
Ultramicronized Palmitoylethanolamide (um-PEA): A New Possible Adjuvant Treatment in COVID-19 patients

 , , , , , ,
, , , , , ,  and
and 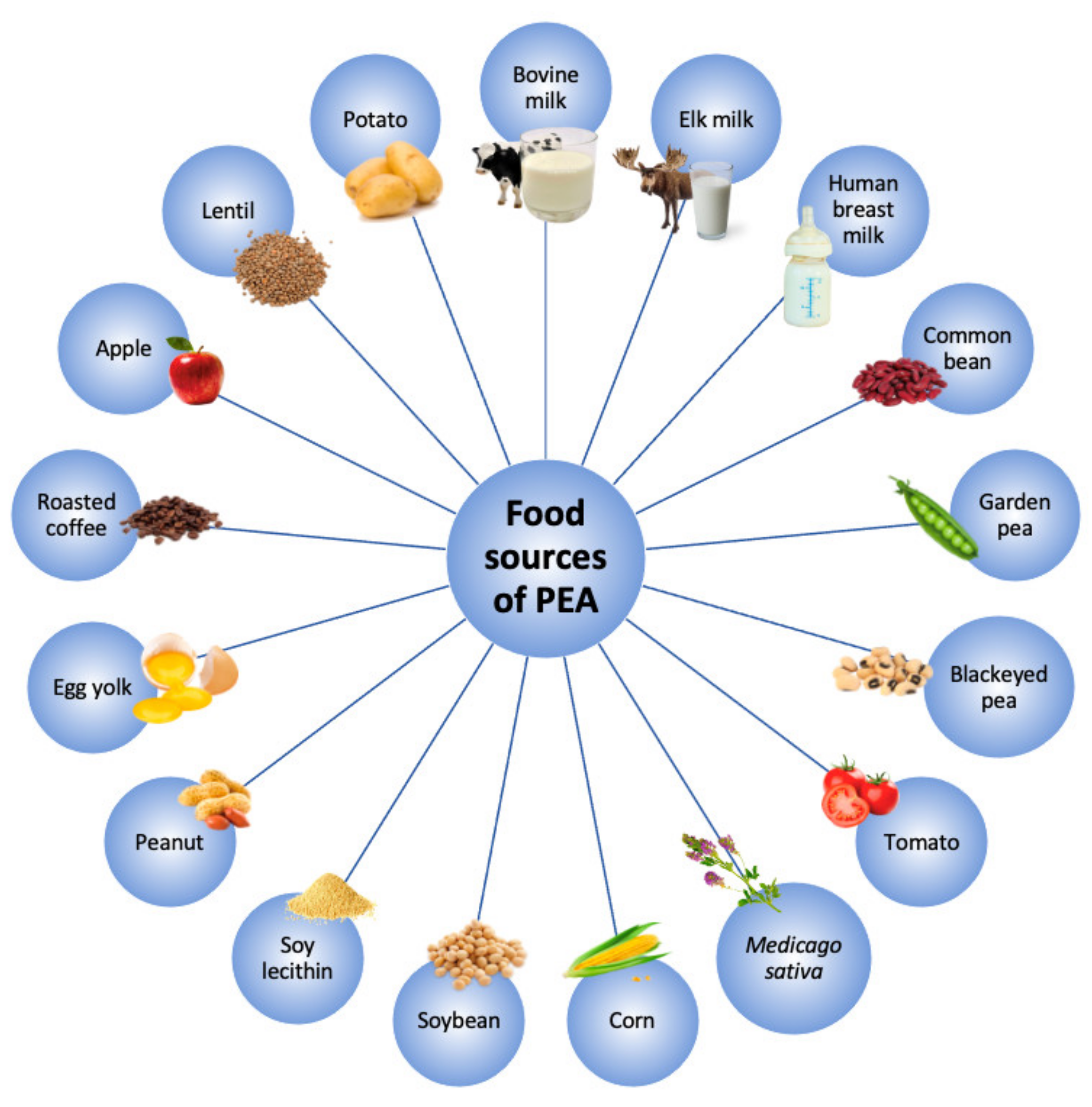{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
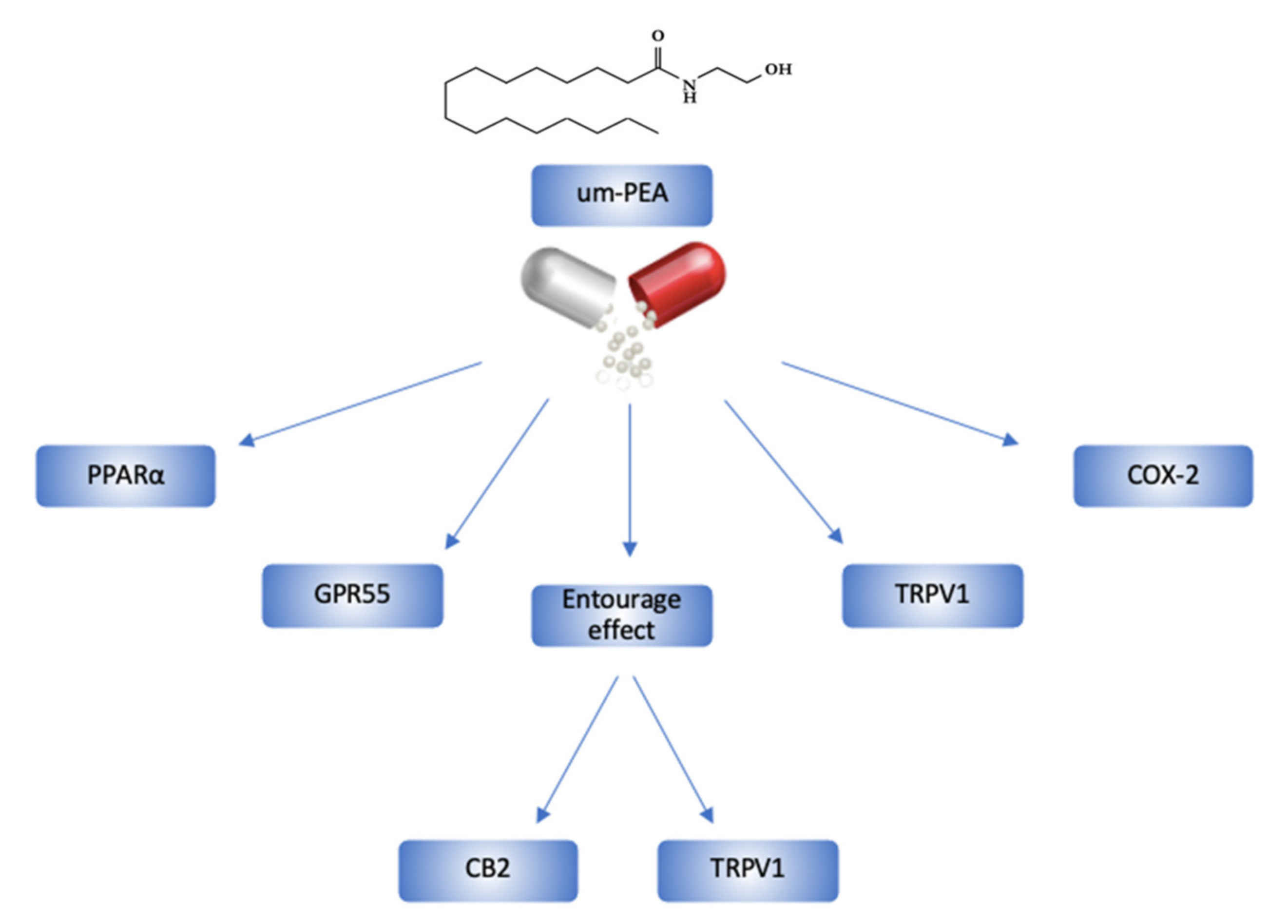
2. Ultramicronized-PEA and its Mechanisms of Action
3. Pathogenic Mechanisms of SARS-CoV-2
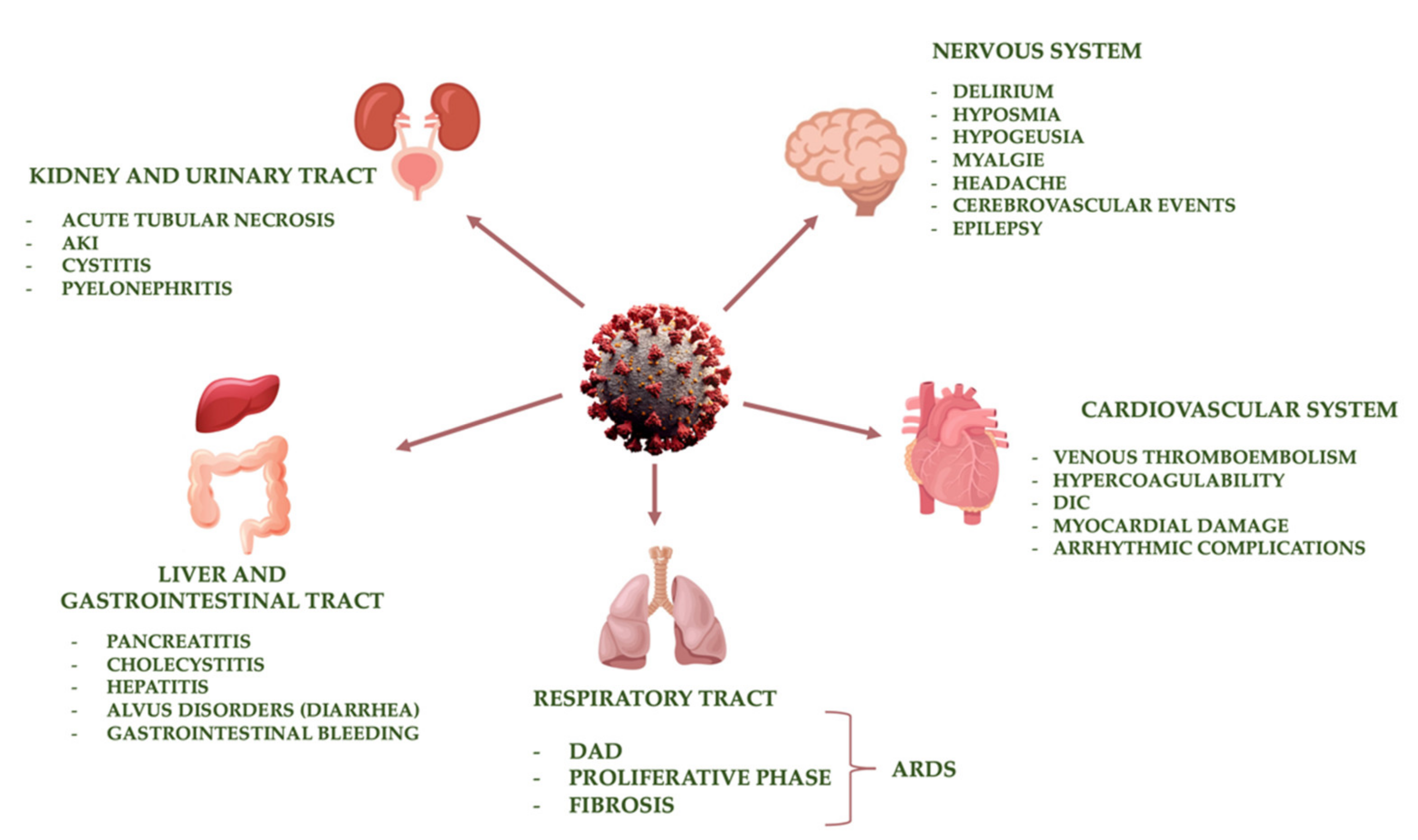
4. Main Organ Damage Induced by SARS-CoV-2
4.1. Respiratory Tract
4.2. Cardiovascular System
4.3. Kidney and Urinary Tract
4.4. Liver and Gastrointestinal Tract
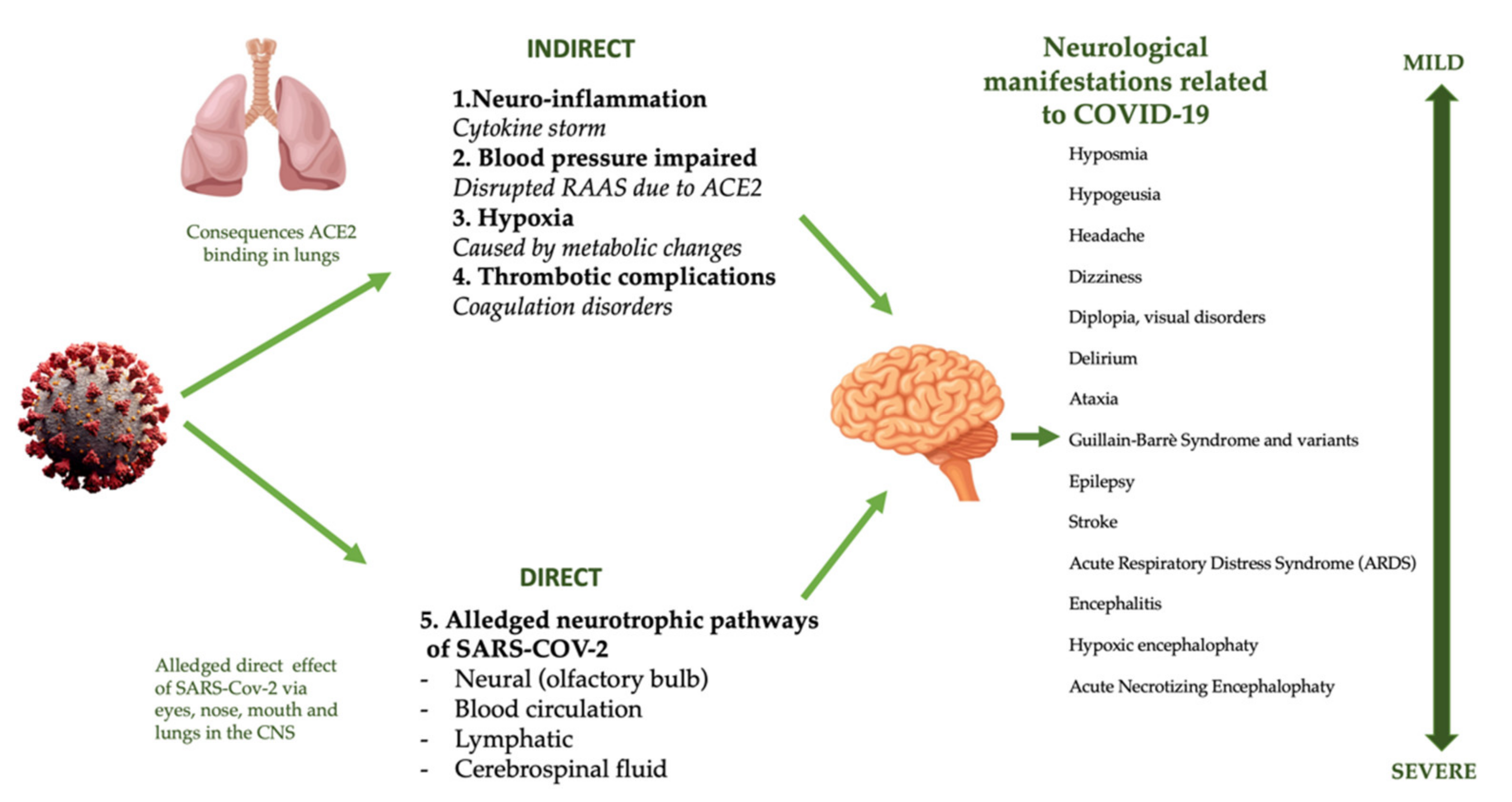
4.5. Nervous System
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations List
| ACE2 | Angiotensin-Converting Enzyme |
| AKI | Acute Kidney Injury |
| AKP | Alkaline Phosphating |
| ALT | Alanine Aminotransferase |
| ARDS | Acute Respiratory Distress Syndrome |
| AST | Aspartate Aminotransferase |
| CCL2 | Chemokine Ligand 2 |
| CGRP | Calcitonin Gene-Related Peptide |
| CNS | Central Nervous System |
| COVID-19 | Coronavirus Disease-19 |
| COX-2 | Cyclooxygenase-2 |
| CSF | Fluid Cerebrospinal Analysis |
| CTSI | Catepsine-1 |
| CXCL10 | C-X-C motif ligand (CXCL)10 |
| DAD | Diffuse Alveolar Damage |
| DIC | Disseminated Intravascular Coagulation |
| FDA | Food and Drug Administration Agency |
| GGT | Gamma-Glutamyl Transferase |
| GM-CSF | Granulocyte-Macrophage Colony Stimulating Factor |
| GPR55 | G-Protein-Coupled Receptors 55 |
| HEV | Porcine Hemagglutinating Encephalomyelitis |
| HIF | Hypoxia-Inducible Factor |
| IKCa | High Conductance Potassium Channels |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| MERS-CoV | Middle East Respiratory Syndrome Coronavirus Infection |
| MOF | Multiorgan Failure |
| NBC | Natural Bioactive Compound |
| Nf-kβ | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| PEA | Palmitoylethanolamide |
| PG | Prostaglandin |
| PPARα | Peroxisome Proliferator-Activated Receptor α |
| PT | Prothrombin Time |
| RT-PCR | Reverse Transcriptase Polymerase Chain Reaction |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus-2 |
| TLRs | Toll-Like Receptors |
| TMPRSS2 | Transmembrane Serine Protease 2 |
| TNF-α | Tumor Necrosis Factor-Α |
| TRP | Transient Receptor Potential |
| um-PEA | Ultramicronized Palmitoylethanolamide |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Noce, A.; Santoro, M.L.; Marrone, G.; D’Agostini, C.; Amelio, I.; Duggento, A.; Tesauro, M.; di Daniele, N. Serological determinants of COVID-19. Biol. Direct 2020, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- WHO. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019?gclid=CjwKCAiAgJWABhArEi-wAmNVTB5aM0iEjG_rAywBeJGuiMooqqGeEMk1ecaYH6Jmy8LNLAZTQJL5U9RoCS2gQAvD_BwE (accessed on 30 January 2021).
- Fu, L.; Wang, B.; Yuan, T.; Chen, X.; Ao, Y.; Fitzpatrick, T.; Li, P.; Zhou, Y.; Lin, Y.F.; Duan, Q.; et al. Clinical characteristics of coronavirus disease 2019 (COVID-19) in China: A systematic review and meta-analysis. J. Infect. 2020, 80, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, O.; Virani, A.; Cheema, T. COVID-19: An update on the epidemiological, clinical, preventive, and therapeutic management of 2019 novel coronavirus disease. Crit. Care Nurs. Q. 2021, 44, 128–137. [Google Scholar] [CrossRef]
- Lai, C.C.; Ko, W.C.; Lee, P.I.; Jean, S.S.; Hsueh, P.R. Extra-respiratory manifestations of COVID-19. Int. J. Antimicrob. Agents 2020, 56, 106024. [Google Scholar] [CrossRef]
- Tahir, F.; Arif, T.B.; Ahmed, J.; Malik, F.; Khalid, M. Cardiac manifestations of coronavirus disease 2019 (COVID-19): A comprehensive review. Cureus 2020, 12, e8021. [Google Scholar] [CrossRef]
- Rokkas, T. Gastrointestinal involvement in COVID-19: A systematic review and meta-analysis. Ann. Gastroenterol. 2020, 33, 355–365. [Google Scholar] [CrossRef]
- Cheong, J.; Bartell, N.; Peeraphatdit, T.; Mosli, M.; Al-Judaibi, B. Gastrointestinal and liver manifestations of COVID-19. Saudi J. Gastroenterol. 2020, 26, 226–232. [Google Scholar] [CrossRef]
- Daneshgaran, G.; Dubin, D.P.; Gould, D.J. Cutaneous manifestations of COVID-19: An evidence-based review. Am. J. Clin. Dermatol. 2020, 21, 627–639. [Google Scholar] [CrossRef]
- Vaira, L.A.; Salzano, G.; Deiana, G.; de Riu, G. Anosmia and ageusia: Common findings in COVID-19 patients. Laryngoscope 2020, 130, 1787. [Google Scholar] [CrossRef]
- Ahmad, I.; Rathore, F.A. Neurological manifestations and complications of COVID-19: A literature review. J. Clin. Neurosci. 2020, 77, 8–12. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef]
- Kolhe, N.V.; Fluck, R.J.; Selby, N.M.; Taal, M.W. Acute kidney injury associated with COVID-19: A retrospective cohort study. PLoS Med. 2020, 17, e1003406. [Google Scholar] [CrossRef]
- Esakandari, H.; Nabi-Afjadi, M.; Fakkari-Afjadi, J.; Farahmandian, N.; Miresmaeili, S.M.; Bahreini, E. A comprehensive review of COVID-19 characteristics. Biol. Proc. Online 2020, 22, 19. [Google Scholar] [CrossRef]
- Petrosino, S.; Iuvone, T.; di Marzo, V. N-palmitoyl-ethanolamine: Biochemistry and new therapeutic opportunities. Biochimie 2010, 92, 724–727. [Google Scholar] [CrossRef]
- Petrosino, S.; di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2017, 174, 1349–1365. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; la Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Gabrielsson, L.; Mattsson, S.; Fowler, C.J. Palmitoylethanolamide for the treatment of pain: Pharmacokinetics, safety and efficacy. Br. J. Clin. Pharmacol. 2016, 82, 932–942. [Google Scholar] [CrossRef]
- Gatti, A.; Lazzari, M.; Gianfelice, V.; di Paolo, A.; Sabato, E.; Sabato, A.F. Palmitoylethanolamide in the treatment of chronic pain caused by different etiopathogenesis. Pain Med. 2012, 13, 1121–1130. [Google Scholar] [CrossRef]
- Kuehl, F.A.; Jacob, T.A.; Ganley, O.H.; Ormond, R.E.; Meisinger, M.A.P. The identification of n-(2-hydroxyethyl)-palmitamide as a naturally occurring anti-inflammatory agent. J. Am. Chem. Soc. 1957, 79, 5577–5578. [Google Scholar] [CrossRef]
- Paladini, A.; Fusco, M.; Cenacchi, T.; Schievano, C.; Piroli, A.; Varrassi, G. Palmitoylethanolamide, a special food for medical purposes, in the treatment of chronic pain: A pooled data meta-analysis. Pain Physician 2016, 19, 11–24. [Google Scholar]
- Cordaro, M.; Cuzzocrea, S.; Crupi, R. An update of palmitoylethanolamide and luteolin effects in preclinical and clinical studies of neuroinflammatory events. Antioxidants (Basel) 2020, 9, 216. [Google Scholar] [CrossRef]
- Kahlich, R.; Klima, J.; Cihla, F.; Frankova, V.; Masek, K.; Rosicky, M.; Matousek, F.; Bruthans, J. Studies on prophylactic efficacy of N-2-hydroxyethyl palmitamide (Impulsin) in acute respiratory infections. Serologically controlled field trials. J. Hyg. Epidemiol. Microbiol. Immunol. 1979, 23, 11–24. [Google Scholar]
- Hesselink, J.M.K.; de Boer, T.; Witkamp, R.F. Palmitoylethanolamide: A natural body-own anti-inflammatory agent, effective and safe against influenza and common cold. Int. J. Inflam. 2013, 2013, 151028. [Google Scholar] [CrossRef]
- Vacondio, F.; Bassi, M.; Silva, C.; Castelli, R.; Carmi, C.; Scalvini, L.; Lodola, A.; Vivo, V.; Flammini, L.; Barocelli, E.; et al. Amino acid derivatives as palmitoylethanolamide prodrugs: Synthesis, in vitro metabolism and in vivo plasma profile in rats. PLoS ONE 2015, 10, e0128699. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Crupi, R.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Micronized/ultramicronized palmitoylethanolamide displays superior oral efficacy compared to nonmicronized palmitoylethanolamide in a rat model of inflammatory pain. J. Neuroinflamm. 2014, 11, 136. [Google Scholar] [CrossRef]
- Petrosino, S.; Schiano Moriello, A.; Cerrato, S.; Fusco, M.; Puigdemont, A.; de Petrocellis, L.; di Marzo, V. The anti-inflammatory mediator palmitoylethanolamide enhances the levels of 2-arachidonoyl-glycerol and potentiates its actions at TRPV1 cation channels. Br. J. Pharmacol. 2016, 173, 1154–1162. [Google Scholar] [CrossRef]
- Esposito, E.; Paterniti, I.; Mazzon, E.; Genovese, T.; di Paola, R.; Galuppo, M.; Cuzzocrea, S. Effects of palmitoylethanolamide on release of mast cell peptidases and neurotrophic factors after spinal cord injury. Brain Behav. Immun. 2011, 25, 1099–1112. [Google Scholar] [CrossRef]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2015, 6, 620. [Google Scholar] [CrossRef]
- Hu, Y.; Jin, Y.; Han, D.; Zhang, G.; Cao, S.; Xie, J.; Xue, J.; Li, Y.; Meng, D.; Fan, X.; et al. Mast cell-induced lung injury in mice infected with H5N1 influenza virus. J. Virol. 2012, 86, 3347–3356. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R.; Skaper, S.D.; dal Toso, R.; Petrelli, L.; Leon, A. Nerve growth factor: From neurotrophin to neurokine. Trends Neurosci. 1996, 19, 514–520. [Google Scholar] [CrossRef]
- Gigante, A.; Aquili, A.; Farinelli, L.; Caraffa, A.; Ronconi, G.; Gallenga, C.E.; Tete, G.; Kritas, S.K.; Conti, P. Sodium chromo-glycate and palmitoylethanolamide: A possible strategy to treat mast cell-induced lung inflammation in COVID-19. Med. Hypotheses 2020, 143, 109856. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Selvakumar, G.P.; Ahmed, M.E.; Raikwar, S.P.; Thangavel, R.; Khan, A.; Zaheer, S.A.; Iyer, S.S.; Burton, C.; James, D.; et al. COVID-19, Mast Cells, Cytokine Storm, Psychological Stress, and Neuroinflammation. Neuroscientist 2020, 26, 402–414. [Google Scholar] [CrossRef]
- Kritas, S.K.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Conti, P. Mast cells contribute to coronavirus-induced inflammation: New anti-inflammatory strategy. J. Biol. Regul. Homeost. Agents 2020, 34, 9–14. [Google Scholar] [CrossRef]
- Magadum, A.; Engel, F.B. PPARbeta/delta: Linking Metabolism to Regeneration. Int. J. Mol. Sci. 2018, 19, 2013. [Google Scholar] [CrossRef]
- Mazzari, S.; Canella, R.; Petrelli, L.; Marcolongo, G.; Leon, A. N-(2-hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol. 1996, 300, 227–236. [Google Scholar] [CrossRef]
- Roviezzo, F.; Rossi, A.; Caiazzo, E.; Orlando, P.; Riemma, M.A.; Iacono, V.M.; Guarino, A.; Ialenti, A.; Cicala, C.; Peritore, A.; et al. Palmitoylethanolamide Supplementation during Sensitization Prevents Airway Allergic Symptoms in the Mouse. Front. Pharmacol. 2017, 8, 857. [Google Scholar] [CrossRef]
- Del Giorno, R.; Skaper, S.; Paladini, A.; Varrassi, G.; Coaccioli, S. Palmitoylethanolamide in Fibromyalgia: Results from Prospective and Retrospective Observational Studies. Pain Ther. 2015, 4, 169–178. [Google Scholar] [CrossRef]
- Fusco, R.; Cordaro, M.; Genovese, T.; Impellizzeri, D.; Siracusa, R.; Gugliandolo, E.; Peritore, A.F.; D’Amico, R.; Crupi, R.; Cuzzocrea, S.; et al. Adelmidrol: A New Promising Antioxidant and Anti-Inflammatory Therapeutic Tool in Pulmonary Fibrosis. Antioxidants (Basel) 2020, 9, 601. [Google Scholar] [CrossRef]
- Fusco, M.; Skaper, S.D.; Coaccioli, S.; Varrassi, G.; Paladini, A. Degenerative Joint Diseases and Neuroinflammation. Pain Pract. 2017, 17, 522–532. [Google Scholar] [CrossRef]
- Iannotti, F.A.; di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef]
- Romero, T.R.; Duarte, I.D. N-palmitoyl-ethanolamine (PEA) induces peripheral antinociceptive effect by ATP-sensitive K+-channel activation. J. Pharmacol. Sci. 2012, 118, 156–160. [Google Scholar] [CrossRef]
- Lowin, T.; Apitz, M.; Anders, S.; Straub, R.H. Anti-inflammatory effects of N-acylethanolamines in rheumatoid arthritis synovial cells are mediated by TRPV1 and TRPA1 in a COX-2 dependent manner. Arthritis Res. Ther. 2015, 17, 321. [Google Scholar] [CrossRef]
- D’Agostino, G.; la Rana, G.; Russo, R.; Sasso, O.; Iacono, A.; Esposito, E.; Raso, G.M.; Cuzzocrea, S.; Loverme, J.; Piomelli, D.; et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF-kappaB nuclear signalling in dorsal root ganglia. Eur. J. Pharmacol. 2009, 613, 54–59. [Google Scholar] [CrossRef]
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? Br. J. Pharmacol. 2007, 152, 984–986. [Google Scholar] [CrossRef]
- Sasso, O.; la Rana, G.; Vitiello, S.; Russo, R.; D’Agostino, G.; Iacono, A.; Russo, E.; Citraro, R.; Cuzzocrea, S.; Piazza, P.V.; et al. Palmitoylethanolamide modulates pentobarbital-evoked hypnotic effect in mice: Involvement of allopregnanolone biosynthesis. Eur. Neuropsychopharmacol. 2010, 20, 195–206. [Google Scholar] [CrossRef]
- Artukoglu, B.B.; Beyer, C.; Zuloff-Shani, A.; Brener, E.; Bloch, M.H. Efficacy of Palmitoylethanolamide for Pain: A Meta-Analysis. Pain Physician 2017, 20, 353–362. [Google Scholar]
- Richardson, J.D.; Vasko, M.R. Cellular mechanisms of neurogenic inflammation. J. Pharmacol. Exp. Ther. 2002, 302, 839–845. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Davis, J.B.; di Marzo, V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001, 506, 253–256. [Google Scholar] [CrossRef]
- Ambrosino, P.; Soldovieri, M.V.; Russo, C.; Taglialatela, M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARalpha agonist palmitoylethanolamide. Br. J. Pharmacol. 2013, 168, 1430–1444. [Google Scholar] [CrossRef]
- Rankin, L.; Fowler, C.J. The Basal Pharmacology of Palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef]
- Sharir, H.; Console-Bram, L.; Mundy, C.; Popoff, S.N.; Kapur, A.; Abood, M.E. The endocannabinoids anandamide and virodhamine modulate the activity of the candidate cannabinoid receptor GPR55. J. Neuroimmune Pharmacol. 2012, 7, 856–865. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Karamloo, F.; Konig, R. SARS-CoV-2 immunogenicity at the crossroads. Allergy 2020, 75, 1822–1824. [Google Scholar] [CrossRef]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut 2014, 63, 1300–1312. [Google Scholar] [CrossRef]
- Pesce, M.; Seguella, L.; Cassarano, S.; Aurino, L.; Sanseverino, W.; Lu, J.; Corpetti, C.; del Re, A.; Vincenzi, M.; Sarnelli, G.; et al. Phytotherapics in COVID19: Why palmitoylethanolamide? Phytother. Res. 2020, 1–9. [Google Scholar] [CrossRef]
- Schonrich, G.; Raftery, M.J.; Samstag, Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar] [CrossRef]
- Wong, S.H.; Lui, R.N.; Sung, J.J. Covid-19 and the digestive system. J. Gastroenterol. Hepatol. 2020, 35, 744–748. [Google Scholar] [CrossRef]
- Alomari, S.O.; Abou-Mrad, Z.; Bydon, A. COVID-19 and the central nervous system. Clin. Neurol. Neurosurg. 2020, 198, 106116. [Google Scholar] [CrossRef] [PubMed]
- Couch, D.G.; Cook, H.; Ortori, C.; Barrett, D.; Lund, J.N.; O’Sullivan, S.E. Palmitoylethanolamide and Cannabidiol Prevent Inflammation-induced Hyperpermeability of the Human Gut In Vitro and In Vivo—A Randomized, Placebo-controlled, Double-blind Controlled Trial. Inflamm. Bowel Dis. 2019, 25, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Raso, G.M.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS health and disease. Pharmacol. Res. 2014, 86, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Raso, G.M.; Esposito, E.; Vitiello, S.; Iacono, A.; Santoro, A.; D’Agostino, G.; Sasso, O.; Russo, R.; Piazza, P.V.; Calignano, A.; et al. Palmitoylethanolamide stimulation induces allopregnanolone synthesis in C6 Cells and primary astrocytes: Involvement of peroxisome-proliferator activated receptor-alpha. J. Neuroendocrinol. 2011, 23, 591–600. [Google Scholar] [CrossRef]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L., Jr.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxid Med. Cell Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; de Bosscher, K. Molecular Actions of PPARalpha in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Steardo, L.; Pompili, E.; Fumagalli, L.; Campolongo, P.; et al. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1419. [Google Scholar] [CrossRef]
- Grippo, A.J.; Scotti, M.A. Stress and neuroinflammation. Mod. Trends Pharm. 2013, 28, 20–32. [Google Scholar] [CrossRef]
- Wojtowicz, S.; Strosznajder, A.K.; Jezyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease 2019 (COVID-19) Situation Report. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200513-covid-19-sitrep-114.pdf?sfvrsn=17ebbbe_4 (accessed on 30 January 2021).
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Gubernatorova, E.O.; Gorshkova, E.A.; Polinova, A.I.; Drutskaya, M.S. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020, 53, 13–24. [Google Scholar] [CrossRef]
- Jacob, C.O. On the genetics and immunopathogenesis of COVID-19. Clin. Immunol. 2020, 220, 108591. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb Haemost. 2020, 18, 2103–2109. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Lippi, G. Which lessons shall we learn from the 2019 novel coronavirus outbreak? Ann. Transl. Med. 2020, 8, 48. [Google Scholar] [CrossRef]
- Renu, K.; Prasanna, P.L.; Gopalakrishnan, A.V. Coronaviruses pathogenesis, comorbidities and multi-organ damage—A review. Life Sci. 2020, 255, 117839. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.I.; Maiolo, C.; Iacopino, L.; Pepe, M.; di Daniele, N.; de Lorenzo, A. The impact of body-weight components on forced spirometry in healthy italians. Lung 2002, 180, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.Y.F.; Lam, H.Y.S.; Fong, A.H.; Leung, S.T.; Chin, T.W.; Lo, C.S.Y.; Lui, M.M.; Lee, J.C.Y.; Chiu, K.W.; Chung, T.W.; et al. Frequency and Distribution of Chest Radiographic Findings in Patients Positive for COVID-19. Radiology 2020, 296, E72–E78. [Google Scholar] [CrossRef]
- Cheung, O.Y.; Chan, J.W.; Ng, C.K.; Koo, C.K. The spectrum of pathological changes in severe acute respiratory syndrome (SARS). Histopathology 2004, 45, 119–124. [Google Scholar] [CrossRef]
- Franks, T.J.; Chong, P.Y.; Chui, P.; Galvin, J.R.; Lourens, R.M.; Reid, A.H.; Selbs, E.; McEvoy, C.P.; Hayden, C.D.; Fukuoka, J.; et al. Lung pathology of severe acute respiratory syndrome (SARS): A study of 8 autopsy cases from Singapore. Hum. Pathol. 2003, 34, 743–748. [Google Scholar] [CrossRef]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef]
- Guo, Y.; Korteweg, C.; McNutt, M.A.; Gu, J. Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008, 133, 4–12. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Mitchell, W.B. Thromboinflammation in COVID-19 acute lung injury. Paediatr. Respir. Rev. 2020, 35, 20–24. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The clinical pathology of severe acute respiratory syndrome (SARS): A report from China. J. Pathol. 2003, 200, 282–289. [Google Scholar] [CrossRef]
- Hwang, D.M.; Chamberlain, D.W.; Poutanen, S.M.; Low, D.E.; Asa, S.L.; Butany, J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod. Pathol. 2005, 18, 1–10. [Google Scholar] [CrossRef]
- Kory, P.; Kanne, J.P. SARS-CoV-2 organising pneumonia: ‘Has there been a widespread failure to identify and treat this prevalent condition in COVID-19?’. BMJ Open Respir. Res. 2020, 7. [Google Scholar] [CrossRef]
- Aiolfi, A.; Bruni, B.; Biraghi, T.; Montisci, A.; Miceli, A.; Baronio, B.; Khor, D.; Cirri, S.; Donatelli, F.; Clemente, C.; et al. Late histological findings in symptomatic COVID-19 patients: A case report. Medicine (Baltimore) 2020, 99, e21046. [Google Scholar] [CrossRef]
- Revzin, M.V.; Raza, S.; Warshawsky, R.; D’Agostino, C.; Srivastava, N.C.; Bader, A.S.; Malhotra, A.; Patel, R.D.; Chen, K.; Kyriakakos, C.; et al. Multisystem Imaging Manifestations of COVID-19, Part 1: Viral Pathogenesis and Pulmonary and Vascular System Complications. Radiographics 2020, 40, 1574–1599. [Google Scholar] [CrossRef]
- Chung, M.; Bernheim, A.; Mei, X.; Zhang, N.; Huang, M.; Zeng, X.; Cui, J.; Xu, W.; Yang, Y.; Fayad, Z.A.; et al. CT Imaging Features of 2019 Novel Coronavirus (2019-nCoV). Radiology 2020, 295, 202–207. [Google Scholar] [CrossRef]
- Samidurai, A.; Das, A. Cardiovascular Complications Associated with COVID-19 and Potential Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 6790. [Google Scholar] [CrossRef]
- Liu, K.; Fang, Y.Y.; Deng, Y.; Liu, W.; Wang, M.F.; Ma, J.P.; Xiao, W.; Wang, Y.N.; Zhong, M.H.; Li, C.H.; et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin. Med. J. (Engl.) 2020, 133, 1025–1031. [Google Scholar] [CrossRef]
- Liu, M.; He, P.; Liu, H.G.; Wang, X.J.; Li, F.J.; Chen, S.; Lin, J.; Chen, P.; Liu, J.H.; Li, C.H. Clinical characteristics of 30 medical workers infected with new coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi 2020, 43, E016. [Google Scholar] [CrossRef]
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 2004, 25, 291–294. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Pfeffer, M.A. Plasma angiotensin-converting enzyme 2: Novel biomarker in heart failure with implications for COVID-19. Eur. Heart J. 2020, 41, 1818–1820. [Google Scholar] [CrossRef]
- Lindner, D.; Fitzek, A.; Brauninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection with SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef]
- Blyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, Y.; Wang, D.W. SARS-CoV-2: A potential novel etiology of fulminant myocarditis. Herz 2020, 45, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Gheibi Hayat, S.M.; Taghizadeh, H.; Akbari, A.; Inabadi, M.; Savardashtaki, A.; Johnston, T.P.; Sahebkar, A. COVID-19 and cardiac injury: Clinical manifestations, biomarkers, mechanisms, diagnosis, treatment, and follow up. Expert Rev. Anti Infect. Ther. 2020, 19, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef]
- Qu, R.; Ling, Y.; Zhang, Y.H.; Wei, L.Y.; Chen, X.; Li, X.M.; Liu, X.Y.; Liu, H.M.; Guo, Z.; Ren, H.; et al. Platelet-to-lymphocyte ratio is associated with prognosis in patients with coronavirus disease-19. J. Med. Virol. 2020, 92, 1533–1541. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tang, J. COVID-19 and Kidney Injury. R. I. Med. J. 2020, 103, 24–28. [Google Scholar]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J., Jr.; Liu, K.D.; Ostermann, M.; Rimmele, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar] [CrossRef]
- Gupta, S.; Hayek, S.S.; Wang, W.; Chan, L.; Mathews, K.S.; Melamed, M.L.; Brenner, S.K.; Leonberg-Yoo, A.; Schenck, E.J.; Radbel, J.; et al. Factors Associated with Death in Critically Ill Patients with Coronavirus Disease 2019 in the US. JAMA Intern. Med. 2020, 180, 1436–1446. [Google Scholar] [CrossRef]
- Li, Z.; Wu, M.; Yao, J.; Guo, J.; Liao, X.; Song, S.; Li, J.; Duan, G.; Zhou, Y.; Wu, X.; et al. Caution on Kidney Dysfunctions of COVID-19 Patients. medRxiv 2020, 25. [Google Scholar] [CrossRef]
- Noce, A.; Marrone, G.; Rovella, V.; Busca, A.; Gola, C.; Ferrannini, M.; di Daniele, N. Fenoldopam mesylate: A narrative review of its use in acute kidney injury. Curr. Pharm. Biotechnol. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Farkash, E.A.; Wilson, A.M.; Jentzen, J.M. Ultrastructural Evidence for Direct Renal Infection with SARS-CoV-2. J. Am. Soc. Nephrol. 2020, 31, 1683–1687. [Google Scholar] [CrossRef]
- Chien, J.Y.; Hsueh, P.R.; Cheng, W.C.; Yu, C.J.; Yang, P.C. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology 2006, 11, 715–722. [Google Scholar] [CrossRef]
- Chu, K.H.; Tsang, W.K.; Tang, C.S.; Lam, M.F.; Lai, F.M.; To, K.F.; Fung, K.S.; Tang, H.L.; Yan, W.W.; Chan, H.W.; et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005, 67, 698–705. [Google Scholar] [CrossRef]
- Min, C.K.; Cheon, S.; Ha, N.Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.Y.; Inn, K.S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef]
- Welch, H.K.; Kellum, J.A.; Kane-Gill, S.L. Drug-Associated Acute Kidney Injury Identified in the United States Food and Drug Administration Adverse Event Reporting System Database. Pharmacotherapy 2018, 38, 785–793. [Google Scholar] [CrossRef]
- Ostermann, M.; Chawla, L.S.; Forni, L.G.; Kane-Gill, S.L.; Kellum, J.A.; Koyner, J.; Murray, P.T.; Ronco, C.; Goldstein, S.L.; ADQI 16 Workgroup. Drug management in acute kidney disease—Report of the Acute Disease Quality Initiative XVI meeting. Br. J. Clin. Pharmacol. 2018, 84, 396–403. [Google Scholar] [CrossRef]
- Noce, A.; Fabrini, R.; Dessi, M.; Bocedi, A.; Santini, S.; Rovella, V.; Pastore, A.; Tesauro, M.; Bernardini, S.; di Daniele, N.; et al. Erythrocyte glutathione transferase activity: A possible early biomarker for blood toxicity in uremic diabetic patients. Acta Diabetol. 2014, 51, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Sargiacomo, C.; Sotgia, F.; Lisanti, M.P. COVID-19 and chronological aging: Senolytics and other anti-aging drugs for the treatment or prevention of corona virus infection? Aging (Albany NY) 2020, 12, 6511–6517. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Chan, V.W.; Chiu, P.K.; Yee, C.H.; Yuan, Y.; Ng, C.F.; Teoh, J.Y. A systematic review on COVID-19: Urological manifestations, viral RNA detection and special considerations in urological conditions. World J. Urol. 2020, 12. [Google Scholar] [CrossRef]
- Mumm, J.N.; Osterman, A.; Ruzicka, M.; Stihl, C.; Vilsmaier, T.; Munker, D.; Khatamzas, E.; Giessen-Jung, C.; Stief, C.; Staehler, M.; et al. Urinary Frequency as a Possibly Overlooked Symptom in COVID-19 Patients: Does SARS-CoV-2 Cause Viral Cystitis? Eur. Urol. 2020, 78, 624–628. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Zhao, D.; Yao, F.; Wang, L.; Zheng, L.; Gao, Y.; Ye, J.; Guo, F.; Zhao, H.; Gao, R. A Comparative Study on the Clinical Features of Coronavirus 2019 (COVID-19) Pneumonia with Other Pneumonias. Clin. Infect. Dis. 2020, 71, 756–761. [Google Scholar] [CrossRef]
- Zhang, H.; Kang, Z.; Gong, H.; Xu, D.; Wang, J.; Li, Z.; Li, Z.; Cui, X.; Xiao, J.; Zhan, J.; et al. Digestive system is a potential route of COVID-19: An analysis of single-cell coexpression pattern of key proteins in viral entry process. Gut 2020, 69, 1010–1018. [Google Scholar] [CrossRef]
- Muus, C.; Luecken, M.D.; Eraslan, G.; Waghray, A.; Heimberg, G.; Sikkema, L.; Kobayashi, Y.; Vaishnav, E.D.; Subramanian, A.; Smilie, C.; et al. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Holshue, M.L.; de Bolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, L.; Du, H.; Zhang, J.; Li, Y.Y.; Qu, J.; Zhang, W.; Wang, Y.; Bao, S.; Li, Y.; et al. SARS-CoV-2 Infection in Children. N. Engl. J. Med. 2020, 382, 1663–1665. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Duan, C.; Zhang, S.; Spiegel, B.; Shi, H.; Wang, W.; Zhang, L.; Lin, R.; Liu, J.; Ding, Z.; et al. Digestive Symptoms in COVID-19 Patients with Mild Disease Severity: Clinical Presentation, Stool Viral RNA Testing, and Outcomes. Am. J. Gastroenterol. 2020, 115, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Alqusairi, R.; Adams, A.; Paul, M.; Kothari, N.; Peters, S.; de Benedet, A.T. SARS-CoV-2 Gastrointestinal Infection Causing Hemorrhagic Colitis: Implications for Detection and Transmission of COVID-19 Disease. Am. J. Gastroenterol. 2020, 115, 942–946. [Google Scholar] [CrossRef]
- Ferrey, A.J.; Choi, G.; Hanna, R.M.; Chang, Y.; Tantisattamo, E.; Ivaturi, K.; Park, E.; Nguyen, L.; Wang, B.; Tonthat, S.; et al. A Case of Novel Coronavirus Disease 19 in a Chronic Hemodialysis Patient Presenting with Gastroenteritis and Developing Severe Pulmonary Disease. Am. J. Nephrol. 2020, 51, 337–342. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffre, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136. [Google Scholar] [CrossRef]
- Dirweesh, A.; Li, Y.; Trikudanathan, G.; Mallery, J.S.; Freeman, M.L.; Amateau, S.K. Clinical Outcomes of Acute Pancreatitis in Patients with Coronavirus Disease 2019. Gastroenterology 2020, 159, 1972–1974. [Google Scholar] [CrossRef]
- Balaphas, A.; Gkoufa, K.; Meyer, J.; Peloso, A.; Bornand, A.; McKee, T.A.; Toso, C.; Popeskou, S.G. COVID-19 can mimic acute cholecystitis and is associated with the presence of viral RNA in the gallbladder wall. J. Hepatol. 2020, 73, 1566–1568. [Google Scholar] [CrossRef]
- Hunt, R.H.; East, J.E.; Lanas, A.; Malfertheiner, P.; Satsangi, J.; Scarpignato, C.; Webb, G.J. COVID-19 and Gastrointestinal Disease. Implications for the Gastroenterologist. Dig. Dis. 2020, 39, 119–139. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Lanas, A.; Dumonceau, J.M.; Hunt, R.H.; Fujishiro, M.; Scheiman, J.M.; Gralnek, I.M.; Campbell, H.E.; Rostom, A.; Villanueva, C.; Sung, J.J.Y. Non-variceal upper gastrointestinal bleeding. Nat. Rev. Dis. Primers 2018, 4, 18020. [Google Scholar] [CrossRef]
- Massironi, S.; Vigano, C.; Dioscoridi, L.; Filippi, E.; Pagliarulo, M.; Manfredi, G.; Conti, C.B.; Signorelli, C.; Redaelli, A.E.; Bonato, G.; et al. Endoscopic Findings in Patients Infected with 2019 Novel Coronavirus in Lombardy, Italy. Clin. Gastroenterol. Hepatol. 2020, 18, 2375–2377. [Google Scholar] [CrossRef]
- Farina, D.; Rondi, P.; Botturi, E.; Renzulli, M.; Borghesi, A.; Guelfi, D.; Ravanelli, M. Gastrointestinal: Bowel ischemia in a suspected coronavirus disease (COVID-19) patient. J. Gastroenterol. Hepatol. 2021, 36, 41. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Artifoni, M.; Danic, G.; Gautier, G.; Gicquel, P.; Boutoille, D.; Raffi, F.; Neel, A.; Lecomte, R. Systematic assessment of venous thromboembolism in COVID-19 patients receiving thromboprophylaxis: Incidence and role of D-dimer as predictive factors. J. Thromb Thrombolysis 2020, 50, 211–216. [Google Scholar] [CrossRef]
- Xu, L.; Liu, J.; Lu, M.; Yang, D.; Zheng, X. Liver injury during highly pathogenic human coronavirus infections. Liver Int. 2020, 40, 998–1004. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Dube, M.; le Coupanec, A.; Wong, A.H.M.; Rini, J.M.; Desforges, M.; Talbot, P.J. Axonal Transport Enables Neuron-to-Neuron Propagation of Human Coronavirus OC43. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Desforges, M.; le Coupanec, A.; Brison, E.; Meessen-Pinard, M.; Talbot, P.J. Neuroinvasive and neurotropic human respiratory coronaviruses: Potential neurovirulent agents in humans. Adv. Exp. Med. Biol. 2014, 807, 75–96. [Google Scholar] [CrossRef]
- Keyhanian, K.; Umeton, R.P.; Mohit, B.; Davoudi, V.; Hajighasemi, F.; Ghasemi, M. SARS-CoV-2 and nervous system: From pathogenesis to clinical manifestation. J. Neuroimmunol. 2020, 350, 577436. [Google Scholar] [CrossRef]
- Prabakaran, P.; Xiao, X.; Dimitrov, D.S. A model of the ACE2 structure and function as a SARS-CoV receptor. Biochem. Biophys. Res. Commun. 2004, 314, 235–241. [Google Scholar] [CrossRef]
- Abboud, H.; Abboud, F.Z.; Kharbouch, H.; Arkha, Y.; el Abbadi, N.; el Ouahabi, A. COVID-19 and SARS-Cov-2 Infection: Pathophysiology and Clinical Effects on the Nervous System. World Neurosurg. 2020, 140, 49–53. [Google Scholar] [CrossRef]
- Xia, H.; Lazartigues, E. Angiotensin-converting enzyme 2: Central regulator for cardiovascular function. Curr. Hypertens. Rep. 2010, 12, 170–175. [Google Scholar] [CrossRef]
- Siso, S.; Jeffrey, M.; Gonzalez, L. Sensory circumventricular organs in health and disease. Acta Neuropathol. 2010, 120, 689–705. [Google Scholar] [CrossRef]
- Bohmwald, K.; Galvez, N.M.S.; Rios, M.; Kalergis, A.M. Neurologic Alterations Due to Respiratory Virus Infections. Front. Cell Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef]
- Poyiadji, N.; Shahin, G.; Noujaim, D.; Stone, M.; Patel, S.; Griffith, B. COVID-19-associated Acute Hemorrhagic Necrotizing Encephalopathy: Imaging Features. Radiology 2020, 296, E119–E120. [Google Scholar] [CrossRef] [PubMed]
- Filatov, A.; Sharma, P.; Hindi, F.; Espinosa, P.S. Neurological Complications of Coronavirus Disease (COVID-19): Encephalopathy. Cureus 2020, 12, e7352. [Google Scholar] [CrossRef] [PubMed]
- Driessen, A.K.; Farrell, M.J.; Mazzone, S.B.; McGovern, A.E. Multiple neural circuits mediating airway sensations: Recent advances in the neurobiology of the urge-to-cough. Respir. Physiol. Neurobiol. 2016, 226, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Audrit, K.J.; Delventhal, L.; Aydin, O.; Nassenstein, C. The nervous system of airways and its remodeling in inflammatory lung diseases. Cell Tissue Res. 2017, 367, 571–590. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef]
- Sasannejad, C.; Ely, E.W.; Lahiri, S. Long-term cognitive impairment after acute respiratory distress syndrome: A review of clinical impact and pathophysiological mechanisms. Crit. Care 2019, 23, 352. [Google Scholar] [CrossRef]
- Xu, J.; Zhong, S.; Liu, J.; Li, L.; Li, Y.; Wu, X.; Li, Z.; Deng, P.; Zhang, J.; Zhong, N.; et al. Detection of severe acute respiratory syndrome coronavirus in the brain: Potential role of the chemokine mig in pathogenesis. Clin. Infect. Dis. 2005, 41, 1089–1096. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Menghini, R.; Campia, U.; Tesauro, M.; Marino, A.; Rovella, V.; Rodia, G.; Schinzari, F.; Tolusso, B.; di Daniele, N.; Federici, M.; et al. Toll-like receptor 4 mediates endothelial cell activation through NF-kappaB but is not associated with endothelial dysfunction in patients with rheumatoid arthritis. PLoS ONE 2014, 9, e99053. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Steardo, L., Jr.; Bronzuoli, M.R.; Iacomino, A.; Esposito, G.; Steardo, L.; Scuderi, C. Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front. Neurosci. 2015, 9, 259. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Rodriguez, J.J.; Steardo, L. Astrogliopathology: A central element of neuropsychiatric diseases? Neuroscientist 2014, 20, 576–588. [Google Scholar] [CrossRef]
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef]
- Jiang, F.; Deng, L.; Zhang, L.; Cai, Y.; Cheung, C.W.; Xia, Z. Review of the Clinical Characteristics of Coronavirus Disease 2019 (COVID-19). J. Gen. Intern. Med. 2020, 35, 1545–1549. [Google Scholar] [CrossRef]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef]
- Calcia, M.A.; Bonsall, D.R.; Bloomfield, P.S.; Selvaraj, S.; Barichello, T.; Howes, O.D. Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology (Berl.) 2016, 233, 1637–1650. [Google Scholar] [CrossRef]
- Klein, R.S.; Garber, C.; Funk, K.E.; Salimi, H.; Soung, A.; Kanmogne, M.; Manivasagam, S.; Agner, S.; Cain, M. Neuroinflammation During RNA Viral Infections. Annu. Rev. Immunol. 2019, 37, 73–95. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Bobker, S.M.; Robbins, M.S. COVID-19 and Headache: A Primer for Trainees. Headache 2020, 60, 1806–1811. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Maccaferri, M.; Ruini, C.; Tomasi, A.; Ozben, T. Biomarkers associated with COVID-19 disease progression. Crit. Rev. Clin. Lab. Sci. 2020, 57, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Kermali, M.; Khalsa, R.K.; Pillai, K.; Ismail, Z.; Harky, A. The role of biomarkers in diagnosis of COVID-19—A systematic review. Life Sci. 2020, 254, 117788. [Google Scholar] [CrossRef]
- Tietjen, G.E.; Khubchandani, J.; Herial, N.; Palm-Meinders, I.H.; Koppen, H.; Terwindt, G.M.; van Buchem, M.A.; Launer, L.J.; Ferrari, M.D.; Kruit, M.C. Migraine and vascular disease biomarkers: A population-based case-control study. Cephalalgia 2018, 38, 511–518. [Google Scholar] [CrossRef]
- Fong, T.G.; Tulebaev, S.R.; Inouye, S.K. Delirium in elderly adults: Diagnosis, prevention and treatment. Nat. Rev. Neurol. 2009, 5, 210–220. [Google Scholar] [CrossRef]
- FSD Pharma. Ultramicronized Palmitoylethanolamide (PEA) Treatment in Hospitalized Participants with COVID-19; NCT04619706; FSD Pharma: Cobourg, Canada, 2020. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noce, A.; Albanese, M.; Marrone, G.; Di Lauro, M.; Pietroboni Zaitseva, A.; Palazzetti, D.; Guerriero, C.; Paolino, A.; Pizzenti, G.; Di Daniele, F.; et al. Ultramicronized Palmitoylethanolamide (um-PEA): A New Possible Adjuvant Treatment in COVID-19 patients. Pharmaceuticals 2021, 14, 336. https://doi.org/10.3390/ph14040336
Noce A, Albanese M, Marrone G, Di Lauro M, Pietroboni Zaitseva A, Palazzetti D, Guerriero C, Paolino A, Pizzenti G, Di Daniele F, et al. Ultramicronized Palmitoylethanolamide (um-PEA): A New Possible Adjuvant Treatment in COVID-19 patients. Pharmaceuticals. 2021; 14(4):336. https://doi.org/10.3390/ph14040336
Chicago/Turabian StyleNoce, Annalisa, Maria Albanese, Giulia Marrone, Manuela Di Lauro, Anna Pietroboni Zaitseva, Daniela Palazzetti, Cristina Guerriero, Agostino Paolino, Giuseppa Pizzenti, Francesca Di Daniele, and et al. 2021. "Ultramicronized Palmitoylethanolamide (um-PEA): A New Possible Adjuvant Treatment in COVID-19 patients" Pharmaceuticals 14, no. 4: 336. https://doi.org/10.3390/ph14040336
APA StyleNoce, A., Albanese, M., Marrone, G., Di Lauro, M., Pietroboni Zaitseva, A., Palazzetti, D., Guerriero, C., Paolino, A., Pizzenti, G., Di Daniele, F., Romani, A., D’Agostini, C., Magrini, A., Mercuri, N. B., & Di Daniele, N. (2021). Ultramicronized Palmitoylethanolamide (um-PEA): A New Possible Adjuvant Treatment in COVID-19 patients. Pharmaceuticals, 14(4), 336. https://doi.org/10.3390/ph14040336