 ± 1.06 | 0.58 ± 0.01 |
| F8 | +1 | −1 | 0 | 76.81 ± 2.8 | 77.09 ± 3.5 | 162.18 ± 6.2 | −30.9 ± 0.62 | 0.41 ± 0.02 |
| F9 | 0 | 0 | 0 | 87.12 ± 0.6 | 87.29 ± 4.1 | 210.12 ± 3.7 | −33.5 ± 0.92 | 0.47 ± 0.01 |
| F10 | −1 | 0 | −1 | 78.84 ± 1.5 | 73.73 ± 1.9 | 399.08 ± 2.4 | −29.7 ± 1.08 | 0.80 ± 0.07 |
| F11 | 0 | 0 | 0 | 86.09 ± 1.1 | 86.26 ± 3.7 | 202.23 ± 5.4 | −28.7 ± 0.62 | 0.92 ± 0.10 |
| F12 | 0 | −1 | 0 | 94.46 ± 0.7 | 99.08 ± 0.4 | 100.13 ± 7.2 | −34.4 ±1.64 | 0.32 ± 0.02 |
| F13 | 0 | 0 | −1 | 88.21 ± 0.2 | 84.26 ± 2.4 | 220.29 ± 5.1 | −25.6±1.13 | 0.58 ± 0.01 |
| F14 | 0 | +1 | −1 | 89.37 ± 1.6 | 62.83 ± 5.1 | 278.84 ± 4.9 | −31.1 ± 0.72 | 0.82 ± 0.04 |
| F15 | 0 | 0 | −1 | 86.31 ± 0.5 | 86.13 ± 2.4 | 198.29 ± 3.4 | −30.4 ± 1.44 | 0.80 ± 0.07 |
| F16 | −1 | 0 | +1 | 87.94 ± 0.2 | 88.07 ± 1.5 | 355.71 ± 0.9 | −32.3 ± 1.61 | 0.47 ± 0.01 |
| F17 | 0 | +1 | +1 | 89.29 ± 1.5 | 87.01 ± 2.8 | 315.25 ± 3.4 | −29.1 ± 0.62 | 0.41 ± 0.02 |
| Parameter | Source | Df | Sum of Squares | Mean of Squares | F-Values | p-Values |
|---|---|---|---|---|---|---|
| %EE | Model | 9 | 714.83 | 79.43 | 50.86 | <0.0001 |
| Residual | 7 | 10.93 | 1.56 | |||
| Lack of Fit | 4 | 10.18 | 2.55 | 10.18 | ||
| Pure Error | 3 | 0.7500 | 0.2500 | |||
| %CDR | Model | 9 | 1143.14 | 127.02 | 54.26 | <0.0001 |
| Residual | 7 | 16.39 | 2.34 | |||
| Lack of Fit | 4 | 15.64 | 3.91 | 15.64 | ||
| Pure Error | 3 | 0.7500 | 0.2500 | |||
| Particle size | Model | 9 | 1.204 × 105 | 13,377.76 | 33.59 | <0.0001 |
| Residual | 7 | 398.30 | ||||
| Lack of Fit | 4 | 645.03 | 9.30 | |||
| Pure Error | 3 | 69.33 |
| Y₁ (%EE) | Y₂ (CDR %) | Y₃ (PARTICLE SIZE) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Model | R² | Adjusted R² | Predicted R² | R² | Adjusted R² | Predicted R² | R² | Adjusted R² | Predicted R² |
| Linear | 0.296 | 0.134 | −0.437 | 0.473 | 0.352 | −0.038 | 0.299 | 0.137 | −0.383 |
| 2FI | 0.810 | 0.697 | 0.172 | 0.806 | 0.691 | 0.269 | 0.416 | 0.067 | −1.646 |
| Quadratic | 0.993 | 0.985 | 0.916 | 0.975 | 0.945 | 0.920 | 0.997 | 0.993 | 0.991 |
| Cubic | 0.891 | 0.645 | 0.812 | 0.654 | 0.726 | 0.564 | |||
| p-value | <0.0001 | <0.0001 | <0.0001 | ||||||
| Pharmacokinetic Parameters | Pure Drug Solution | Marketed Formulation | Optimized SLNs |
|---|---|---|---|
| Intercept | 2.378 | 2.481 | 2.582 |
| Slope | 0.0019 | 0.0076 | 0.011 |
| Co (mcg/mL) | 238.850 | 303.024 | 382.68 |
| K (h−1) | 0.0044 | 0.0175 | 0.027 |
| Dose (mg) | 100 | 100 | 100 |
| Dose (mcg) | 100,000 | 100,000 | 100,000 |
| Vd (mL) | 41.86 | 33.00 | 26.13 |
| Vd (L) | 0.041 | 0.033 | 0.026 |
| t1/2 (h) | 155.26 | 39.54 | 25.20 |
| Cl (L/h) | 0.0001 | 0.0005 | 0.0007 |
| AUCo-t (mcg.h/mL) | 59.83 | 54.23 | 95.79 |
| AUC1-t (mcg.h/mL) | 8531.975 | 12,744.44 | 18,036.93 |
| AUC1-inf (mcg.h/mL) | 31,425.20 | 12,349.3 | 12,885.67 |
| AUCtotal (mcg.h/mL) | 22,833.39 | 449.326 | 5247.046 |
| Cmax (mcg/mL/h) | 834.26 | 621.57 | 1258.37 |
| Tmax (mL/min) | 12.1 | 5.97 | 12.06 |
| Months | Temperature (°C) | EE (%) | CDR (%) | Drug Content (%) | Vesicle Size (nm) | Zeta Value (mv) |
|---|---|---|---|---|---|---|
| 1st Month | Refrigeration temperature (4 ± 2 °C) | 94.46 ± 0.7 | 99.08 ± 0.4 | 98.49 ± 2.1 | 150.37 ± 4.6 | −34.4 ± 1.64 |
| 2nd Month | 92.34 ± 2.1 | 96.45 ± 0.1 | 98.12 ± 1.1 | 167.42 ± 3.8 | −32.1 ± 1.1 | |
| 3rd Month | 89.26 ± 0.5 | 92.46 ± 0.4 | 97.89 ± 1.3 | 180.64 ± 2.4 | −30.9 ± 1.4 | |
| 1st Month | Room temperature (30 ± 2 °C) | 94.46 ± 0.7 | 99.08 ± 0.4 | 98.49 ± 2.1 | 150.37 ± 4.6 | −34.4 ± 1.64 |
| 2nd Month | 94.13 ± 0.1 | 99.02 ± 0.2 | 98.15 ± 1.1 | 155.26 ± 2.5 | −33.1 ± 0.38 | |
| 3rd Month | 93.46 ± 0.4 | 98.19 ± 0.1 | 97.89 ± 0.3 | 160.45 ± 1.4 | −32.41 ± 0.26 |
| Parameter | Units | Low (−1) | High (+1) |
|---|---|---|---|
| X1- Polymer concentration | % (w/v) | 1 | 5 |
| X2- Surfactant | % (w/v) | 2 | 2.5 |
| X3- Stirring Speed | min | 2 | 5 |
| Dependent Variables | Low | High | |
| Y1- %EE | 68.26 ± 0.2 | 94.46 ± 0.7 | |
| Y2- %CDR | 62.83 ± 5.1 | 99.08 ± 0.4 | |
| Y3- Particle Size | 100.13 ± 7.2 | 399.08 ± 2.4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unnisa, A.; Chettupalli, A.K.; Al Hagbani, T.; Khalid, M.; Jandrajupalli, S.B.; Chandolu, S.; Hussain, T. Development of Dapagliflozin Solid Lipid Nanoparticles as a Novel Carrier for Oral Delivery: Statistical Design, Optimization, In-Vitro and In-Vivo Characterization, and Evaluation. Pharmaceuticals 2022, 15, 568. https://doi.org/10.3390/ph15050568
Unnisa A, Chettupalli AK, Al Hagbani T, Khalid M, Jandrajupalli SB, Chandolu S, Hussain T. Development of Dapagliflozin Solid Lipid Nanoparticles as a Novel Carrier for Oral Delivery: Statistical Design, Optimization, In-Vitro and In-Vivo Characterization, and Evaluation. Pharmaceuticals. 2022; 15(5):568. https://doi.org/10.3390/ph15050568
Chicago/Turabian StyleUnnisa, Aziz, Ananda K. Chettupalli, Turki Al Hagbani, Mohammad Khalid, Suresh B. Jandrajupalli, Swarnalatha Chandolu, and Talib Hussain. 2022. "Development of Dapagliflozin Solid Lipid Nanoparticles as a Novel Carrier for Oral Delivery: Statistical Design, Optimization, In-Vitro and In-Vivo Characterization, and Evaluation" Pharmaceuticals 15, no. 5: 568. https://doi.org/10.3390/ph15050568
APA StyleUnnisa, A., Chettupalli, A. K., Al Hagbani, T., Khalid, M., Jandrajupalli, S. B., Chandolu, S., & Hussain, T. (2022). Development of Dapagliflozin Solid Lipid Nanoparticles as a Novel Carrier for Oral Delivery: Statistical Design, Optimization, In-Vitro and In-Vivo Characterization, and Evaluation. Pharmaceuticals, 15(5), 568. https://doi.org/10.3390/ph15050568