
HIV-1 Integrase Strand Transfer Inhibitors and Neurodevelopment


Abstract
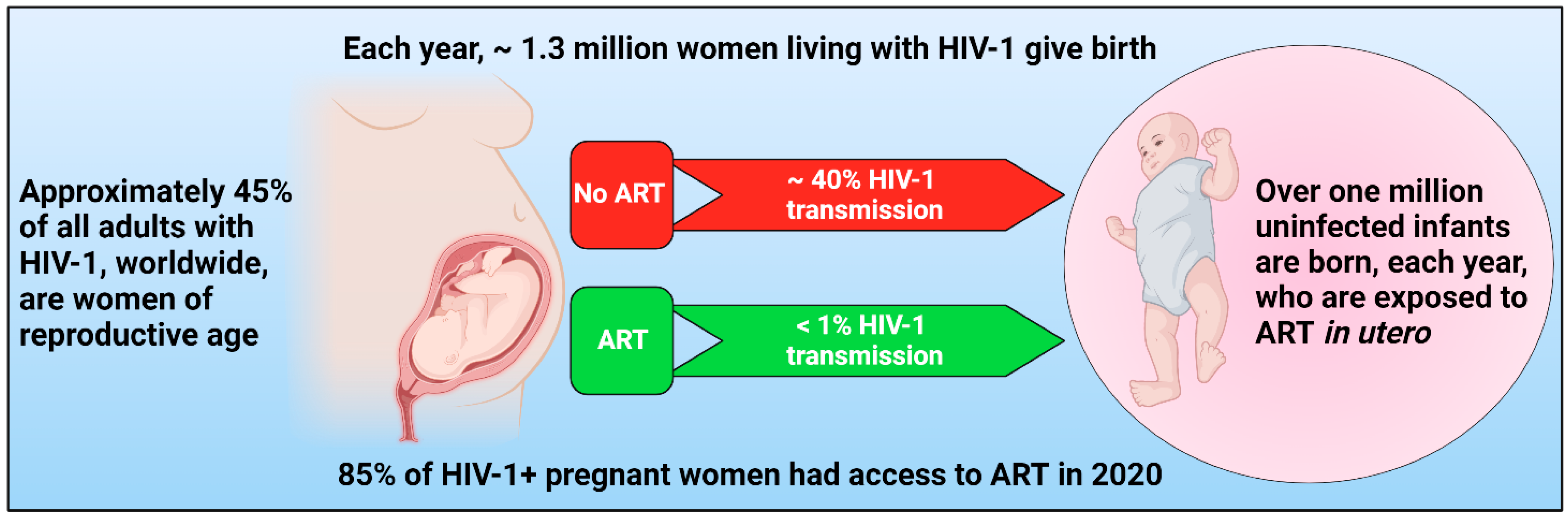
1. Introduction
2. Fetal Neurodevelopment
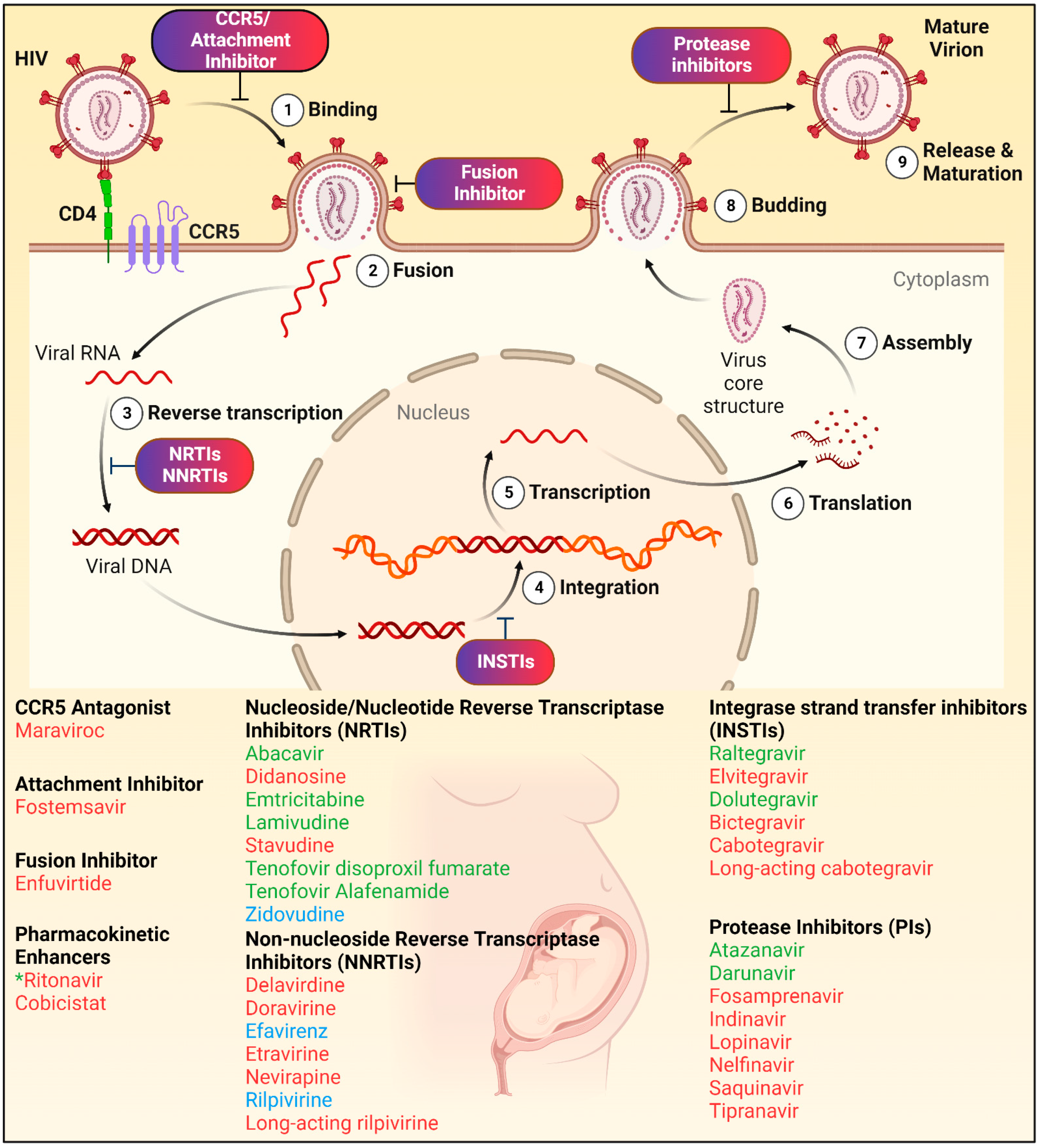
3. Antiretroviral Drugs (ARVs)
4. ARVs during Pregnancy
5. INSTI PK Profiles during Pregnancy
5.1. Raltegravir (RAL)
5.2. Elvitegravir (EVG)
5.3. Dolutegravir (DTG)
5.4. Bictegravir (BIC)
5.5. Cabotegravir (CAB)
6. INSTI Adverse Events
6.1. Neuropsychiatric Adverse Events
6.2. Weight Gain
6.3. Diabetes Mellitus
7. Known Side Effects of INSTIs during Development
7.1. Neural Tube Defects (NTDs)
7.2. Postnatal Neurodevelopmental Deficits
8. Potential Mechanisms Affecting Neurodevelopment
8.1. Maternal Mental Health
8.2. Maternal Weight Gain
8.3. Folate Levels
8.4. Neurotoxicity
8.5. Bilirubin Neurotoxicity
8.6. Inhibition of Matrix Metalloproteinase (MMPs) Activity
9. Long-Acting ARV Formulations and Neuroprotection
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection: Recommendations for a Public Health Approach—Second Edition. Available online: https://www.who.int/hiv/pub/arv/chapter4.pdf?ua=1 (accessed on 27 February 2022).
- Department of Health and Human Services (DHHS), Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. Available online: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/AdultandAdolescentGL.pdf (accessed on 5 August 2022).
- Zash, R.; Makhema, J.; Shapiro, R.L. Neural-Tube Defects with Dolutegravir Treatment from the Time of Conception. N. Engl. J. Med. 2018, 379, 979–981. [Google Scholar] [CrossRef] [PubMed]
- The U.S. Department of Health and Human Services. Recommendations for the Use of Antiretroviral Drugs in Pregnant Women with HIV Infection and Interventions to Reduce Perinatal HIV Transmission in the United States. Available online: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/Perinatal_GL.pdf (accessed on 5 August 2022).
- World Health Organization (WHO). Update of Recommendations on First- and Second-Line Antiretroviral Regimens. Available online: https://apps.who.int/iris/bitstream/handle/10665/325892/WHO-CDS-HIV-19.15-eng.pdf (accessed on 1 October 2022).
- Centers for Disease Control and Prevention. HIV and Pregnant Women, Infants, and Children. Available online: https://www.cdc.gov/hiv/group/gender/pregnantwomen/index.html (accessed on 7 April 2022).
- Peters, H.; Francis, K.; Sconza, R.; Horn, A.; Peckham, C.S.; Tookey, P.A.; Thorne, C. UK Mother-to-Child HIV Transmission Rates Continue to Decline: 2012–2014. Clin. Infect. Dis. 2017, 64, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Schnoll, J.G.; Temsamrit, B.; Zhang, D.; Song, H.; Ming, G.L.; Christian, K.M. Evaluating Neurodevelopmental Consequences of Perinatal Exposure to Antiretroviral Drugs: Current Challenges and New Approaches. J. Neuroimmune Pharmacol. 2019, 16, 113–129. [Google Scholar] [CrossRef]
- Rasi, V.; Peters, H.; Sconza, R.; Francis, K.; Bukasa, L.; Thorne, C.; Cortina-Borja, M. Trends in antiretroviral use in pregnancy in the UK and Ireland, 2008–2018. HIV Med. 2022, 23, 397–405. [Google Scholar] [CrossRef] [PubMed]
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). Start Free, Stay Free, AIDS Free Final Report on 2020 Targets. Available online: https://www.unaids.org/en/resources/documents/2021/start-free-stay-free-aids-free-final-report-on-2020-targets (accessed on 7 April 2022).
- Crowell, C.S.; Williams, P.L.; Yildirim, C.; Van Dyke, R.B.; Smith, R.; Chadwick, E.G.; Seage, G.R., 3rd; Diperna, A.; Hazra, R.; the Pediatric HIV/AIDS Cohort Study. Safety of in-utero antiretroviral exposure: Neurologic outcomes in children who are HIV-exposed but uninfected. AIDS 2020, 34, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Ramokolo, V.; Goga, A.E.; Slogrove, A.L.; Powis, K.M. Unmasking the vulnerabilities of uninfected children exposed to HIV. BMJ 2019, 366, l4479. [Google Scholar] [CrossRef] [PubMed]
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). Global HIV & AIDS Statistics Fact Sheet—2021; The Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2021. [Google Scholar]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. Integrase Strand Transfer Inhibitors Are Effective Anti-HIV Drugs. Viruses 2021, 13, 205. [Google Scholar] [CrossRef]
- The Lancet HIV. End resistance to dolutegravir roll-out. Lancet HIV 2020, 7, e593. [Google Scholar] [CrossRef]
- Dorward, J.; Lessells, R.; Drain, P.K.; Naidoo, K.; de Oliveira, T.; Pillay, Y.; Abdool Karim, S.S.; Garrett, N. Dolutegravir for first-line antiretroviral therapy in low-income and middle-income countries: Uncertainties and opportunities for implementation and research. Lancet HIV 2018, 5, E400–E404. [Google Scholar] [CrossRef]
- Hill, A.; Clayden, P.; Thorne, C.; Christie, R.; Zash, R. Safety and pharmacokinetics of dolutegravir in HIV-positive pregnant women: A systematic review. J. Virus Erad. 2018, 4, 66–71. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Dolutegravir (DTG) and the Fixed Dose Combination (FDC) of Tenofovir/Lamivudine/Dolutegravir (TLD): Briefing Note. Available online: http://www.who.int/hiv/pub/arv/DTG-TLD-arv_briefing_2018.pdf (accessed on 1 October 2022).
- World Health Organization (WHO). HIV Drug Resistance Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Schepanski, S.; Buss, C.; Hanganu-Opatz, I.L.; Arck, P.C. Prenatal Immune and Endocrine Modulators of Offspring’s Brain Development and Cognitive Functions Later in Life. Front. Immunol. 2018, 9, 2186. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Wedderburn, C.J.; Evans, C.; Yeung, S.; Gibb, D.M.; Donald, K.A.; Prendergast, A.J. Growth and Neurodevelopment of HIV-Exposed Uninfected Children: A Conceptual Framework. Curr. HIV/AIDS Rep. 2019, 16, 501–513. [Google Scholar] [CrossRef]
- Bade, A.N.; McMillan, J.M.; Liu, Y.; Edagwa, B.J.; Gendelman, H.E. Dolutegravir Inhibition of Matrix Metalloproteinases Affects Mouse Neurodevelopment. Mol. Neurobiol. 2021, 58, 5703–5721. [Google Scholar] [CrossRef]
- Cabrera, R.M.; Souder, J.P.; Steele, J.W.; Yeo, L.; Tukeman, G.; Gorelick, D.A.; Finnell, R.H. The antagonism of folate receptor by dolutegravir: Developmental toxicity reduction by supplemental folic acid. AIDS 2019, 33, 1967–1976. [Google Scholar] [CrossRef]
- Mohan, H.; Lenis, M.G.; Laurette, E.Y.; Tejada, O.; Sanghvi, T.; Leung, K.Y.; Cahill, L.S.; Sled, J.G.; Delgado-Olguin, P.; Greene, N.D.E.; et al. Dolutegravir in pregnant mice is associated with increased rates of fetal defects at therapeutic but not at supratherapeutic levels. EBioMedicine 2020, 63, 103167. [Google Scholar] [CrossRef]
- Zash, R.; Holmes, L.; Diseko, M.; Jacobson, D.L.; Brummel, S.; Mayondi, G.; Isaacson, A.; Davey, S.; Mabuta, J.; Mmalane, M.; et al. Neural-Tube Defects and Antiretroviral Treatment Regimens in Botswana. N. Engl. J. Med. 2019, 381, 827–840. [Google Scholar] [CrossRef]
- Vasung, L.; Abaci Turk, E.; Ferradal, S.L.; Sutin, J.; Stout, J.N.; Ahtam, B.; Lin, P.Y.; Grant, P.E. Exploring early human brain development with structural and physiological neuroimaging. Neuroimage 2019, 187, 226–254. [Google Scholar] [CrossRef]
- Tau, G.Z.; Peterson, B.S. Normal development of brain circuits. Neuropsychopharmacology 2010, 35, 147–168. [Google Scholar] [CrossRef]
- Monier, A.; Evrard, P.; Gressens, P.; Verney, C. Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J. Comp. Neurol. 2006, 499, 565–582. [Google Scholar] [CrossRef]
- Schafer, D.P.; Stevens, B. Microglia Function in Central Nervous System Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 7, a020545. [Google Scholar] [CrossRef] [PubMed]
- Budday, S.; Steinmann, P.; Kuhl, E. Physical biology of human brain development. Front. Cell. Neurosci. 2015, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, A.C.; Crim, M.J.; Franklin, C.L. A brief history of animal modeling. Mo. Med. 2013, 110, 201–205. [Google Scholar] [PubMed]
- Kala, S.; Watson, B.; Zhang, J.G.; Papp, E.; Guzman Lenis, M.; Dennehy, M.; Cameron, D.W.; Harrigan, P.R.; Serghides, L. Improving the clinical relevance of a mouse pregnancy model of antiretroviral toxicity; a pharmacokinetic dosing-optimization study of current HIV antiretroviral regimens. Antiviral Res. 2018, 159, 45–54. [Google Scholar] [CrossRef]
- German Advisory Committee Blood (Arbeitskreis Blut), Subgroup ‘Assessment of Pathogens Transmissible by Blood’. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemother. 2016, 43, 203–222. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef]
- Ramdas, P.; Sahu, A.K.; Mishra, T.; Bhardwaj, V.; Chande, A. From Entry to Egress: Strategic Exploitation of the Cellular Processes by HIV-1. Front. Microbiol. 2020, 11, 559792. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Aquaro, S.; Borrajo, A.; Pellegrino, M.; Svicher, V. Mechanisms underlying of antiretroviral drugs in different cellular reservoirs with a focus on macrophages. Virulence 2020, 11, 400–413. [Google Scholar] [CrossRef]
- Jeong, H.; Choi, S.; Song, J.W.; Chen, H.; Fischer, J.H. Regulation of UDP-glucuronosyltransferase (UGT) 1A1 by progesterone and its impact on labetalol elimination. Xenobiotica 2008, 38, 62–75. [Google Scholar] [CrossRef]
- Van der Galien, R.; Ter Heine, R.; Greupink, R.; Schalkwijk, S.J.; van Herwaarden, A.E.; Colbers, A.; Burger, D.M. Pharmacokinetics of HIV-Integrase Inhibitors During Pregnancy: Mechanisms, Clinical Implications and Knowledge Gaps. Clin. Pharmacokinet. 2019, 58, 309–323. [Google Scholar] [CrossRef]
- Colbers, A.; Greupink, R.; Burger, D. Pharmacological considerations on the use of antiretrovirals in pregnancy. Curr. Opin. Infect. Dis. 2013, 26, 575–588. [Google Scholar] [CrossRef]
- Schalkwijk, S.; Greupink, R.; Colbers, A.P.; Wouterse, A.C.; Verweij, V.G.; van Drongelen, J.; Teulen, M.; van den Oetelaar, D.; Burger, D.M.; Russel, F.G. Placental transfer of the HIV integrase inhibitor dolutegravir in an ex vivo human cotyledon perfusion model. J. Antimicrob. Chemother. 2016, 71, 480–483. [Google Scholar] [CrossRef]
- Watts, D.H.; Stek, A.; Best, B.M.; Wang, J.; Capparelli, E.V.; Cressey, T.R.; Aweeka, F.; Lizak, P.; Kreitchmann, R.; Burchett, S.K.; et al. Raltegravir pharmacokinetics during pregnancy. J. Acquir. Immune Defic. Syndr. 2014, 67, 375–381. [Google Scholar] [CrossRef]
- Blonk, M.I.; Colbers, A.P.; Hidalgo-Tenorio, C.; Kabeya, K.; Weizsacker, K.; Haberl, A.E.; Molto, J.; Hawkins, D.A.; van der Ende, M.E.; Gingelmaier, A.; et al. Raltegravir in HIV-1-Infected Pregnant Women: Pharmacokinetics, Safety, and Efficacy. Clin. Infect. Dis. 2015, 61, 809–816. [Google Scholar] [CrossRef]
- Zheng, Y.; Hirt, D.; Delmas, S.; Lui, G.; Benaboud, S.; Lechedanec, J.; Treluyer, J.M.; Chenevier-Gobeaux, C.; Arezes, E.; Gelley, A.; et al. Effect of Pregnancy on Unbound Raltegravir Concentrations in the ANRS 160 RalFe Trial. Antimicrob. Agents Chemother. 2020, 64, e00759-20. [Google Scholar] [CrossRef]
- Momper, J.D.; Best, B.M.; Wang, J.; Capparelli, E.V.; Stek, A.; Barr, E.; Badell, M.L.; Acosta, E.P.; Purswani, M.; Smith, E.; et al. Elvitegravir/cobicistat pharmacokinetics in pregnant and postpartum women with HIV. AIDS 2018, 32, 2507–2516. [Google Scholar] [CrossRef]
- Bukkems, V.; Necsoi, C.; Tenorio, C.H.; Garcia, C.; Rockstroh, J.; Schwarze-Zander, C.; Lambert, J.S.; Burger, D.; Konopnicki, D.; Colbers, A. Clinically Significant Lower Elvitegravir Exposure During the Third Trimester of Pregnant Patients Living With Human Immunodeficiency Virus: Data From the Pharmacokinetics of ANtiretroviral agents in HIV-infected pregNAnt women (PANNA) Network. Clin. Infect. Dis. 2020, 71, e714–e717. [Google Scholar] [CrossRef]
- Marzolini, C.; Decosterd, L.; Winterfeld, U.; Tissot, F.; Francini, K.; Buclin, T.; Livio, F. Free and total plasma concentrations of elvitegravir/cobicistat during pregnancy and postpartum: A case report. Br. J. Clin. Pharmacol. 2017, 83, 2835–2838. [Google Scholar] [CrossRef]
- Mulligan, N.; Best, B.M.; Wang, J.; Capparelli, E.V.; Stek, A.; Barr, E.; Buschur, S.L.; Acosta, E.P.; Smith, E.; Chakhtoura, N.; et al. Dolutegravir pharmacokinetics in pregnant and postpartum women living with HIV. AIDS 2018, 32, 729–737. [Google Scholar] [CrossRef]
- Waitt, C.; Orrell, C.; Walimbwa, S.; Singh, Y.; Kintu, K.; Simmons, B.; Kaboggoza, J.; Sihlangu, M.; Coombs, J.A.; Malaba, T.; et al. Safety and pharmacokinetics of dolutegravir in pregnant mothers with HIV infection and their neonates: A randomised trial (DolPHIN-1 study). PLoS Med. 2019, 16, e1002895. [Google Scholar] [CrossRef] [PubMed]
- Bollen, P.; Freriksen, J.; Konopnicki, D.; Weizsacker, K.; Hidalgo Tenorio, C.; Molto, J.; Taylor, G.; Alba-Alejandre, I.; van Crevel, R.; Colbers, A.; et al. The Effect of Pregnancy on the Pharmacokinetics of Total and Unbound Dolutegravir and Its Main Metabolite in Women Living With Human Immunodeficiency Virus. Clin. Infect. Dis. 2021, 72, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Bukkems, V.E.; Hidalgo-Tenorio, C.; Garcia, C.; van Hulzen, A.G.W.; Richel, O.; Burger, D.M.; Colbers, A.P. First pharmacokinetic data of bictegravir in pregnant women living with HIV. AIDS 2021, 35, 2405–2406. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.F.; Acosta, E.P.; Rizk, M.L.; Bryson, Y.J.; Spector, S.A.; Mofenson, L.M.; Handelsman, E.; Teppler, H.; Welebob, C.; Persaud, D.; et al. Raltegravir pharmacokinetics in neonates following maternal dosing. J. Acquir. Immune Defic. Syndr. 2014, 67, 310–315. [Google Scholar] [CrossRef]
- Kassahun, K.; McIntosh, I.; Cui, D.; Hreniuk, D.; Merschman, S.; Lasseter, K.; Azrolan, N.; Iwamoto, M.; Wagner, J.A.; Wenning, L.A. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab. Dispos. 2007, 35, 1657–1663. [Google Scholar] [CrossRef]
- Podany, A.T.; Scarsi, K.K.; Fletcher, C.V. Comparative Clinical Pharmacokinetics and Pharmacodynamics of HIV-1 Integrase Strand Transfer Inhibitors. Clin. Pharmacokinet. 2017, 56, 25–40. [Google Scholar] [CrossRef]
- Barau, C.; Furlan, V.; Yazdanpanah, Y.; Fagard, C.; Molina, J.M.; Taburet, A.M.; Barrail-Tran, A. Characterization of binding of raltegravir to plasma proteins. Antimicrob. Agents Chemother. 2013, 57, 5147–5150. [Google Scholar] [CrossRef]
- Vinot, C.; Treluyer, J.M.; Giraud, C.; Gavard, L.; Peytavin, G.; Mandelbrot, L. Bidirectional Transfer of Raltegravir in an Ex Vivo Human Cotyledon Perfusion Model. Antimicrob. Agents Chemother. 2016, 60, 3112–3114. [Google Scholar] [CrossRef]
- McKeown, D.A.; Rosenvinge, M.; Donaghy, S.; Sharland, M.; Holt, D.W.; Cormack, I.; Hay, P.; Sadiq, S.T. High neonatal concentrations of raltegravir following transplacental transfer in HIV-1 positive pregnant women. AIDS 2010, 24, 2416–2418. [Google Scholar] [CrossRef]
- Clavel-Osorio, C.; Cazassus, F.; Stegmann, S.; Huc-Anais, P.; Lecam, D.; Peytavin, G. One-month transplacental pharmacokinetics of raltegravir in a premature newborn after short-course treatment of the HIV-1-infected mother. Antimicrob. Agents Chemother. 2013, 57, 6393–6394. [Google Scholar] [CrossRef]
- Custodio, J.M.; Rhee, M.; Shen, G.; Ling, K.H.; Kearney, B.P.; Ramanathan, S. Pharmacokinetics and safety of boosted elvitegravir in subjects with hepatic impairment. Antimicrob. Agents Chemother. 2014, 58, 2564–2569. [Google Scholar] [CrossRef]
- Schalkwijk, S.; Colbers, A.; Konopnicki, D.; Greupink, R.; Russel, F.G.; Burger, D.; network, P. First reported use of elvitegravir and cobicistat during pregnancy. AIDS 2016, 30, 807–808. [Google Scholar] [CrossRef]
- Faure-Bardon, V.; Mandelbrot, L.; Duro, D.; Dussaux, C.; Le, M.; Peytavin, G. Placental transfer of elvitegravir and cobicistat in an ex-vivo human cotyledon double perfusion model. AIDS 2018, 32, 321–325. [Google Scholar] [CrossRef]
- Castellino, S.; Moss, L.; Wagner, D.; Borland, J.; Song, I.; Chen, S.; Lou, Y.; Min, S.S.; Goljer, I.; Culp, A.; et al. Metabolism, excretion, and mass balance of the HIV-1 integrase inhibitor dolutegravir in humans. Antimicrob. Agents Chemother. 2013, 57, 3536–3546. [Google Scholar] [CrossRef]
- Reese, M.J.; Savina, P.M.; Generaux, G.T.; Tracey, H.; Humphreys, J.E.; Kanaoka, E.; Webster, L.O.; Harmon, K.A.; Clarke, J.D.; Polli, J.W. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab. Dispos. 2013, 41, 353–361. [Google Scholar] [CrossRef]
- Song, I.H.; Borland, J.; Savina, P.M.; Chen, S.; Patel, P.; Wajima, T.; Peppercorn, A.F.; Piscitelli, S.C. Pharmacokinetics of Single-Dose Dolutegravir in HIV-Seronegative Subjects With Moderate Hepatic Impairment Compared to Healthy Matched Controls. Clin. Pharmacol. Drug Dev. 2013, 2, 342–348. [Google Scholar] [CrossRef]
- Mandelbrot, L.; Ceccaldi, P.F.; Duro, D.; Le, M.; Pencole, L.; Peytavin, G. Placental transfer and tissue accumulation of dolutegravir in the ex vivo human cotyledon perfusion model. PLoS ONE 2019, 14, e0220323. [Google Scholar] [CrossRef]
- Pain, J.B.; Le, M.P.; Caseris, M.; Amiel, C.; Lassel, L.; Charpentier, C.; Desnoyer, A.; Farnoux, C.; Pialoux, G.; Descamps, D.; et al. Pharmacokinetics of dolutegravir in a premature neonate after HIV treatment intensification during pregnancy. Antimicrob. Agents Chemother. 2015, 59, 3660–3662. [Google Scholar] [CrossRef]
- Pham, H.T.; Mesplede, T. Bictegravir in a fixed-dose tablet with emtricitabine and tenofovir alafenamide for the treatment of HIV infection: Pharmacology and clinical implications. Expert Opin. Pharmacother. 2019, 20, 385–397. [Google Scholar] [CrossRef]
- Pencole, L.; Le, M.P.; Bouchet-Crivat, F.; Duro, D.; Peytavin, G.; Mandelbrot, L. Placental transfer of the integrase strand inhibitors cabotegravir and bictegravir in the ex-vivo human cotyledon perfusion model. AIDS 2020, 34, 2145–2149. [Google Scholar] [CrossRef]
- Le, M.P.; Ferre, V.M.; Mazy, F.; Bourgeois-Moine, A.; Damond, F.; Matheron, S.; Descamps, D.; Ghosn, J.; Peytavin, G. Bictegravir pharmacokinetics in a late-presenting HIV-1-infected pregnant woman: A case report. J. Antimicrob. Chemother. 2022, 77, 851–853. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration (FDA). FDA Approves Cabenuva and Vocabria for the Treatment of HIV-1 Infection. Available online: https://www.fda.gov/drugs/human-immunodeficiency-virus-hiv/fda-approves-cabenuva-and-vocabria-treatment-hiv-1-infection (accessed on 27 February 2022).
- US Food and Drug Administration (FDA). FDA Approves First Injectable Treatment for HIV Pre-Exposure Prevention. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-injectable-treatment-hiv-pre-exposure-prevention (accessed on 7 April 2022).
- Trezza, C.; Ford, S.L.; Spreen, W.; Pan, R.; Piscitelli, S. Formulation and pharmacology of long-acting cabotegravir. Curr. Opin. HIV AIDS 2015, 10, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Scheper, H.; van Holten, N.; Hovens, J.; de Boer, M. Severe depression as a neuropsychiatric side effect induced by dolutegravir. HIV Med. 2018, 19, e58–e59. [Google Scholar] [CrossRef] [PubMed]
- Fettiplace, A.; Stainsby, C.; Winston, A.; Givens, N.; Puccini, S.; Vannappagari, V.; Hsu, R.; Fusco, J.; Quercia, R.; Aboud, M.; et al. Psychiatric Symptoms in Patients Receiving Dolutegravir. J. Acquir. Immune Defic. Syndr. 2017, 74, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Llibre, J.M. Neuropsychiatric Adverse Events with Dolutegravir and Other Integrase Strand Transfer Inhibitors. AIDS Rev. 2019, 21, 4–10. [Google Scholar] [CrossRef]
- O’Halloran, J.A.; Cooley, S.A.; Strain, J.F.; Boerwinkle, A.; Paul, R.; Presti, R.M.; Ances, B.M. Altered neuropsychological performance and reduced brain volumetrics in people living with HIV on integrase strand transfer inhibitors. AIDS 2019, 33, 1477–1483. [Google Scholar] [CrossRef]
- O’Halloran, J.A.; Wang, K.; Spence, A.B.; Williams, D.W.; Dastgheyb, R.; Fitzgerald, K.C.; Kamkwalala, A.R.; Maki, P.M.; Sharma, A.; Gustafson, D.R.; et al. Integrase Strand Transfer Inhibitor Start or Switch Impacts Learning in Women With HIV. J. Acquir. Immune Defic. Syndr. 2021, 86, 593–599. [Google Scholar] [CrossRef]
- Prats, A.; Martinez-Zalacain, I.; Mothe, B.; Negredo, E.; Perez-Alvarez, N.; Garolera, M.; Domenech-Puigcerver, S.; Coll, P.; Meulbroek, M.; Chamorro, A.; et al. Effects of integrase inhibitor-based antiretroviral therapy on brain outcomes according to time since acquisition of HIV-1 infection. Sci. Rep. 2021, 11, 11289. [Google Scholar] [CrossRef]
- Cuzin, L.; Pugliese, P.; Katlama, C.; Bani-Sadr, F.; Ferry, T.; Rey, D.; Lourenco, J.; Bregigeon, S.; Allavena, C.; Reynes, J.; et al. Integrase strand transfer inhibitors and neuropsychiatric adverse events in a large prospective cohort. J. Antimicrob. Chemother. 2019, 74, 754–760. [Google Scholar] [CrossRef]
- Hoffmann, C.; Welz, T.; Sabranski, M.; Kolb, M.; Wolf, E.; Stellbrink, H.J.; Wyen, C. Higher rates of neuropsychiatric adverse events leading to dolutegravir discontinuation in women and older patients. HIV Med. 2017, 18, 56–63. [Google Scholar] [CrossRef]
- Elzi, L.; Erb, S.; Furrer, H.; Cavassini, M.; Calmy, A.; Vernazza, P.; Gunthard, H.; Bernasconi, E.; Battegay, M.; the Swiss HIV Cohort Study Group. Adverse events of raltegravir and dolutegravir. AIDS 2017, 31, 1853–1858. [Google Scholar] [CrossRef]
- Penafiel, J.; de Lazzari, E.; Padilla, M.; Rojas, J.; Gonzalez-Cordon, A.; Blanco, J.L.; Blanch, J.; Marcos, M.A.; Lonca, M.; Martinez-Rebollar, M.; et al. Tolerability of integrase inhibitors in a real-life setting. J. Antimicrob. Chemother. 2017, 72, 1752–1759. [Google Scholar] [CrossRef]
- Llibre, J.M.; Montoliu, A.; Miro, J.M.; Domingo, P.; Riera, M.; Tiraboschi, J.; Curran, A.; Homar, F.; Ambrosioni, J.; Abdulghani, N.; et al. Discontinuation of dolutegravir, elvitegravir/cobicistat and raltegravir because of toxicity in a prospective cohort. HIV Med. 2019, 20, 237–247. [Google Scholar] [CrossRef]
- Lepik, K.J.; Yip, B.; Ulloa, A.C.; Wang, L.; Toy, J.; Akagi, L.; Lima, V.D.; Guillemi, S.; Montaner, J.S.G.; Barrios, R. Adverse drug reactions to integrase strand transfer inhibitors. AIDS 2018, 32, 903–912. [Google Scholar] [CrossRef]
- Yombi, J.C. Dolutegravir Neuropsychiatric Adverse Events: Specific Drug Effect or Class Effect. AIDS Rev. 2018, 20, 14–26. [Google Scholar]
- Kanai, O.; Fujita, K.; Mio, T. An implicit threat: Dolutegravir-induced schizophrenic brief psychotic disorder and persistent cenesthopathy. AIDS 2018, 32, 2853–2854. [Google Scholar] [CrossRef]
- Menard, A.; Montagnac, C.; Solas, C.; Meddeb, L.; Dhiver, C.; Tomei, C.; Ravaux, I.; Tissot-Dupont, H.; Mokhtari, S.; Colson, P.; et al. Neuropsychiatric adverse effects on dolutegravir: An emerging concern in Europe. AIDS 2017, 31, 1201–1203. [Google Scholar] [CrossRef]
- Cid-Silva, P.; Llibre, J.M.; Fernandez-Bargiela, N.; Margusino-Framinan, L.; Balboa-Barreiro, V.; Pernas-Souto, B.; Martin-Herranz, I.; Castro-Iglesias, A.; Poveda, E. Clinical Experience with the Integrase Inhibitors Dolutegravir and Elvitegravir in HIV-infected Patients: Efficacy, Safety and Tolerance. Basic Clin. Pharmacol. Toxicol. 2017, 121, 442–446. [Google Scholar] [CrossRef]
- De Boer, M.G.; van den Berk, G.E.; van Holten, N.; Oryszcyn, J.E.; Dorama, W.; Moha, D.A.; Brinkman, K. Intolerance of dolutegravir-containing combination antiretroviral therapy regimens in real-life clinical practice. AIDS 2016, 30, 2831–2834. [Google Scholar] [CrossRef]
- Borghetti, A.; Baldin, G.; Capetti, A.; Sterrantino, G.; Rusconi, S.; Latini, A.; Giacometti, A.; Madeddu, G.; Picarelli, C.; De Marco, R.; et al. Efficacy and tolerability of dolutegravir and two nucleos(t)ide reverse transcriptase inhibitors in HIV-1-positive, virologically suppressed patients. AIDS 2017, 31, 457–459. [Google Scholar] [CrossRef]
- Yagura, H.; Watanabe, D.; Kushida, H.; Tomishima, K.; Togami, H.; Hirano, A.; Takahashi, M.; Hirota, K.; Ikuma, M.; Kasai, D.; et al. Impact of UGT1A1 gene polymorphisms on plasma dolutegravir trough concentrations and neuropsychiatric adverse events in Japanese individuals infected with HIV-1. BMC Infect. Dis. 2017, 17, 622. [Google Scholar] [CrossRef]
- Letendre, S.L.; Mills, A.M.; Tashima, K.T.; Thomas, D.A.; Min, S.S.; Chen, S.; Song, I.H.; Piscitelli, S.C.; the extended ING116070 study team. ING116070: A study of the pharmacokinetics and antiviral activity of dolutegravir in cerebrospinal fluid in HIV-1-infected, antiretroviral therapy-naive subjects. Clin. Infect. Dis. 2014, 59, 1032–1037. [Google Scholar] [CrossRef]
- Kheloufi, F.; Boucherie, Q.; Blin, O.; Micallef, J. Neuropsychiatric events and dolutegravir in HIV patients: A worldwide issue involving a class effect. AIDS 2017, 31, 1775–1777. [Google Scholar] [CrossRef]
- Eiden, C.; Peyriere, H.; Peytavin, G.; Reynes, J. Severe insomnia related to high concentrations of raltegravir. AIDS 2011, 25, 725–727. [Google Scholar] [CrossRef]
- Harris, M.; Larsen, G.; Montaner, J.S. Exacerbation of depression associated with starting raltegravir: A report of four cases. AIDS 2008, 22, 1890–1892. [Google Scholar] [CrossRef]
- Gallant, J.; Lazzarin, A.; Mills, A.; Orkin, C.; Podzamczer, D.; Tebas, P.; Girard, P.M.; Brar, I.; Daar, E.S.; Wohl, D.; et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection (GS-US-380-1489): A double-blind, multicentre, phase 3, randomised controlled non-inferiority trial. Lancet 2017, 390, 2063–2072. [Google Scholar] [CrossRef]
- Sax, P.E.; Pozniak, A.; Montes, M.L.; Koenig, E.; DeJesus, E.; Stellbrink, H.J.; Antinori, A.; Workowski, K.; Slim, J.; Reynes, J.; et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380-1490): A randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet 2017, 390, 2073–2082. [Google Scholar] [CrossRef]
- Eckard, A.R.; McComsey, G.A. Weight gain and integrase inhibitors. Curr. Opin. Infect. Dis. 2020, 33, 10–19. [Google Scholar] [CrossRef]
- Sax, P.E.; Erlandson, K.M.; Lake, J.E.; McComsey, G.A.; Orkin, C.; Esser, S.; Brown, T.T.; Rockstroh, J.K.; Wei, X.; Carter, C.C.; et al. Weight Gain Following Initiation of Antiretroviral Therapy: Risk Factors in Randomized Comparative Clinical Trials. Clin. Infect. Dis. 2020, 71, 1379–1389. [Google Scholar] [CrossRef]
- Chen, Y.W.; Anderson, D.; Pericone, C.D.; Donga, P. Real-World Assessment of Weight Change in African American Females and Hispanics with HIV-1 After Initiating Integrase Strand-Transfer Inhibitors or Protease Inhibitors. J. Health Econ. Outcomes Res. 2022, 9, 1–10. [Google Scholar] [CrossRef]
- Kerchberger, A.M.; Sheth, A.N.; Angert, C.D.; Mehta, C.C.; Summers, N.A.; Ofotokun, I.; Gustafson, D.; Weiser, S.D.; Sharma, A.; Adimora, A.A.; et al. Weight Gain Associated With Integrase Stand Transfer Inhibitor Use in Women. Clin. Infect. Dis. 2020, 71, 593–600. [Google Scholar] [CrossRef]
- Bourgi, K.; Jenkins, C.A.; Rebeiro, P.F.; Palella, F.; Moore, R.D.; Altoff, K.N.; Gill, J.; Rabkin, C.S.; Gange, S.J.; Horberg, M.A.; et al. Weight gain among treatment-naive persons with HIV starting integrase inhibitors compared to non-nucleoside reverse transcriptase inhibitors or protease inhibitors in a large observational cohort in the United States and Canada. J. Int. AIDS Soc. 2020, 23, e25484. [Google Scholar] [CrossRef]
- Venter, W.D.F.; Moorhouse, M.; Sokhela, S.; Fairlie, L.; Mashabane, N.; Masenya, M.; Serenata, C.; Akpomiemie, G.; Qavi, A.; Chandiwana, N.; et al. Dolutegravir plus Two Different Prodrugs of Tenofovir to Treat HIV. N. Engl. J. Med. 2019, 381, 803–815. [Google Scholar] [CrossRef]
- Group, N.A.S.; Kouanfack, C.; Mpoudi-Etame, M.; Omgba Bassega, P.; Eymard-Duvernay, S.; Leroy, S.; Boyer, S.; Peeters, M.; Calmy, A.; Delaporte, E. Dolutegravir-Based or Low-Dose Efavirenz-Based Regimen for the Treatment of HIV-1. N. Engl. J. Med. 2019, 381, 816–826. [Google Scholar] [CrossRef]
- Yeoh, D.K.; Campbell, A.J.; Bowen, A.C. Increase in Body Mass Index in Children With HIV, Switched to Tenofovir Alafenamide Fumarate or Dolutegravir Containing Antiretroviral Regimens. Pediatr. Infect. Dis. J. 2021, 40, e215–e216. [Google Scholar] [CrossRef]
- Eifa, B.A.; Ketema, W. Could a Dolutegravir-Based Antiretroviral Therapy Lead to Clinical Obesity? A Retrospective Cohort Study Conducted at Hawassa University Comprehensive Specialized Hospital in Hawassa, Sidama, Ethiopia. AIDS Res. Treat 2022, 2022, 2965325. [Google Scholar] [CrossRef]
- Esber, A.L.; Chang, D.; Iroezindu, M.; Bahemana, E.; Kibuuka, H.; Owuoth, J.; Singoei, V.; Maswai, J.; Dear, N.F.; Crowell, T.A.; et al. Weight gain during the dolutegravir transition in the African Cohort Study. J. Int. AIDS Soc. 2022, 25, e25899. [Google Scholar] [CrossRef]
- Alhassan, Y.; Twimukye, A.; Malaba, T.; Myer, L.; Waitt, C.; Lamorde, M.; Colbers, A.; Reynolds, H.; Khoo, S.; Taegtmeyer, M. "It’s only fatness, it doesn’t kill": A qualitative study on perceptions of weight gain from use of dolutegravir-based regimens in women living with HIV in Uganda. BMC Womens Health 2022, 22, 246. [Google Scholar] [CrossRef]
- Lake, J.E.; Wu, K.; Bares, S.H.; Debroy, P.; Godfrey, C.; Koethe, J.R.; McComsey, G.A.; Palella, F.J.; Tassiopoulos, K.; Erlandson, K.M. Risk Factors for Weight Gain Following Switch to Integrase Inhibitor-Based Antiretroviral Therapy. Clin. Infect. Dis. 2020, 71, e471–e477. [Google Scholar] [CrossRef]
- Mallon, P.W.; Brunet, L.; Hsu, R.K.; Fusco, J.S.; Mounzer, K.C.; Prajapati, G.; Beyer, A.P.; Wohlfeiler, M.B.; Fusco, G.P. Weight gain before and after switch from TDF to TAF in a U.S. cohort study. J. Int. AIDS Soc. 2021, 24, e25702. [Google Scholar] [CrossRef]
- Summers, N.A.; Lahiri, C.D.; Angert, C.D.; Aldredge, A.; Mehta, C.C.; Ofotokun, I.; Kerchberger, A.M.; Gustafson, D.; Weiser, S.D.; Kassaye, S.; et al. Metabolic Changes Associated With the Use of Integrase Strand Transfer Inhibitors Among Virally Controlled Women. J. Acquir. Immune Defic. Syndr. 2020, 85, 355–362. [Google Scholar] [CrossRef]
- Ursenbach, A.; Max, V.; Maurel, M.; Bani-Sadr, F.; Gagneux-Brunon, A.; Garraffo, R.; Ravaux, I.; Robineau, O.; Makinson, A.; Rey, D.; et al. Incidence of diabetes in HIV-infected patients treated with first-line integrase strand transfer inhibitors: A French multicentre retrospective study. J. Antimicrob. Chemother. 2020, 75, 3344–3348. [Google Scholar] [CrossRef]
- O’Halloran, J.A.; Sahrmann, J.; Parra-Rodriguez, L.; Vo, D.T.; Butler, A.M.; Olsen, M.A.; Powderly, W.G. Integrase Strand Transfer Inhibitors are Associated with Incident Diabetes Mellitus in People with HIV. Clin. Infect. Dis. 2022, ciac355. [Google Scholar] [CrossRef]
- Nolan, N.S.; Adamson, S.; Reeds, D.; O’Halloran, J.A. Bictegravir-Based Antiretroviral Therapy-Associated Accelerated Hyperglycemia and Diabetes Mellitus. Open Forum Infect. Dis. 2021, 8, ofab077. [Google Scholar] [CrossRef]
- Cahn, P.; Pozniak, A.L.; Mingrone, H.; Shuldyakov, A.; Brites, C.; Andrade-Villanueva, J.F.; Richmond, G.; Buendia, C.B.; Fourie, J.; Ramgopal, M.; et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: Week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 2013, 382, 700–708. [Google Scholar] [CrossRef]
- Castagna, A.; Maggiolo, F.; Penco, G.; Wright, D.; Mills, A.; Grossberg, R.; Molina, J.M.; Chas, J.; Durant, J.; Moreno, S.; et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J. Infect. Dis. 2014, 210, 354–362. [Google Scholar] [CrossRef]
- Raffi, F.; Rachlis, A.; Stellbrink, H.J.; Hardy, W.D.; Torti, C.; Orkin, C.; Bloch, M.; Podzamczer, D.; Pokrovsky, V.; Pulido, F.; et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet 2013, 381, 735–743. [Google Scholar] [CrossRef]
- Walmsley, S.L.; Antela, A.; Clumeck, N.; Duiculescu, D.; Eberhard, A.; Gutierrez, F.; Hocqueloux, L.; Maggiolo, F.; Sandkovsky, U.; Granier, C.; et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N. Engl. J. Med. 2013, 369, 1807–1818. [Google Scholar] [CrossRef]
- Fong, P.S.; Flynn, D.M.; Evans, C.D.; Korthuis, P.T. Integrase strand transfer inhibitor-associated diabetes mellitus: A case report. Int. J. STD AIDS 2017, 28, 626–628. [Google Scholar] [CrossRef]
- Hailu, W.; Tesfaye, T.; Tadesse, A. Hyperglycemia After Dolutegravir-Based Antiretroviral Therapy. Int. Med. Case Rep. J. 2021, 14, 503–507. [Google Scholar] [CrossRef]
- Hirigo, A.T.; Gutema, S.; Eifa, A.; Ketema, W. Experience of dolutegravir-based antiretroviral treatment and risks of diabetes mellitus. SAGE Open Med. Case Rep. 2022, 10, 2050313X221079444. [Google Scholar] [CrossRef]
- Horikawa, M.; Toyoda, M.; Saito, N.; Kimura, M.; Kobayashi, T.; Takagi, A.; Fukagawa, M. Raltegravir-associated Diabetic Ketoacidosis in a Patient with HIV Infection: A Case Report. Tokai J. Exp. Clin. Med. 2018, 43, 19–23. [Google Scholar]
- McLaughlin, M.; Walsh, S.; Galvin, S. Dolutegravir-induced hyperglycaemia in a patient living with HIV. J. Antimicrob. Chemother. 2018, 73, 258–260. [Google Scholar] [CrossRef]
- Shah, S.; Hill, A. Risks of metabolic syndrome and diabetes with integrase inhibitor-based therapy. Curr. Opin. Infect. Dis. 2021, 34, 16–24. [Google Scholar] [CrossRef]
- Raesima, M.M.; Ogbuabo, C.M.; Thomas, V.; Forhan, S.E.; Gokatweng, G.; Dintwa, E.; Petlo, C.; Motswere-Chirwa, C.; Rabold, E.M.; Tinker, S.C.; et al. Dolutegravir Use at Conception—Additional Surveillance Data from Botswana. N. Engl. J. Med. 2019, 381, 885–887. [Google Scholar] [CrossRef]
- Grayhack, C.; Sheth, A.; Kirby, O.; Davis, J.; Sibliss, K.; Nkwihoreze, H.; Aaron, E.; Alleyne, G.; Laguerre, R.; Rana, A.; et al. Evaluating outcomes of mother-infant pairs using dolutegravir for HIV treatment during pregnancy. AIDS 2018, 32, 2017–2021. [Google Scholar] [CrossRef]
- Kreitchmann, R.; Oliveira, F.R.; Sprinz, E. Two cases of neural tube defects with dolutegravir use at conception in south Brazil. Braz. J. Infect. Dis. 2021, 25, 101572. [Google Scholar] [CrossRef]
- Vannappagari, V.; Thorne, C.; APR and EPPICC. Pregnancy and Neonatal Outcomes Following Prenatal Exposure to Dolutegravir. J. Acquir. Immune Defic. Syndr. 2019, 81, 371–378. [Google Scholar] [CrossRef]
- Money, D.; Lee, T.; O’Brien, C.; Brophy, J.; Bitnun, A.; Kakkar, F.; Boucoiran, I.; Alimenti, A.; Vaudry, W.; Singer, J.; et al. Congenital anomalies following antenatal exposure to dolutegravir: A Canadian surveillance study. BJOG 2019, 126, 1338–1345. [Google Scholar] [CrossRef]
- Pereira, G.F.M.; Kim, A.; Jalil, E.M.; Fernandes Fonseca, F.; Shepherd, B.E.; Veloso, V.G.; Rick, F.; Ribeiro, R.; Pimenta, M.C.; Beber, A.; et al. Dolutegravir and pregnancy outcomes in women on antiretroviral therapy in Brazil: A retrospective national cohort study. Lancet HIV 2021, 8, e33–e41. [Google Scholar] [CrossRef]
- Posobiec, L.M.; Chapman, S.P.; Murzyn, S.F.; Rendemonti, J.E.; Stanislaus, D.J.; Romach, E.H. No developmental toxicity observed with dolutegravir in rat whole embryo culture. Birth Defects Res. 2021, 113, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Stanislaus, D.J.; Posobiec, L.M.; Laffan, S.B.; Solomon, H.M.; Ziejewski, M.K.; Romach, E.H. Absence of developmental and reproductive toxicity in animals exposed to dolutegravir. Birth Defects Res. 2020, 112, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood-Johnson, L.; Katayama, N.; Marikawa, Y. Dolutegravir Impairs Stem Cell-Based 3D Morphogenesis Models in a Manner Dependent on Dose and Timing of Exposure: An Implication for Its Developmental Toxicity. Toxicol. Sci. 2021, 184, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Amusan, P.; Power, C.; Gill, M.J.; Gomez, D.; Johnson, E.; Rubin, L.H.; Fujiwara, E. Lifetime antiretroviral exposure and neurocognitive impairment in HIV. J. Neurovirol. 2020, 26, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Montenegro-Burke, J.R.; Woldstad, C.J.; Fang, M.; Bade, A.N.; McMillan, J.; Edagwa, B.; Boska, M.D.; Gendelman, H.E.; Siuzdak, G. Nanoformulated Antiretroviral Therapy Attenuates Brain Metabolic Oxidative Stress. Mol. Neurobiol. 2019, 56, 2896–2907. [Google Scholar] [CrossRef]
- Smith, A.S.; Ankam, S.; Farhy, C.; Fiengo, L.; Basa, R.C.B.; Gordon, K.L.; Martin, C.T.; Terskikh, A.V.; Jordan-Sciutto, K.L.; Price, J.H.; et al. High-content analysis and Kinetic Image Cytometry identify toxicity and epigenetic effects of HIV antiretrovirals on human iPSC-neurons and primary neural precursor cells. J. Pharmacol. Toxicol. Methods 2022, 114, 107157. [Google Scholar] [CrossRef]
- Chang, J.J.; Pien, G.W.; Duntley, S.P.; Macones, G.A. Sleep deprivation during pregnancy and maternal and fetal outcomes: Is there a relationship? Sleep Med. Rev. 2010, 14, 107–114. [Google Scholar] [CrossRef]
- Drury, S.S.; Scaramella, L.; Zeanah, C.H. The Neurobiological Impact of Postpartum Maternal Depression: Prevention and Intervention Approaches. Child Adolesc. Psychiatr. Clin. N. Am. 2016, 25, 179–200. [Google Scholar] [CrossRef]
- Kingston, D.; Tough, S.; Whitfield, H. Prenatal and postpartum maternal psychological distress and infant development: A systematic review. Child Psychiatry Hum. Dev. 2012, 43, 683–714. [Google Scholar] [CrossRef]
- Sethna, V.; Siew, J.; Gudbrandsen, M.; Pote, I.; Wang, S.; Daly, E.; Deprez, M.; Pariante, C.M.; Seneviratne, G.; Murphy, D.G.M.; et al. Maternal depression during pregnancy alters infant subcortical and midbrain volumes. J. Affect. Disord. 2021, 291, 163–170. [Google Scholar] [CrossRef]
- Tuovinen, S.; Lahti-Pulkkinen, M.; Girchenko, P.; Lipsanen, J.; Lahti, J.; Heinonen, K.; Reynolds, R.M.; Hamalainen, E.; Kajantie, E.; Laivuori, H.; et al. Maternal depressive symptoms during and after pregnancy and child developmental milestones. Depress Anxiety 2018, 35, 732–741. [Google Scholar] [CrossRef]
- Motoki, N.; Inaba, Y.; Shibazaki, T.; Misawa, Y.; Ohira, S.; Kanai, M.; Kurita, H.; Tsukahara, T.; Nomiyama, T.; Japan, E.; et al. Insufficient maternal gestational weight gain and infant neurodevelopment at 12 months of age: The Japan Environment and Children’s Study. Eur. J. Pediatr. 2022, 181, 921–931. [Google Scholar] [CrossRef]
- Van Lieshout, R.J. Role of maternal adiposity prior to and during pregnancy in cognitive and psychiatric problems in offspring. Nutr. Rev. 2013, 71 (Suppl. 1), S95–S101. [Google Scholar] [CrossRef]
- Caniglia, E.C.; Shapiro, R.; Diseko, M.; Wylie, B.J.; Zera, C.; Davey, S.; Isaacson, A.; Mayondi, G.; Mabuta, J.; Luckett, R.; et al. Weight gain during pregnancy among women initiating dolutegravir in Botswana. EClinicalMedicine 2020, 29–30, 100615. [Google Scholar] [CrossRef]
- Botto, L.D.; Moore, C.A.; Khoury, M.J.; Erickson, J.D. Neural-tube defects. N. Engl. J. Med. 1999, 341, 1509–1519. [Google Scholar] [CrossRef]
- Fekete, K.; Berti, C.; Cetin, I.; Hermoso, M.; Koletzko, B.V.; Decsi, T. Perinatal folate supply: Relevance in health outcome parameters. Matern. Child Nutr. 2010, 6 (Suppl. 2), 23–38. [Google Scholar] [CrossRef]
- Steele, J.W.; Kim, S.E.; Finnell, R.H. One-carbon metabolism and folate transporter genes: Do they factor prominently in the genetic etiology of neural tube defects? Biochimie 2020, 173, 27–32. [Google Scholar] [CrossRef]
- Pitkin, R.M. Folate and neural tube defects. Am. J. Clin. Nutr. 2007, 85, 285S–288S. [Google Scholar] [CrossRef]
- Kancherla, V.; Wagh, K.; Johnson, Q.; Oakley, G.P., Jr. A 2017 global update on folic acid-preventable spina bifida and anencephaly. Birth Defects Res. 2018, 110, 1139–1147. [Google Scholar] [CrossRef]
- Matok, I.; Gorodischer, R.; Koren, G.; Landau, D.; Wiznitzer, A.; Levy, A. Exposure to folic acid antagonists during the first trimester of pregnancy and the risk of major malformations. Br. J. Clin. Pharmacol. 2009, 68, 956–962. [Google Scholar] [CrossRef]
- Chandiwana, N.C.; Chersich, M.; Venter, W.F.; Akpomiemie, G.; Hill, A.; Simmons, B.; Lockman, S.; Serenata, C.M.; Fairlie, L.; Moorhouse, M.A. Unexpected interactions between dolutegravir and folate: Randomised trial evidence from South Africa. AIDS 2020, 35, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Zhang, X.; Mudunuru, J.; Du, Y.; Chen, J.L.; Taskar, K.S.; Huang, J.; Huang, Y.; Romach, E.H. Clinical Extrapolation of the Effects of Dolutegravir and Other HIV Integrase Inhibitors on Folate Transport Pathways. Drug Metab. Dispos. 2019, 47, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, J.C.; Hoque, M.T.; Dai, W.; Mohan, H.; Dunk, C.; Serghides, L.; Bendayan, R. Interaction between dolutegravir and folate transporters and receptor in human and rodent placenta. EBioMedicine 2022, 75, 103771. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.L.; Lee, R.N.; Panvelker, N.; Li, J.; Harowitz, J.; Jordan-Sciutto, K.L.; Akay-Espinoza, C. Differential Effects of Antiretroviral Drugs on Neurons In Vitro: Roles for Oxidative Stress and Integrated Stress Response. J. Neuroimmune Pharmacol. 2018, 13, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.M.; Zidane, B.; Festa, L.; Putatunda, R.; Romer, M.; Monnerie, H.; Jordan-Sciutto, K.L.; Grinspan, J.B. Differential effects of integrase strand transfer inhibitors, elvitegravir and raltegravir, on oligodendrocyte maturation: A role for the integrated stress response. Glia 2021, 69, 362–376. [Google Scholar] [CrossRef]
- Latronico, T.; Pati, I.; Ciavarella, R.; Fasano, A.; Mengoni, F.; Lichtner, M.; Vullo, V.; Mastroianni, C.M.; Liuzzi, G.M. In vitro effect of antiretroviral drugs on cultured primary astrocytes: Analysis of neurotoxicity and matrix metalloproteinase inhibition. J. Neurochem. 2018, 144, 271–284. [Google Scholar] [CrossRef]
- Jacobs, T.G.; van Aerde, K.J.; Colbers, A.; Burger, D.M. Raltegravir-based Postnatal HIV Prophylaxis Therapy in a Neonate After in Utero Dolutegravir Exposure. Pediatr. Infect. Dis. J. 2022, 41, 131–132. [Google Scholar] [CrossRef]
- Miyagi, S.J.; Collier, A.C. The development of UDP-glucuronosyltransferases 1A1 and 1A6 in the pediatric liver. Drug Metab. Dispos. 2011, 39, 912–919. [Google Scholar] [CrossRef]
- Wusthoff, C.J.; Loe, I.M. Impact of bilirubin-induced neurologic dysfunction on neurodevelopmental outcomes. Semin. Fetal Neonatal Med. 2015, 20, 52–57. [Google Scholar] [CrossRef]
- Ayoub, A.E.; Cai, T.Q.; Kaplan, R.A.; Luo, J. Developmental expression of matrix metalloproteinases 2 and 9 and their potential role in the histogenesis of the cerebellar cortex. J. Comp. Neurol. 2005, 481, 403–415. [Google Scholar] [CrossRef]
- Bednarek, N.; Clement, Y.; Lelievre, V.; Olivier, P.; Loron, G.; Garnotel, R.; Gressens, P. Ontogeny of MMPs and TIMPs in the murine neocortex. Pediatr. Res. 2009, 65, 296–300. [Google Scholar] [CrossRef]
- Larsen, P.H.; DaSilva, A.G.; Conant, K.; Yong, V.W. Myelin formation during development of the CNS is delayed in matrix metalloproteinase-9 and -12 null mice. J. Neurosci. 2006, 26, 2207–2214. [Google Scholar] [CrossRef]
- Ulrich, R.; Gerhauser, I.; Seeliger, F.; Baumgartner, W.; Alldinger, S. Matrix metalloproteinases and their inhibitors in the developing mouse brain and spinal cord: A reverse transcription quantitative polymerase chain reaction study. Dev. Neurosci. 2005, 27, 408–418. [Google Scholar] [CrossRef]
- Reinhard, S.M.; Razak, K.; Ethell, I.M. A delicate balance: Role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Front. Cell. Neurosci. 2015, 9, 280. [Google Scholar] [CrossRef]
- Aujla, P.K.; Huntley, G.W. Early postnatal expression and localization of matrix metalloproteinases-2 and -9 during establishment of rat hippocampal synaptic circuitry. J. Comp. Neurol. 2014, 522, 1249–1263. [Google Scholar] [CrossRef]
- Vaillant, C.; Didier-Bazes, M.; Hutter, A.; Belin, M.F.; Thomasset, N. Spatiotemporal expression patterns of metalloproteinases and their inhibitors in the postnatal developing rat cerebellum. J. Neurosci. 1999, 19, 4994–5004. [Google Scholar] [CrossRef]
- Beroun, A.; Mitra, S.; Michaluk, P.; Pijet, B.; Stefaniuk, M.; Kaczmarek, L. MMPs in learning and memory and neuropsychiatric disorders. Cell. Mol. Life Sci. 2019, 76, 3207–3228. [Google Scholar] [CrossRef]
- Fujioka, H.; Dairyo, Y.; Yasunaga, K.; Emoto, K. Neural functions of matrix metalloproteinases: Plasticity, neurogenesis, and disease. Biochem. Res. Int. 2012, 2012, 789083. [Google Scholar] [CrossRef]
- Small, C.D.; Crawford, B.D. Matrix metalloproteinases in neural development: A phylogenetically diverse perspective. Neural. Regen. Res. 2016, 11, 357–362. [Google Scholar] [CrossRef]
- Agrawal, S.M.; Lau, L.; Yong, V.W. MMPs in the central nervous system: Where the good guys go bad. Semin. Cell Dev. Biol. 2008, 19, 42–51. [Google Scholar] [CrossRef]
- De Stefano, M.E.; Herrero, M.T. The multifaceted role of metalloproteinases in physiological and pathological conditions in embryonic and adult brains. Prog. Neurobiol. 2017, 155, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Ethell, I.M.; Ethell, D.W. Matrix metalloproteinases in brain development and remodeling: Synaptic functions and targets. J. Neurosci. Res. 2007, 85, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Shimamura, R.; Koizumi-Kitajima, M.; Okano, H. Degradation of Extracellular Matrix by Matrix Metalloproteinase 2 Is Essential for the Establishment of the Blood-Brain Barrier in Drosophila. iScience 2019, 16, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Loffek, S.; Schilling, O.; Franzke, C.W. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef]
- Shinotsuka, N.; Yamaguchi, Y.; Nakazato, K.; Matsumoto, Y.; Mochizuki, A.; Miura, M. Caspases and matrix metalloproteases facilitate collective behavior of non-neural ectoderm after hindbrain neuropore closure. BMC Dev. Biol. 2018, 18, 17. [Google Scholar] [CrossRef]
- Van Hinsbergh, V.W.; Koolwijk, P. Endothelial sprouting and angiogenesis: Matrix metalloproteinases in the lead. Cardiovasc. Res. 2008, 78, 203–212. [Google Scholar] [CrossRef]
- Vafadari, B.; Salamian, A.; Kaczmarek, L. MMP-9 in translation: From molecule to brain physiology, pathology, and therapy. J. Neurochem. 2016, 139 (Suppl. 2), 91–114. [Google Scholar] [CrossRef]
- Luo, C.; Li, Q.; Gao, Y.; Shen, X.; Ma, L.; Wu, Q.; Wang, Z.; Zhang, M.; Zhao, Z.; Chen, X.; et al. Poloxamer 188 Attenuates Cerebral Hypoxia/Ischemia Injury in Parallel with Preventing Mitochondrial Membrane Permeabilization and Autophagic Activation. J. Mol. Neurosci. 2015, 56, 988–998. [Google Scholar] [CrossRef]
- Luo, C.L.; Chen, X.P.; Li, L.L.; Li, Q.Q.; Li, B.X.; Xue, A.M.; Xu, H.F.; Dai, D.K.; Shen, Y.W.; Tao, L.Y.; et al. Poloxamer 188 attenuates in vitro traumatic brain injury-induced mitochondrial and lysosomal membrane permeabilization damage in cultured primary neurons. J. Neurotrauma 2013, 30, 597–607. [Google Scholar] [CrossRef]
- Serbest, G.; Horwitz, J.; Jost, M.; Barbee, K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006, 20, 308–310. [Google Scholar] [CrossRef]
- Cadichon, S.B.; Le Hoang, M.; Wright, D.A.; Curry, D.J.; Kang, U.; Frim, D.M. Neuroprotective effect of the surfactant poloxamer 188 in a model of intracranial hemorrhage in rats. J. Neurosurg. 2007, 106, 36–40. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Surfactant poloxamer 188-related decreases in inflammation and tissue damage after experimental brain injury in rats. J. Neurosurg. 2004, 101, 91–96. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Poloxamer 188 volumetrically decreases neuronal loss in the rat in a time-dependent manner. Neurosurgery 2004, 55, 943–948, discussion 948–949. [Google Scholar] [CrossRef]
- Dalal, N.D.; Lee, R.C. Treatment of burn injury by cellular repair. J. Craniofac. Surg. 2008, 19, 903–906. [Google Scholar] [CrossRef]
- Moloughney, J.G.; Weisleder, N. Poloxamer 188 (p188) as a membrane resealing reagent in biomedical applications. Recent Pat. Biotechnol. 2012, 6, 200–211. [Google Scholar] [CrossRef]
- Sillman, B.; Bade, A.N.; Dash, P.K.; Bhargavan, B.; Kocher, T.; Mathews, S.; Su, H.; Kanmogne, G.D.; Poluektova, L.Y.; Gorantla, S.; et al. Creation of a long-acting nanoformulated dolutegravir. Nat. Commun. 2018, 9, 443. [Google Scholar] [CrossRef]
- Deodhar, S.; Sillman, B.; Bade, A.N.; Avedissian, S.N.; Podany, A.T.; McMillan, J.M.; Gautam, N.; Hanson, B.; Dyavar Shetty, B.L.; Szlachetka, A.; et al. Transformation of dolutegravir into an ultra-long-acting parenteral prodrug formulation. Nat. Commun. 2022, 13, 3226. [Google Scholar] [CrossRef]
- Singh-Joy, S.D.; McLain, V.C. Safety assessment of poloxamers 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, and 407, poloxamer 105 benzoate, and poloxamer 182 dibenzoate as used in cosmetics. Int. J. Toxicol. 2008, 27 (Suppl. 2), 93–128. [Google Scholar] [CrossRef]
- Williams, P.E.; Crauwels, H.M.; Basstanie, E.D. Formulation and pharmacology of long-acting rilpivirine. Curr. Opin. HIV AIDS 2015, 10, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, N.; Kaitu’u-Lino, T.; Harris, L.; Tong, S.; Hannan, N. Nanoparticles in pregnancy: The next frontier in reproductive therapeutics. Hum. Reprod. Update 2021, 27, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liang, R.; Zheng, M.; Cai, L.; Fan, X. Surface-Functionalized Nanoparticles as Efficient Tools in Targeted Therapy of Pregnancy Complications. Int. J. Mol. Sci. 2019, 20, 3642. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Class | Drug name | Acronyms | US FDA Approval Year | Recommendations for Pregnant Women Who Are ART Naïve | Recommendations for Continuation of ART in Women Who Become Pregnant | Recommendations for Women Planning to Become Pregnant |
|---|---|---|---|---|---|---|
| Tenofovir Alafenamide | TAF | 2016 | Preferred | Continue | Preferred | |
| Nucleoside Reverse Transcriptase Inhibitors (NRTIs) | Emtricitabine Tenofovir disoproxil fumarate Abacavir Lamivudine | FTC TDF ABC 3TC | 2003 2001 1998 1995 | Preferred Preferred Preferred Preferred | Continue Continue Continue Continue | Preferred Preferred Preferred Preferred |
| Zidovudine | ZDV | 1987 | Alternative | Continue | Alternative | |
| Long-acting rilpivirine | LA-RPV | 2021 | Not recommended | Insufficient data | Insufficient data | |
| Doravirine | DOR | 2018 | Insufficient data | Insufficient data | Insufficient data | |
| Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) | Rilpivirine Etravirine Efavirenz | RPV ETR EFV | 2011 2008 1998 | Alternative Not recommended Alternative | Continue Continue Continue | Alternative Not recommended, except in special circumstances Alternative |
| Nevirapine | NVP | 1996 | Not recommended | Continue | Not recommended, except in special circumstances | |
| Darunavir | DRV | 2006 | Preferred | Continue | Preferred | |
| Tipranavir | TPV | 2005 | Do not use | Switch | Do not use | |
| Fosamprenavir | FPV | 2003 | Do not use | Switch | Do not use | |
| Protease Inhibitors (PIs) | Atazanavir | ATV | 2003 | Preferred | Continue | Preferred |
| Lopinavir | LPV | 2000 | Not recommended, except in special circumstances | Continue | Not recommended, except in special circumstances | |
| Saquinavir | SQV | 1995 | Do not use | Switch | Do not use | |
| Fusion Inhibitor | Enfuviritide | T-20 | 2003 | Not recommended | Continue | Not recommended, except in special circumstances |
| CCR5 Antagonist | Maraviroc | MVC | 2007 | Not recommended | Continue | Not recommended, except in special circumstances |
| Cabotegravir | CAB | 2021 | Not recommended | Insufficient data | Insufficient data | |
| Long-acting cabotegravir | LA-CAB | 2021 | Not recommended | Insufficient data | Insufficient data | |
| Integrase Strand Transfer | Bictegravir | BIC | 2018 | Insufficient data | Insufficient data | Insufficient data |
| Inhibitors (INSTIs) | Elvitegravir | EVG | 2014 | Not recommended | Continue with viral monitoring or consider switching | Not recommended |
| Dolutegravir | DTG | 2013 | Preferred | Continue | Preferred | |
| Raltegravir | RAL | 2007 | Preferred | Continue | Preferred | |
| Attachment Inhibitor | Fostemsavir | FTR | 2020 | Insufficient data | Insufficient data | Insufficient data |
| Pharmacokinetic Enhancers | Cobicistat | COBI, c | 2014 | Not recommended | Not recommended | Not recommended |
| Ritonavir | RTV | 1996 | Depends on Protease Inhibitor | Depends on Protease Inhibitor | Depends on Protease Inhibitor |
| Raltegravir | Elvitegravir/Cobicistat | Dolutegravir | Bictegravir | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Route of administration | Oral | Oral | Oral | Oral | ||||||
| Dosing | 400 mg twice daily | 150/150 mg once daily | 50 mg once daily | 50 mg once daily | ||||||
| Metabolism | Primarily UGT1A1 | Primarily CYP3A4 Minor UGT1A1 | Primarily UGT1A1 Minor CYP3A4 | Equal contribution by CYP3A4 and UGT1A1 | ||||||
| Protein Binding | 76–83% | Elvitegravir 98–99% | Approximately 99% | Approximately 99.7% | ||||||
| AUC0-12 or 0–24 (µg*h/mL) | [43] ad 2nd trimester: 6.6 (2.1–18.5) 3rd trimester: 5.4 (1.4–35.6) Postpartum: 11.6 (1.6–39.9) | [44] bd 3rd trimester: 5.0 (3.56–7.01) Postpartum: 7.11 (4.91–10.30) | [45] ad 3rd trimester: 3.921 (0.699–14.706) Postpartum: 6.770 (1.506–21.859) | [46] ae 2nd trimester: 15.283 (11.939–19.038) 3rd trimester: 14.004 (9.119–18.798) Postpartum: 21.039 (13.532–32.788) | [47] ace 3rd trimester: 14.1 (39) Postpartum: 21.7 (29) | [48] f 3rd trimester: 14.339 Postpartum: 15.356 | [49] ae 2nd trimester: 47.6 (33.4 – 63.7) 3rd trimester: 49.2 (36.4 –62.0) Postpartum: 65.0 (47.8–88.4) | [50] be 3rd trimester: 35.322 (19.196–67.922) Postpartum: 40.127 (22.795–59.633) | [51] ace 3rd trimester: 40.8 (35) Postpartum: 47.0 (42) | [52] e 3rd trimester: 37.9 Postpartum: 58 |
| Cmax (µg/mL) | 2nd trimester: 2.250 (0.365–5.960) 3rd trimester: 1.770 (0.315–7.820) Postpartum: 3.035 (0.312–12.600) | 3rd trimester: 1.43 (0.93–2.22) Postpartum: 1.76 (1.10–2.80) | 3rd trimester: NR Postpartum: NR | 2nd trimester: 1.447 (1.133–1.579) 3rd trimester: 1.432 (0.705–1.570) Postpartum: 1.713 (0.955–2.284) | 3rd trimester: 1.4 (42) Postpartum: 1.8 (26) | 3rd trimester: 1.270 Postpartum: 1.352 | 2nd trimester: 3.62 (2.57–4.63) 3rd trimester: 3.54 (2.66–4.24) Postpartum: 4.85 (3.83–5.97) | 3rd trimester: 2.534 (1.462–3.986) Postpartum: 2.899 (1.397–4.224) | 3rd trimester: 3.15 (31) Postpartum: 3.34 (32) | 3rd trimester: 3.82 Postpartum: 4.7 |
| Ch or trough (µg/mL) | 2nd trimester: 0.0621 (0.0128–0.438) 3rd trimester: 0.064 (0.0114–0.607) Postpartum: 0.0797 (0.0199–1.340) | 3rd trimester: 0.077 (0.043–0.137) Postpartum: 0.120 (0.074–0.193) | 3rd trimester: 0.057 (0.003–0.277) Postpartum: 0.069 (0.005–0.248) | 2nd trimester: 0.025 (0.017–0.067) 3rd trimester: 0.048 (0.014–0.075) Postpartum: 0.377 (0.228.5–0.568) | 3rd trimester: NR Postpartum: NR | 3rd trimester: 0.018 Postpartum: 0.068 | 2nd trimester: 0.73 (0.63–1.34) 3rd trimester: 0.93 (0.68–1.34) Postpartum: 1.28 (0.80–1.95) | 3rd trimester: 0.642 (0.188–0.3088) Postpartum: 0.777 (0.348–0.1210) | 3rd trimester: 0.68 (84) Postpartum: 1.03 (68) | 3rd trimester: 0.63 Postpartum: 1.23 |
| Tmax (h) | 2nd trimester: 4.0 (1.0–8.0) 3rd trimester: 2.0 (0–12.0) Postpartum: 2.0 (0–8.0) | 3rd trimester: 1.98 (0–11.3) Postpartum: 2.03 (0–7.97) | 3rd trimester: NR Postpartum: NR | 2nd trimester: 4 (2–4) 3rd trimester: 4 (2–6) Postpartum: 4 (2–6) | 3rd trimester: 4 (2.0–6.0) Postpartum: 5 (3.0–7.8) | 3rd trimester: 2.9 Postpartum: 3.0 | 2nd trimester: 2 (2–4) 3rd trimester: 3 (2–4) Postpartum: 2 (2–4) | 3rd trimester: NR Postpartum: NR | 3rd trimester: 3.0 (1.0–4.5) Postpartum: 3.8 (0.5–8.0) | 3rd trimester: NR Postpartum: NR |
| T1/2 (h) | 2nd trimester: 2.9 (1.2–85.6) 3rd trimester: 3.7 (1.1–211.7) Postpartum: 3.6 (1.1–30.5) | 3rd trimester: 2.55 (1.88–3.45) Postpartum: 2.53 (1.91–3.36) | 3rd trimester: NR Postpartum: NR | 2nd trimester: 3.1 (2.6–3.9) 3rd trimester: 3.4 (2.7–4.7) Postpartum: 8.8 (7.0–13.2) | 3rd trimester: 4.3 (37) Postpartum: 7.7 (30) | 3rd trimester: 2.6 Postpartum: 3.7 | 2nd trimester: 11.0 (8.9–13.1) 3rd trimester: 12.2 (10.4–15.0) Postpartum: 13.5 (10.6–18.6) | 3rd trimester: NR Postpartum: NR | 3rd trimester: 10.5 (49) Postpartum: 14.4 (46) | 3rd trimester: NR Postpartum: NR |
| Cord blood:maternal blood ratio | 1.5 | 1.21 | NR | 0.91 | 0.75 | 0.64 | 1.25 | 1.21 | 1.29 | 1.49 |
| Placental Transfer | High | High | High | High | ||||||
| T1/2 (h) in infants post delivery | 26.6 (9.3–184) a [53] | 7.6 (6.3–10.2) a [46] | 32.8 (25.9–35.9) a [49] | NR | ||||||
| Dosing recommendations in pregnancy | No change in dose indicated | Insufficient data to make dosing recommendations | No change in dose indicated | Insufficient data to make dosing recommendations | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foster, E.G.; Gendelman, H.E.; Bade, A.N. HIV-1 Integrase Strand Transfer Inhibitors and Neurodevelopment. Pharmaceuticals 2022, 15, 1533. https://doi.org/10.3390/ph15121533
Foster EG, Gendelman HE, Bade AN. HIV-1 Integrase Strand Transfer Inhibitors and Neurodevelopment. Pharmaceuticals. 2022; 15(12):1533. https://doi.org/10.3390/ph15121533
Chicago/Turabian StyleFoster, Emma G., Howard E. Gendelman, and Aditya N. Bade. 2022. "HIV-1 Integrase Strand Transfer Inhibitors and Neurodevelopment" Pharmaceuticals 15, no. 12: 1533. https://doi.org/10.3390/ph15121533
APA StyleFoster, E. G., Gendelman, H. E., & Bade, A. N. (2022). HIV-1 Integrase Strand Transfer Inhibitors and Neurodevelopment. Pharmaceuticals, 15(12), 1533. https://doi.org/10.3390/ph15121533