
Chromatin Alterations in Neurological Disorders and Strategies of (Epi)Genome Rescue


Abstract
:1. Epigenetic Mechanisms Driving Cell Identity and Disease
1.1. Mammalian DNA Cis-Regulatory Elements Classification and Chromatin Signature
1.2. DNA Methylation and Hydroxy-Methylation
1.3. Histone Modifications
1.3.1. Histone Acetylation
1.3.2. Histone Methylation
1.4. Nucleosome Positioning
1.5. Three-Dimensional Structure of Chromatin–The Role of CTCF, Cohesins and Phase Separation in the Buildup of Genome Topology
2. Genome Activity in Neurological Disorders
2.1. NDs Frequently Feature Defective Machinery Establishing and Reading Histone and DNA Modifications
2.2. Chromatin Topology Is Related to Neuronal Function and Can Be Affected in NDs
2.3. Next Generation Brain Regulome Maps to Link Genetic Variants to Essential Loci and Identify New Genes Underlying ND
2.3.1. Comparison of Wild Type and ND Chromatin Can Help Identifying Essential Pathways Related to NDs
2.3.2. From GWAS to the Causal Gene
2.3.3. Towards High Resolution Functional Genomics of NDs
2.4. Targetting Global Epigenetic Signature in NDs
3. Precision Medicine for (Epi)genome Engineering and Their Possible Application to Treat NDs
3.1. CRISPR-Cas-Based Genome Engineering Principles and Considerations
3.2. From Genome to (Epi)genome Edition
3.2.1. dCas9-Orchestrated Base Edition in the Genome
3.2.2. Site-Specific Epigenome Rewiring to Enhance Gene Expression
3.2.3. Approaches for Locus-Specific Gene Silencing
3.2.4. Complex Designs to Simultaneously Activate and Repress Distinct Sets of Genes
3.2.5. Engineering Genome Topology to Orchestrate Gene Expression
3.3. From Mutation to Precise Target CRISPR-Cas-Mediated In Vivo (Epi)genome Editing to Target NDs
Essential Considerations When Designing Epigenetic Engineering Strategies
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waddington, C.H. The Epigenotype. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Felsenfeld, G. A Brief History of Epigenetics. Cold Spring Harb. Perspect. Biol. 2014, 6, a018200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allis, D.C.; Caparros, M.-L.; Jenuwein, T.; Reinberg, D.; Lachlan, M. (Eds.) Epigenetics, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2015; ISBN 978-1-936113-59-0. [Google Scholar]
- Roeder, R.G. 50+ years of eukaryotic transcription: An expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol. 2019, 26, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Olins, A.L.; Olins, D.E. Spheroid Chromatin Units (ngr Bodies). Science 1974, 183, 330–332. [Google Scholar] [CrossRef]
- Woodcock, C.; Safer, J.; Stanchfield, J. Structural repeating units in chromatin: I. Evidence for their general occurrence. Exp. Cell Res. 1976, 97, 101–110. [Google Scholar] [CrossRef]
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 2017, 357, eaag0025. [Google Scholar] [CrossRef] [Green Version]
- Fussner, E.; Strauss, M.; Djuric, U.; Li, R.; Ahmed, K.; Hart, M.; Ellis, J.; Bazett-Jones, D.P. Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep. 2012, 13, 992–996. [Google Scholar] [CrossRef] [Green Version]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Resolution Core Particle at 2.8 A Resolution. Nature 1997, 389, 251–259. [Google Scholar] [CrossRef]
- Ricci, M.A.; Manzo, C.; Garcia-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin Fibers Are Formed by Heterogeneous Groups of Nucleosomes In vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef] [Green Version]
- Knezetic, J.A.; Luse, D.S. The presence of nucleosomes on a DNA template prevents initiation by RNA polymerase II in vitro. Cell 1986, 45, 95–104. [Google Scholar] [CrossRef]
- Lorch, Y.; Lapointe, J.W.; Kornberg, R.D. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell 1987, 49, 203–210. [Google Scholar] [CrossRef]
- Han, M.; Grunstein, M. Nucleosome loss activates yeast downstream promoters in vivo. Cell 1988, 55, 1137–1145. [Google Scholar] [CrossRef]
- Kieffer-Kwon, K.-R.; Nimura, K.; Rao, S.S.; Xu, J.; Jung, S.; Pekowska, A.; Dose, M.; Stevens, E.; Mathe, E.; Dong, P.; et al. Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Mol. Cell 2017, 67, 566–578.e10. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S. Pioneer Transcription Factors Initiating Gene Network Changes. Annu. Rev. Genet. 2020, 54, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P. Organization and regulation of gene transcription. Nat. Cell Biol. 2019, 573, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Hannah, A. Localization and Function of Heterochromatin in Drosophila Melanogaster. Adv. Genet. 1951, 4, 87–125. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.J. Types of visible variations induced by X-rays inDrosophila. J. Genet. 1930, 22, 299–334. [Google Scholar] [CrossRef]
- Dorer, D.R. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell 1994, 77, 993–1002. [Google Scholar] [CrossRef]
- Schultz, J. Variegation in Drosophila and the Intert Chromosome Regions. Genetics 1936, 22, 27–33. [Google Scholar]
- Klosin, A.; Casas, E.; Hidalgo-Carcedo, C.; Vavouri, T.; Lehner, B. Transgenerational transmission of environmental information inC. elegans. Science 2017, 356, 320–323. [Google Scholar] [CrossRef] [Green Version]
- Spitz, F.; Furlong, E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.H.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieffer-Kwon, K.-R.; Tang, Z.; Mathe, E.; Qian, J.; Sung, M.-H.; Li, G.; Resch, W.; Baek, S.; Pruett, N.; Grøntved, L.; et al. Interactome Maps of Mouse Gene Regulatory Domains Reveal Basic Principles of Transcriptional Regulation. Cell 2013, 155, 1507–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshavsky, A.J.; Sundin, O.; Bohn, M. A stretch of “late” SV40 viral DNA about 400 bp long which includes the origin of replication is specifically exposed in SV40 minichromosomes. Cell 1979, 16, 453–466. [Google Scholar] [CrossRef]
- Wu, C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nat. Cell Biol. 1980, 286, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Almer, A.; Hörz, W. Nuclease Hypersensitive Regions with Adjacent Positioned Nucleosomes Mark the Gene Boundaries of the PHO5/PHO3 Locus in Yeast. EMBO J. 1986, 5, 2681–2687. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.-Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic Regulation of Nucleosome Positioning in the Human Genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2010, 470, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Pekowska, A.; Benoukraf, T.; Cabeza, J.Z.; Belhocine, M.; Koch, F.; Holota, H.; Imbert, J.; Andrau, J.-C.; Ferrier, P.; Spicuglia, S. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011, 30, 4198–4210. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The Protein CTCF Is Required for the Enhancer Blocking Activity of Vertebrate Insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Doskočil, J.; Šorm, F. Distribution of 5-methylcytosine in pyrimidine sequences of deoxyribonucleic acids. Biochim. Biophys. Acta (BBA) Bioenerg. 1962, 55, 953–959. [Google Scholar] [CrossRef]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Bird, A.; Taggart, M.; Frommer, M.; Miller, O.J.; Macleod, D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 1985, 40, 91–99. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.F.M. CpG Islands in Vertebrate Genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Iglesias, A.D. Epigenetic defects, assisted reproductive technology, and clinical practice: A call for clinicians and genetic counselors. Clin. Genet. 2004, 66, 481–482. [Google Scholar] [CrossRef]
- Riggs, A. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef]
- Cattanach, B.M.; Kirk, M. Differential activity of maternally and paternally derived chromosome regions in mice. Nat. Cell Biol. 1985, 315, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Compere, S.J.; Palmiter, R.D. DNA methylation controls the inducibility of the mouse metallothionein-I gene in lymphoid cells. Cell 1981, 25, 233–240. [Google Scholar] [CrossRef]
- Yoder, J.; Soman, N.S.; Verdine, G.L.; Bestor, T.H. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. studies with a mechanism-based probe. J. Mol. Biol. 1997, 270, 385–395. [Google Scholar] [CrossRef]
- Pradhan, S.; Chin, H.G.; Jacobsen, S.E. SET7/9 Mediated Methylation of Non-Histone Proteins in Mammalian Cells. Proteins 2009, 4, 383–387. [Google Scholar] [CrossRef]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L Cooperates with the Dnmt3 Family of de Novo DNA Methyltransferases to Establish Maternal Imprints in Mice. Development 2002, 129, 1983–1993. [Google Scholar] [CrossRef]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.-P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, M.; Hermann, A.; Gowher, H.; Jeltsch, A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. JBIC J. Biol. Inorg. Chem. 2002, 269, 4981–4984. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Iurlaro, M.; Ficz, G.; Oxley, D.; Raiber, E.-A.; Bachman, M.; Booth, M.J.; Andrews, S.; Balasubramanian, S.; Reik, W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013, 14, R119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruijt, C.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Muenzel, M.; Wagner, M.; Müller, M.; Khan, F.; et al. Dynamic Readers for 5-(Hydroxy)Methylcytosine and Its Oxidized Derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, A.; Drohat, A.C. Thymine DNA Glycosylase Can Rapidly Excise 5-Formylcytosine and 5-Carboxylcytosine: POTENTIAL IMPLICATIONS FOR ACTIVE DEMETHYLATION OF CpG SITES. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nat. Cell Biol. 2019, 570, 395–399. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Mirsky, A.E. Evidence for The Complete Dna-Dependence of Rna Synthesis in Isolated Thymus Nuclei. Proc. Natl. Acad. Sci. USA 1962, 48, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Allfrey, G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena Histone Acetyltransferase A: A Homolog to Yeast Gcn5p Linking Histone Acetylation to Gene Activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.H.; Zhou, J.; Jambeck, P.; Churchill, M.E.; Allis, C.D. Histone Acetyltransferase Activity of Yeast Gcn5p Is Required for the Activation of Target Genes in vivo. Genes Dev. 1998, 12, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Ogryzko, V.; Schiltz, R.; Russanova, V.; Howard, B.H.; Nakatani, Y. The Transcriptional Coactivators p300 and CBP Are Histone Acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Rundlett, S.E.; Carmen, A.A.; Kobayashi, R.; Bavykin, S.; Turner, B.M.; Grunstein, M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 14503–14508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, M.; Gaber, R.F. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 6317–6327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar] [CrossRef] [Green Version]
- Heinzel, T.; Lavinsky, R.M.; Mullen, T.-M.; Soderstrom, M.; Laherty, C.D.; Torchia, J.A.; Yang, W.-M.; Brard, G.; Ngo, S.D.; Davie, J.; et al. A complex containing N-CoR, mSln3 and histone deacetylase mediates transcriptional repression. Nat. Cell Biol. 1997, 387, 43–48. [Google Scholar] [CrossRef]
- Alland, L.; Muhle, R.A.; Hou, H.; Potes, J.; Chin, L.; Schreiber-Agus, N.; Depinho, R.A. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nat. Cell Biol. 1997, 387, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Kao, H.-Y.; Chakravarti, D.; Lin, R.J.; Hassig, C.A.; Ayer, D.; Schreiber, S.L.; Evans, R.M. Nuclear Receptor Repression Mediated by a Complex Containing SMRT, mSin3A, and Histone Deacetylase. Cell 1997, 89, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Merika, M.; Williams, A.J.; Chen, G.; Collins, T.; Thanos, D. Recruitment of CBP/P300 by the IFNβ Enhanceosome Is Required for Synergistic Activation of Transcription. Mol. Cell 1998, 1, 277–287. [Google Scholar] [CrossRef]
- Visel, A.; Blow, M.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.; Zhang, W.; Jiang, J.; et al. Integration of External Signaling Pathways with the Core Transcriptional Network in Embryonic Stem Cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, T.-Y.; Cuddapah, S.; Cui, K.; Zhao, K. The genomic landscape of histone modifications in human T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15782–15787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Herskowitz, I. Characterization of the yeast SWI1, SWI2, and SWI3 genes, which encode a global activator of transcription. Cell 1992, 68, 573–583. [Google Scholar] [CrossRef]
- Chiba, H.; Muramatsu, M.; Nomoto, A.; Kato, H. Two human homologues ofSaccharomyces cerevisiae SWI2/SNF2andDrosophila brahmaare transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic Acids Res. 1994, 22, 1815–1820. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nat. Cell Biol. 1994, 370, 477–481. [Google Scholar] [CrossRef]
- Wang, W.; Côté, J.; Xue, Y.; Zhou, S.; Khavari, P.A.; Biggar, S.R.; Muchardt, C.; Kalpana, G.V.; Goff, S.P.; Yaniv, M.; et al. Pu-rification and Biochemical Heterogeneity of the Mammalian SWI-SNF Complex. EMBO J. 1996, 15, 5370–5382. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Daniel, C.; Tamkun, J.; Wu, C. ISWI, a member of the SWl2/SNF2 ATPase family, encodes the 140 kDa subunit of the nucleosome remodeling factor. Cell 1995, 83, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Tsukiyama, T.; Wu, C. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 1995, 83, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Varga-Weisz, P.D.; Wilm, M.; Bonte, E.; Dumas, K.; Mann, M.; Becker, P. Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nat. Cell Biol. 1997, 388, 598–602. [Google Scholar] [CrossRef]
- Ito, T.; Bulger, M.; Pazin, M.; Kobayashi, R.; Kadonaga, J.T. ACF, an ISWI-Containing and ATP-Utilizing Chromatin Assembly and Remodeling Factor. Cell 1997, 90, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Aihara, T.; Miyoshi, Y.; Koyama, K.; Suzuki, M.; Takahashi, E.; Monden, M.; Nakamura, Y. Cloning and mapping of SMARCA5 encoding hSNF2H, a novel human homologue of Drosophila ISWI. Cytogenet. Cell Genet. 1998, 81, 191–193. [Google Scholar] [CrossRef]
- Lazzaro, M.A.; Picketts, D.J. Cloning and characterization of the murine Imitation Switch (ISWI) genes: Differential expression patterns suggest distinct developmental roles for Snf2h and Snf2l. J. Neurochem. 2001, 77, 1145–1156. [Google Scholar] [CrossRef]
- Tolstorukov, M.Y.; Sansam, C.; Lu, P.; Koellhoffer, E.; Helming, K.C.; Alver, B.; Tillman, E.; Evans, J.A.; Wilson, B.G.; Park, P.J.; et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. USA 2013, 110, 10165–10170. [Google Scholar] [CrossRef] [Green Version]
- Yen, K.; Vinayachandran, V.; Batta, K.; Koerber, R.T.; Pugh, B.F. Genome-wide Nucleosome Specificity and Directionality of Chromatin Remodelers. Cell 2012, 149, 1461–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Schones, D.E.; Cui, K.; Ybarra, R.; Northrup, D.; Tang, Q.; Gattinoni, L.; Restifo, N.P.; Huang, S.; Zhao, K. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res. 2011, 21, 1650–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Bossen, C.; Murre, C.S.; Chang, A.N.; Mansson, R.; Rodewald, H.-R.; Murre, C. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 2015, 16, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.; Hota, S.; He, D.; Thomas, S.; Ho, L.; Pennacchio, L.A.; Bruneau, B.G. Brg1 modulates enhancer activation in mesoderm lineage commitment. Development 2015, 142, 1418–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Z.; El Hassan, M.A.; Xu, Z.; Yu, T.; Bremner, R. The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat. Immunol. 2008, 9, 785–793. [Google Scholar] [CrossRef]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Chen, Y.; Kim, B.; Wang, H.; Zhao, C.; He, X.; Liu, L.; Liu, W.; Wu, L.M.N.; Mao, M.; et al. Olig2 Targets Chromatin Remodelers to Enhancers to Initiate Oligodendrocyte Differentiation. Cell 2013, 152, 248–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alver, B.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W.M. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, R.T.; Pulice, J.L.; Valencia, A.; McBride, M.; McKenzie, Z.M.; Gillespie, M.A.; Ku, W.L.; Teng, M.; Cui, K.; Williams, R.; et al. SMARCB1 is required for widespread BAF complex–mediated activation of enhancers and bivalent promoters. Nat. Genet. 2017, 49, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.Y.; Grisan, V.; Jang, B.; Herbert, J.; Badenhorst, P. Genome-Wide Mapping Targets of the Metazoan Chromatin Remodeling Factor NURF Reveals Nucleosome Remodeling at Enhancers, Core Promoters and Gene Insulators. PLoS Genet. 2016, 12, e1005969. [Google Scholar] [CrossRef] [Green Version]
- Barisic, D.; Stadler, M.B.; Iurlaro, M.; Schübeler, D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nat. Cell Biol. 2019, 569, 136–140. [Google Scholar] [CrossRef]
- Qiu, Z.; Song, C.; Malakouti, N.; Murray, D.; Hariz, A.; Zimmerman, M.; Gygax, D.; Alhazmi, A.; Landry, J.W. Functional Interactions between NURF and Ctcf Regulate Gene Expression. Mol. Cell. Biol. 2015, 35, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Wiechens, N.; Singh, V.; Gkikopoulos, T.; Schofield, P.; Rocha, S.; Owen-Hughes, T. The Chromatin Remodelling Enzymes SNF2H and SNF2L Position Nucleosomes adjacent to CTCF and Other Transcription Factors. PLoS Genet. 2016, 12, e1005940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, L.F.; Ward, J.M.; Archer, T.K. Tumor suppressor SMARCB1 suppresses super-enhancers to govern hESC lineage determination. eLife 2019, 8, e45672. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, I.; Rando, O.; Delrow, J.; Tsukiyama, T. Chromatin remodelling at promoters suppresses antisense transcription. Nature 2007, 450, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, H.C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Geerdink, N.; Rotteveel, J.J.; Lammens, M.; Sistermans, E.; Heikens, G.T.; Gabreëls, F.J.M.; Mullaart, R.A.; Hamel, B.C.J. MECP2 Mutation in a Boy with Severe Neonatal Encephalopathy: Clinical, Neuropathological and Molecular Findings. Neuropediatrics 2002, 33, 33–36. [Google Scholar] [CrossRef]
- Bourdon, V.; Philippe, C.; Labrune, O.; Amsallem, D.; Arnould, C.; Jonveaux, P. A detailed analysis of the MECP2 gene: Prevalence of recurrent mutations and gross DNA rearrangements in Rett syndrome patients. Qual. Life Res. 2001, 108, 43–50. [Google Scholar] [CrossRef]
- Xiang, F.; Buervenich, S.; Nicolao, P.; Bailey, M.E.S.; Zhang, Z.; Anvret, M. Mutation screening in Rett syndrome patients. J. Med. Genet. 2000, 37, 250–255. [Google Scholar] [CrossRef]
- Bienvenu, T.; Carrié, A.; de Roux, N.; Vinet, M.-C.; Jonveaux, P.; Couvert, P.; Villard, L.; Arzimanoglou, A.; Beldjord, C.; Fontes, M.; et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum. Mol. Genet. 2000, 9, 1377–1384. [Google Scholar] [CrossRef]
- Klein, C.J.; Botuyan, M.-V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J.; Bird, T.; Ertekin-Taner, N.; Lincoln, S.; Hjorth, R.; Wu, Y.; Kwok, J.; Mer, G.; Dyck, P.J.; Nicholson, G.A. DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 2013, 80, 824–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmann, J.; Lin, L.; Schormair, B.; Kornum, B.R.; Faraco, J.; Plazzi, G.; Melberg, A.; Cornelio, F.; Urban, A.E.; Pizza, F.; et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 2012, 21, 2205–2210. [Google Scholar] [CrossRef] [Green Version]
- Tatton-Brown, K.; Childhood Overgrowth Consortium; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Duarte, S.D.V.; Zachariou, A.; Hanks, S.; O’Brien, E.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Heeley, J.M.; Carlston, C.M.; Acuna-Hidalgo, R.; Nillesen, W.M.; Dent, K.M.; Douglas, G.V.; Levine, K.L.; Bayrak-Toydemir, P.; Marcelis, C.L.; et al. The spectrum of DNMT3A variants in Tatton-Brown-Rahman syndrome overlaps with that in hematologic malignancies. Am. J. Med. Genet. 2017, 173, 3022–3028. [Google Scholar] [CrossRef]
- Kosaki, R.; Terashima, H.; Kubota, M.; Kosaki, K. Acute myeloid leukemia-associatedDNMT3Ap.Arg882His mutation in a patient with Tatton-Brown-Rahman overgrowth syndrome as a constitutional mutation. Am. J. Med. Genet. 2017, 173, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Heyn, P.; Logan, C.V.; Fluteau, A.; Challis, R.C.; Auchynnikava, T.; Martin, C.-A.; Marsh, J.A.; Taglini, F.; Kilanowski, F.; Parry, D.A.; et al. Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat. Genet. 2019, 51, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, J.H.; White, S.J.; Ariyürek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; Dunnen, J.T.D.; van Ommen, G.-J.B.; Breuning, M.H.; et al. Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, M.J.; Newbury-Ecob, R.; Holder-Espinasse, M.; Yau, S.; Lillis, S.; Hurst, J.A.; Clement, E.; Reardon, W.; Joss, S.; Hobson, E.; et al. Rubinstein–Taybi syndrome type 2: Report of nine new cases that extend the phenotypic and genotypic spectrum. Clin. Dysmorphol. 2016, 25, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Foley, P.; Bunyan, D.; Stratton, J.; Dillon, M.; Lynch, S.A. Further case of Rubinstein-Taybi syndrome due to a deletion in EP300. Am. J. Med. Genet. 2009, 149, 997–1000. [Google Scholar] [CrossRef]
- Menke, L.A.; Gardeitchik, T.; Hammond, P.; Heimdal, K.R.; Houge, G.; Hufnagel, S.B.; Ji, J.; Johansson, S.; Kant, S.G.; Kinning, E.; et al. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am. J. Med. Genet. 2018, 176, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Kalkhoven, E.; Roelfsema, J.H.; Teunissen, H.; den Boer, A.; Ariyurek, Y.; Zantema, A.; Breuning, M.H.; Hennekam, R.C.M.; Peters, D.J.M. Loss of CBP Acetyltransferase Activity by PHD Finger Mutations in Rubinstein-Taybi Syndrome. Human Mol. Genet. 2003, 12, 441–450. [Google Scholar] [CrossRef]
- Petrij, F.; Dauwerse, H.G.; Blough, R.I.; Giles, R.H.; Van Der Smagt, J.J.; Wallerstein, R.; Maaswinkel-Mooy, P.D.; van Karnebeek, C.; Van Ommen, G.-J.B.; Van Haeringen, A.; et al. Diagnostic analysis of the Rubinstein-Taybi syndrome: Five cosmids should be used for microdeletion detection and low number of protein truncating mutations. J. Med. Genet. 2000, 37, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Coupry, I.; Roudaut, C.; Stef, M.; Delrue, M.A.; Marche, M.; Burgelin, I.; Taine, L.; Cruaud, C.; Lacombe, D.; Arveiler, B. Molecular Analysis of the CBP Gene in 60 Patients with Rubinstein-Taybi Syndrome. J. Med. Genet. 2002, 39, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Menke, L.A.; van Belzen, M.J.; Alders, M.; Cristofoli, F.; Ehmke, N.; Fergelot, P.; Foster, A.; Gerkes, E.H.; Hoffer, M.J.V.; Horn, D.; et al. CREBBPmutations in individuals without Rubinstein-Taybi syndrome phenotype. Am. J. Med. Genet. 2016, 170, 2681–2693. [Google Scholar] [CrossRef] [PubMed]
- Angius, A.; Uva, P.; Oppo, M.; Persico, I.; Onano, S.; Olla, S.; Pes, V.; Perria, C.; Cuccuru, G.; Atzeni, R.; et al. Confirmation of a new phenotype in an individual with a variant in the last part of exon 30 of CREBBP. Am. J. Med. Genet. 2019, 179, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Goudie, D.; Blair, E.; Chandler, K.; Joss, S.; McKay, V.; Green, A.; Armstrong, R.; Lees, M.; Kamien, B.; et al. KAT6A Syndrome: Genotype–phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 2019, 21, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Arboleda, V.; Lee, H.; Dorrani, N.; Zadeh, N.; Willis, M.; MacMurdo, C.F.; Manning, M.A.; Kwan, A.; Hudgins, L.; Barthelemy, F.; et al. De Novo Nonsense Mutations in KAT6A, a Lysine Acetyl-Transferase Gene, Cause a Syndrome Including Microcephaly and Global Developmental Delay. Am. J. Hum. Genet. 2015, 96, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Millan, F.; Cho, M.T.; Retterer, K.; Monaghan, K.G.; Bai, R.; Vitazka, P.; Everman, D.B.; Smith, B.; Angle, B.; Roberts, V.; et al. Whole exome sequencing reveals de novo pathogenic variants inKAT6Aas a cause of a neurodevelopmental disorder. Am. J. Med. Genet. 2016, 170, 1791–1798. [Google Scholar] [CrossRef]
- Tham, E.; Lindstrand, A.; Santani, A.; Malmgren, H.; Nesbitt, A.; Dubbs, H.A.; Zackai, E.H.; Parker, M.J.; Millan, F.; Rosenbaum, K.; et al. Dominant Mutations in KAT6A Cause Intellectual Disability with Recognizable Syndromic Features. Am. J. Hum. Genet. 2015, 96, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Szakszon, K.; Salpietro, C.; Kakar, N.; Knegt, A.C.; Oláh, É.; Dallapiccola, B.; Borck, G. De novo mutations of the gene encoding the histone acetyltransferase KAT6B in two patients with Say-Barber/Biesecker/Young-Simpson syndrome. Am. J. Med. Genet. Part A 2013, 161, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Szakszon, K.; Berényi, E.; Jakab, A.; Bessenyei, B.; Balogh, E.; Köbling, T.; Szilvássy, J.; Knegt, A.C.; Oláh, É. Blepharophimosis mental retardation syndrome Say-Barber/Biesecker/Young-Simpson type—New findings with neuroimaging. Am. J. Med. Genet. Part A 2011, 155, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Yates, T.M.; Langley, C.L.; Grozeva, D.; Raymond, F.L.; Johnson, D.S.; DDD Study. Novel KAT6B proximal familial variant expands genotypic and phenotypic spectrum. Clin. Genet. 2019, 95, 334–335. [Google Scholar] [CrossRef]
- Clayton-Smith, J.; O’Sullivan, J.; Daly, S.; Bhaskar, S.; Day, R.; Anderson, B.; Voss, A.K.; Thomas, T.; Biesecker, L.G.; Smith, P.; et al. Whole-Exome-Sequencing Identifies Mutations in Histone Acetyltransferase Gene KAT6B in Individuals with the Say-Barber-Biesecker Variant of Ohdo Syndrome. Am. J. Hum. Genet. 2011, 89, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Campeau, P.M.; Kim, J.C.; Lu, J.T.; Schwartzentruber, J.A.; Abdul-Rahman, O.A.; Schlaubitz, S.; Murdock, D.M.; Jiang, M.-M.; Lammer, E.J.; Enns, G.M.; et al. Mutations in KAT6B, Encoding a Histone Acetyltransferase, Cause Genitopatellar Syndrome. Am. J. Hum. Genet. 2012, 90, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.; Deshpande, C.; Dafou, D.; Vissers, L.; Woollard, W.J.; Holder, S.E.; Gillessen-Kaesbach, G.; Derks, R.; White, S.M.; Cohen-Snuijf, R.; et al. De Novo Mutations of the Gene Encoding the Histone Acetyltransferase KAT6B Cause Genitopatellar Syndrome. Am. J. Hum. Genet. 2012, 90, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Haarhuis, J.H.I.; Cacciatore, Á.S.; Oldenkamp, R.; Van Ruiten, M.S.; Willems, L.; Teunissen, H.; Muir, K.W.; De Wit, E.; Rowland, B.D.; et al. The structural basis for cohesin–CTCF-anchored loops. Nat. Cell Biol. 2020, 578, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Laloo, B.; Barillot, M.; Barnetche, T.; Blanchard, C.; Rooryck, C.; Marche, M.; Burgelin, I.; Coupry, I.; Chassaing, N.; et al. A mutation in the 3′-UTR of the HDAC6 gene abolishing the post-transcriptional regulation mediated by hsa-miR-433 is linked to a new form of dominant X-linked chondrodysplasia. Hum. Mol. Genet. 2010, 19, 2015–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassaing, N.; Siani, V.; Carles, D.; Delezoide, A.L.; Alberti, E.M.; Battin, J.; Chateil, J.-F.; Gilbert-Dussardier, B.; Coupry, I.; Arveiler, B.; et al. X-linked dominant chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia. Am. J. Med. Genet. 2005, 136, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Harakalova, M.; Boogaard, M.-J.V.D.; Sinke, R.; Van Lieshout, S.; Van Tuil, M.C.; Duran, K.; Renkens, I.; Terhal, P.A.; de Kovel, C.; Nijman, I.; et al. X-exome sequencing identifies aHDAC8variant in a large pedigree with X-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J. Med. Genet. 2012, 49, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef]
- Libert, S.; Pointer, K.; Bell, E.L.; Das, A.; Cohen, D.E.; Asara, J.M.; Kapur, K.; Bergmann, S.; Preisig, M.; Otowa, T.; et al. SIRT1 Activates MAO-A in the Brain to Mediate Anxiety and Exploratory Drive. Cell 2011, 147, 1459–1472. [Google Scholar] [CrossRef] [Green Version]
- Wolff, D.; Endele, S.; Azzarello-Burri, S.; Hoyer, J.; Zweier, M.; Schanze, I.; Schmitt, B.; Rauch, A.; Reis, A.; Zweier, C. In-Frame Deletion and Missense Mutations of the C-Terminal Helicase Domain of SMARCA2 in Three Patients with Nicolaides-Baraitser Syndrome. Mol. Syndr. 2012, 2, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Van Houdt, J.K.J.; Nowakowska, B.A.; Sousa, S.B.; Van Schaik, B.D.C.; Seuntjens, E.; Avonce, N.; Sifrim, A.; Abdul-Rahman, O.A.; Boogaard, M.-J.H.V.D.; Bottani, A.; et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat. Genet. 2012, 44, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Ishiguro, H.; Yazaki, S.; Horiuchi, Y.; Arai, M.; Niizato, K.; Iritani, S.; Itokawa, M.; Inada, T.; Iwata, N.; et al. Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum. Mol. Genet. 2009, 18, 2483–2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurusaki, Y.; Okamoto, N.; Ohashi, H.; Kosho, T.; Imai, Y.; Hibi-Ko, Y.; Kaname, T.; Naritomi, K.; Kawame, H.; Wakui, K.; et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 2012, 44, 376–378. [Google Scholar] [CrossRef]
- Sévenet, N.; Sheridan, E.; Amram, D.; Schneider, P.; Handgretinger, R.; Delattre, O. Constitutional Mutations of the hSNF5/INI1 Gene Predispose to a Variety of Cancers. Am. J. Hum. Genet. 1999, 65, 1342–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.D.; Gokgoz, N.; Andrulis, I.L.; Mainprize, T.G.; Drake, J.M.; Rutka, J.T. Familial Posterior Fossa Brain Tumors of Infancy Secondary to Germline Mutation of the hSNF5 Gene. Am. J. Hum. Genet. 2000, 66, 1403–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiaans, I.; Kenter, S.B.; Brink, H.C.; Van Os, T.A.M.; Baas, F.; Munckhof, P.V.D.; Kidd, A.M.J.; Hulsebos, T.J.M. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J. Med. Genet. 2010, 48, 93–97. [Google Scholar] [CrossRef]
- Hulsebos, T.J.; Plomp, A.S.; Wolterman, R.A.; Robanus-Maandag, E.C.; Baas, F.; Wesseling, P. Germline Mutation of INI1/SMARCB1 in Familial Schwannomatosis. Am. J. Hum. Genet. 2007, 80, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Sestini, R.; Bacci, C.; Provenzano, A.; Genuardi, M.; Papi, L. Evidence of a four-hit mechanism involvingSMARCB1andNF2in schwannomatosis-associated schwannomas. Hum. Mutat. 2008, 29, 227–231. [Google Scholar] [CrossRef]
- Hadfield, K.D.; Newman, W.; Bowers, N.L.; Wallace, A.; Bolger, C.; Colley, A.; McCann, E.; Trump, D.; Prescott, T.; Evans, G. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J. Med. Genet. 2008, 45, 332–339. [Google Scholar] [CrossRef]
- Machol, K.; Rousseau, J.; Ehresmann, S.; Garcia, T.; Nguyen, T.T.M.; Spillmann, R.C.; Sullivan, J.A.; Shashi, V.; Jiang, Y.-H.; Stong, N.; et al. Expanding the Spectrum of BAF-Related Disorders: De Novo Variants in SMARCC2 Cause a Syndrome with Intellectual Disability and Developmental Delay. Am. J. Hum. Genet. 2019, 104, 164–178. [Google Scholar] [CrossRef] [Green Version]
- Nixon, K.C.; Rousseau, J.; Stone, M.H.; Sarikahya, M.; Ehresmann, S.; Mizuno, S.; Matsumoto, N.; Miyake, N.; Baralle, D.; McKee, S.; et al. A Syndromic Neurodevelopmental Disorder Caused by Mutations in SMARCD1, a Core SWI/SNF Subunit Needed for Context-Dependent Neuronal Gene Regulation in Flies. Am. J. Hum. Genet. 2019, 104, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; O’Sullivan, J.; Bhaskar, S.S.; Hadfield, K.D.; Poke, G.; Caird, J.; Sharif, S.; Eccles, D.M.; FitzPatrick, D.R.; Rawluk, D.; et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat. Genet. 2013, 45, 295–298. [Google Scholar] [CrossRef]
- Wieczorek, D.; Bögershausen, N.; Beleggia, F.; Steiner-Haldenstätt, S.; Pohl, E.; Li, Y.; Milz, E.; Martin, M.; Thiele, H.; Altmüller, J.; et al. A comprehensive molecular study on Coffin–Siris and Nicolaides–Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 2013, 22, 5121–5135. [Google Scholar] [CrossRef]
- Karaca, E.; Harel, T.; Pehlivan, D.; Jhangiani, S.N.; Gambin, T.; Akdemir, Z.C.; Gonzaga-Jauregui, C.; Erdin, S.; Bayram, Y.; Campbell, I.; et al. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Fichera, M.; Failla, P.; Saccuzzo, L.; Miceli, M.; Salvo, E.; Castiglia, L.; Galesi, O.; Grillo, L.; Cali, F.; Greco, D.; et al. Mutations in ACTL6B, coding for a subunit of the neuron-specific chromatin remodeling complex nBAF, cause early onset severe developmental and epileptic encephalopathy with brain hypomyelination and cerebellar atrophy. Qual. Life Res. 2019, 138, 187–198. [Google Scholar] [CrossRef]
- Maddirevula, S.; Bs, F.A.; Al-Owain, M.; Al Muhaizea, M.A.; Kayyali, H.R.; Alhashem, A.; Rahbeeni, Z.; Al-Otaibi, M.; Alzaidan, H.I.; Ms, A.B.; et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 2019, 21, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, S.; Rousseau, J.; Peng, H.; Aouabed, Z.; Priam, P.; Theroux, J.-F.; Jefri, M.; Tanti, A.; Wu, H.; Kolobova, I.; et al. Mutations in ACTL6B Cause Neurodevelopmental Deficits and Epilepsy and Lead to Loss of Dendrites in Human Neurons. Am. J. Hum. Genet. 2019, 104, 815–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santen, G.W.; Aten, E.; Silfhout, A.T.V.-V.; Pottinger, C.; Van Bon, B.W.; Van Minderhout, I.J.; Snowdowne, R.; Van Der Lans, C.A.; Boogaard, M.; Linssen, M.M.; et al. Coffin-Siris Syndrome and the BAF Complex: Genotype-Phenotype Study in 63 Patients. Hum. Mutat. 2013, 34, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Zweier, M.; Peippo, M.M.; Pöyhönen, M.; Kääriäinen, H.; Begemann, A.; Joset, P.; Oneda, B.; Rauch, A. The HHID Syndrome of Hypertrichosis, Hyperkeratosis, Abnormal Corpus Callosum, Intellectual Disability, and Minor Anomalies Is Caused by Mutations in ARID1B. Am. J. Med. Genet. 2017, 173, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyer, J.; Ekici, A.B.; Endele, S.; Popp, B.; Zweier, C.; Wiesener, A.; Wohlleber, E.; Dufke, A.; Rossier, E.; Petsch, C.; et al. Haploinsufficiency of ARID1B, a Member of the SWI/SNF-A Chromatin-Remodeling Complex, Is a Frequent Cause of Intellectual Disability. Am. J. Hum. Genet. 2012, 90, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Van Paemel, R.; De Bruyne, P.; Van Der Straaten, S.; D’Hondt, M.; Fränkel, U.; Dheedene, A.; Menten, B.; Callewaert, B. Confirmation of an ARID2 defect in SWI/SNF-related intellectual disability. Am. J. Med. Genet. 2017, 173, 3104–3108. [Google Scholar] [CrossRef]
- Shang, L.; Cho, M.T.; Retterer, K.; Folk, L.; Humberson, J.; Rohena, L.; Sidhu, A.; Saliganan, S.; Iglesias, A.; Vitazka, P.; et al. Mutations in ARID2 are associated with intellectual disabilities. Neurogenetics 2015, 16, 307–314. [Google Scholar] [CrossRef]
- Bramswig, N.C.; Caluseriu, O.; Lüdecke, H.-J.; Bolduc, F.V.; Noel, N.C.L.; Wieland, T.; Surowy, H.; Christen, H.-J.; Engels, H.; Strom, T.M.; et al. Heterozygosity for ARID2 loss-of-function mutations in individuals with a Coffin–Siris syndrome-like phenotype. Qual. Life Res. 2017, 136, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Gearing, M.; Juncos, J.L.; Procaccio, V.; Gutekunst, C.-A.; Marino-Rodriguez, E.M.; Gyure, K.A.; Ono, S.; Santoianni, R.; Krawiecki, N.S.; Wallace, D.C.; et al. Aggregation of actin and cofilin in identical twins with juvenile-onset dystonia. Ann. Neurol. 2002, 52, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Procaccio, V.; Salazar, G.; Ono, S.; Styers, M.L.; Gearing, M.; Davila, A.; Jimenez, R.; Juncos, J.; Gutekunst, C.-A.; Meroni, G.; et al. A Mutation of β-Actin That Alters Depolymerization Dynamics Is Associated with Autosomal Dominant Developmental Malformations, Deafness, and Dystonia. Am. J. Hum. Genet. 2006, 78, 947–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Donato, N.; Rump, A.; Koenig, R.; Der Kaloustian, V.M.; Halal, F.; Sonntag, K.; Krause, C.; Hackmann, K.; Hahn, G.; Schrock, E.; et al. Severe forms of Baraitser–Winter syndrome are caused by ACTB mutations rather than ACTG1 mutations. Eur. J. Hum. Genet. 2013, 22, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Rivière, J.-B.; Van Bon, B.W.M.; Hoischen, A.; Kholmanskikh, S.S.; O’Roak, B.; Gilissen, C.; Gijsen, S.; Sullivan, C.T.; Christian, S.L.; Abdul-Rahman, O.A.; et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012, 44, 440–444. [Google Scholar] [CrossRef] [Green Version]
- Vasileiou, G.; Vergarajauregui, S.; Endele, S.; Popp, B.; Büttner, C.; Ekici, A.B.; Gerard, M.; Bramswig, N.C.; Albrecht, B.; Clayton-Smith, J.; et al. Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome. Am. J. Hum. Genet. 2018, 102, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Dias, C.; Estruch, S.B.; Graham, S.; McRae, J.; Sawiak, S.; Hurst, J.A.; Joss, S.K.; Holder, S.E.; Morton, J.E.; Turner, C.; et al. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am. J. Hum. Genet. 2016, 99, 253–274. [Google Scholar] [CrossRef] [Green Version]
- Lessel, D.; Gehbauer, C.; Bramswig, N.C.; Schluth-Bolard, C.; Venkataramanappa, S.; van Gassen, K.L.I.; Hempel, M.; Haack, T.B.; Baresic, A.; Genetti, C.A.; et al. BCL11B mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain 2018, 141, 2299–2311. [Google Scholar] [CrossRef] [Green Version]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’Ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.-F.; Stevens, C.; Wang, L.-S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- Borck, G.; Hög, F.; Dentici, M.L.; Tan, P.L.; Sowada, N.; Medeira, A.; Gueneau, L.; Thiele, H.; Kousi, M.; Lepri, F.; et al. BRF1mutations alter RNA polymerase III–dependent transcription and cause neurodevelopmental anomalies. Genome Res. 2015, 25, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Stankiewicz, P.; Khan, T.; Szafranski, P.; Slattery, L.; Streff, H.; Vetrini, F.; Bernstein, J.A.; Brown, C.W.; Rosenfeld, J.A.; Rednam, S.; et al. Haploinsufficiency of the Chromatin Remodeler BPTF Causes Syndromic Developmental and Speech Delay, Postnatal Microcephaly, and Dysmorphic Features. Am. J. Hum. Genet. 2017, 101, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Homann, O.R.; Misura, K.; Lamas, E.; Sandrock, R.W.; Nelson, P.; McDonough, S.I.; DeLisi, L.E. Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol. Psychiatry 2016, 21, 1690–1695. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.; Barbosa, M.; Ameur, A.; Soares, G.; De Sá, J.; Dias, A.I.; Oliveira, G.; Cabral, P.; Temudo, T.; Calado, E.; et al. Identification of novel genetic causes of Rett syndrome-likephenotypes. J. Med. Genet. 2016, 53, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaghlool, A.; Halvardson, J.; Zhao, J.J.; Etemadikhah, M.; Kalushkova, A.; Konska, K.; Jernberg-Wiklund, H.; Thuresson, A.-C.; Feuk, L. A Role for the Chromatin-Remodeling Factor BAZ1A in Neurodevelopment. Hum. Mutat. 2016, 37, 964–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilarowski, G.; Vernon, H.; Applegate, C.D.; Boukas, L.; Cho, M.T.; Gurnett, C.; Benke, P.J.; Beaver, E.; Heeley, J.M.; Medne, L.; et al. Missense variants in the chromatin remodeler CHD1 are associated with neurodevelopmental disability. J. Med. Genet. 2018, 55, 561–566. [Google Scholar] [CrossRef]
- Suls, A.; Jaehn, J.A.; Kecskés, A.; Weber, Y.; Weckhuysen, S.; Craiu, D.; Siekierska, A.; Djémié, T.; Afrikanova, T.; Gormley, P.; et al. De Novo Loss-of-Function Mutations in CHD2 Cause a Fever-Sensitive Myoclonic Epileptic Encephalopathy Sharing Features with Dravet Syndrome. Am. J. Hum. Genet. 2013, 93, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef]
- Petersen, A.K.; Streff, H.; Tokita, M.; Bostwick, B.L. The first reported case of an inherited pathogenic CHD2 variant in a clinically affected mother and daughter. Am. J. Med. Genet. 2018, 176, 1667–1669. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.M.; O’Roak, B.J.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eising, E.; Carrion-Castillo, A.; Vino, A.; Strand, E.A.; Jakielski, K.J.; Scerri, T.S.; Hildebrand, M.S.; Webster, R.; Ma, A.; Mazoyer, B.; et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 2019, 24, 1065–1078. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Sifrim, A.; Hitz, M.-P.; Wilsdon, A.; Breckpot, J.; Al Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef]
- Gabriele, M.; Silfhout, A.T.V.-V.; Germain, P.-L.; Vitriolo, A.; Kumar, R.; Douglas, E.; Haan, E.; Kosaki, K.; Takenouchi, T.; Rauch, A.; et al. YY1 Haploinsufficiency Causes an Intellectual Disability Syndrome Featuring Transcriptional and Chromatin Dysfunction. Am. J. Hum. Genet. 2017, 100, 907–925. [Google Scholar] [CrossRef] [Green Version]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Hood, R.L.; Lines, M.A.; Nikkel, S.M.; Schwartzentruber, J.; Beaulieu, C.; Nowaczyk, M.J.; Allanson, J.; Kim, C.; Wieczorek, D.; Moilanen, J.; et al. Mutations in SRCAP, Encoding SNF2-Related CREBBP Activator Protein, Cause Floating-Harbor Syndrome. Am. J. Hum. Genet. 2012, 90, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Musio, A.; Selicorni, A.; Focarelli, M.L.; Gervasini, C.; Milani, D.; Russo, S.; Vezzoni, P.; Larizza, L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006, 38, 528–530. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in Cohesin Complex Members SMC3 and SMC1A Cause a Mild Variant of Cornelia de Lange Syndrome with Predominant Mental Retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruszka, P.; Berger, S.; Casa, V.; Dekker, M.R.; Gaesser, J.; Weiss, K.; Martinez, A.F.; Murdock, D.R.; Louie, R.J.; Prijoles, E.J.; et al. Cohesin complex-associated holoprosencephaly. Brain 2019, 142, 2631–2643. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.; Joss, S.; Metcalfe, K.A.; Somarathi, S.; Cruden, J.; Devlin, A.M.; Donaldson, A.; Di Donato, N.; FitzPatrick, D.R.; Kaiser, F.J.; et al. Heterozygous truncation mutations of theSMC1Agene cause a severe early onset epilepsy with cluster seizures in females: Detailed phenotyping of 10 new cases. Epilepsia 2017, 58, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.H.; Tim-Aroon, T.; Shieh, J.; Merrill, M.; Deeb, K.K.; Zhang, S.; Bass, N.E.; Bedoyan, J.K. Novel SMC1A frameshift mutations in children with developmental delay and epilepsy. Eur. J. Med. Genet. 2015, 58, 562–568. [Google Scholar] [CrossRef]
- Lebrun, N.; Lebon, S.; Jeannet, P.-Y.; Jacquemont, S.; Billuart, P.; Bienvenu, T. Early-onset encephalopathy with epilepsy associated with a novel splice site mutation inSMC1A. Am. J. Med. Genet. 2015, 167, 3076–3081. [Google Scholar] [CrossRef]
- Jansen, S.; Kleefstra, T.; Willemsen, M.H.; De Vries, P.; Pfundt, R.; Hehir-Kwa, J.Y.; Gilissen, C.; Veltman, J.A.; De Vries, B.B.A.; Vissers, L.E.L.M. De novoloss-of-function mutations in X-linkedSMC1Acause severe ID and therapy-resistant epilepsy in females: Expanding the phenotypic spectrum. Clin. Genet. 2016, 90, 413–419. [Google Scholar] [CrossRef]
- Ansari, M.; Poke, G.; Ferry, Q.; Williamson, K.; Aldridge, R.; Meynert, A.M.; Bengani, H.; Chan, C.Y.; Kayserili, H.; Avci, Ş.; et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet. 2014, 51, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Gil-Rodríguez, M.C.; Deardorff, M.A.; Ansari, M.; Tan, C.A.; Parenti, I.; Baquero-Montoya, C.; Ousager, L.B.; Puisac, B.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E.; et al. De NovoHeterozygous Mutations inSMC3Cause a Range of Cornelia de Lange Syndrome-Overlapping Phenotypes. Hum. Mutat. 2015, 36, 454–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, M.; Jespersgaard, C.; Nazaryan, L.; Bisgaard, A.-M.; Tümer, Z. A novelRAD21variant associated with intrafamilial phenotypic variation in Cornelia de Lange syndrome—Review of the literature. Clin. Genet. 2016, 91, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Wilde, J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Mönnich, M.; Yan, Y.; Xu, W.; Gil-Rodríguez, M.C.; et al. RAD21 Mutations Cause a Human Cohesinopathy. Am. J. Hum. Genet. 2012, 90, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.; Parasivam, G. Another case of holoprosencephaly associated with RAD21 loss-of-function variant. Brain 2020, 143, e64. [Google Scholar] [CrossRef]
- Lehalle, D.; Mosca-Boidron, A.-L.; Begtrup, A.; Boute-Benejean, O.; Charles, P.; Cho, M.T.; Clarkson, A.; Devinsky, O.; Duffourd, Y.; Duplomb-Jego, L.; et al. STAG1mutations cause a novel cohesinopathy characterised by unspecific syndromic intellectual disability. J. Med. Genet. 2017, 54, 479–488. [Google Scholar] [CrossRef]
- Yuan, B.; DDD study; Neira, J.; Pehlivan, D.; Santiago-Sim, T.; Song, X.; Rosenfeld, J.; Posey, J.; Patel, V.; Jin, W.; et al. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet. Med. 2018, 21, 663–675. [Google Scholar] [CrossRef]
- Soardi, F.C.; Machado-Silva, A.; Linhares, N.; Zheng, G.; Qu, Q.; Pena, H.B.; Martins, T.M.D.M.; Vieira, H.G.S.; Pereira, N.B.; Melo-Minardi, R.C.; et al. Familial STAG2 germline mutation defines a new human cohesinopathy. NPJ Genom. Med. 2017, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Mullegama, S.V.; Klein, S.D.; Mulatinho, M.V.; Senaratne, T.N.; Singh, K.; Nguyen, D.C.; Gallant, N.M.; Strom, S.P.; Ghahremani, S.; Rao, N.P.; et al. De novo loss-of-function variants inSTAG2are associated with developmental delay, microcephaly, and congenital anomalies. Am. J. Med. Genet. 2017, 173, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Gillis, L.A.; McCallum, J.; Kaur, M.; DeScipio, C.; Yaeger, D.; Mariani, A.; Kline, A.D.; Li, H.-H.; Devoto, M.; Jackson, L.G.; et al. NIPBL Mutational Analysis in 120 Individuals with Cornelia de Lange Syndrome and Evaluation of Genotype-Phenotype Correlations. Am. J. Hum. Genet. 2004, 75, 610–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkin, E.; Wang, T.-J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Gregor, A.; Oti, M.; Kouwenhoven, E.N.; Hoyer, J.; Sticht, H.; Ekici, A.B.; Kjaergaard, S.; Rauch, A.; Stunnenberg, H.G.; Uebe, S.; et al. De Novo Mutations in the Genome Organizer CTCF Cause Intellectual Disability. Am. J. Hum. Genet. 2013, 93, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.-A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Williamson, I.; Lettice, L.A.; Hill, R.E.; Bickmore, W.A. Shh and ZRS enhancer co-localisation is specific to the zone of polarizing activity. Development 2016, 143, 2994–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudelaar, A.M.; Higgs, D.R. The relationship between genome structure and function. Nat. Rev. Genet. 2021, 22, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Larochelle, S.; Batliner, J.; Gamble, M.J.; Barboza, N.M.; Kraybill, B.C.; Blethrow, J.D.; Shokat, K.M.; Fisher, R.P. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat. Struct. Mol. Biol. 2005, 13, 55–62. [Google Scholar] [CrossRef]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Symmons, O.; Pan, L.; Remeseiro, S.; Aktas, T.; Klein, F.; Huber, W.; Spitz, F. The Shh Topological Domain Facilitates the Action of Remote Enhancers by Reducing the Effects of Genomic Distances. Dev. Cell 2016, 39, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Symmons, O.; Uslu, V.V.; Tsujimura, T.; Ruf, S.; Nassari, S.; Schwarzer, W.; Ettwiller, L.; Spitz, F. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014, 24, 390–400. [Google Scholar] [CrossRef] [Green Version]
- De Laat, W.; Duboule, D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 2013, 502, 499–506. [Google Scholar] [CrossRef]
- Kalhor, R.; Tjong, H.; Jayathilaka, N.; Alber, F.; Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 2012, 30, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasmyth, K. Disseminating the Genome: Joining, Resolving, and Separating Sister Chromatids During Mitosis and Meiosis. Annu. Rev. Genet. 2001, 35, 673–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alipour, E.; Marko, J.F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 2012, 40, 11202–11212. [Google Scholar] [CrossRef] [Green Version]
- Ganji, M.; Shaltiel, I.A.; Bisht, S.; Kim, E.; Kalichava, A.; Haering, C.H.; Dekker, C. Real-time imaging of DNA loop extrusion by condensin. Science 2018, 360, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.-C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.; Huber, W.; Haering, C.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.; Huang, S.-C.; Hilaire, B.G.S.; Engreitz, J.M.; Perez, E.; Kieffer-Kwon, K.-R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e24. [Google Scholar] [CrossRef] [Green Version]
- Wutz, G.; Várnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef]
- Haarhuis, J.H.; van der Weide, R.H.; Blomen, V.A.; Yáñez-Cuna, J.O.; Amendola, M.; Van Ruiten, M.S.; Krijger, P.; Teunissen, H.; Medema, R.; Van Steensel, B.; et al. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 2017, 169, 693–707.e14. [Google Scholar] [CrossRef] [Green Version]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vian, L.; Pekowska, A.; Rao, S.S.; Kieffer-Kwon, K.-R.; Jung, S.; Baranello, L.; Huang, S.-C.; el Khattabi, L.; Dose, M.; Pruett, N.; et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 2018, 173, 1165–1178.e20. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Shi, Z.; Zhang, H.; Finkelstein, I.J.; Yu, H. Human cohesin compacts DNA by loop extrusion. Science 2019, 366, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.-M. DNA loop extrusion by human cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef]
- Gutierrez-Escribano, P.; Newton, M.D.; Llauró, A.; Huber, J.; Tanasie, L.; Davy, J.; Aly, I.; Aramayo, R.; Montoya, A.; Kramer, H.; et al. A conserved ATP- and Scc2/4-dependent activity for cohesin in tethering DNA molecules. Sci. Adv. 2019, 5, eaay6804. [Google Scholar] [CrossRef] [Green Version]
- De Wit, E.; Vos, E.; Holwerda, S.J.; Valdes-Quezada, C.; Verstegen, M.J.; Teunissen, H.; Splinter, E.; Wijchers, P.J.; Krijger, P.; de Laat, W. CTCF Binding Polarity Determines Chromatin Looping. Mol. Cell 2015, 60, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Choi, J.-M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Feric, M.; Misteli, T. Phase separation in genome organization across evolution. Trends Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.B.; Rohatgi, R. In vivo Formation of Vacuolated Multi-phase Compartments Lacking Membranes. Cell Rep. 2016, 16, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.-K.; Spille, J.-H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wu, T.; Gutman, O.; Lu, H.; Zhou, Q.; Henis, Y.I.; Luo, K. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat. Cell Biol. 2020, 22, 453–464. [Google Scholar] [CrossRef]
- Nair, S.J.; Yang, L.; Meluzzi, D.; Oh, S.; Yang, F.; Friedman, M.J.; Wang, S.; Suter, T.; Alshareedah, I.; Gamliel, A.; et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 2019, 26, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M.L. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic Heritability and Shared Environmental Factors Among Twin Pairs With Autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- The Brainstorm Consortium; Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.H.; Anttila, V.; Won, H.; Feng, Y.-C.A.; Rosenthal, J.; Zhu, Z.; Tucker-Drob, E.M.; Nivard, M.; Grotzinger, A.D.; Posthuma, D.; et al. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 2019, 179, 1469–1482.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Tipping the Balance of Chromatin States. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef] [Green Version]
- Kleefstra, T.; Schenck, A.; Kramer, J.; van Bokhoven, H. The genetics of cognitive epigenetics. Neuropharmacology 2014, 80, 83–94. [Google Scholar] [CrossRef]
- Garcia-Dominguez, M.; Reyes, J.C. SUMO association with repressor complexes, emerging routes for transcriptional control. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1789, 451–459. [Google Scholar] [CrossRef]
- Vitriolo, A.; Gabriele, M.; Testa, G. From enhanceropathies to the epigenetic manifold underlying human cognition. Hum. Mol. Genet. 2019, 28, R226–R234. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Gabriele, M.; Tobon, A.L.; D’Agostino, G.A.; Testa, G. The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 84, 306–327. [Google Scholar] [CrossRef]
- Pohodich, A.E.; Zoghbi, H.Y. Rett syndrome: Disruption of epigenetic control of postnatal neurological functions. Hum. Mol. Genet. 2015, 24, R10–R16. [Google Scholar] [CrossRef]
- Berson, A.; Sartoris, A.; Nativio, R.; Van Deerlin, V.; Toledo, J.; Porta, S.; Liu, S.; Chung, C.-Y.; Garcia, B.A.; Lee, V.M.-Y.; et al. TDP-43 Promotes Neurodegeneration by Impairing Chromatin Remodeling. Curr. Biol. 2017, 27, 3579–3590.e6. [Google Scholar] [CrossRef] [Green Version]
- Ernst, C. Proliferation and Differentiation Deficits are a Major Convergence Point for Neurodevelopmental Disorders. Trends Neurosci. 2016, 39, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Béna, F.; Breckpot, J.; Philip, N.; Menten, B.; Van Esch, H.; Scalais, E.; Salamone, J.M.; Fong, C.-T.; Kussmann, J.L.; et al. Duplications of the critical Rubinstein-Taybi deletion region on chromosome 16p13.3 cause a novel recognisable syndrome. J. Med. Genet. 2009, 47, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Marangi, G.; Leuzzi, V.; Orteschi, D.; Grimaldi, M.E.; Lecce, R.; Neri, G.; Zollino, M. Duplication of the Rubinstein-Taybi region on 16p13.3 is associated with a distinctive phenotype. Am. J. Med. Genet. 2008, 146, 2313–2317. [Google Scholar] [CrossRef] [PubMed]
- Bahari-Javan, S.; Varbanov, H.; Halder, R.; Benito, E.; Kaurani, L.; Burkhardt, S.; Anderson-Schmidt, H.; Anghelescu, I.; Budde, M.; Stilling, R.; et al. HDAC1 links early life stress to schizophrenia-like phenotypes. Proc. Natl. Acad. Sci. USA 2017, 114, E4686–E4694. [Google Scholar] [CrossRef] [Green Version]
- Benes, F.M.; Lim, B.; Matzilevich, D.; Walsh, J.P.; Subburaju, S.; Minns, M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. USA 2007, 104, 10164–10169. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.P.; Grayson, D.R.; Gavin, D.P. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: Analysis of the National Brain Databank microarray collection. Schizophr. Res. 2008, 98, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. α-Synuclein Sequesters Dnmt1 from the Nucleus. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [Green Version]
- Toker, L.; Tran, G.T.; Sundaresan, J.; Tysnes, O.-B.; Alves, G.; Haugarvoll, K.; Nido, G.S.; Dölle, C.; Tzoulis, C. Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson’s disease brain. Mol. Neurodegener. 2021, 16, 1–20. [Google Scholar] [CrossRef]
- Corpión, I.H.; Guiretti, D.; Alcaraz-Iborra, M.; Olivares, R.; Campos-Caro, A.; Barco, A.; Valor, L.M. Early alteration of epigenetic-related transcription in Huntington’s disease mouse models. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.-Z.; Gohler, H.; Wanker, E.; Bates, G.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef] [Green Version]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-Wide DNA Methylation Differences Between Late-Onset Alzheimer’s Disease and Cognitively Normal Controls in Human Frontal Cortex. J. Alzheimer’s Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef]
- Marzi, S.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.S.; Keleshian, V.L.; Klein, S.; Rapoport, S.I. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2012, 2, e132. [Google Scholar] [CrossRef]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar] [CrossRef] [Green Version]
- Furlong, E.E.M.; Levine, M. Developmental enhancers and chromosome topology. Science 2018, 361, 1341–1345. [Google Scholar] [CrossRef] [Green Version]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef] [Green Version]
- Hyle, J.; Zhang, Y.; Wright, S.; Xu, B.; Shao, Y.; Easton, J.; Tian, L.; Feng, R.; Xu, P.; Li, C. Acute depletion of CTCF directly affects MYC regulation through loss of enhancer–promoter looping. Nucleic Acids Res. 2019, 47, 6699–6713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuijers, J.; Manteiga, J.C.; Weintraub, A.S.; Day, D.S.; Zamudio, A.V.; Hnisz, D.; Lee, T.I.; Young, R.A. Transcriptional Dysregulation of MYC Reveals Common Enhancer-Docking Mechanism. Cell Rep. 2018, 23, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushiki, A.; University of Washington Center for Mendelian Genomics; Zhang, Y.; Xiong, C.; Zhao, J.; Georgakopoulos-Soares, I.; Kane, L.; Jamieson, K.; Bamshad, M.J.; Nickerson, D.A.; et al. Deletion of CTCF sites in the SHH locus alters enhancer–promoter interactions and leads to acheiropodia. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Tarusawa, E.; Yoshimura, Y.; Galjart, N.; Yagi, T. CTCF Is Required for Neural Development and Stochastic Expression of Clustered Pcdh Genes in Neurons. Cell Rep. 2012, 2, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Sams, D.S.; Nardone, S.; Getselter, D.; Raz, D.; Tal, M.; Rayi, P.R.; Kaphzan, H.; Hakim, O.; Elliott, E. Neuronal CTCF Is Necessary for Basal and Experience-Dependent Gene Regulation, Memory Formation, and Genomic Structure of BDNF and Arc. Cell Rep. 2016, 17, 2418–2430. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Yang, Y.; Valnegri, P.; Juric, I.; Abnousi, A.; Markwalter, K.H.; Guthrie, A.N.; Godec, A.; Oldenborg, A.; Hu, M.; et al. Sensory experience remodels genome architecture in neural circuit to drive motor learning. Nature 2019, 569, 708–713. [Google Scholar] [CrossRef]
- Beagan, J.A.; Pastuzyn, E.D.; Fernandez, L.R.; Guo, M.H.; Feng, K.; Titus, K.; Chandrashekar, H.; Shepherd, J.D.; Phillips-Cremins, J.E. Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat. Neurosci. 2020, 23, 707–717. [Google Scholar] [CrossRef]
- Sofueva, S.; Yaffe, E.; Chan, W.-C.; Georgopoulou, D.; Rudan, M.V.; Mira-Bontenbal, H.; Pollard, S.; Schroth, G.P.; Tanay, A.; Hadjur, S. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013, 32, 3119–3129. [Google Scholar] [CrossRef] [Green Version]
- Seitan, V.; Faure, A.; Zhan, Y.; McCord, R.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar] [CrossRef] [Green Version]
- Nora, E.P.; Goloborodko, A.; Valton, A.-L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.J.A.; Ye, Z.; Kolovos, P.; Brouwer, R.W.W.; van de Corput, M.P.C.; van de Werken, H.J.G.; Knoch, T.A.; van Ijcken, W.F.J.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casiño, C.; Powell, S.; Yashaswini, C.; Lamarca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, eaat4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Liu, X.; Huang, W.-K.; Giusti-Rodríguez, P.; Cui, J.; Zhang, S.; Xu, W.; Wen, Z.; Ma, S.; Rosen, J.D.; et al. Robust Hi-C Maps of Enhancer-Promoter Interactions Reveal the Function of Non-coding Genome in Neural Development and Diseases. Mol. Cell 2020, 79, 521–534.e15. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Posavi, M.; Huang, P.; Unger, T.L.; Berlyand, Y.; Gruenewald, A.L.; Chesi, A.; Manduchi, E.; Wells, A.D.; Grant, S.F.; et al. A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. Am. J. Hum. Genet. 2017, 101, 643–663. [Google Scholar] [CrossRef] [Green Version]
- Hillary, R.F.; McCartney, D.L.; Harris, S.E.; Stevenson, A.; Seeboth, A.; Zhang, Q.; Liewald, D.C.; Evans, K.L.; Ritchie, C.W.; Tucker-Drob, E.M.; et al. Genome and epigenome wide studies of neurological protein biomarkers in the Lothian Birth Cohort 1936. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurano, M.T.; Haugen, E.; Sandstrom, R.; Vierstra, J.; Shafer, A.; Kaul, R.; Stamatoyannopoulos, J.A. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015, 47, 1393–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Van Der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.; Steinberg, S.; Sealock, J.; Karlsson, I.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Hannon, E.; Marzi, S.J.; Schalkwyk, L.S.; Mill, J. Genetic risk variants for brain disorders are enriched in cortical H3K27ac domains. Mol. Brain 2019, 12, 1–6. [Google Scholar] [CrossRef]
- Adolfsson, R. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Ki Sung GTEx Consortium Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [CrossRef] [PubMed]
- Kikuchi, M.; Hara, N.; Hasegawa, M.; Miyashita, A.; Kuwano, R.; Ikeuchi, T.; Nakaya, A. Enhancer variants associated with Alzheimer’s disease affect gene expression via chromatin looping. BMC Med. Genom. 2019, 12, 128. [Google Scholar] [CrossRef] [Green Version]
- Vermunt, M.W.; Reinink, P.; Korving, J.; De Bruijn, E.; Creyghton, P.M.; Basak, O.; Geeven, G.; Toonen, P.W.; Lansu, N.; Meunier, C.; et al. Large-Scale Identification of Coregulated Enhancer Networks in the Adult Human Brain. Cell Rep. 2014, 9, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Li, S.; Liu, J.; Li, X.; Luo, X.-J. Functional genomics reveal gene regulatory mechanisms underlying schizophrenia risk. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bryois, J.; Garrett, M.E.; Song, L.; Safi, A.; Giusti-Rodriguez, P.; Johnson, G.D.; Shieh, A.W.; Buil, A.; Fullard, J.F.; Roussos, P.; et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, C.; Li, W.; Yang, Y.; Li, X.; Liu, J.; Wang, J.; Li, S.; Liu, Y.; Li, K.; et al. A missense variant in NDUFA6 confers schizophrenia risk by affecting YY1 binding and NAGA expression. Mol. Psychiatry 2021, 1–16. [Google Scholar] [CrossRef]
- Kumasaka, N.; Knights, A.J.; Gaffney, D.J. High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat. Genet. 2019, 51, 128–137. [Google Scholar] [CrossRef]
- Cano-Gamez, E.; Trynka, G. From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front. Genet. 2020, 11, 424. [Google Scholar] [CrossRef]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; De Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.; Zhang, H.; Moy, W.; McGowan, H.; Leites, C.; Dionisio, L.; Xu, Z.; Shi, J.; Sanders, A.R.; Greenleaf, W.J.; et al. Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci. Cell Stem Cell 2017, 21, 305–318.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikov, A.; Murugan, A.; Zhang, X.; Tesileanu, T.; Wang, L.; Rogov, P.; Feizi, S.; Gnirke, A.; Jr, C.G.C.; Kinney, J.; et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012, 30, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Van Arensbergen, J.; Pagie, L.; Fitzpatrick, V.D.; De Haas, M.; Baltissen, M.P.; Comoglio, F.; Van Der Weide, R.H.; Teunissen, H.; Võsa, U.; Franke, L.; et al. High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet. 2019, 51, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Tewhey, R.; Kotliar, D.; Park, D.; Liu, B.; Winnicki, S.; Reilly, S.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F.; et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noordermeer, D.; de Wit, E.; Klous, P.; Van De Werken, H.; Simonis, M.; Lopez-Jones, M.; Eussen, B.; De Klein, A.; Singer, R.H.; de Laat, W. Variegated gene expression caused by cell-specific long-range DNA interactions. Nat. Cell Biol. 2011, 13, 944–951. [Google Scholar] [CrossRef] [Green Version]
- Vernimmen, D.; De Gobbi, M.; Sloane-Stanley, J.A.; Wood, W.G.; Higgs, D.R. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 2007, 26, 2041–2051. [Google Scholar] [CrossRef]
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; de Wit, E.; Van Steensel, B.; de Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat. Genet. 2006, 38, 1348–1354. [Google Scholar] [CrossRef]
- Baù, D.; Sanyal, A.; Lajoie, B.R.; Capriotti, E.; Byron, M.; Lawrence, J.B.; Dekker, J.; Marti-Renom, M.A. The three-dimensional folding of the α-globin gene domain reveals formation of chromatin globules. Nat. Struct. Mol. Biol. 2010, 18, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Palstra, R.-J.; Tolhuis, B.; Splinter, E.; Nijmeijer, R.; Grosveld, F.; de Laat, W. The β-globin nuclear compartment in development and erythroid differentiation. Nat. Genet. 2003, 35, 190–194. [Google Scholar] [CrossRef]
- Nott, A.; Holtman, I.R.; Coufal, N.G.; Schlachetzki, J.C.M.; Yu, M.; Hu, R.; Han, C.Z.; Pena, M.; Xiao, J.; Wu, Y.; et al. Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science 2019, 366, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; De La Torre-Ubieta, L.; Stein, J.; Parikshak, N.; Huang, J.; Opland, C.K.; Gandal, M.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Hauberg, M.E.; The CommonMind Consortium; Fullard, J.F.; Zhu, L.; Cohain, A.T.; Giambartolomei, C.; Misir, R.; Reach, S.; Johnson, J.; Wang, M.; et al. Differential activity of transcribed enhancers in the prefrontal cortex of 537 cases with schizophrenia and controls. Mol. Psychiatry 2019, 24, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Ruderfer, D.M.; Ripke, S.; McQuillin, A.; Boocock, J.; Stahl, E.A.; Pavlides, J.M.W.; Mullins, N.; Charney, A.W.; Ori, A.P.; Loohuis, L.M.O.; et al. Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell 2018, 173, 1705–1715.e16. [Google Scholar] [CrossRef] [Green Version]
- Parikshak, N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, S.E.; Liao, M.; Smith, R.V.; White, C.; Lagomarsino, V.N.; Xu, J.; Taga, M.; Bennett, D.A.; De Jager, P.L.; Young-Pearse, T. Candidate-based screening via gene modulation in human neurons and astrocytes implicates FERMT2 in Aβ and TAU proteostasis. Hum. Mol. Genet. 2018, 28, 718–735. [Google Scholar] [CrossRef]
- Delaneau, O.; Zazhytska, M.; Borel, C.; Giannuzzi, G.; Rey, G.; Howald, C.; Kumar, S.; Ongen, H.; Popadin, K.; Marbach, D.; et al. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science 2019, 364, eaat8266. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.; Daza, R.; Adey, A.; Pliner, H.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Armand, E.J.; Li, J.; Xie, F.; Luo, C.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Sanes, J.R. Neuronal cell-type classification: Challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017, 18, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.-L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef] [Green Version]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70–80. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Gephart, M.G.H.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanov, R.; Zeisel, A.; Bakker, J.; Girach, F.; Hellysaz, A.; Tomer, R.; Alpár, A.; Mulder, J.; Clotman, F.; Keimpema, E.; et al. Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci. 2017, 20, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Tasic, B.; Yao, Z.; Graybuck, L.T.; Smith, K.A.; Nguyen, T.N.; Bertagnolli, D.; Goldy, J.; Garren, E.; Economo, M.N.; Viswanathan, S.; et al. Shared and distinct transcriptomic cell types across neocortical areas. Nat. Cell Biol. 2018, 563, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Ii, J.L.C.; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 2021, 24, 425–436. [Google Scholar] [CrossRef]
- Ortiz, C.; Navarro, J.F.; Jurek, A.; Märtin, A.; Lundeberg, J.; Meletis, K. Molecular atlas of the adult mouse brain. Sci. Adv. 2020, 6, eabb3446. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Chen, W.-T.; Lu, A.; Craessaerts, K.; Pavie, B.; Frigerio, C.S.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Renthal, W.; Boxer, L.; Hrvatin, S.; Li, E.; Silberfeld, A.; Nagy, M.A.; Griffith, E.; Vierbuchen, T.; Greenberg, M.E. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci. 2018, 21, 1670–1679. [Google Scholar] [CrossRef]
- Nagy, C.; Maitra, M.; Tanti, A.; Suderman, M.; Théroux, J.-F.; Davoli, M.A.; Perlman, K.; Yerko, V.; Wang, Y.C.; Tripathy, S.J.; et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 2020, 23, 771–781. [Google Scholar] [CrossRef]
- Lee, D.-S.; Luo, C.; Zhou, J.; Chandran, S.; Rivkin, A.; Bartlett, A.; Nery, J.R.; Fitzpatrick, C.; O’Connor, C.; Dixon, J.R.; et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods 2019, 16, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, Y.; Zhang, Y.; Kubo, N.; Yu, M.; Fang, R.; Kellis, M.; Ren, B. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 2019, 16, 991–993. [Google Scholar] [CrossRef]
- Luo, C.; Liu, H.; Xie, F.; Armand, E.J.; Siletti, K.; Bakken, T.E.; Fang, R.; Doyle, W.I.; Hodge, R.D.; Hu, L.; et al. Single Nucleus Multi-Omics Links Human Cortical Cell Regulatory Genome Diversity to Disease Risk Variants. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Butler, R.; Bates, G. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat. Rev. Neurosci. 2006, 7, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.-H. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Agis-Balboa, R.C.; Walter, J.; Sananbenesi, F.; Fischer, A. Sodium Butyrate Improves Memory Function in an Alzheimer’s Disease Mouse Model When Administered at an Advanced Stage of Disease Progression. J. Alzheimer’s Dis. 2011, 26, 187–197. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Marco, S.; Pérez-Otaño, I.; García-Osta, A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 2012, 22, 1040–1050. [Google Scholar] [CrossRef]
- Gardian, G.; Browne, S.E.; Choi, D.-K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective Effects of Phenylbutyrate in the N171-82Q Transgenic Mouse Model of Huntington’s Disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-Y.; Schluesener, H.J. Oral Administration of Histone Deacetylase Inhibitor MS-275 Ameliorates Neuroinflammation and Cerebral Amyloidosis and Improves Behavior in a Mouse Model. J. Neuropathol. Exp. Neurol. 2013, 72, 178–185. [Google Scholar] [CrossRef]
- Sung, Y.M.; Lee, T.; Yoon, H.; DiBattista, A.; Song, J.M.; Sohn, Y.; Moffat, E.I.; Turner, R.; Jung, M.; Kim, J.; et al. Mercaptoacetamide-based class II HDAC inhibitor lowers Aβ levels and improves learning and memory in a mouse model of Alzheimer’s disease. Exp. Neurol. 2013, 239, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado-Tejedor, M.; Garciabarroso, C.; Sanzhez-Arias, J.; Mederos, S.; Rabal, O.; Ugarte, A.; Franco, R.; Pascuallucas, M.; Segura, V.; Perea, G.; et al. Concomitant histone deacetylase and phosphodiesterase 5 inhibition synergistically prevents the disruption in synaptic plasticity and it reverses cognitive impairment in a mouse model of Alzheimer’s disease. Clin. Epigenet. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sanchez-Arias, J.A.; Rabal, O.; González, M.P.; Mederos, S.; Ugarte, A.; Franco, R.; Segura, V.; Perea, G.; et al. A First-in-Class Small-Molecule that Acts as a Dual Inhibitor of HDAC and PDE5 and that Rescues Hippocampal Synaptic Impairment in Alzheimer’s Disease Mice. Neuropharmacology 2017, 42, 524–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabal, O.; Sanchez-Arias, J.A.; Cuadrado-Tejedor, M.; de Miguel, I.; González, M.P.; García-Barroso, C.; Ugarte, A.; de Mendoza, A.E.-H.; Sáez, E.; Espelosin, M.; et al. Design, Synthesis, and Biological Evaluation of First-in-Class Dual Acting Histone Deacetylases (HDACs) and Phosphodiesterase 5 (PDE5) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2016, 59, 8967–9004. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-P.; Xie, J.-W.; Wang, C.-Y.; Wang, T.; Wang, X.; Wang, S.-L.; Teng, W.-P.; Wang, Z.-Y. Valproate reduces tau phosphorylation via cyclin-dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res. Bull. 2011, 85, 194–200. [Google Scholar] [CrossRef]
- Green, K.N.; Steffan, J.S.; Martínez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide Restores Cognition in Alzheimer’s Disease Transgenic Mice via a Mechanism Involving Sirtuin Inhibition and Selective Reduction of Thr231-Phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S.; et al. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dompierre, J.P.; Godin, J.; Charrin, B.C.; Cordelières, F.; King, S.J.; Humbert, S.; Saudou, F. Histone Deacetylase 6 Inhibition Compensates for the Transport Deficit in Huntington’s Disease by Increasing Tubulin Acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.S.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone Deacetylase Inhibition by Sodium Butyrate Chemotherapy Ameliorates the Neurodegenerative Phenotype in Huntington’s Disease Mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate Up-regulates the DJ-1 Protein and Protects Neurons in Cell Culture and in Animal Models of Parkinson Disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.-Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nat. Cell Biol. 2001, 413, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Saft, C.; Lauter, T.; Kraus, P.H.; Przuntek, H.; Andrich, J.E. Dose-dependent improvement of myoclonic hyperkinesia due to Valproic acid in eight Huntington’s Disease patients: A case series. BMC Neurol. 2006, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Kast, R.J.; Steffan, J.S.; Thomas, E.A. Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: Implications for the ubiquitin–proteasomal and autophagy systems. Hum. Mol. Genet. 2012, 21, 5280–5293. [Google Scholar] [CrossRef]
- Collins, L.; Adriaanse, L.J.; Theratile, S.D.; Hegarty, S.V.; Sullivan, A.; O’Keeffe, G.W. Class-IIa Histone Deacetylase Inhibition Promotes the Growth of Neural Processes and Protects Them Against Neurotoxic Insult. Mol. Neurobiol. 2014, 51, 1432–1442. [Google Scholar] [CrossRef]
- Pan, Y.; Daito, T.; Sasaki, Y.; Chung, Y.H.; Xing, X.; Pondugula, S.; Swamidass, S.J.; Wang, T.; Kim, A.H.; Yano, H. Inhibition of DNA Methyltransferases Blocks Mutant Huntingtin-Induced Neurotoxicity. Sci. Rep. 2016, 6, 31022. [Google Scholar] [CrossRef]
- Monti, B.; Gatta, V.; Piretti, F.; Raffaelli, S.S.; Virgili, M.; Contestabile, A. Valproic Acid is Neuroprotective in the Rotenone Rat Model of Parkinson’s Disease: Involvement of α-Synuclein. Neurotox. Res. 2010, 17, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Jr, R.H.B.; et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Camelo, S.; Iglesias, A.H.; Hwang, D.; Due, B.; Ryu, H.; Smith, K.; Gray, S.; Imitola, J.; Duran, G.; Assaf, B.; et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 164, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef]
- Sales, A.; Biojone, C.; Terceti, M.S.; Guimarães, F.S.; Gomes, M.V.M.; Joca, S.R.L. Antidepressant-like effect induced by systemic and intra-hippocampal administration of DNA methylation inhibitors. Br. J. Pharmacol. 2011, 164, 1711–1721. [Google Scholar] [CrossRef] [Green Version]
- Covington, H.E.; Vialou, V.; LaPlant, Q.; Ohnishi, Y.N.; Nestler, E.J. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 2011, 493, 122–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Geng, X.; Dang, W.; Wu, B.; Dai, Z.; Li, Y.; Yang, Y.; Zhang, H.; Shi, J. Molecular mechanisms associated with the antidepressant effects of the class I histone deacetylase inhibitor MS-275 in the rat ventrolateral orbital cortex. Brain Res. 2012, 1447, 119–125. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.; Akbarian, S. Antidepressant-Like Effects of the Histone Deacetylase Inhibitor, Sodium Butyrate, in the Mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef]
- Yamawaki, Y.; Fuchikami, M.; Morinobu, S.; Segawa, M.; Matsumoto, T.; Yamawaki, S. Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. World J. Biol. Psychiatry 2011, 13, 458–467. [Google Scholar] [CrossRef]
- Zádori, D.; Geisz, A.; Vámos, E.; Vécsei, L.; Klivényi, P. Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol. Biochem. Behav. 2009, 94, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Rane, P.; Shields, J.; Heffernan, M.; Guo, Y.; Akbarian, S.; King, J.A. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 2012, 62, 2409–2412. [Google Scholar] [CrossRef]
- Jovanova, O.S.; (Nedeljkovic), I.P.; Spieler, D.; Walker, R.; Liu, C.; Luciano, M.; Bressler, J.; Brody, J.; Drake, A.J.; Evans, K.; et al. DNA Methylation Signatures of Depressive Symptoms in Middle-aged and Elderly Persons. JAMA Psychiatry 2018, 75, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Atef, A.; Piatek, A.; Ali, Z.; Piatek, M.; Aouida, M.; Sharakuu, A.; Mahjoub, A.; Wang, G.; Khan, M.S.; et al. Characterization and DNA-Binding Specificities of Ralstonia TAL-Like Effectors. Mol. Plant 2013, 6, 1318–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morbitzer, R.; Römer, P.; Boch, J.; Lahaye, T. Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 21617–21622. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.; McLachlan, A.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.; Arlotta, P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2010, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.; Lo, T.-W.; Zeitler, B.; Pickle, C.S.; Ralston, E.J.; Lee, A.H.; Amora, R.; Miller, J.C.; Leung, E.; Meng, X.; et al. Targeted Genome Editing Across Species Using ZFNs and TALENs. Science 2011, 333, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, A.; Wleklinski, M.J.; Spurgat, M.C.; Heiderscheit, E.A.; Kropornicka, A.S.; Vu, C.K.; Bhimsaria, D.; Swanson, S.A.; Stewart, R.; Ramanathan, P.; et al. Reprogramming cell fate with a genome-scale library of artificial transcription factors. Proc. Natl. Acad. Sci. USA 2016, 113, E8257–E8266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, A.W.; Rios, X.; Chari, R.; Yang, L.; Zhang, F.; Mali, P.; Church, G. Iterative capped assembly: Rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012, 40, e117. [Google Scholar] [CrossRef] [Green Version]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005, 151, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojica, F.J.M.; Rodriguez-Valera, F. The discovery of CRISPR in archaea and bacteria. FEBS J. 2016, 283, 3162–3169. [Google Scholar] [CrossRef]
- Tang, T.-H.; Bachellerie, J.-P.; Rozhdestvensky, T.; Bortolin, M.-L.; Huber, H.; Drungowski, M.; Elge, T.; Brosius, J.; Hüttenhofer, A. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc. Natl. Acad. Sci. USA 2002, 99, 7536–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nat. Cell Biol. 2019, 566, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Nishimasu, H.; Ran, F.A.; Hsu, P.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Jore, M.M.; Lundgren, M.; Van Duijn, E.; Bultema, J.B.; Westra, E.R.; Waghmare, S.P.; Wiedenheft, B.; Pul, Ü.; Wurm, R.; Wagner, R.; et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol. 2011, 18, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Charpentier, E.; Marraffini, L.A. Harnessing CRISPR-Cas9 immunity for genetic engineering. Curr. Opin. Microbiol. 2014, 19, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Kim, Y.; Cheong, S.-A.; Lee, J.G.; Lee, S.-W.; Lee, M.S.; Baek, I.-J.; Sung, Y.H. Generation of knockout mice by Cpf1-mediated gene targeting. Nat. Biotechnol. 2016, 34, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.K.; Kim, K.; Been, K.W.; Baek, G.; Ye, S.; Hur, J.W.; Ryu, S.-M.; Lee, Y.S.; Kim, J.-S. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol. 2016, 34, 807–808. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.; Aryee, Z.R.M.M.J.; Joung, B.P.K.S.Q.T.M.S.P.N.T.N.M.M.W.J.M.L.Z.R.M.M.J.A.J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.K.J.K.K.W.B.S.-H.Y.J.-S.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.-H.; Kim, J.-S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Tóth, E.; Weinhardt, N.; Bencsura, P.; Huszár, K.; Kulcsár, P.I.; Tálas, A.; Fodor, E.; Welker, E. Cpf1 nucleases demonstrate robust activity to induce DNA modification by exploiting homology directed repair pathways in mammalian cells. Biol. Direct 2016, 11, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Esvelt, K.M.; Church, G. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef]
- Koike-Yusa, H.; Li, Y.; Tan, E.-P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sanjana, N.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.H.; Ziebell, F.; Joberty, G.; Zinn, N.; Mueller, W.F.; Clauder-Münster, S.; Eberhard, D.; Savitski, M.F.; Grandi, P.; Jakob, P.; et al. Biological plasticity rescues target activity in CRISPR knock outs. Nat. Methods 2019, 16, 1087–1093. [Google Scholar] [CrossRef]
- Bae, S.; Kweon, J.; Kim, H.S.; Kim, J.-S. Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods 2014, 11, 705–706. [Google Scholar] [CrossRef]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nat. Cell Biol. 2018, 563, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Suresh, S.; Guo, D.; Daman, K.; Chen, J.C.J.; Liu, P.; Zieger, M.; Luk, K.; Roscoe, B.P.; Mueller, C.; et al. Precise therapeutic gene correction by a simple nuclease-induced double-stranded break. Nat. Cell Biol. 2019, 568, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Kim, Y.B.; Komor, A.; Levy, J.M.; Packer, M.S.; Zhao, K.; Liu, Y.B.K.A.C.K.J.M.L.M.S.P.K.T.Z.D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- Rees, H.A.; Komor, A.; Yeh, W.-H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, W.-H.; Chiang, H.; Rees, H.A.; Edge, A.S.B.; Liu, D.R. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, I.; Ma, J.; Triezenberg, S.; Ptashne, M. GAL4-VP16 is an unusually potent transcriptional activator. Nature 1988, 335, 563–564. [Google Scholar] [CrossRef]
- Liu, P.-Q.; Rebar, E.J.; Zhang, L.; Liu, Q.; Jamieson, A.C.; Liang, Y.; Qi, H.; Li, P.-X.; Chen, B.; Mendel, M.C.; et al. Regulation of an Endogenous Locus Using a Panel of Designed Zinc Finger Proteins Targeted to Accessible Chromatin Regions. J. Biol. Chem. 2001, 276, 11323–11334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Spratt, S.K.; Liu, Q.; Johnstone, B.; Qi, H.; Raschke, E.E.; Jamieson, A.C.; Rebar, E.J.; Wolffe, A.P.; Case, C.C. Synthetic Zinc Finger Transcription Factor Action at an Endogenous Chromosomal Site. J. Biol. Chem. 2000, 275, 33850–33860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerli, R.R.; Segal, D.; Dreier, B.; Barbas, C.F. Toward controlling gene expression at will: Specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. USA 1998, 95, 14628–14633. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Linder, S.J.; Reyon, D.; Angstman, J.F.; Fu, Y.; Sander, J.D.; Joung, J.K. Robust, synergistic regulation of human gene expression using TALE activators. Nat. Chem. Biol. 2013, 10, 243–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pinera, P.; Ousterout, D.G.; Brunger, J.M.; Farin, A.M.; Glass, K.A.; Guilak, F.; Crawford, G.E.; Hartemink, A.J.; Gersbach, C.A. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat. Methods 2013, 10, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Dreier, B.; Barbas, C.F. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA 2000, 97, 1495–1500. [Google Scholar] [CrossRef] [Green Version]
- Tanenbaum, M.E.; Gilbert, L.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.D.; Pruitt, B.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Kanamori, M.; Someya, K.; Nakatsukasa, H.; Yoshimura, A. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenet. Chromatin 2017, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Shan, L.; Han, L.; Ma, S.; Zhang, Y.; Hao, B.; Lin, Y.; Rong, Z. Gene activation in human cells using CRISPR/Cpf1-p300 and CRISPR/Cpf1-SunTag systems. Protein Cell 2018, 9, 380–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajwan, S.; Mannervik, M. Gene activation by dCas9-CBP and the SAM system differ in target preference. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.; Habib, N.; Gootenberg, J.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.C.; Chen, W.; Gasperini, M.; Shendure, J. Identifying Novel Enhancer Elements with CRISPR-Based Screens. ACS Chem. Biol. 2018, 13, 326–332. [Google Scholar] [CrossRef]
- Yan, J.; Chen, S.-A.A.; Local, A.; Liu, T.; Qiu, Y.; Dorighi, K.M.; Preissl, S.; Rivera, C.M.; Wang, C.; Ye, Z.; et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018, 28, 204–220. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, D.C.; Gjaltema, R.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.J.; Rots, D.C.-R.R.A.F.G.L.J.J.P.J.J.D.-F.M.H.J.R.M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Gregory, D.J.; Zhang, Y.; Kobzik, L.; Fedulov, A.V. Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics 2013, 8, 1205–1212. [Google Scholar] [CrossRef] [Green Version]
- Huisman, C.; van der Wijst, M.; Schokker, M.; Blancafort, P.; Terpstra, M.M.; Kok, K.; van der Zee, A.G.J.; Schuuring, E.; Wisman, G.B.A.; Rots, M.G. Re-expression of Selected Epigenetically Silenced Candidate Tumor Suppressor Genes in Cervical Cancer by TET2-directed Demethylation. Mol. Ther. 2016, 24, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Kazemier, H.G.; De Groote, M.L.; Ruiters, M.H.J.; Xu, G.-L.; Rots, M.G. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014, 42, 1563–1574. [Google Scholar] [CrossRef] [Green Version]
- Sgro, A.; Blancafort, P. Epigenome engineering: New technologies for precision medicine. Nucleic Acids Res. 2020, 48, 12453–12482. [Google Scholar] [CrossRef]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, K.A.; Berchtold, N.C.; Malvaez, M.; Carlos, A.J.; McQuown, S.C.; Cunningham, M.J.; Wood, M.A.; Cotman, C.W. Exercise and Sodium Butyrate Transform a Subthreshold Learning Event into Long-Term Memory via a Brain-Derived Neurotrophic factor-Dependent Mechanism. Neuropsychopharmacology 2013, 38, 2027–2034. [Google Scholar] [CrossRef]
- Sartor, G.C.; Malvezzi, A.M.; Kumar, A.; Andrade, N.S.; Wiedner, H.; Vilca, S.; Janczura, K.J.; Bagheri, A.; Al-Ali, H.; Powell, S.; et al. Enhancement of BDNF Expression and Memory by HDAC Inhibition Requires BET Bromodomain Reader Proteins. J. Neurosci. 2019, 39, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Margolin, J.F.; Friedman, J.; Meyer, W.K.; Vissing, H.; Thiesen, H.J.; Rauscher, F.J. Kruppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. USA 1994, 91, 4509–4513. [Google Scholar] [CrossRef] [Green Version]
- Sgouras, D.N.; Athanasiou, M.A.; Beal, G.J.J.; Fisher, R.J.; Blair, D.G.; Mavrothalassitis, G.J. ERF: An ETS Domain Protein with Strong Transcriptional Repressor Activity, Can Suppress Ets-Associated Tumorigenesis and Is Regulated by Phosphorylation during Cell Cycle and Mitogenic Stimulation. EMBO J. 1995, 14, 4781–4793. [Google Scholar] [CrossRef]
- Ayer, D.E.; Laherty, C.D.; Lawrence, Q.A.; Armstrong, A.P.; Eisenman, R.N. Mad proteins contain a dominant transcription repression domain. Mol. Cell. Biol. 1996, 16, 5772–5781. [Google Scholar] [CrossRef] [Green Version]
- O’Geen, H.; Ren, C.; Nicolet, C.M.; Perez, A.; Halmai, J.; Le, V.M.; Mackay, J.; Farnham, P.J.; Segal, D.J. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017, 45, 9901–9916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenet. Chromatin 2019, 12, 1–20. [Google Scholar] [CrossRef]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.-C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Bintu, L.; Yong, J.; Antebi, Y.; McCue, K.; Kazuki, Y.; Uno, N.; Oshimura, M.; Elowitz, M.B. Dynamics of epigenetic regulation at the single-cell level. Science 2016, 351, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A.L. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [Green Version]
- Siddique, A.N.; Nunna, S.; Rajavelu, A.; Zhang, Y.; Jurkowska, R.Z.; Reinhardt, R.; Rots, M.G.; Ragozin, S.; Jurkowski, T.; Jeltsch, A. Targeted Methylation and Gene Silencing of VEGF-A in Human Cells by Using a Designed Dnmt3a–Dnmt3L Single-Chain Fusion Protein with Increased DNA Methylation Activity. J. Mol. Biol. 2013, 425, 479–491. [Google Scholar] [CrossRef]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 Corepressor Functions To Coordinate the Assembly of De Novo HP1-Demarcated Microenvironments of Heterochromatin Required for KRAB Zinc Finger Protein-Mediated Transcriptional Repression. Mol. Cell. Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolzenburg, S.; Rots, M.G.; Beltran, A.S.; Rivenbark, A.G.; Yuan, X.; Qian, H.; Strahl, B.D.; Blancafort, P. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 2012, 40, 6725–6740. [Google Scholar] [CrossRef]
- Cong, L.; Zhou, R.; Kuo, Y.-C.; Cunniff, M.M.; Zhang, F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mlambo, T.; Nitsch, S.; Hildenbeutel, M.; Romito, M.; Müller, M.; Bossen, C.; Diederichs, S.; Cornu, T.I.; Cathomen, T.; Mussolino, C. Designer epigenome modifiers enable robust and sustained gene silencing in clinically relevant human cells. Nucleic Acids Res. 2018, 46, 4456–4468. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guo, R.; Du, Z.; Bai, L.; Li, L.; Cui, J.; Li, W.; Hoffman, A.R.; Hu, J.-F. Epigenetic Targeting of Granulin in Hepatoma Cells by Synthetic CRISPR dCas9 Epi-suppressors. Mol. Ther. Nucleic Acids 2018, 11, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Josipović, G.; Tadić, V.; Klasić, M.; Zanki, V.; Bečeheli, I.; Chung, F.; Ghantous, A.; Keser, T.; Madunić, J.; Bošković, M.; et al. Antagonistic and synergistic epigenetic modulation using orthologous CRISPR/dCas9-based modular system. Nucleic Acids Res. 2019, 47, 9637–9657. [Google Scholar] [CrossRef] [Green Version]
- Zalatan, J.; Lee, M.E.; Almeida, R.; Gilbert, L.; Whitehead, E.H.; La Russa, M.; Tsai, J.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.W.; Jillette, N.; Lee, P.; Plaskon, D.; Fujiwara, Y.; Wang, W.; Taghbalout, A.; Wang, H. Casilio: A versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016, 26, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Nolis, I.K.; McKay, D.J.; Mantouvalou, E.; Lomvardas, S.; Merika, M.; Thanos, D. Transcription factors mediate long-range enhancer-promoter interactions. Proc. Natl. Acad. Sci. USA 2009, 106, 20222–20227. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Rupon, J.W.; Krivega, I.; Breda, L.; Motta, I.; Jahn, K.S.; Reik, A.; Gregory, P.; Rivella, S.; Dean, A.; et al. Reactivation of Developmentally Silenced Globin Genes by Forced Chromatin Looping. Cell 2014, 158, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Hao, N.; Shearwin, K.; Dodd, I. Programmable DNA looping using engineered bivalent dCas9 complexes. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Rege, M.; Valeri, J.; Dunagin, M.C.; Metzger, A.; Titus, K.; Gilgenast, T.G.; Gong, W.; Beagan, J.A.; Raj, A.; et al. LADL: Light-activated dynamic looping for endogenous gene expression control. Nat. Methods 2019, 16, 633–639. [Google Scholar] [CrossRef]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laganiere, J.; Kells, A.P.; Lai, J.T.; Guschin, D.; Paschon, D.E.; Meng, X.; Fong, L.K.; Yu, Q.; Rebar, E.J.; Gregory, P.; et al. An Engineered Zinc Finger Protein Activator of the Endogenous Glial Cell Line-Derived Neurotrophic Factor Gene Provides Functional Neuroprotection in a Rat Model of Parkinson’s Disease. J. Neurosci. 2010, 30, 16469–16474. [Google Scholar] [CrossRef] [Green Version]
- Baxter, P.S.; Márkus, N.M.; Dando, O.; He, X.; Al-Mubarak, B.R.; Qiu, J.; Hardingham, G.E. Targeted de-repression of neuronal Nrf2 inhibits α-synuclein accumulation. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Bustos, F.; Ampuero, E.; Jury, N.; Aguilar, R.; Falahi, F.; Toledo, J.; Ahumada, J.; Lata, J.; Cubillos, P.; Henríquez, B.; et al. Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer’s disease mice. Brain 2017, 140, 3252–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- György, B.; Lööv, C.; Zaborowski, M.; Takeda, S.; Kleinstiver, B.; Commins, C.; Kastanenka, K.; Mu, D.; Volak, A.; Giedraitis, V.; et al. CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer’s Disease. Mol. Ther. Nucleic Acids 2018, 11, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Carlson-Stevermer, J.; Das, U.; Shen, M.; Delenclos, M.; Snead, A.M.; Koo, S.Y.; Wang, L.; Qiao, D.; Loi, J.; et al. CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Oh, J.; Shim, G.; Cho, B.; Chang, Y.; Kim, S.; Baek, S.; Kim, H.; Shin, J.; Choi, H.; et al. In vivo neuronal gene editing via CRISPR–Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 524–528. [Google Scholar] [CrossRef]
- Garriga-Canut, M.; Agustín-Pavón, C.; Herrmann, F.; Sánchez, A.; Dierssen, M.; Fillat, C.; Isalan, M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. USA 2012, 109, E3136–E3145. [Google Scholar] [CrossRef] [Green Version]
- Merienne, N.; Vachey, G.; de Longprez, L.; Meunier, C.; Zimmer, V.; Perriard, G.; Canales, M.; Mathias, A.; Herrgott, L.; Beltraminelli, T.; et al. The Self-Inactivating KamiCas9 System for the Editing of CNS Disease Genes. Cell Rep. 2017, 20, 2980–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In vitro and In vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Zeitler, B.; Froelich, S.; Marlen, K.; Shivak, D.A.; Yu, Q.; Li, D.; Pearl, J.R.; Miller, J.C.; Zhang, L.; Paschon, D.E.; et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019, 25, 1131–1142. [Google Scholar] [CrossRef]
- Yu, B.; Yuan, B.; Dai, J.-K.; Cheng, T.-L.; Xia, S.-N.; He, L.-J.; Yuan, Y.-T.; Zhang, Y.-F.; Xu, H.-T.; Xu, F.-Q.; et al. Reversal of Social Recognition Deficit in Adult Mice with MECP2 Duplication via Normalization of MeCP2 in the Medial Prefrontal Cortex. Neurosci. Bull. 2020, 36, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.H.; Tran, N.T.; Dao, T.M.L.; Nguyen, D.D.; Do, H.D.; Ha, T.L.; Kühn, R.; Nguyen, T.L.; Rajewsky, K.; Chu, V.T. Efficient and Precise CRISPR/Cas9-Mediated MECP2 Modifications in Human-Induced Pluripotent Stem Cells. Front. Genet. 2019, 10, 625. [Google Scholar] [CrossRef] [PubMed]
- Croci, S.; Carriero, M.L.; Capitani, K.; Daga, S.; Donati, F.; Frullanti, E.; Lamacchia, V.; Tita, R.; Giliberti, A.; Valentino, F.; et al. High rate of HDR in gene editing of p.(Thr158Met) MECP2 mutational hotspot. Eur. J. Hum. Genet. 2020, 28, 1231–1242. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.E.; Gersbach, C.A. Engineering Delivery Vehicles for Genome Editing. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 637–662. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Kauffman, K.J.; Anderson, H.Y.K.J.K.D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Ates, I.; Rathbone, T.; Stuart, C.; Bridges, P.H.; Cottle, R.N. Delivery Approaches for Therapeutic Genome Editing and Challenges. Genes 2020, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Tagliafierro, L.; Ilich, E.; Moncalvo, M.; Gu, J.; Sriskanda, A.; Grenier, C.; Murphy, S.K.; Chiba-Falek, O.; Kantor, B. Lentiviral Vector Platform for the Efficient Delivery of Epigenome-editing Tools into Human Induced Pluripotent Stem Cell-derived Disease Models. J. Vis. Exp. 2019, 10, e59241. [Google Scholar] [CrossRef]
- Van Haasteren, J.; Li, J.; Scheideler, O.J.; Murthy, N.; Schaffer, D.V. The delivery challenge: Fulfilling the promise of therapeutic genome editing. Nat. Biotechnol. 2020, 38, 845–855. [Google Scholar] [CrossRef]
- Li, R.; Qu, Y.; Liu, Y.; Noor, A.F.; Tran, J. Characteristics and advantages of adeno-associated virus vector-mediated gene therapy for neurodegenerative diseases. Neural Regen. Res. 2019, 14, 931–938. [Google Scholar] [CrossRef]
- Fuentes, C.M.; Schaffer, D.V. Adeno-associated virus-mediated delivery of CRISPR-Cas9 for genome editing in the central nervous system. Curr. Opin. Biomed. Eng. 2018, 7, 33–41. [Google Scholar] [CrossRef]
- Hanafy, A.S.; Schoch, S.; Lamprecht, A. CRISPR/Cas9 Delivery Potentials in Alzheimer’s Disease Management: A Mini Review. Pharmaceutics 2020, 12, 801. [Google Scholar] [CrossRef] [PubMed]
- Van Alstyne, M.; Tattoli, I.; Delestrée, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.S.; Zhang, C.; Mentis, G.Z.; Pellizzoni, L. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat. Neurosci. 2021, 24, 1–11. [Google Scholar] [CrossRef]
- Tong, S.; Moyo, B.; Lee, C.M.; Leong, K.; Bao, G. Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater. 2019, 4, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Song, C.-Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016, 34, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Smirnov, V.; Volchkova, E.; Lukashev, A.; Chulanov, V. Gene Editing by Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 7362. [Google Scholar] [CrossRef]
- Huang, H.; Zou, X.; Zhong, L.; Hou, Y.; Zhou, J.; Zhang, Z.; Xing, X.; Sun, J. CRISPR/dCas9-mediated activation of multiple endogenous target genes directly converts human foreskin fibroblasts into Leydig-like cells. J. Cell. Mol. Med. 2019, 23, 6072–6084. [Google Scholar] [CrossRef] [Green Version]
- Fidanza, A.; Lopez-Yrigoyen, M.; Romanò, N.; Jones, R.; Taylor, A.H.; Forrester, L.M. An all-in-one UniSam vector system for efficient gene activation. Sci. Rep. 2017, 7, 6394. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, X.; Wong, S.; Charles, E.J.; Lim, W.A.; Qi, L.S. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 2016, 13, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.-S.; Ho, W.Q.; Crabtree, G.R. Engineering the ABA Plant Stress Pathway for Regulation of Induced Proximity. Sci. Signal. 2011, 4, rs2. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, T.; DeRose, R.; Suarez, A.; Ueno, T.; Chen, M.; Sun, T.-P.; Wolfgang, M.J.; Mukherjee, C.; Meyers, D.J.; Inoue, T. Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nat. Chem. Biol. 2012, 8, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Kirkland, J.G.; Chory, E.J.; Husmann, D.; Calarco, J.P.; Crabtree, G.R. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.M.; Brigham, M.D.; Trevino, A.E.; Hsu, P.; Heidenreich, M.; Cong, L.; Platt, R.; Scott, D.A.; Church, G.; Zhang, F. Optical control of mammalian endogenous transcription and epigenetic states. Nature 2013, 500, 472–476. [Google Scholar] [CrossRef]
- Polstein, L.R.; Gersbach, C.A. Light-Inducible Spatiotemporal Control of Gene Activation by Customizable Zinc Finger Transcription Factors. J. Am. Chem. Soc. 2012, 134, 16480–16483. [Google Scholar] [CrossRef]
- Singh, R.; Kuscu, C.; Quinlan, A.; Qi, Y.; Adli, M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015, 43, e118. [Google Scholar] [CrossRef]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Cradick, T.; Fine, E.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Aach, J.; Stranges, P.; Esvelt, K.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Root, D.E. Optimized SgRNA Design to Maximize Activity and Minimize Off-Target Effects of CRISPR-Cas9 Synthesis of an Arrayed SgRNA Library Targeting the Human Genome. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Senís, E.; Fatouros, C.; Große, S.; Wiedtke, E.; Niopek, D.; Mueller, A.-K.; Börner, K.; Grimm, D. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014, 9, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Oiwa, R.; Kumita, W.; Henry, R.; Sakuma, T.; Ito, R.; Nozu, R.; Inoue, T.; Katano, I.; Sato, K.; et al. Generation of a Nonhuman Primate Model of Severe Combined Immunodeficiency Using Highly Efficient Genome Editing. Cell Stem Cell 2016, 19, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schacker, M.; Seimetz, D. From fiction to science: Clinical potentials and regulatory considerations of gene editing. Clin. Transl. Med. 2019, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
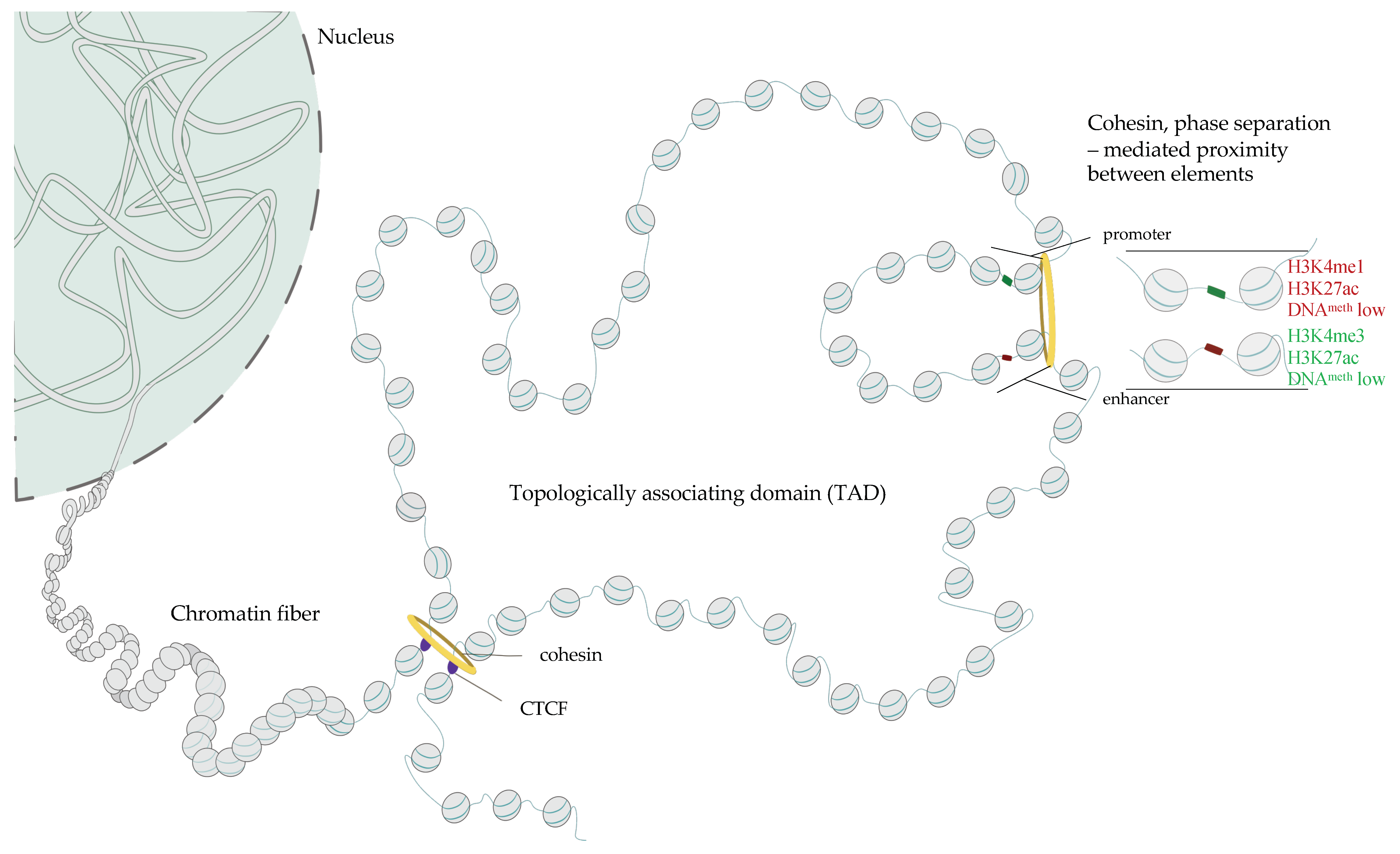
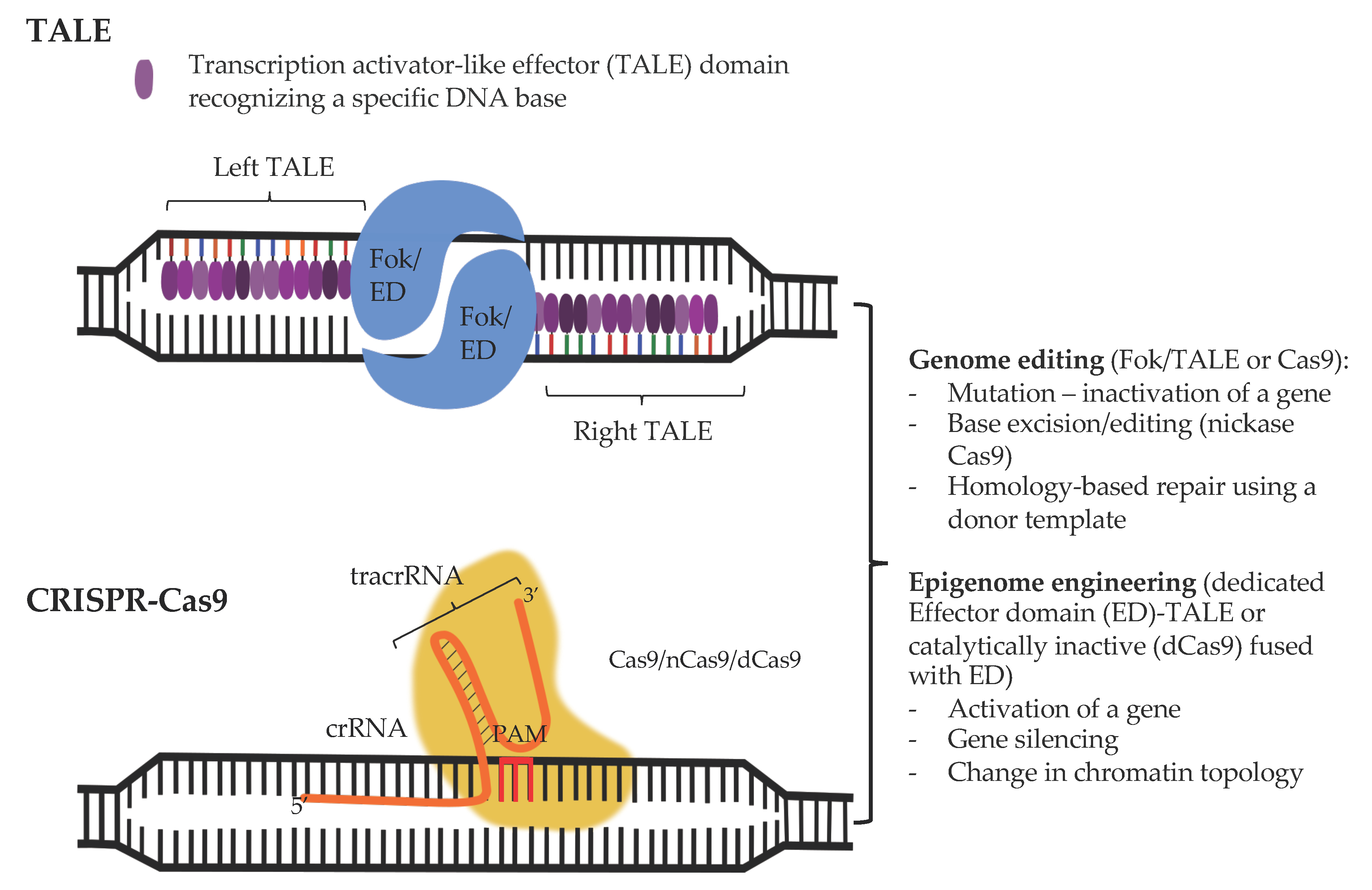
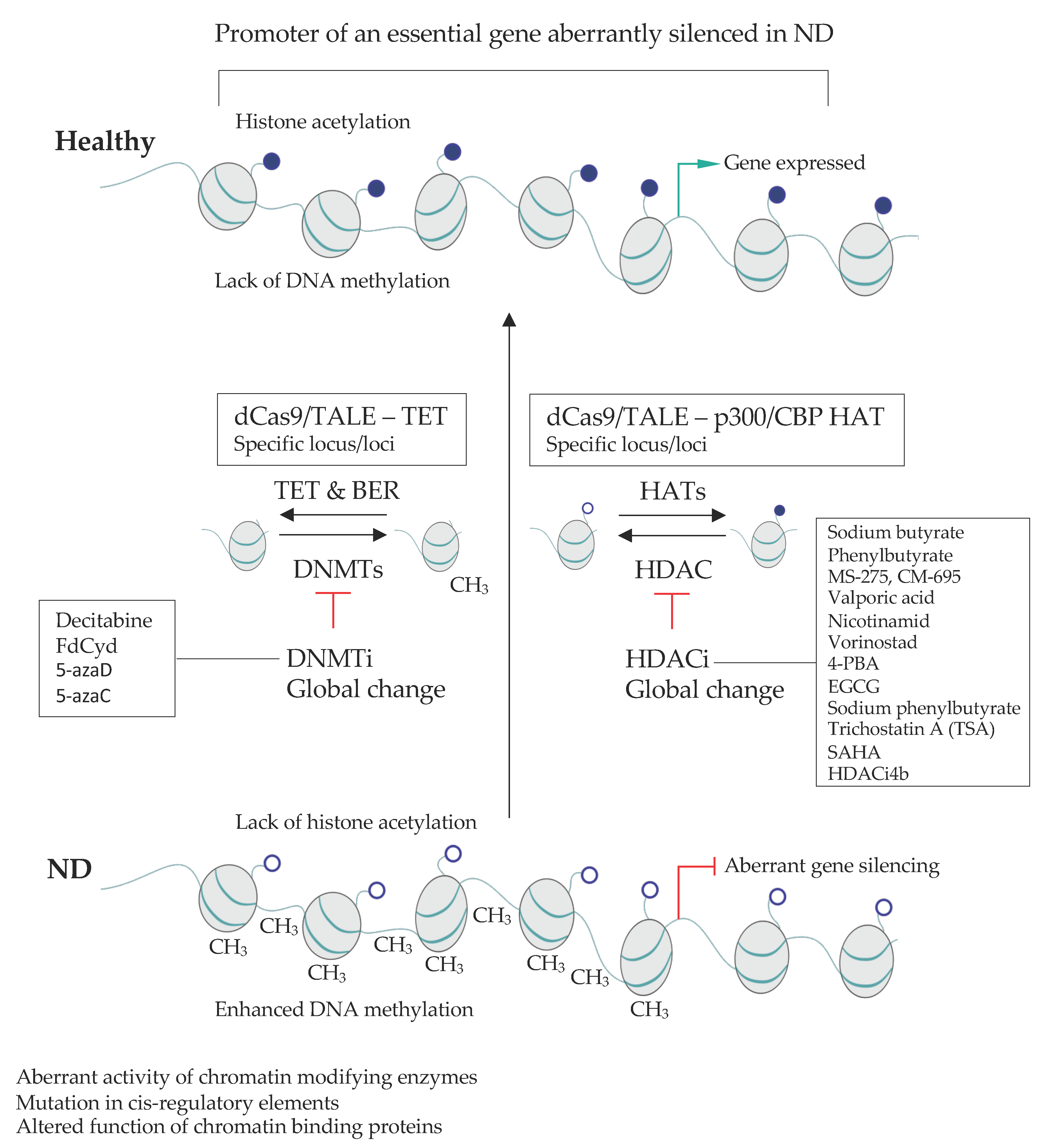
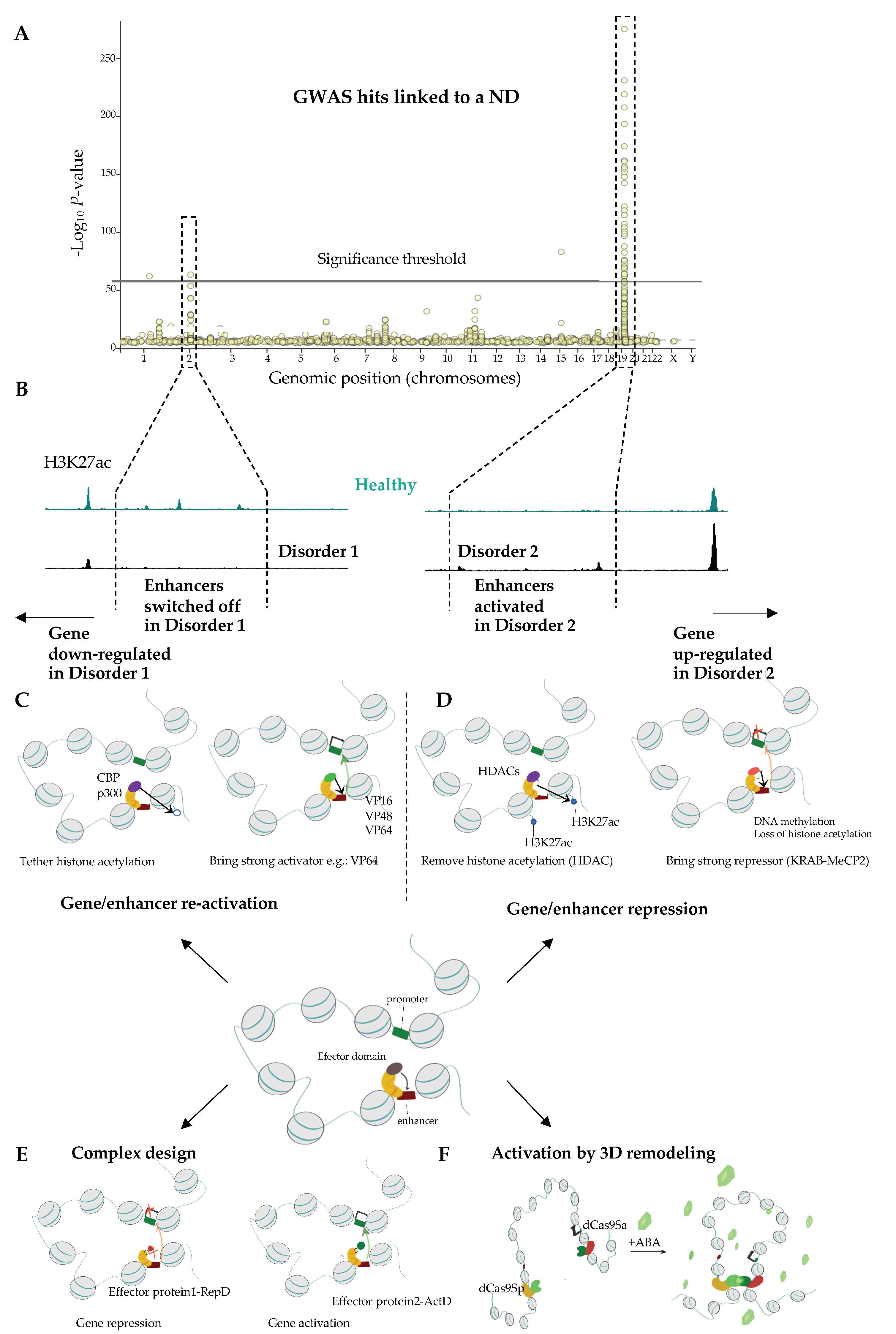

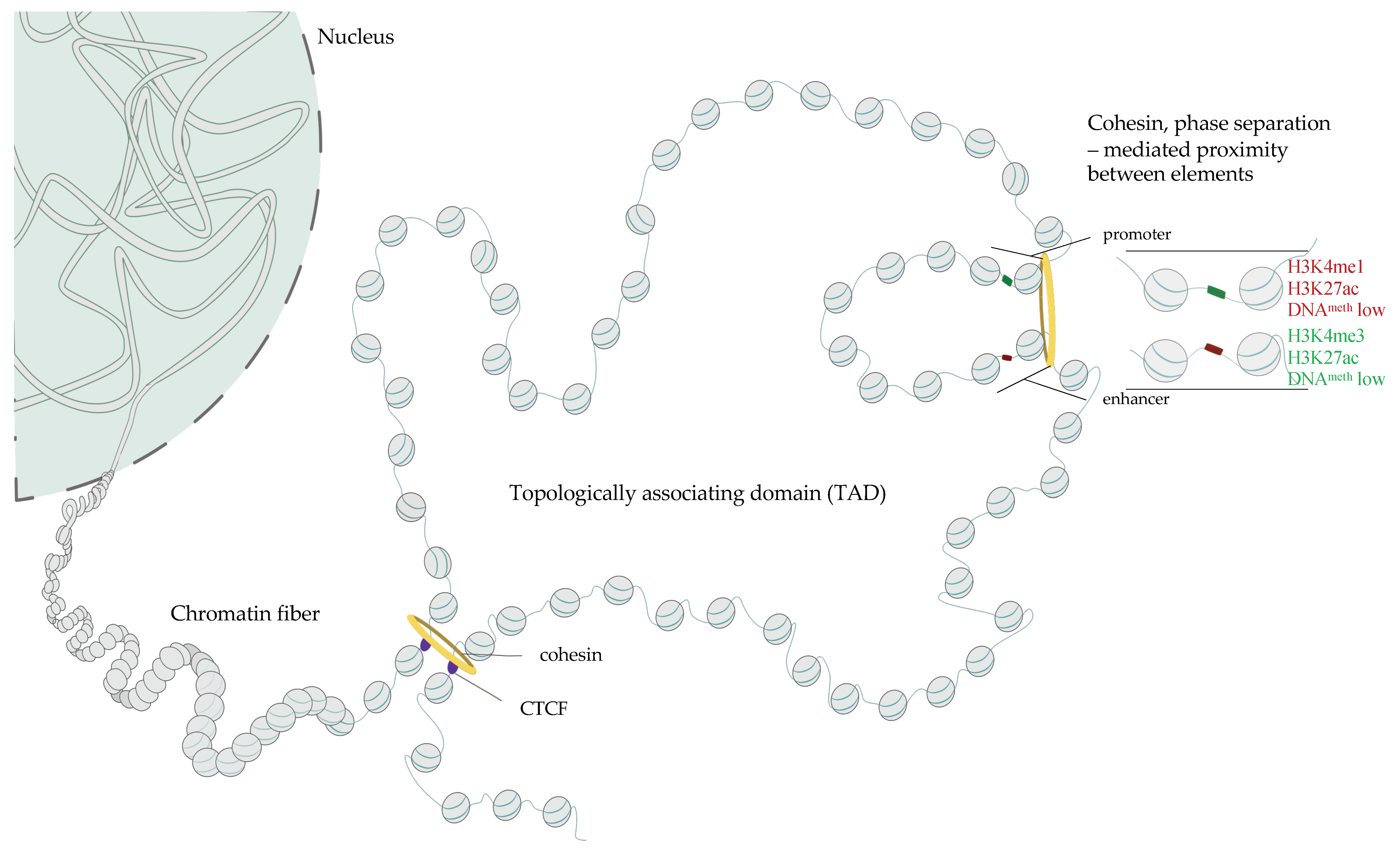{kind=link}
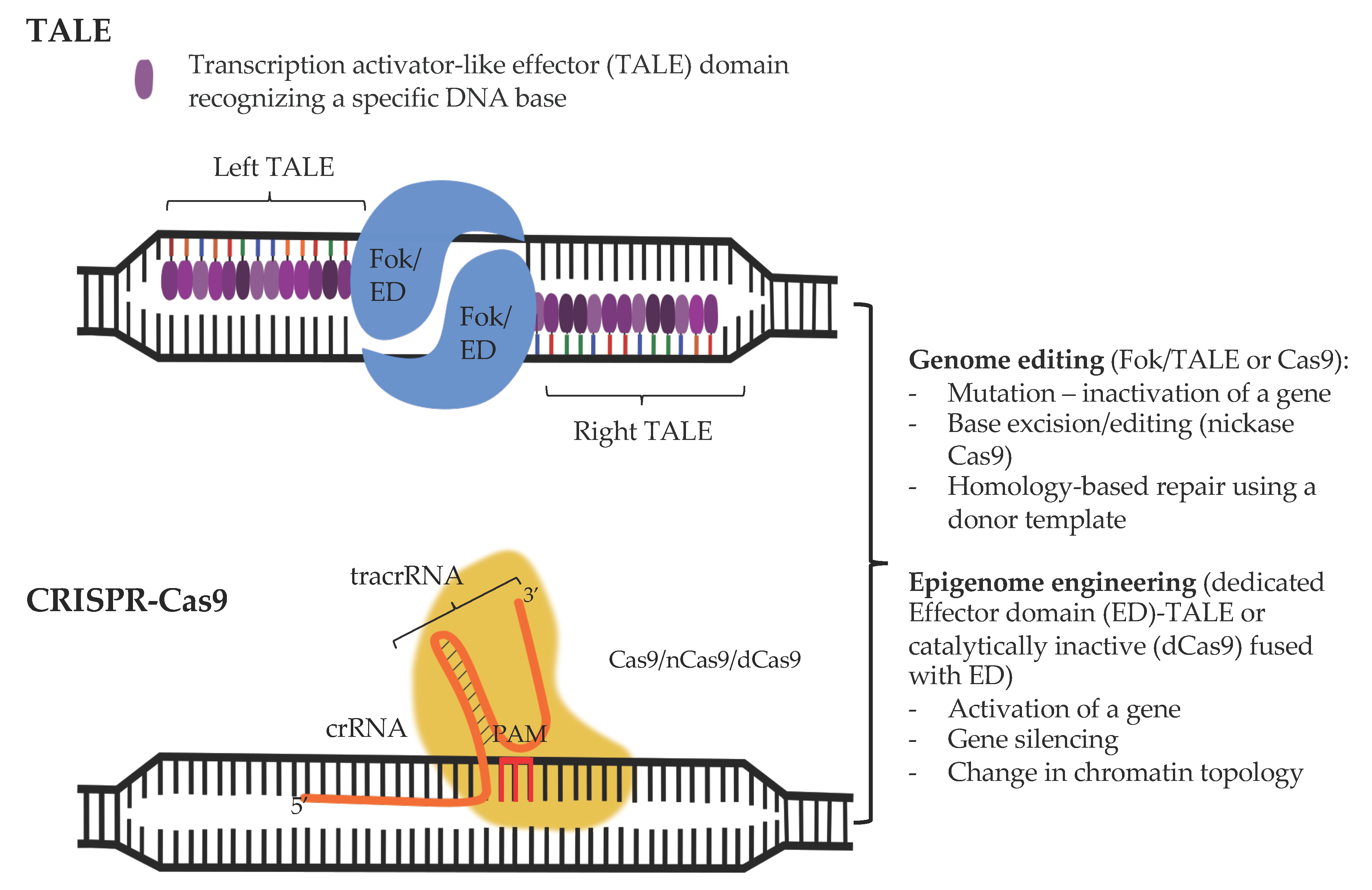{kind=link}
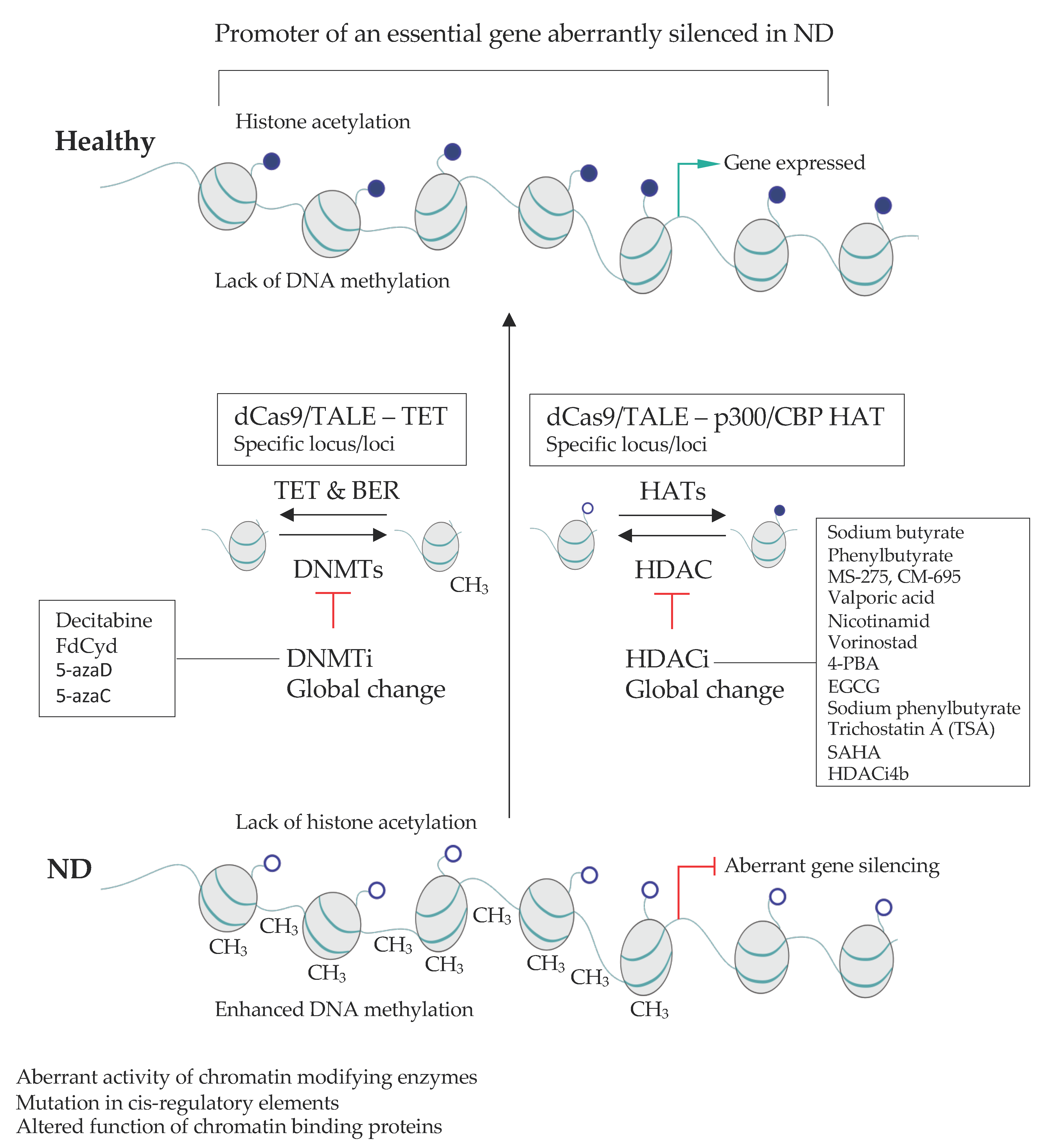{kind=link}
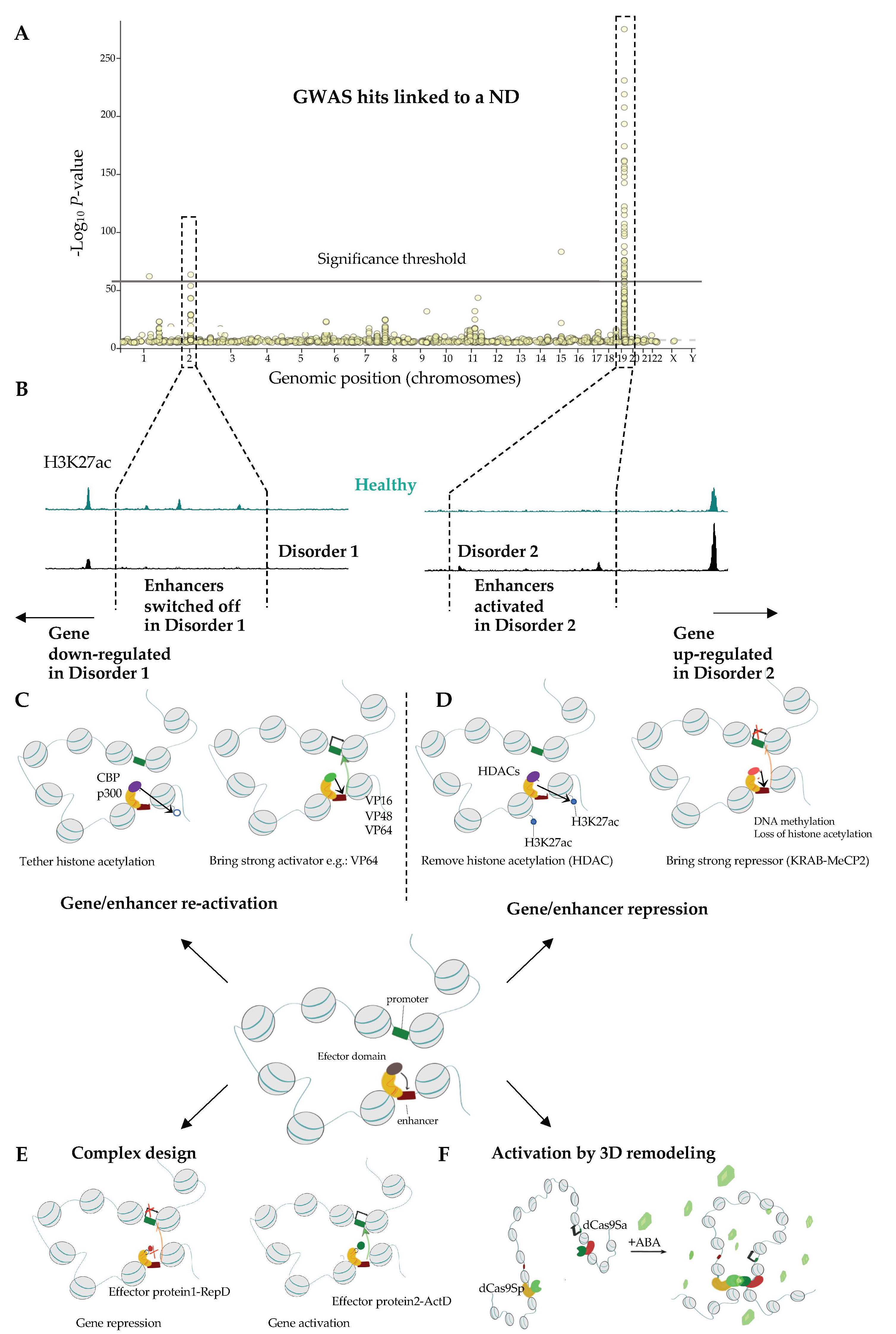{kind=link}
| Protein | Disease | Phenotype | Reference |
|---|---|---|---|
| DNA Methylases and DNA Methylation-Related Machinery | |||
| MECP2 | Rett syndrome | Encephalopathy, intellectual disability (ID), epilepsy | [116,117,118,119] |
| DNMT1 | Hereditary Sensory Neuropathy Type IE—heterozygous mutation | [120] | |
| autonomic neuropathies with dementia and hearing loss (HSANIE)—heterozygous mutation | [121] | ||
| cerebellar ataxia with deafness and narcolepsy | cerebellar ataxia, narcolepsy/cataplexy, sensorineural deafness, and dementia | [122] | |
| DNMT3a | Tatton-Brown-Rahman syndrome—heterozygous mutation | intellectual disability | [123,124,125] |
| Heyn-Sproul-Jackson syndrome—heterozygous mutation | microcephalic dwarfism | [126] | |
| Histone Acetylases | |||
| P300 | Rubinstein-Taybi syndrome—heterozygous mutation | microcephaly, mental retardation | [127,128,129] |
| Menke-Hennekam syndrome—heterozygous mutation | microcephaly, mental retardation, autistic behavior | [128,130] | |
| CBP | Rubinstein-Taybi syndrome | microcephaly, mental retardation, postnatal growth deficiency | [131,132,133] |
| Menke-Hennekam syndrome | microcephaly, Intellectual impairment, autistic behavior | [130,134,135] | |
| MOZ/MYST3/KAT6A | Arboleda-Tham syndrome—heterozygous mutation | microcephaly, impaired intellectual development | [136,137,138,139] |
| MORF/MYST4/KAT6B | SBBYSS syndrome | mental retardation | [140,141,142,143] |
| Genitopatellar syndrome | microcephaly, severe psychomotor retardation | [144,145] | |
| MOF/MYST1/KAT8 | Li-Ghorbani-Weisz-Hubshman syndrome—heterozygous mutation | developmental delay, impaired intellectual development | [146] |
| Histone deacetylases | |||
| HDAC6 | dominant X-linked chondrodyspasia | Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, microphthalmia | [147,148] |
| HDAC8 | Cornelia de Lange syndrome | dysmorphic facial features, cleft palate, distal limb defects, growth retardation, developmental delay | [149,150] |
| SIRT1 | anxiety disorder, panic disorder and social phobia | feeling persistent fear, panic attacks, fear of social situations | [151] |
| Chromatin remodelers | |||
| SWI/SNF complex | |||
| BRM/SMARCA2 | Nicolaides-Baraitser syndrome—heterozygous mutation | severe mental retardation, early-onset seizures, short stature, dysmorphic facial features | [152,153] |
| Schizophrenia | delusions, disorganized thinking, hallucinations | [154] | |
| Coffin-Siris syndrome 4 | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [155] | |
| BRG1/SMARCA4 | Rhabdoid Tumor Predisposition syndrome 2; RTPS1—heterozygous mutation | brain, spinal cord and kidney tumors | [156,157] |
| BAF47/SMARCB1 | Schwannomatosis 1—heterozygous mutation | multiple cutaneous neurilemmomas, spinal schwannomas | [158,159,160,161] |
| Multiple meningiomas | meningeal tumor | [158] | |
| Coffin-Siris syndrome | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [155] | |
| BAF170/SMARCC2 | Coffin-Siris syndrome—heterozygous mutation | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [162] |
| BAF60A/SMARCD1 | Coffin-Siris syndrome—heterozygous mutation | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [163] |
| BAF57/SMARCE1 | Meningioma—heterozygous mutation | spinal tumors | [164] |
| Coffin-Siris syndrome—heterozygous mutation | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [155,165] | |
| BAF53B/ACTL6B | Developmental and Epileptic Encephalopathy | delayed global development, hypotonia, peripheral spasticity, mainly cerebral atrophy and delayed myelination | [166,167,168,169] |
| Intellectual Developmental Disorder With Severe Speech And Ambulation Defects—heterozygous mutation | global developmental delay, impaired intellectual, dysmorphic features: prominent forehead and wide mouth | [169] | |
| BAF250A/ARID1A | Coffin-Siris syndrome—heterozygous mutation | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [155,170] |
| BAF250B/ARID1B | Coffin-Siris syndrome—heterozygous mutation | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [155,171,172] |
| BAF200/ARID2 | Coffin-Siris syndrome—heterozygous mutation | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [173,174,175] |
| ACTB | Juvenile-Onset Dystonia—homozygous mutation | eosinophilic, rod-like cytoplasmic inclusions in neocortical and thalamic neurons; abundant eosinophilic spherical structures in the striatum that were strongly actin-and actin depolarizing factor/cofilin-positive | [176,177] |
| Baraitser-Winter syndrome—heterozygous mutation | microcephaly, rare lissencephaly or neuronal heterotopia | [178,179] | |
| BAF45D/DPF2 | Coffin-Siris syndrome—heterozygous mutation | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [180] |
| BCL11A | Intellectual developmental disorder with persistence of fetal hemoglobin—heterozygous mutation | delayed psychomotor development, intellectual disability, microcephaly, downslanting palpebral fissures, strabismus, external ear abnormalities, and asymptomatic persistence of fetal hemoglobin (HbF) | [181] |
| BCL11B | Intellectual Developmental Disorder with Speech Delay, Dysmorphic Facies, and T-Cell Abnormalities—heterozygous mutation | delayed psychomotor development with intellectual disability and speech delay. included autistic features, attention deficit-hyperactivity disorder, anxiety, and other behavioral abnormalities | [182] |
| BAF180/PBRM1 | Autism spectrum disorders | difficulties with social interaction and communication, restricted and repetitive behavior | [183] |
| ISWI complex | |||
| BRF1 | cerebellofaciodental syndrome (CFDS)—homozygous mutation | delayed development, intellectual disability, abnormal facial and dental findings, cerebellar hypoplasia | [184] |
| BPTF | neurodevelopmental disorder with dysmorphic facies and distal limb anomalies (NEDDFL)—heterozygous mutation | delayed psychomotor development and intellectual disability | [185] |
| SNF2L/SMARCA1 | Schizophrenia syndrome disorders | delusions, disorganized thinking, hallucinations | [186] |
| Rett syndrome-like phenotype | Encephalopathy, severe intellectual disability, autistic features, epilepsy | [187] | |
| Coffin-Siris syndrome-like phenotype | developmental delay, intellectual disability, coarse facial features, feeding difficulties | [166] | |
| BAZ1A/ACF1 | Intelectual disability, epilepsy | [188] | |
| CHD protein family | |||
| CHD1 | Pilarowski-Bjornsson syndrome—heterozygous mutation | delayed development, intellectual disability, autistic features, speech apraxia, mild dysmorphic features. | [189] |
| CHD2 | childhood-onset epileptic encephalopathy (EEOC)—heterozygous mutation | severe intellectual disability, epilepsy characterized by onset of multiple seizure | [190,191,192,193] |
| CHD3 | Snijders Blok-Campeau syndrome (SNIBCPS)—heterozygous mutation | global developmental delay, impaired intellectual development. Macrocephaly, prominent forehead and hypertelorism, hypotonia, and joint laxity. | [194] |
| CHD4 | Sifrim-Hitz-Weiss syndrome | intellectual developmental disorder | [195,196] |
| CHD8 | Autism spectrum disorders | difficulties with social interaction and communication, restricted and repetitive behavior | [183] |
| INO80 complex | |||
| YY1 | Gabriele-de Vries syndrome—heterozygous mutation | delayed psychomotor development, variable cognitive impairment, behavioral problems, feeding problems | [197] |
| INO80E | microcephaly, intelectual disability | [198] | |
| SRCAP | Floating-harbor sydrome—heterozygous mutation | short stature, delayed bone age, delayed speech development, facial features | [199] |
| Cohesin complex | |||
| SMC1 | Cornelia de Lange syndrome | intelectual disability, microcephaly | [200,201] |
| Developmental and Epileptic Encephalopathy 85 with or without Midline Brain Defects—heterozygous mutation | severe seizures in the first year of life, global developmental delay, impaired intellectual development, poor/absent speech, dysmorphic facial features. | [202,203,204,205,206] | |
| SMC3 | Cornelia de Lange syndrome—heterozygous mutation | intelectual disability, microcephaly | [207,208] |
| RAD21 | Cornelia de Lange syndrome—heterozygous mutation | Intelectual disability, microcephaly | [202,209,210,211] |
| STAG1 | autosomal dominant mental retardation-47 (MRD47)—heterozygous mutation | delayed psychomotor development, mild to moderate intellectual disability | [212] |
| STAG2 | Mullegama-Klein-Martinez syndrome—heterozygous mutation | impaired intellectual development, speech delay, hypotonia, microcephaly | [213,214,215] |
| X-Linked Holoprosencephaly—heterozygous mutation | incomplete division of the embryonic forebrain | [202] | |
| NIPBL | Cornelia de Lange syndrome | Intelectual disability, microcephaly | [216,217,218] |
| CTCF | mental retardation-21 (MRD21) | mild intellectual disability, short stature, microcephaly | [219] |
| Disease | Epidrug class | Active compound | Model Organism | Remarks | References |
|---|---|---|---|---|---|
| Alzeimer’s disease | HDACi | Sodium Butyrate | CK-p25 Tg mice | Improved learning and memory | [371] |
| APPPS1-21 mice | Increased histone acetylation, upregulation of neuronal plasticity-related genes, improved associative memory | [372] | |||
| Phenylbutyrate | Tg2576 mice | Improved memory, increased dendritic spines density, Increased histone acetylation | [373] | ||
| N171-82Q mice | Increased H3 and h4 acetylation, mice survival, reduces brain gross and neuronal atrophy | [374] | |||
| MS-275 | APPPS1-21 mice | Reduced behavioral impairment, reduced number of amyloid plaques in cortex, enhanced microglia activation in cortex | [375] | ||
| Mercaptoacetamide-based HDACi (W2 and I2) | 3xTg AD mice | Decreased alpha amyloid level in vitro, Alpha amyloid synthesis genes downregulation/degradation genes upregulation, decreased Tau protein phosphorylation, improved learning and memory | [376] | ||
| CM-695 | Tg2576 Mice | Improved memory, Decreased alpha amyloid level | [377,378,379] | ||
| Valproic acid | APP23 mice | Improved memory, Decreased tau and alpha amyloid | [380] | ||
| Clinical trials | ClinicalTrials.gov Identifier: NCT01729598; NCT00088387 | ||||
| Nicotinamide | Triple Knock-in mice: presenilin PS1M146V, Amyloid Precursor protein (APPKM670/671NL), Tau (tauP301L) | Prevented cognitive deficits, decreased phosphorylation of tau protein | [381] | ||
| Clinical trials | ClinicalTrials.gov Identifier: NTC00580931; TC03061474 | ||||
| Vorinostat | Clinical trials | ClinicalTrials.gov Identifier: NCT03056495 | |||
| 4-PBA | Clinical trials | ClinicalTrials.gov Identifier: NCT03533257 | |||
| epigallocatechin-3-gallate (EGCG) | Clinical trials | ClinicalTrials.gov Identifier: NCT00951834 | |||
| Huntington’s disease | HDACi | TSA/SAHA | Hdh-Q109 mice | Increased BDNF transport, improved neuron survival | [382,383] |
| SAHA | R6/2 HD mouse | improved motor skills | [384] | ||
| Sodium Butyrate | R6/2 HD mice | Improved survival of R6/2 HD mice | [385] | ||
| Sodium butyrate/phenylbutyrate | Human Y39C α-Synuclein Transgenic mouse | Increased expression of DJ1 gene, protection against α-Synuclein Induced Toxicity, reduce α-Synuclein level in mice brain, Prevents Age-related Motor and Cognitive Decline | [386] | ||
| Sodium butyrate, SAHA | D.melanogaster Q48 | Decreased neurodegeneration | [387] | ||
| valproic acid | PD patients | Improved myoclonic hyperkinesia | [388] | ||
| HDACi 4b | N171-82Q mice | Decreased Huntingtin aggregation in brain’s cells nuclei, ameliorated cognitive functions | [389] | ||
| MC1568 | SHSY5Y Cell line | Enhanced neurite growth of Dopaminergic and Sympathetic Neurons, Increased histone acetylation, Enhanced protection against MPP+ induced neurotoxicity | [390] | ||
| DMNTi | decitabine, FdCyd | In vitro cultured neurons | Reduced cytotoxicity, Increased BDNF expression | [391] | |
| Parkinson’s Disease | HDACi | Sodium phenylbutyrate | Clinical trials | ClinicalTrials.gov Identifier: NCT02046434 | |
| Valproic acid | Rotenone-treated rats | Protection against neurotoxicity | [392] | ||
| phenylbutyrate | SOD1 (G93A) mice | Increased histone acetylation, increased motoneuron survival | [132,393] | ||
| Amyotrophic lateral sclerosis | HDACi | trichostatin A | myelin oligodendrocyte glycoprotein peptide-treated WT mice | improved neuronal survival and enhance anti-inflammatory pathway action | [394] |
| Vorinostat | myelin oligodendrocyte glycoprotein peptide-treated WT mice | CNS inflammation and demyelination inhibition | [395] | ||
| Multiple sclerosis | HDACi | Sodium butyrate | WT rats | Histone 4 acetylation in promotor region of Transthyretin (Ttr), improved performance in FST, TST tests | [396] |
| MS-275 | WT mice | Increased H3K27 acetylation in accumbens nucleus, decreased expression of stress induced genes | [397] | ||
| Depressive disorders | HDACi | MS-275 | WT mice | Increased CREB and BDNF expression | [398] |
| DNMTi | 5-azaD 5-azaC | WT mice | BDNF expression, improved performance in FST test | [396] |
| Disease | (Epi)genome Modification Strategy and Delivery System | Remarks | References |
|---|---|---|---|
| Parkinson ‘s Disease (PD) | CRISPR-dCas9- DNMT3A targeting α-synuclein Lentiviral vector delivery. | PD patient iPS cell-derived neurons. Silencing of SNCA (α-synuclein) leads to a reduction of disease phenotype downmodulation of ROS * production and an increase in cellular viability. | [512] |
| ZF-p65 targeting glial cell line-derived neurotrophic factor (GDNF). AAV2 delivery. | Rat model of PD. Striatal GDNF activation increases neuronal survival. | [513] | |
| Lewy Body Dementia | Combined dCas9-VP64 and sgRNA-MS2-p65 targeting Nrf2 promoter. | In vitro mouse neurons Nrf2 overexpression indued by HDACi and dCas9 ActD helps to clear the toxic deposits of α-synuclein | [514] |
| Alzheimer’s disease (AD) | ZF fused to G9a, Suvdel76, SKD) or VP64 Dlg4/PSD95. | Mouse. Activation of PSD95 leads to memory rescue in aged and Alzheimer’s disease mice. | [515] |
| CRISPR-Cas9 deletion of the mutated allele coding the amyloid precursor protein (APP). | Patient-derived fibroblasts and mouse. Selective silencing of the mutated amyloid precursor protein (APP); 60% reduction in secreted Aβ | [516] | |
| CRISPR-Cas9-mediated truncation of the C-terminus of the APP AAV9–mediated delivery of CRISPR-Cas9 into mouse hippocampus. | iPS cell-derived neurons, cultured hippocampal neurons, mouse. Removal of the C-terminus of APP results in upregulation of the neuroprotective α-cleavage pathway. | [517] | |
| CRISPR-Cas9 nanocomplexes Disruption of Bace1 and Th genes in neurons. | 5XFAD and App knock-in mice mouse. Reduction in Aβ plaque accumulation and Aβ42 secretion. Reduction of Bace1 lead to an increased associative learning and improved spatial working memory and reduced deficit in alternation performance compared with 5XFAD controls. Significant reduction of Aβ42 plaque accumulation and secretion at 8- and 12-weeks post Cas9 injection. | [518] | |
| Huntington‘s Disease | ZFs to block mutant HTT expression AAV2/1 intra-striatal injection. | Cell lines and the R6/2 mouse model. Reduction of diseased clasping behavior, enhanced motor coordination. | [519] |
| KAMI-Cas9 targeting HTT gene lentiviral-mediated in vivo delivery. | Neuronal/glial cultures, striatum of mice, and patient-specific iPSC neuronal cells. Decreased HTT aggregation, reduced neuronal dysfunction both in vitro and in vivo. | [520] | |
| CRISPR-Cas9. rAAV delivery of Cas9. | HD-patient-derived fibroblasts and BacHD mice. Allele-specific removal of mutated HTT gene | [521] | |
| CRISPR-dCas9MeCP2 downregulation of human HTT AAV-mediated delivery. | Adult brain (striatum) of the mouse HD model (HD140Q-KI). Silencing of the HTT led to reduced neuropathology and increased performance in rotarod, balance beam, and grip strength tests. | [522] | |
| ZF-KRAB, AAV-mediated delivery. | Patient-derived fibroblasts and neurons, HD mouse models (intra-striatal injection). Improvement in neuronal functions and alleviation of motoneuron symptoms. | [523] | |
| MeCP2 Duplication syndrome | CRISPR-Cas9.AAV-mediated delivery. | Transgenic mouse expressing human MeCP2. Knockout of the human MeCP2. Reversal of the social aversion deficit, yet no impact on the reduced locomotor activity, the heightened anxiety-like behaviors, and the fear generalization phenotype. | [524] |
| Rett syndrome | CRISPR-Cas9 | HR in human iPS cells to correct disease variants. | [525,526] |
| Fragile X syndrome | CRISPR-dCas9Tet1 demethylation of the FMR1 gene Lentiviral delivery. | iPS cell model of the Fragile X syndrome Demethylation of the FMR1 promoter accompanied by a gain of H3K27ac and transcriptional onset in post mitotic neurons derived from the iPS cells | [527] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janowski, M.; Milewska, M.; Zare, P.; Pękowska, A. Chromatin Alterations in Neurological Disorders and Strategies of (Epi)Genome Rescue. Pharmaceuticals 2021, 14, 765. https://doi.org/10.3390/ph14080765
Janowski M, Milewska M, Zare P, Pękowska A. Chromatin Alterations in Neurological Disorders and Strategies of (Epi)Genome Rescue. Pharmaceuticals. 2021; 14(8):765. https://doi.org/10.3390/ph14080765
Chicago/Turabian StyleJanowski, Marcin, Małgorzata Milewska, Peyman Zare, and Aleksandra Pękowska. 2021. "Chromatin Alterations in Neurological Disorders and Strategies of (Epi)Genome Rescue" Pharmaceuticals 14, no. 8: 765. https://doi.org/10.3390/ph14080765
APA StyleJanowski, M., Milewska, M., Zare, P., & Pękowska, A. (2021). Chromatin Alterations in Neurological Disorders and Strategies of (Epi)Genome Rescue. Pharmaceuticals, 14(8), 765. https://doi.org/10.3390/ph14080765