
Update on PET Tracer Development for Muscarinic Acetylcholine Receptors

 , ,
, ,  , and
, and
Abstract
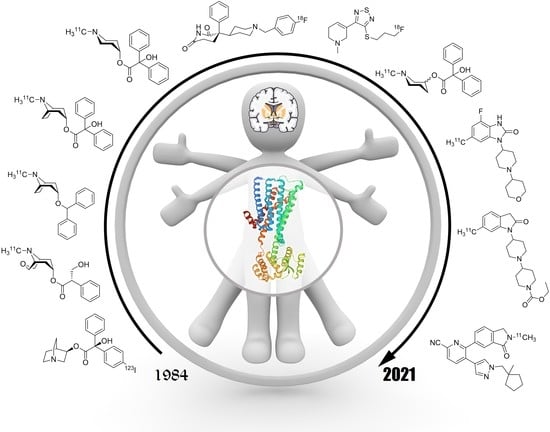
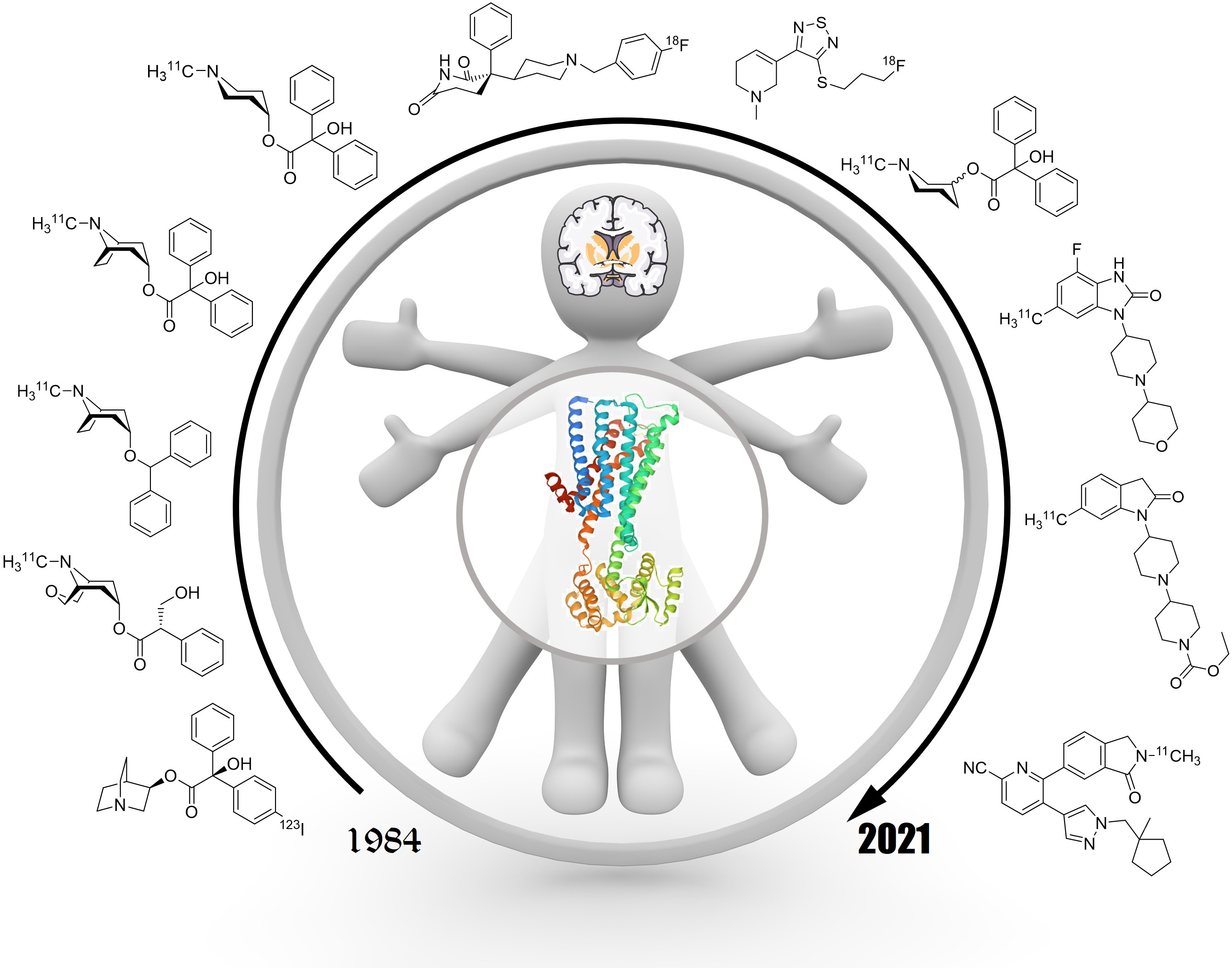
1. Introduction
1.1. Muscarinic Acetylcholine Receptors
1.2. The Role of PET in Diagnosis and Therapeutic Drug Development
1.3. Designing Small Molecules as PET Tracers for the CNS
1.4. Involvement of Computational Approaches in PET Tracer Design
2. Development of PET Tracers for mAChRs
2.1. Development of mAChR Ligands
2.2. PET Tracer Development for In Vivo Muscarinic Imaging of the CNS
2.3. PET Tracer Development for In Vivo Muscarinic Imaging of the PNS
3. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moran, S.P.; Maksymetz, J.; Conn, P.J. Targeting Muscarinic Acetylcholine Receptors for the Treatment of Psychiatric and Neurological Disorders. Trends Pharmacol. Sci. 2019, 40, 1006–1020. [Google Scholar] [CrossRef]
- Fernández de Sevilla, D.; Núñez, A.; Buño, W. Muscarinic Receptors, from Synaptic Plasticity to Its Role in Network Activity. Neuroscience 2021, 456, 60–70. [Google Scholar] [CrossRef]
- Chen, J.; Cheuk, I.W.Y.; Shin, V.Y.; Kwong, A. Acetylcholine Receptors: Key Players in Cancer Development. Surg. Oncol. 2019, 31, 46–53. [Google Scholar] [CrossRef]
- Sales, M.E.; Español, A.J.; Salem, A.R.; Pulido, P.M.; Sanchez, Y.; Sanchez, F. Role of Muscarinic Acetylcholine Receptors in Breast Cancer: Design of Metronomic Chemotherapy. Curr. Clin. Pharmacol. 2019, 14, 91–100. [Google Scholar] [CrossRef]
- Eckelman, W.C. Imaging of Muscarinic Receptors in the Central Nervous System. Curr. Pharm. Des. 2006, 12, 3901–3913. [Google Scholar] [CrossRef]
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic Acetylcholine Receptors: Novel Opportunities for Drug Development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, Z.; Gentry, P.R.; Berizzi, A.E.; Hirata, K.; Varghese, S.; Thompson, G.; van der Westhuizen, E.T.; Burger, W.A.C.; Rahmani, R.; Valant, C.; et al. Crystal Structure of the M5 Muscarinic Acetylcholine Receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 26001–26007. [Google Scholar] [CrossRef] [PubMed]
- Abrams, P.; Andersson, K.-E.; Buccafusco, J.J.; Chapple, C.; de Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; et al. Muscarinic Receptors: Their Distribution and Function in Body Systems, and the Implications for Treating Overactive Bladder. Br. J. Pharmacol. 2006, 148, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.-M.; Yuan, Z. PET/SPECT Molecular Imaging in Clinical Neuroscience: Recent Advances in the Investigation of CNS Diseases. Quant. Imaging Med. Surg. 2015, 5, 433–447. [Google Scholar] [CrossRef]
- Grimwood, S.; Hartig, P.R. Target Site Occupancy: Emerging Generalizations from Clinical and Preclinical Studies. Pharmacol. Ther. 2009, 122, 281–301. [Google Scholar] [CrossRef] [PubMed]
- Honer, M.; Gobbi, L.; Martarello, L.; Comley, R.A. Radioligand Development for Molecular Imaging of the Central Nervous System with Positron Emission Tomography. Drug Discov. Today 2014, 19, 1936–1944. [Google Scholar] [CrossRef]
- Czernin, J.; Sonni, I.; Razmaria, A.; Calais, J. The Future of Nuclear Medicine as an Independent Specialty. J. Nucl. Med. 2019, 60, 3S–12S. [Google Scholar] [CrossRef]
- Zhang, L.; Villalobos, A. Strategies to Facilitate the Discovery of Novel CNS PET Ligands. EJNMMI Radiopharm. Chem. 2017, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Kolanko, M.A.; Win, Z.; Loreto, F.; Patel, N.; Carswell, C.; Gontsarova, A.; Perry, R.J.; Malhotra, P.A. Amyloid PET Imaging in Clinical Practice. Pract. Neurol. 2020, 20, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Colom, M.; Vidal, B.; Zimmer, L. Is There a Role for GPCR Agonist Radiotracers in PET Neuroimaging? Front. Mol. Neurosci. 2019, 12, 255. [Google Scholar] [CrossRef]
- Bergström, M.; Grahnén, A.; Långström, B. Positron Emission Tomography Microdosing: A New Concept with Application in Tracer and Early Clinical Drug Development. Eur. J. Clin. Pharmacol. 2003, 59, 357–366. [Google Scholar] [CrossRef]
- Lee, K.-S. Future Directions in Overactive Bladder Treatment: Personalized Medicine Can Be Applied? Korean J. Urol. 2015, 56, 671. [Google Scholar] [CrossRef][Green Version]
- Calzetta, L.; Puxeddu, E.; Rogliani, P. Gender-Related Responsiveness to the Pharmacological Treatment of COPD: A First Step Towards the Personalized Medicine. EBioMedicine 2017, 19, 14–15. [Google Scholar] [CrossRef]
- Roy, R.; Niccolini, F.; Pagano, G.; Politis, M. Cholinergic Imaging in Dementia Spectrum Disorders. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1376–1386. [Google Scholar] [CrossRef]
- McCluskey, S.P.; Plisson, C.; Rabiner, E.A.; Howes, O. Advances in CNS PET: The State-of-the-Art for New Imaging Targets for Pathophysiology and Drug Development. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 451–489. [Google Scholar] [CrossRef] [PubMed]
- Suridjan, I.; Comley, R.A.; Rabiner, E.A. The Application of Positron Emission Tomography (PET) Imaging in CNS Drug Development. Brain Imaging Behav. 2019, 13, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.M.; Rabiner, E.A.; Passchier, J.; Gunn, R.N. Positron Emission Tomography Molecular Imaging for Drug Development: PET for Drug Development. Br. J. Clin. Pharmacol. 2012, 73, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, R.J.; Rabiner, E.A. Translational PET Imaging Research. Neurobiol. Dis. 2014, 61, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, R. The Role of Molecular Imaging in Drug Discovery and Development. Clin. Pharmacol. Ther. 2008, 83, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.E.; Andrés, J.I. The Value of PET Ligand Discovery to CNS Drug Development. Future Med. Chem. 2017, 9, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Hern, J.A.; Baig, A.H.; Mashanov, G.I.; Birdsall, B.; Corrie, J.E.T.; Lazareno, S.; Molloy, J.E.; Birdsall, N.J.M. Formation and Dissociation of M1 Muscarinic Receptor Dimers Seen by Total Internal Reflection Fluorescence Imaging of Single Molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
- Maier-Peuschel, M.; Frölich, N.; Dees, C.; Hommers, L.G.; Hoffmann, C.; Nikolaev, V.O.; Lohse, M.J. A Fluorescence Resonance Energy Transfer-Based M2 Muscarinic Receptor Sensor Reveals Rapid Kinetics of Allosteric Modulation*. J. Biol. Chem. 2010, 285, 8793–8800. [Google Scholar] [CrossRef]
- Valuskova, P.; Farar, V.; Forczek, S.; Krizova, I.; Myslivecek, J. Autoradiography of 3H-Pirenzepine and 3H-AFDX-384 in Mouse Brain Regions: Possible Insights into M1, M2, and M4 Muscarinic Receptors Distribution. Front. Pharmacol. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Jositsch, G.; Papadakis, T.; Haberberger, R.V.; Wolff, M.; Wess, J.; Kummer, W. Suitability of Muscarinic Acetylcholine Receptor Antibodies for Immunohistochemistry Evaluated on Tissue Sections of Receptor Gene-Deficient Mice. Naunyn-Schmied. Arch. Pharmacol. 2009, 379, 389–395. [Google Scholar] [CrossRef]
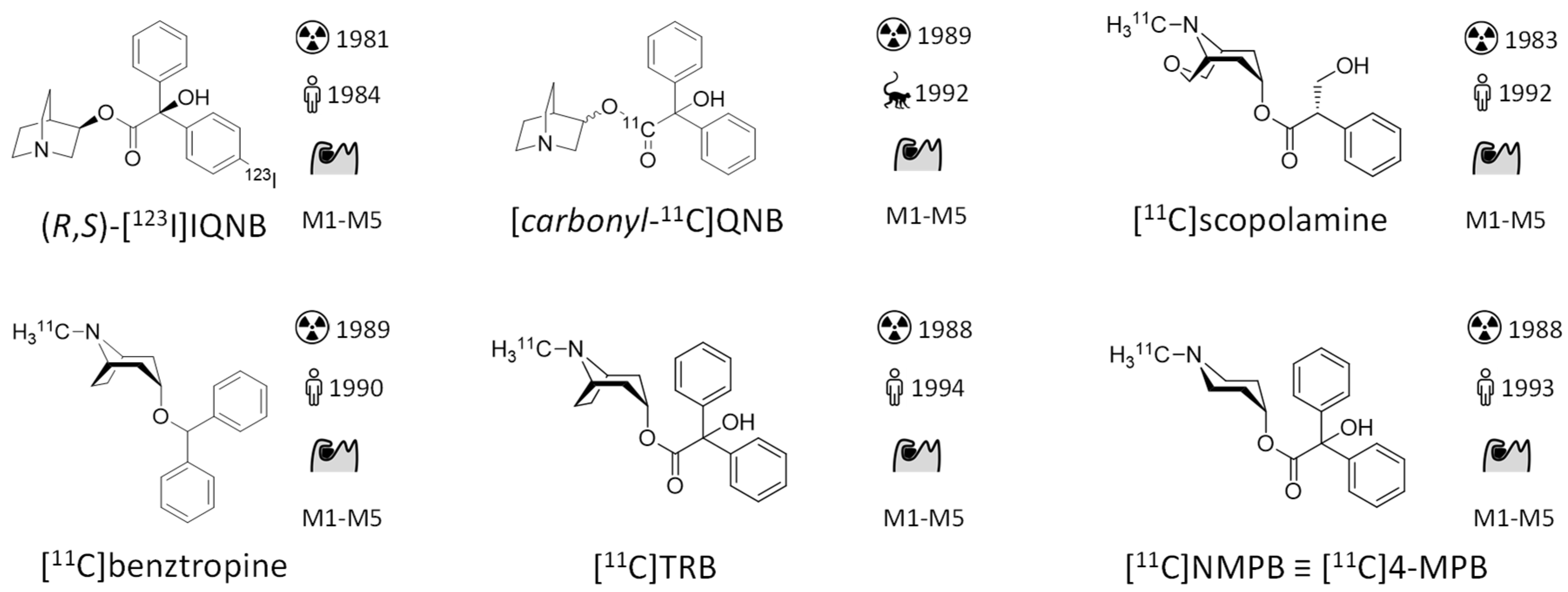
- Eckelman, W.; Reba, R.; Rzeszotarski, W.; Gibson, R.; Hill, T.; Holman, B.; Budinger, T.; Conklin, J.; Eng, R.; Grissom, M. External Imaging of Cerebral Muscarinic Acetylcholine Receptors. Science 1984, 223, 291–293. [Google Scholar] [CrossRef]
- Müller-Gärtner, H.W.; Wilson, A.A.; Dannals, R.F.; Wagner, H.N.; Frost, J.J. Imaging Muscarinic Cholinergic Receptors in Human Brain in Vivo with SPECT, [ 123 I]4-Iododexetimide, and [ 123 I]4-Iodolevetimide. J. Cereb. Blood Flow Metab. 1992, 12, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D.; Furst, A.J.; O’Neil, J.P.; Racine, C.A.; Mormino, E.C.; Baker, S.L.; Chetty, S.; Patel, P.; Pagliaro, T.A.; Klunk, W.E.; et al. 11C-PIB PET Imaging in Alzheimer Disease and Frontotemporal Lobar Degeneration. Neurology 2007, 68, 1205–1212. [Google Scholar] [CrossRef]
- Hirvonen, J.; Aalto, S.; Lumme, V.; Nagren, K.; Kajander, J.; Vilkman, H.; Hagelberg, N.; Oikonen, V.; Hietala, J. Measurement of Striatal and Thalamic Dopamine D2 Receptor Binding with 11C-Raclopride: Nucl. Med. Commun. 2003, 24, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Leskinen-Kallio, S.; Någren, K.; Lehikoinen, P.; Ruotsalainen, U.; Teräs, M.; Joensuu, H. Carbon-11-Methionine and PET Is an Effective Method To Image Head and Neck Cancer. J. Nucl. Med. 1992, 33, 691–695. [Google Scholar] [PubMed]
- Kim, J.S.; Ichise, M.; Sangare, J.; Innis, R.B. PET Imaging of Serotonin Transporters with [11C]DASB: Test-Retest Reproducibility Using a Multilinear Reference Tissue Parametric Imaging Method. J. Nucl. Med. 2006, 47, 208–214. [Google Scholar]
- Gee, A.D.; Bongarzone, S.; Wilson, A.A. Small Molecules as Radiopharmaceutical Vectors. In Radiopharmaceutical Chemistry; Lewis, J.S., Windhorst, A.D., Zeglis, B.M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 119–136. ISBN 978-3-319-98946-4. [Google Scholar]
- Eckelman, W.C.; Kilbourn, M.R.; Mathis, C.A. Discussion of Targeting Proteins in Vivo: In Vitro Guidelines. Nucl. Med. Biol. 2006, 33, 449–451. [Google Scholar] [CrossRef]
- Patel, S.; Gibson, R. In Vivo Site-Directed Radiotracers: A Mini-Review. Nucl. Med. Biol. 2008, 35, 805–815. [Google Scholar] [CrossRef]
- Zeeberg, B.R. Pharmacokinetic Computer Simulations of the Relationship between in Vivo and in Vitro Neuroreceptor Subtype Selectivity of Radioligands. Nucl. Med. Biol. 1999, 26, 803–809. [Google Scholar] [CrossRef]
- Pichler, V.; Berroterán-Infante, N.; Philippe, C.; Vraka, C.; Klebermass, E.-M.; Balber, T.; Pfaff, S.; Nics, L.; Mitterhauser, M.; Wadsak, W. An Overview of PET Radiochemistry, Part 1: The Covalent Labels 18 F, 11 C, and 13 N. J. Nucl. Med. 2018, 59, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Vraka, C.; Nics, L.; Wagner, K.-H.; Hacker, M.; Wadsak, W.; Mitterhauser, M. Log P, a Yesterday’s Value? Nucl. Med. Biol. 2017, 50, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van Assema, D.M.E.; Lubberink, M.; Boellaard, R.; Schuit, R.C.; Windhorst, A.D.; Scheltens, P.; Lammertsma, A.A.; van Berckel, B.N.M. P-Glycoprotein Function at the Blood–Brain Barrier: Effects of Age and Gender. Mol. Imaging Biol. 2012, 14, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Verdurand, M.; Levigoureux, E.; Zeinyeh, W.; Berthier, L.; Mendjel-Herda, M.; Cadarossanesaib, F.; Bouillot, C.; Iecker, T.; Terreux, R.; Lancelot, S.; et al. In Silico, in Vitro, and in Vivo Evaluation of New Candidates for α-Synuclein PET Imaging. Mol. Pharm. 2018, 15, 3153–3166. [Google Scholar] [CrossRef]
- Goud, N.S.; Ghouse, M.S.; Nagaraju, C.; Bharath, R.D.; Alvala, M.; Kumar, P. Automated Radiosynthesis and Molecular Docking Studies of Coumarin-Triazole Hybrid with Fluorine-18: A Feasibility Study. Curr. Radiopharm. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rühl, T.; Deuther-Conrad, W.; Fischer, S.; Günther, R.; Hennig, L.; Krautscheid, H.; Brust, P. Cannabinoid Receptor Type 2 (CB2)-Selective N-Aryl-Oxadiazolyl-Propionamides: Synthesis, Radiolabelling, Molecular Modelling and Biological Evaluation. Org. Med. Chem. Lett. 2012, 2, 32. [Google Scholar] [CrossRef]
- Qi, Y.; Li, Y.; Fang, Y.; Gao, H.; Qiang, B.; Wang, S.; Zhang, H. Design, Synthesis, Biological Evaluation, and Molecular Docking of 2,4-Diaminopyrimidine Derivatives Targeting Focal Adhesion Kinase as Tumor Radiotracers. Mol. Pharm. 2021, 18, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, C.; Adasme, F.; Alzate-Morales, J.H.; Vergara-Jaque, A.; Kniess, T.; Caballero, J. Study of Differences in the VEGFR2 Inhibitory Activities between Semaxanib and SU5205 Using 3D-QSAR, Docking, and Molecular Dynamics Simulations. J. Mol. Graph. Model. 2012, 32, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Ozenil, M.; Pacher, K.; Balber, T.; Vraka, C.; Roller, A.; Holzer, W.; Spreitzer, H.; Mitterhauser, M.; Wadsak, W.; Hacker, M.; et al. Enhanced Arecoline Derivatives as Muscarinic Acetylcholine Receptor M1 Ligands for Potential Application as PET Radiotracers. Eur. J. Med. Chem. 2020, 204, 112623. [Google Scholar] [CrossRef]
- Ozenil, M.; Aronow, J.; Piljak, D.; Vraka, C.; Holzer, W.; Spreitzer, H.; Wadsak, W.; Hacker, M.; Pichler, V. Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting MAChR M1. Pharmaceuticals 2020, 13, 437. [Google Scholar] [CrossRef]
- Gobbi, L.; Mercier, J.; Bang-Andersen, B.; Nicolas, J.; Reilly, J.; Wagner, B.; Whitehead, D.; Briard, E.; Maguire, R.P.; Borroni, E.; et al. A Comparative Study of In Vitro Assays for Predicting the Nonspecific Binding of PET Imaging Agents In Vivo. ChemMedChem 2020, 15, 585–592. [Google Scholar] [CrossRef]
- Dickson, C.J.; Gee, A.D.; Bennacef, I.; Gould, I.R.; Rosso, L. Further Evaluation of Quantum Chemical Methods for the Prediction of Non-Specific Binding of Positron Emission Tomography Tracers. Phys. Chem. Chem. Phys. 2011, 13, 21552. [Google Scholar] [CrossRef]
- Rosso, L.; Gee, A.D.; Gould, I.R. Ab Initio Computational Study of Positron Emission Tomography Ligands Interacting with Lipid Molecule for the Prediction of Nonspecific Binding. J. Comput. Chem. 2008, 29, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal Structures of the M1 and M4 Muscarinic Acetylcholine Receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef]
- Bolden, C.; Cusack, B.; Richelson, E. Antagonism by Antimuscarinic and Neuroleptic Compounds at the Five Cloned Human Muscarinic Cholinergic Receptors Expressed in Chinese Hamster Ovary Cells. J. Pharmacol. Exp. Ther. 1992, 260, 576–580. [Google Scholar] [PubMed]
- Maslinski, W.; Grabczewska, E.; Bartfai, T.; Ryzewski, J.; Larsen, S.; Wilgocki, M.; Wold, S. Muscarinic Antagonist Binding to Intact Rat Thymocytes. Acta Chem. Scand. 1990, 44, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.P.; Nahorski, S.R.; Challiss, R.A.J. Constitutive Activity and Inverse Agonism at the M 2 Muscarinic Acetylcholine Receptor. J. Pharmacol. Exp. Ther. 2006, 316, 279–288. [Google Scholar] [CrossRef] [PubMed]
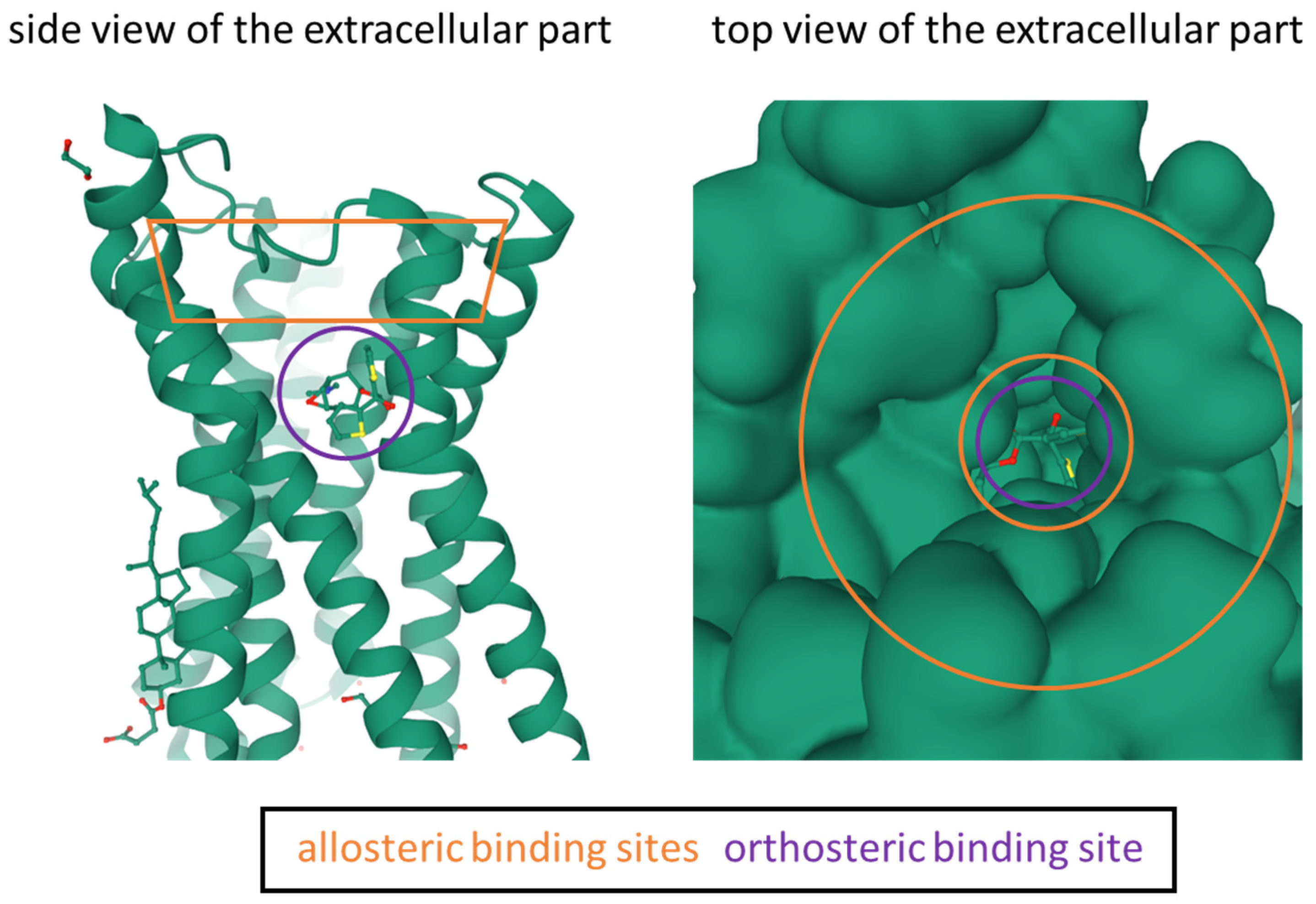
- Jakubik, J.; El-Fakahany, E.E. Current Advances in Allosteric Modulation of Muscarinic Receptors. Biomolecules 2020, 10, 325. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Seager, M.A.; Wittmann, M.; Jacobson, M.; Bickel, D.; Burno, M.; Jones, K.; Graufelds, V.K.; Xu, G.; Pearson, M.; et al. Selective Activation of the M 1 Muscarinic Acetylcholine Receptor Achieved by Allosteric Potentiation. PNAS 2009, 106, 15950–15955. [Google Scholar] [CrossRef]
- Steinfeld, T.; Mammen, M.; Smith, J.A.M.; Wilson, R.D.; Jasper, J.R. A Novel Multivalent Ligand That Bridges the Allosteric and Orthosteric Binding Sites of the M 2 Muscarinic Receptor. Mol. Pharmacol. 2007, 72, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Leach, K.; Loiacono, R.E.; Felder, C.C.; McKinzie, D.L.; Mogg, A.; Shaw, D.B.; Sexton, P.M.; Christopoulos, A. Molecular Mechanisms of Action and In Vivo Validation of an M4 Muscarinic Acetylcholine Receptor Allosteric Modulator with Potential Antipsychotic Properties. Neuropsychopharmacology 2010, 35, 855–869. [Google Scholar] [CrossRef]
- Sehnal, D.; Rose, A.; Koca, J.; Burley, S.; Velankar, S. Mol*: Towards a Common Library and Tools for Web Molecular Graphics. Workshop Mol. Graph. Vis. Anal. Mol. Data 2018, 29–33. [Google Scholar] [CrossRef]
- Raedler, T.J.; Knable, M.B.; Jones, D.W.; Urbina, R.A.; Gorey, J.G.; Lee, K.S.; Egan, M.F.; Coppola, R.; Weinberger, D.R. In Vivo Determination of Muscarinic Acetylcholine Receptor Availability in Schizophrenia. Am. J. Psychiatry 2003, 160, 118–127. [Google Scholar] [CrossRef]
- Raedler, T.J. Comparison of the In-Vivo Muscarinic Cholinergic Receptor Availability in Patients Treated with Clozapine and Olanzapine. Int. J. Neuropsychopharm. 2007, 10, 275. [Google Scholar] [CrossRef]
- Toyohara, J.; Sakata, M.; Ishiwata, K. Human Brain Imaging of Acetylcholine Receptors. In Imaging of the Human Brain in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2014; pp. 113–160. [Google Scholar]
- Prenant, C.; Barre, L.; Crouzel, C. Synthesis of [11C]-3-Quinuclidinylbenzilate (QNB). J. Label. Compd. Radiopharm. 1989, 27, 1257–1265. [Google Scholar] [CrossRef]
- Frey, K.A.; Koeppe, R.A.; Mulholland, G.K.; Jewett, D.; Hichwa, R.; Ehrenkaufer, R.L.E.; Carey, J.E.; Wieland, D.M.; Kuhl, D.E.; Agranoff, B.W. In Vivo Muscarinic Cholingeric Receptor Imaging in Human Brain with [11C]Scopolamine and Positron Emission Tomography. J. Cereb. Blood Flow Metab. 1992, 12, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Dewey, S.L.; Macgregor, R.R.; Brodie, J.D.; Bendriem, B.; King, P.T.; Volkow, N.D.; Schlyer, D.J.; Fowler, J.S.; Wolf, A.P.; Gatley, S.J.; et al. Mapping Muscarinic Receptors in Human and Baboon Brain Using [N-11C-Methyl]-Benztropine. Synapse 1990, 5, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, R.A.; Frey, K.A.; Mulholland, G.K.; Kilbourn, M.R.; Buck, A.; Lee, K.S.; Kuhl, D.E. [11C]Tropanyl Benzilate—Binding to Muscarinic Cholinergic Receptors: Methodology and Kinetic Modeling Alternatives. J. Cereb. Blood Flow Metab. 1994, 14, 85–99. [Google Scholar] [CrossRef]
- Suhara, T.; Inoue, O.; Kobayashi, K.; Suzuki, K.; Tateno, Y. Age-Related Changes in Human Muscarinic Acetylcholine Receptors Measured by Positron Emission Tomography. Neurosci. Lett. 1993, 149, 225–228. [Google Scholar] [CrossRef]
- Quirion, R. Cholinergic Markers in Alzheimer Disease and the Autoregulation of Acetylcholine Release. J. Psychiatry NeuroSci. 1993, 18, 226–234. [Google Scholar]
- Seeman, P.; Madras, B. Imaging of the Human Brain in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Nishiyama, S.; Sato, K.; Harada, N.; Kakiuchi, T.; Tsukada, H. Development and Evaluation of Muscarinic Cholinergic Receptor Ligands N-[11c]Ethyl-4-Piperidyl Benzilate and n-[11c]Propyl-4-Piperidyl Benzilate: A Pet Study in Compsarison with n-[11c]Methyl-4-Piperidyl Benzilate in the Conscious Monkey Brain. Nucl. Med. Biol. 2000, 27, 733–740. [Google Scholar] [CrossRef]
- Tsukada, H.; Takahashi, K.; Miura, S.; Nishiyama, S.; Kakiuchi, T.; Ohba, H.; Sato, K.; Hatazawa, J.; Okudera, T. Evaluation of Novel PET Ligands (+)N-[11C]Methyl-3-Piperidyl Benzilate ([11C](+)3-MPB) and Its Stereoisomer [11C](-)3-MPB for Muscarinic Cholinergic Receptors in the Conscious Monkey Brain: A PET Study in Comparison with [11C]4-MPB. Synapse 2001, 39, 182–192. [Google Scholar] [CrossRef]
- Yamamoto, S.; Ouchi, Y.; Nakatsuka, D.; Tahara, T.; Mizuno, K.; Tajima, S.; Onoe, H.; Yoshikawa, E.; Tsukada, H.; Iwase, M.; et al. Reduction of [11C](+)3-MPB Binding in Brain of Chronic Fatigue Syndrome with Serum Autoantibody against Muscarinic Cholinergic Receptor. PLoS ONE 2012, 7, e51515. [Google Scholar] [CrossRef]
- Hwang, D.-R.; Dence, C.S.; McKinnon, Z.A.; Mathias, C.J.; Welch, M.J. Positron Labeled Muscarinic Acetylcholine Receptor Antagonist: 2- and 4-[18F]Fluorodexetimide. Syntheses and Biodistribution. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1991, 18, 247–252. [Google Scholar] [CrossRef]
- Wilson, A.A.; Scheffel, U.A.; Dannals, R.F.; Stathis, M.; Ravert, H.T.; Wagner, H.N. In Vivo Biodistribution of Two [18F]-Labelled Muscarinic Cholinergic Receptor Ligands: 2-[18F]- and 4-[18F]-Fluorodexetimide. Life Sci. 1991, 48, 1385–1394. [Google Scholar] [CrossRef]
- Rowe, C.; Dean, B.; Ackermann, U.; Goh, R.; Guzman, R.; Kanaan, R.; Chong, L.; Dore, V.; Bozinovski, S.; Masters, C.; et al. In Vivo Imaging of Brain Muscarinic Receptors with 18F-Flurobenzyl Dexetimide: First in Human Studies. J. Nucl. Med. 2019, 60, 1470. [Google Scholar]
- Pain, C.D.; O’Keefe, G.J.; Ackermann, U.; Dore, V.; Villemagne, V.L.; Rowe, C.C. Human Biodistribution and Internal Dosimetry of 4-[18F]Fluorobenzyl-Dexetimide: A PET Radiopharmaceutical for Imaging Muscarinic Acetylcholine Receptors in the Brain and Heart. EJNMMI Res. 2020, 10, 61. [Google Scholar] [CrossRef]
- Flier, J.S.; Underhill, L.H.; Goyal, R.K. Muscarinic Receptor Subtypes. N. Engl. J. Med. 1989, 321, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Elhusseiny, A.; Cohen, Z.; Olivier, A.; Stanimirović, D.B.; Hamel, E. Functional Acetylcholine Muscarinic Receptor Subtypes in Human Brain Microcirculation: Identification and Cellular Localization. J. Cereb. Blood Flow Metab. 1999, 19, 794–802. [Google Scholar] [CrossRef]
- Jagoda, E.M.; Kiesewetter, D.O.; Shimoji, K.; Ravasi, L.; Yamada, M.; Gomeza, J.; Wess, J.; Eckelman, W.C. Regional Brain Uptake of the Muscarinic Ligand, [18F]FP-TZTP, Is Greatly Decreased in M2 Receptor Knockout Mice but Not in M1, M3 and M4 Receptor Knockout Mice. Neuropharmacology 2003, 44, 653–661. [Google Scholar] [CrossRef]
- Podruchny, T.A.; Connolly, C.; Bokde, A.; Herscovitch, P.; Eckelman, W.C.; Kiesewetter, D.O.; Sunderland, T.; Carson, R.E.; Cohen, R.M. In Vivo Muscarinic 2 Receptor Imaging in Cognitively Normal Young and Older Volunteers. Synapse 2003, 48, 39–44. [Google Scholar] [CrossRef]
- Ravasi, L.; Kiesewetter, D.O.; Shimoji, K.; Lucignani, G.; Eckelman, W.C. Why Does the Agonist [18F]FP-TZTP Bind Preferentially to the M2 Muscarinic Receptor? Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 292–300. [Google Scholar] [CrossRef]
- Mirza, N.R.; Peters, D.; Sparks, R.G. Xanomeline and the Antipsychotic Potential of Muscarinic Receptor Subtype Selective Agonists. CNS Drug Rev. 2006, 9, 159–186. [Google Scholar] [CrossRef]
- Farde, L.; Suhara, T.; Halldin, C.; Nybäck, H.; Nakashima, Y.; Swahn, C.-G.; Karlsson, P.; Ginovart, N.; Bymaster, F.; Shannon, H.; et al. PET Study of the M1-Agonists [11C]Xanomeline and [11C]Butylthio-TZTP in Monkey and Man. Dement. Geriatr. Cogn. Disord. 1996, 7, 187–195. [Google Scholar] [CrossRef]
- Montani, C.; Canella, C.; Schwarz, A.J.; Li, J.; Gilmour, G.; Galbusera, A.; Wafford, K.; Gutierrez-Barragan, D.; McCarthy, A.; Shaw, D.; et al. The M1/M4 Preferring Muscarinic Agonist Xanomeline Modulates Functional Connectivity and NMDAR Antagonist-Induced Changes in the Mouse Brain. Neuropsychopharmacology 2020. [Google Scholar] [CrossRef]
- Ridler, K.; Cunningham, V.; Huiban, M.; Martarello, L.; Pampols-Maso, S.; Passchier, J.; Gunn, R.N.; Searle, G.; Abi-Dargham, A.; Slifstein, M.; et al. An Evaluation of the Brain Distribution of [ 11 C]GSK1034702, a Muscarinic-1 (M 1) Positive Allosteric Modulator in the Living Human Brain Using Positron Emission Tomography. EJNMMI Res. 2014, 4, 66. [Google Scholar] [CrossRef][Green Version]
- Mogg, A.J.; Eessalu, T.; Johnson, M.; Wright, R.; Sanger, H.E.; Xiou, H.; Crabtree, M.; Smith, A.; Colvin, E.; Schober, D.; et al. In Vitro Pharmacological Characterization and in Vivo Validation of LSN3172176 a Novel M1 Selective Muscarinic Receptor Agonist Tracer Molecule for Positron Emission Tomography (PET). J. Pharmacol. Exp. Ther. 2018, 365, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.J.; Molloy, C.; Bundgaard, C.; Mogg, A.J.; Thompson, K.J.; Dwomoh, L.; Sanger, H.E.; Crabtree, M.D.; Brooke, S.M.; Sexton, P.M.; et al. Bitopic Binding Mode of an M1 Muscarinic Acetylcholine Receptor Agonist Associated with Adverse Clinical Trial Outcomes. Mol. Pharmacol. 2018, 93, 645–656. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Neumaier, F.; Rüngeler, T.; Drewes, B.; Kolks, N.; Neumaier, B. Preparation of a First 18F-Labeled Agonist for M1 Muscarinic Acetylcholine Receptors. Molecules 2020, 25, 2880. [Google Scholar] [CrossRef] [PubMed]
- Buiter, H.J.C.; Leysen, J.E.; Schuit, R.C.; Fisher, A.; Lammertsma, A.A.; Windhorst, A.D. Radiosynthesis and Biological Evaluation of the M1 Muscarinic Acetylcholine Receptor Agonist Ligand [11C]AF150(S). J. Label. Compd. Radiopharm. 2012, 55, 264–273. [Google Scholar] [CrossRef]
- Buiter, H.J.; Windhorst, A.D.; Huisman, M.C.; Yaqub, M.; Knol, D.L.; Fisher, A.; Lammertsma, A.A.; Leysen, J.E. [11C]AF150(S), an Agonist PET Ligand for M1 Muscarinic Acetylcholine Receptors. EJNMMI Res. 2013, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Malmquist, J.; Varnäs, K.; Svedberg, M.; Vallée, F.; Albert, J.S.; Finnema, S.J.; Schou, M. Discovery of a Novel Muscarinic Receptor PET Radioligand with Rapid Kinetics in the Monkey Brain. ACS Chem. Neurosci. 2017, 9, 224–229. [Google Scholar] [CrossRef]
- Nabulsi, N.B.; Holden, D.; Zheng, M.-Q.; Bois, F.; Lin, S.-F.; Najafzadeh, S.; Gao, H.; Ropchan, J.; Lara-Jaime, T.; Labaree, D.; et al. Evaluation of 11C-LSN3172176 as a Novel PET Tracer for Imaging M1 Muscarinic Acetylcholine Receptors in Nonhuman Primates. J. Nucl. Med. 2019, 60, 1147–1153. [Google Scholar] [CrossRef]
- Naganawa, M.; Nabulsi, N.B.; Henry, S.; Matuskey, D.; Lin, S.; Slieker, L.; Schwarz, A.J.; Kant, N.; Jesudason, C.; Ruley, K.; et al. First in Human Assessment of the Novel M1 Muscarinic Acetylcholine Receptor PET Radiotracer 11C-LSN3172176. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Wood, M.R.; Noetzel, M.J.; Melancon, B.J.; Poslusney, M.S.; Nance, K.D.; Hurtado, M.A.; Luscombe, V.B.; Weiner, R.L.; Rodriguez, A.L.; Lamsal, A.; et al. Discovery of VU0467485/AZ13713945: An M4 PAM Evaluated as a Preclinical Candidate for the Treatment of Schizophrenia. ACS Med. Chem. Lett. 2017, 8, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Hatori, A.; Chen, Z.; Kumata, K.; Shao, T.; Zhang, X.; Yamasaki, T.; Hu, K.; Yu, Q.; Ma, L.; et al. Synthesis and Preliminary Evaluation of 11C-Labeled VU0467485/AZ13713945 and Its Analogues for Imaging Muscarinic Acetylcholine Receptor Subtype 4. ChemMedChem 2019, 14, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Li, W.; Lo, M.M.-C.; Gao, X.; Wai, J.M.-C.; Rudd, M.; Tellers, D.; Joshi, A.; Zeng, Z.; Miller, P.; et al. Discovery of [11C]MK-6884: A Positron Emission Tomography (PET) Imaging Agent for the Study of M4Muscarinic Receptor Positive Allosteric Modulators (PAMs) in Neurodegenerative Diseases. J. Med. Chem. 2020, 63, 2411–2425. [Google Scholar] [CrossRef]
- Third International Symposium on Radiopharmaceutical Chemistry. J. Label. Compd. Radiopharm. 1981, 18, 79–158. [CrossRef]
- Vora, M.M.; Finn, R.D.; Boothe, T.E.; Liskwosky, D.R.; Potter, L.T. [N-Methyl-11C]-Scopolamine: Synthesis and Distribution in Rat Brain. J. Label. Compd. Radiopharm. 1983, 20, 1229–1236. [Google Scholar] [CrossRef]
- Dewey, S.L.; MacGregor, R.R.; Bendriem, B.; Fowler, J.S.; King, P.T.; Schlyer, D.J.; Wolf, A.P.; Volkow, N.D.; Christman, D.R.; Brodie, J.D.; et al. [11C]-Benztropine as a Possible Muscarinic Cholinergic Receptor Ligand for Use in PET. J. Nucl. Med. 1989, 30, 741–748. [Google Scholar]
- Mulholland, G.K.; Jewett, D.M.; Otto, C.A.; Kilbourn, M.R.; Sherman, P.S.; Koeppe, R.A.; Wieland, D.M.; Frey, K.A.; Kuhl, D.E. Synthesis and Preliminary Evaluation of [C-11]-(+)-2-Tropanyl Benzilate (C-11TRB) as a Iigand for the Muscarinic Receptor. J. Nucl. Med. 1988, 29, 932–940. [Google Scholar]
- Mulholland, G.K.; Jewett, D.M.; Otto, C.A.; Kilbourn, M.R.; Sherman, P.S.; Kuhl, D.E. Synthesis and Regional Brain Distribution of [C-11]N-Methyl-4- Piperidyl Benzilate ([C-11]NMPB) in the Rat. J. Nucl. Med. 1988, 29, 768. [Google Scholar]
- Halldin, C.; Müller, L.; Foged, C.; Sauerberg, P.; Karlsson, P.; Hall, H.; Farde, L. Preparation of Three Carbon-11 Labelled Novel Functional M1 Selective Muscarinic Agonists. J. Nucl. Med. 1993, 34, P7. [Google Scholar]
- Kiesewetter, D.O.; Lee, J.; Lang, L.; Park, S.G.; Paik, C.H.; Eckelman, W.C. Preparation of 18F-Labeled Muscarinic Agonist with M2 Selectivity. J. Med. Chem. 1995, 38, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Murakami, M.; Miura, S.; Iida, H.; Kanno, I.; Uemura, K. Synthesis and Autoradiographic Localization of Muscarinic Cholinergic Antagonist (+)N-[11C]Methyl-3-Piperidyl Benzilate as a Potent Radioligand for Positron Emission Tomography. Appl. Radiat. Isot. 1999, 50, 521–525. [Google Scholar] [CrossRef]
- Huiban, M.; Pampols-Maso, S.; Passchier, J. Fully Automated Synthesis of the M1 Receptor Agonist [11C]GSK1034702 for Clinical Use on an Eckert & Ziegler Modular Lab System. Appl. Radiat. Isot. 2011, 69, 1390–1394. [Google Scholar] [CrossRef]
- Nabulsi, N.; Holden, D.; Zheng, M.-Q.; Slieker, L.; Barth, V.; Lin, S.; Kant, N.; Jesudason, C.; Labaree, D.; Shirali, A. Evaluation of a Novel, Selective M1 Muscarinic Acetylcholine Receptor Ligand 11C-LSN3172176 in Non-Human Primates. J. Nucl. Med. 2017, 58, 275. [Google Scholar]
- Varastet, M.; Brouillet, E.; Chavoix, C.; Prenant, C.; Crouzel, C.; Stulzaft, O.; Bottlaender, M.; Cayla, J.; Mazière, B.; Mazière, M. In Vivo Visualization of Central Muscarinic Receptors Using [11C]Quinuclidinyl Benzilate and Positron Emission Tomography in Baboons. Eur. J. Pharmacol. 1992, 213, 275–284. [Google Scholar] [CrossRef]
- Masdeu, J.; Pascual, B.; Zanotti-Fregonara, P.; Yu, M.; Funk, Q.; Arbones, V.; Rockers, E.; Wang, Y.; Li, W.; Cheng, A.; et al. [11C]MK-6884 PET Tracer for M4 Muscarinic Cholinergic Receptors in Alzheimer’s Disease: Comparison with [18F]FDG PET (2640). Neurology 2020, 94, 2640. [Google Scholar]
- Zeeberg, B.R.; Gitler, M.S.; Baumgold, J.; de la Cruz, R.A.; Reba, R.C. Binding of Radioiodinated SPECT Ligands to Transfected Cell Membranes Expressing Single Muscarinic Receptor Subtypes. Biochem. Biophys. Res. Commun. 1991, 179, 768–775. [Google Scholar] [CrossRef]
- Stanton, T.; Bolden-Watson, C.; Cusack, B.; Richelson, E. Antagonism of the Five Cloned Human Muscarinic Cholinergic Receptors Expressed in CHO-K1 Cells by Antidepressants and Antihistaminics. Biochem. Pharmacol. 1993, 45, 2352–2354. [Google Scholar] [CrossRef]
- Zheng, G.; Smith, A.M.; Huang, X.; Subramanian, K.L.; Siripurapu, K.B.; Deaciuc, A.; Zhan, C.-G.; Dwoskin, L.P. Structural Modifications to Tetrahydropyridine-3-Carboxylate Esters En Route to the Discovery of M5-Preferring Muscarinic Receptor Orthosteric Antagonists. J. Med. Chem. 2013, 56, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.Z.; Kameyama, K.; Rinken, A.; Haga, T. Ligand Binding Properties of Muscarinic Acetylcholine Receptor Subtypes (M1–M5) Expressed in Baculovirus-Infected Insect Cells. J. Pharmacol. Exp. Ther. 1995, 274, 378–384. [Google Scholar]
- Otto, C.A.; Mulholland, G.K.; Perry, S.E.; Combs, R.; Sherman, P.S.; Fisher, S.J. In Vitro and Ex Vivo Evaluation of Cyclic Aminoalkyl Benzilates as Potential Emission Tomography Ligands for the Muscarinic Receptor. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1989, 16, 51–55. [Google Scholar] [CrossRef]
- Kloog, Y.; Egozi, Y.; Sokolovsky, M. Characterization of Muscarinic Acetylcholine Receptors from Mouse Brain: Evidence for Regional Heterogeneity and Isomerization. Mol. Pharmacol. 1979, 15, 545–558. [Google Scholar]
- Wilson, A.A.; Dannals, R.F.; Ravert, H.T.; Frost, J.J.; Wagner, H.N. Synthesis and Biological Evaluation of Iodine-125- and Iodine-123-4-Iododexetimide, a Potent Muscarinic Cholinergic Receptor Antagonist. J. Med. Chem. 1989, 32, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, A.; Yano, H.; Del Bello, F.; Farande, A.; Quaglia, W.; Petrelli, R.; Matucci, R.; Nesi, M.; Vistoli, G.; Ferré, S.; et al. Synthesis and Biological Evaluation of a Novel Series of Heterobivalent Muscarinic Ligands Based on Xanomeline and 1-[3-(4-Butylpiperidin-1-Yl)Propyl]-1,2,3,4-Tetrahydroquinolin-2-One (77-LH-28-1). J. Med. Chem. 2014, 57, 9065–9077. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, H.; Nishiyama, S.; Takahashi, K. Imaging of Muscarinic Receptors in the Central Nervous System. In Muscarinic Receptor: From Structure to Animal Models; Myslivecek, J., Jakubik, J., Eds.; Neuromethods; Springer: New York, NY, USA, 2016; Volume 107, pp. 181–203. ISBN 978-1-4939-2857-6. [Google Scholar]
- Fisher, A.; Heldman, E.; Gurwitz, D.; Haring, R.; Meshulam, H.; Brandeis, R.; Pittel, Z.; Marciano, D.; Sapir, M.; Barak, D.; et al. New M1 Agonists: Selective Signaling, Neurotrophic-Like and Cognitive Effects—Implications in the Treatment of Alzheimer’s Disease. In Alzheimer’s and Parkinson’s Diseases. Advances in Behavioral Biology; Hanin, I., Yoshida, M., Fisher, A., Eds.; Springer: Boston, MA, USA, 1995; Volume 44, pp. 449–455. ISBN 978-1-4757-9147-1. [Google Scholar]
- Caldwell, J.H.; Link, J.M. Imaging Left Ventricular Muscarinic Receptor Heterogeneity: A Tool to Evaluate Individuals at Risk for Sudden Death? Circ. Cardiovasc. Imaging 2009, 2, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Delforge, J.; Le Guludec, D.; Syrota, A.; Bendriem, B.; Crouzel, C.; Slama, M.; Merlet, P. Quantification of Myocardial Muscarinic Receptors with PET in Humans. J. Nucl. Med. 1993, 34, 981–991. [Google Scholar] [PubMed]
- Cselényi, Z.; Jucaite, A.; Kristensson, C.; Stenkrona, P.; Ewing, P.; Varrone, A.; Johnström, P.; Schou, M.; Vazquez-Romero, A.; Moein, M.M.; et al. Quantification and Reliability of [11C]VC-002 Binding to Muscarinic Acetylcholine Receptors in the Human Lung—A Test-Retest PET Study in Control Subjects. EJNMMI Res. 2020, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. Whole-Body PET Scanner Produces 3D Images in Seconds. Nature 2019, 570, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Khurana, S.; Cheng, K.; Raufman, J.-P. Muscarinic Receptors and Ligands in Cancer. Am. J. Physiol. Cell Physiol. 2009, 296, C221–C232. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, J.; Yao, L.; Lang, Y.; Liang, Y.; Chen, L.; Zhang, J.; Wang, F.; Wang, Y.; Chen, H.; et al. High Expression of M3 Muscarinic Acetylcholine Receptor Is a Novel Biomarker of Poor Prognostic in Patients with Non-Small Cell Lung Cancer. Tumour Biol. 2013, 34, 3939–3944. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.-Q.; Xu, L.-H.; Zhang, M.; Xu, C. Muscarinic Acetylcholine Receptor M1 Mediates Prostate Cancer Cell Migration and Invasion through Hedgehog Signaling. Asian J. Androl. 2018, 20, 608–614. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
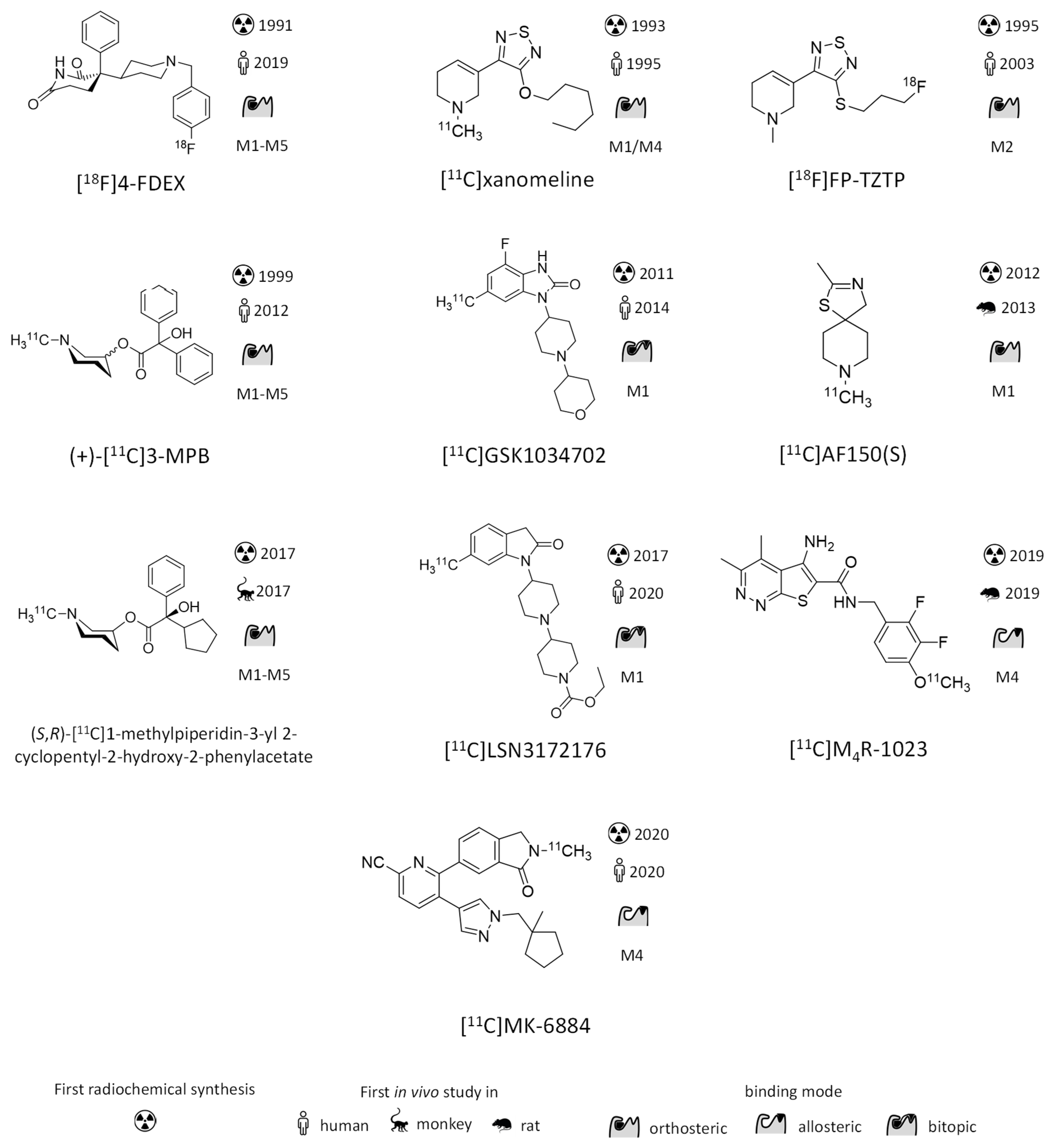{kind=link}
| Design Criteria | Test Criteria |
|---|---|
|
|
| Compound | Binding Site | M1 | M2 | M3 | M4 | M5 | lit. |
|---|---|---|---|---|---|---|---|
| Scopolamine | orthosteric | 1.1 | 2.0 | 0.44 | 0.8 | 2.07 | [55] |
| BQCA | allosteric | 845 | >100,000 | >100,000 | >100,000 | >100,000 | [59] |
| Trivial Name | Systematic Name | M1 | M2 | M3 | M4 | M5 | Method |
|---|---|---|---|---|---|---|---|
| (R,S)-IQNB | (R)-quinuclidin-3-yl (S)-2-hydroxy-2-(4-iodophenyl)-2-phenylacetate | 0.49 | - | 1.27 | - | - | KD on transfected A9 L cell membranes using (R,S)-[125I]IQNB [112] |
| QNB | quinuclidin-3-yl 2-hydroxy-2,2-diphenylacetate | 0.044 | 0.030 | 0.080 | 0.037 | 0.065 | KD on transfected CHO-K1 cell membranes using [3H]QNB [113] |
| scopolamine | (1R,2R,4S,5S,7s)-9-Methyl-3-oxa-9-azatricyclo[3.3.1.02,4]non-7-yl (2S)-3-hydroxy-2-phenylpropanoate | 7.5 | 9.5 | 6.5 | 36.9 | 17.6 | Ki on transfected CHO-K1 cell membranes using [3H]NMS [114] |
| benztropine | (1R,3r,5S)-3-(benzhydryloxy)-8-methyl-8-azabicyclo[3.2.1]octane | 6.8 | 14.1 | 11.2 | 22.9 | 4.6 | Ki on transfected Sf9 cell membranes using [3H]NMS [115] |
| TRB | (1R,3r,5S)-8-methyl-8-azabicyclo[3.2.1]octan-3-yl 2-hydroxy-2,2-diphenylacetate | 0.7, subtypes were not discriminated | IC50 by [3H]QNB competitive binding on mouse brain homogenates [116] | ||||
| NMPB | 1-methylpiperidin-4-yl 2-hydroxy-2,2-diphenylacetate | 0.41, subtypes were not discriminated | KD on mouse cortex [117] | ||||
| 4-FDEX | (S)-1’-(4-fluorobenzyl)-3-phenyl-[3,4’-bipiperidine]-2,6-dione | 98, subtypes were not discriminated | IC50 by [3H]NMS competitive binding on rat brain homogenates [118] | ||||
| xanomeline | 3-[4-(hexyloxy)-1,2,5-thiadiazol-3-yl]-1,2,5,6-tetrahydro-1-methylpyridine oxalate | 7.9 | 8.1 | 7.8 | 11.2 | 9.3 | Ki on transfected CHO-K1 cell membranes using [3H]NMS [119] |
| FP-TZTP | 3-(3-fluoropropylsulfanyl)-4-(1-methyl-3,6-dihydro-2H-pyridin-5-yl)-1,2,5-thiadiazole | 7.4 | 2.2 | 79.7 | - | - | Ki on different tissues with different radioligands [106] |
| (+)-3-MPB | 1-methylpiperidin-3-yl 2-hydroxy-2,2-diphenylacetate | 1.7 no significant selectivity | Ki on rat neocortex with [3H]QNB. No significant subtype selectivity was observed on transfected CHO-K1 cell membranes using a direct radioligand binding assay [120]. | ||||
| GSK1034702 | 4-fluoro-6-methyl-1-(1-(tetrahydro-2H-pyran-4-yl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one | 7.9 | >790 | >790 | >790 | >790 | EC50 of FLIPR assay of stably transfected CHO cells [88] |
| AF150(S) | 2-methyl-8-methyl-1-thia-3,8-diazaspiro[4.5]dec-2-ene | 390 | 22,000 | - | - | - | Ki on rat cerebral cortex using [3H]pirenzepine (M1) or rat cerebellum using [3H]QNB (M2) [121]. |
| - | (S,R)-1-methylpiperidin-3-yl 2-cyclopentyl-2-hydroxy-2-phenylacetate | 3.5 | - | - | - | - | Ki of “high affinity human mAChR M1 assay”. A degree of M1 selectivity was evident from partial blocking of the radioligand with pirenzepine in autoradiography using human brain slices [94]. |
| LSN3172176 | ethyl 4-(6-methyl-2-oxoindolin-1-yl)-[1,4’-bipiperidine]-1’-carboxylate | 8.9 | 63.8 | 3031 | 41.4 | 55.6 | Ki on transfected CHO-K1 cell membranes using [3H]NMS [89]. |
| M4R-1023 | 5-amino-N-(2,3-difluoro-4-methoxybenzyl)-3,4-dimethylthieno[2,3-c]pyridazine-6-carboxamide | 43.4 | >104 | >104 | >104 | >104 | EC50 of calcium release assay on stably transfected CHO cells [97]. |
| MK-6884 | 6-(2-methyl-3-oxoisoindolin-5-yl)-5-(1-((1-methylcyclopentyl)methyl)-1H-pyrazol-4-yl)picolinonitrile | - | - | - | 0.19 | - | Ki on transfected CHO-K1 cell membranes using a tritiated compound of similar chemotype [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozenil, M.; Aronow, J.; Millard, M.; Langer, T.; Wadsak, W.; Hacker, M.; Pichler, V. Update on PET Tracer Development for Muscarinic Acetylcholine Receptors. Pharmaceuticals 2021, 14, 530. https://doi.org/10.3390/ph14060530
Ozenil M, Aronow J, Millard M, Langer T, Wadsak W, Hacker M, Pichler V. Update on PET Tracer Development for Muscarinic Acetylcholine Receptors. Pharmaceuticals. 2021; 14(6):530. https://doi.org/10.3390/ph14060530
Chicago/Turabian StyleOzenil, Marius, Jonas Aronow, Marlon Millard, Thierry Langer, Wolfgang Wadsak, Marcus Hacker, and Verena Pichler. 2021. "Update on PET Tracer Development for Muscarinic Acetylcholine Receptors" Pharmaceuticals 14, no. 6: 530. https://doi.org/10.3390/ph14060530
APA StyleOzenil, M., Aronow, J., Millard, M., Langer, T., Wadsak, W., Hacker, M., & Pichler, V. (2021). Update on PET Tracer Development for Muscarinic Acetylcholine Receptors. Pharmaceuticals, 14(6), 530. https://doi.org/10.3390/ph14060530