
Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis

 and
and
Abstract
1. Introduction
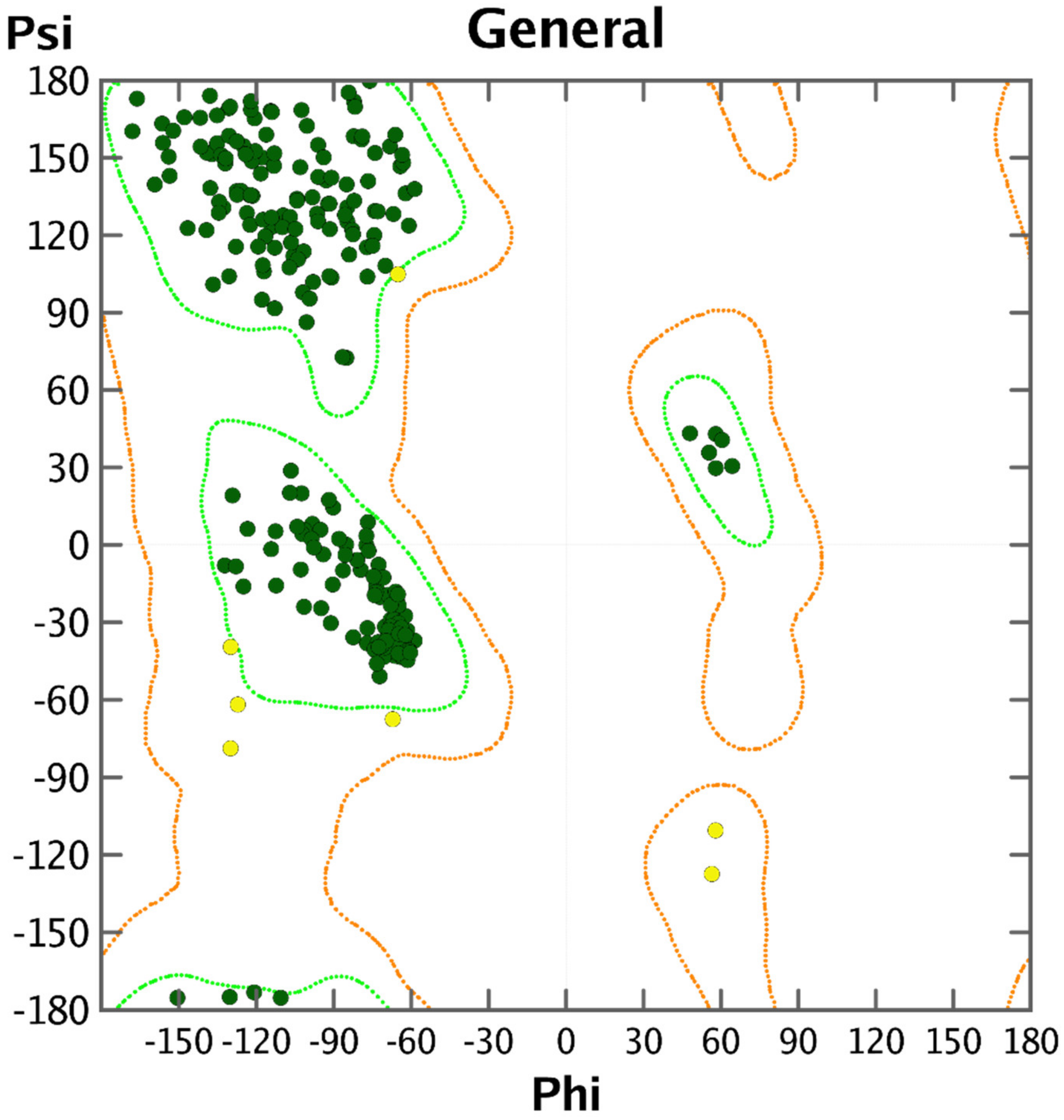
2. Results
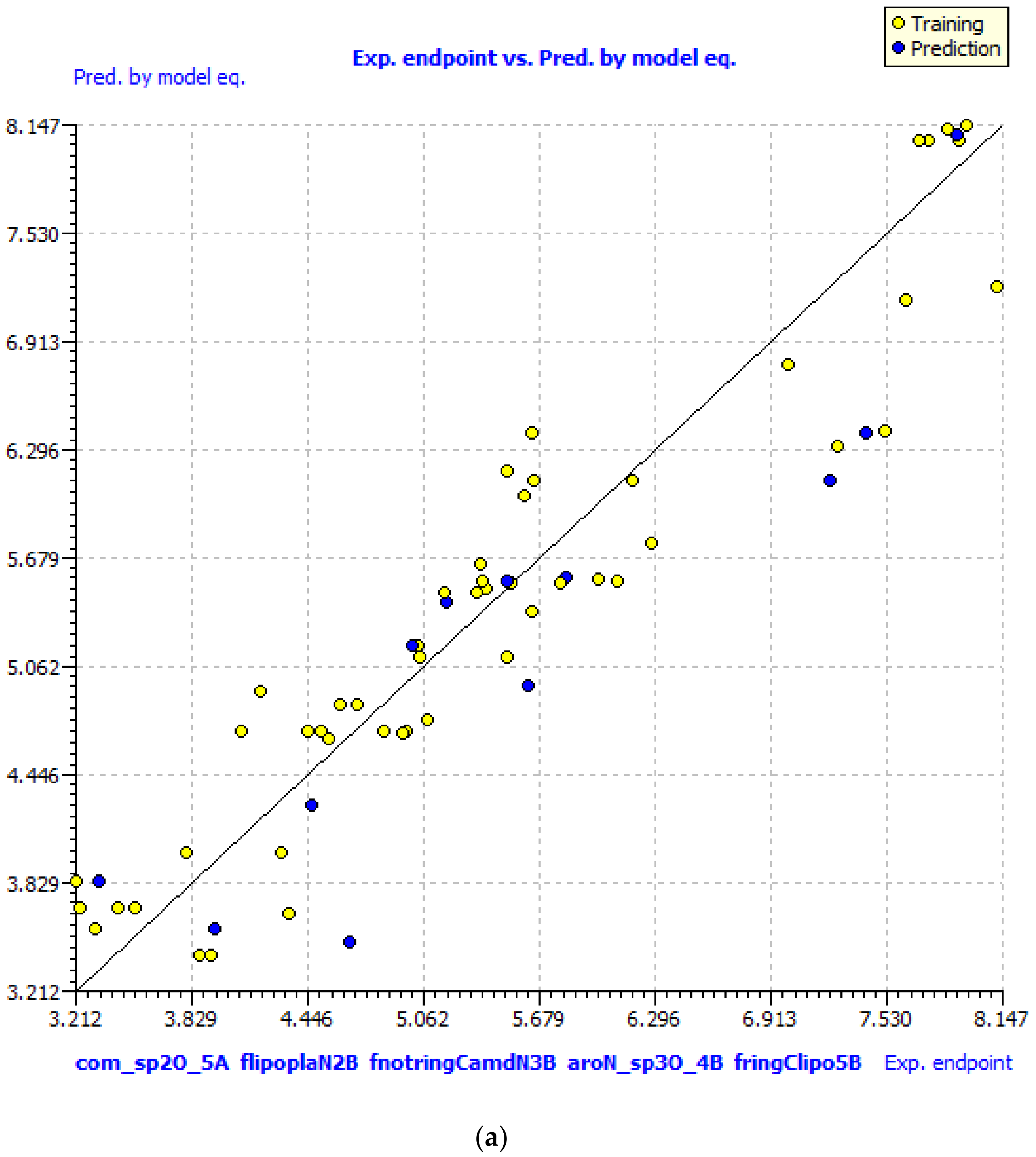
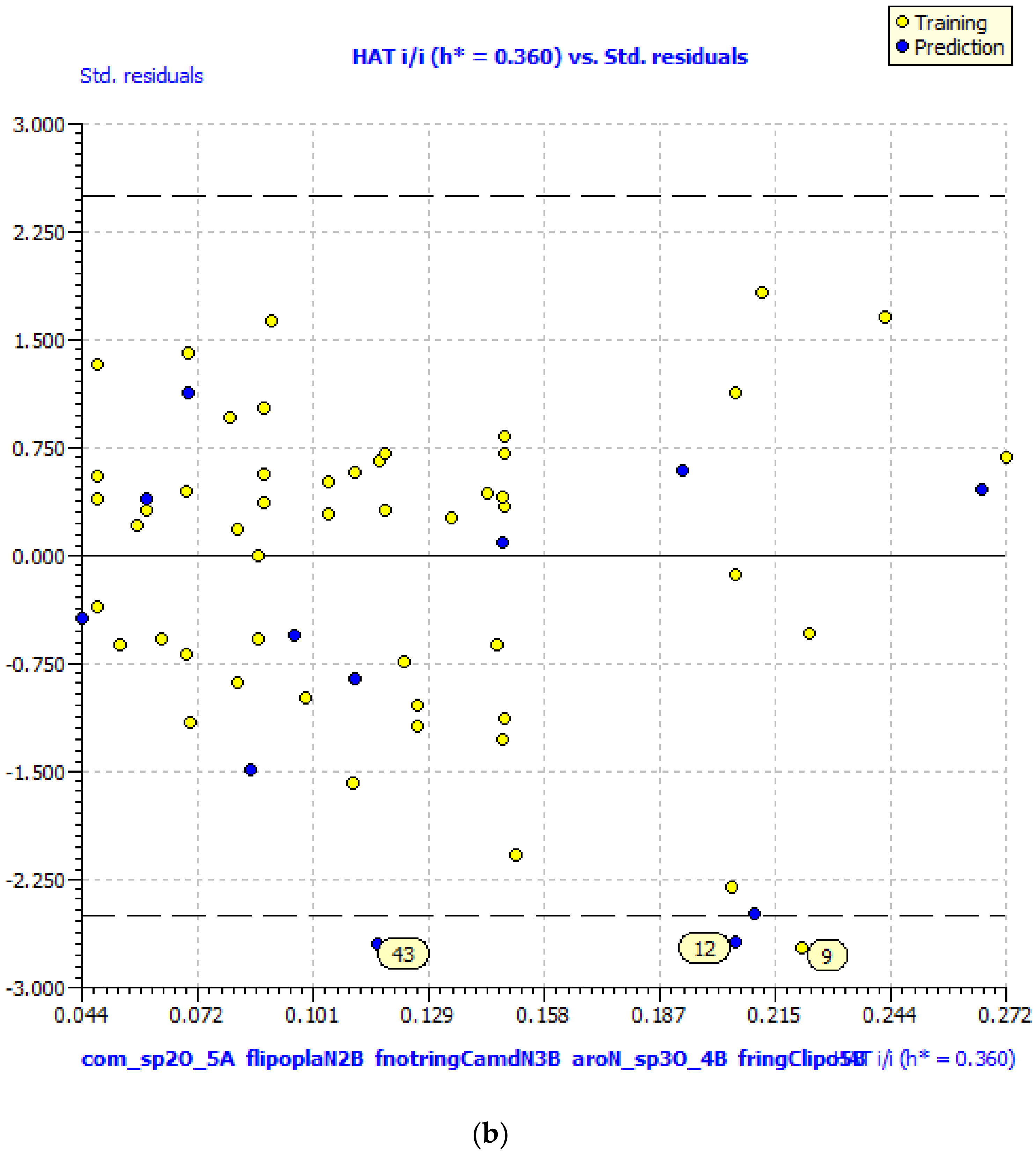
GA–MLR QSAR Model
3. Discussion
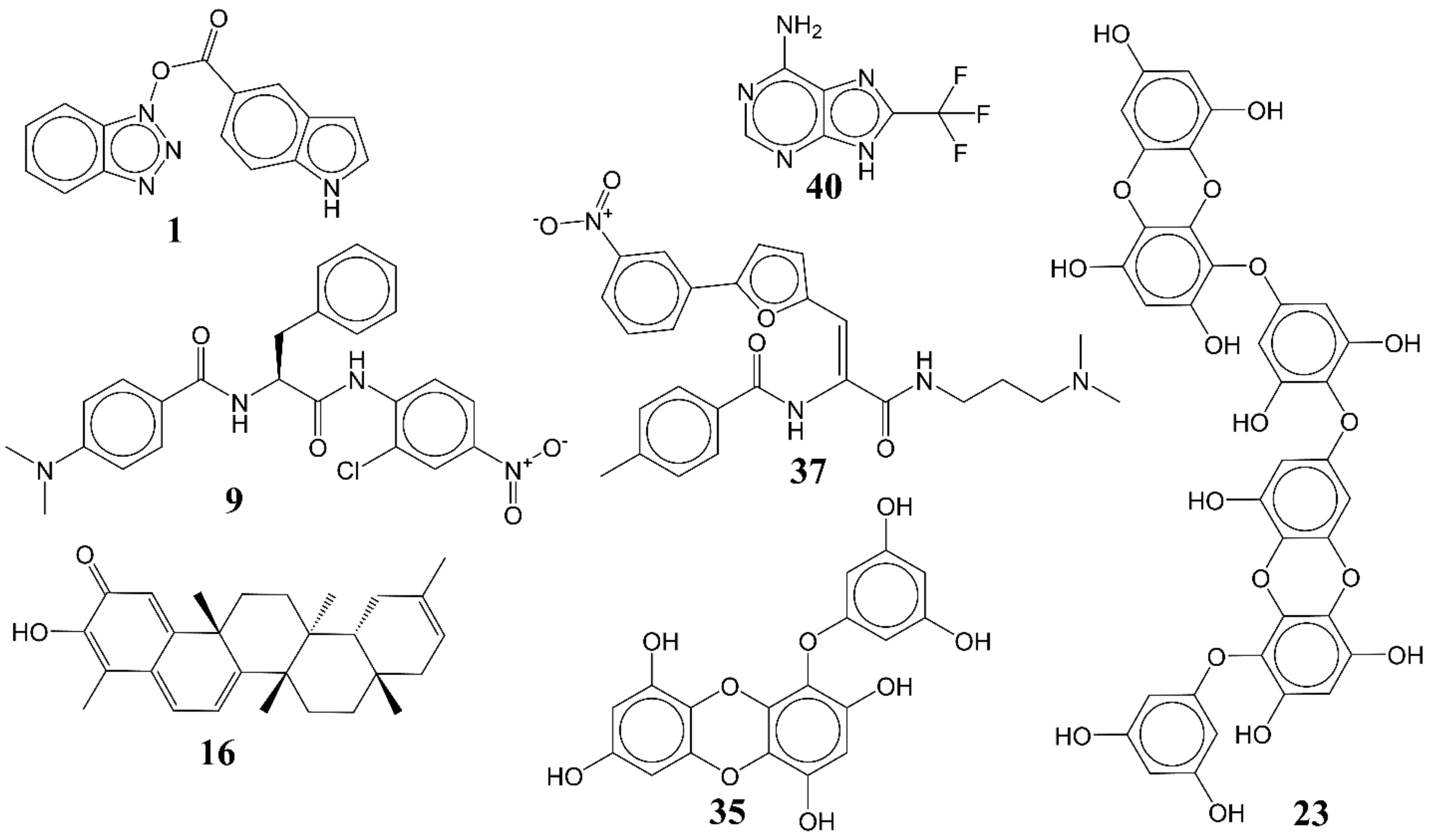
3.1. QSAR-Based Virtual Screening
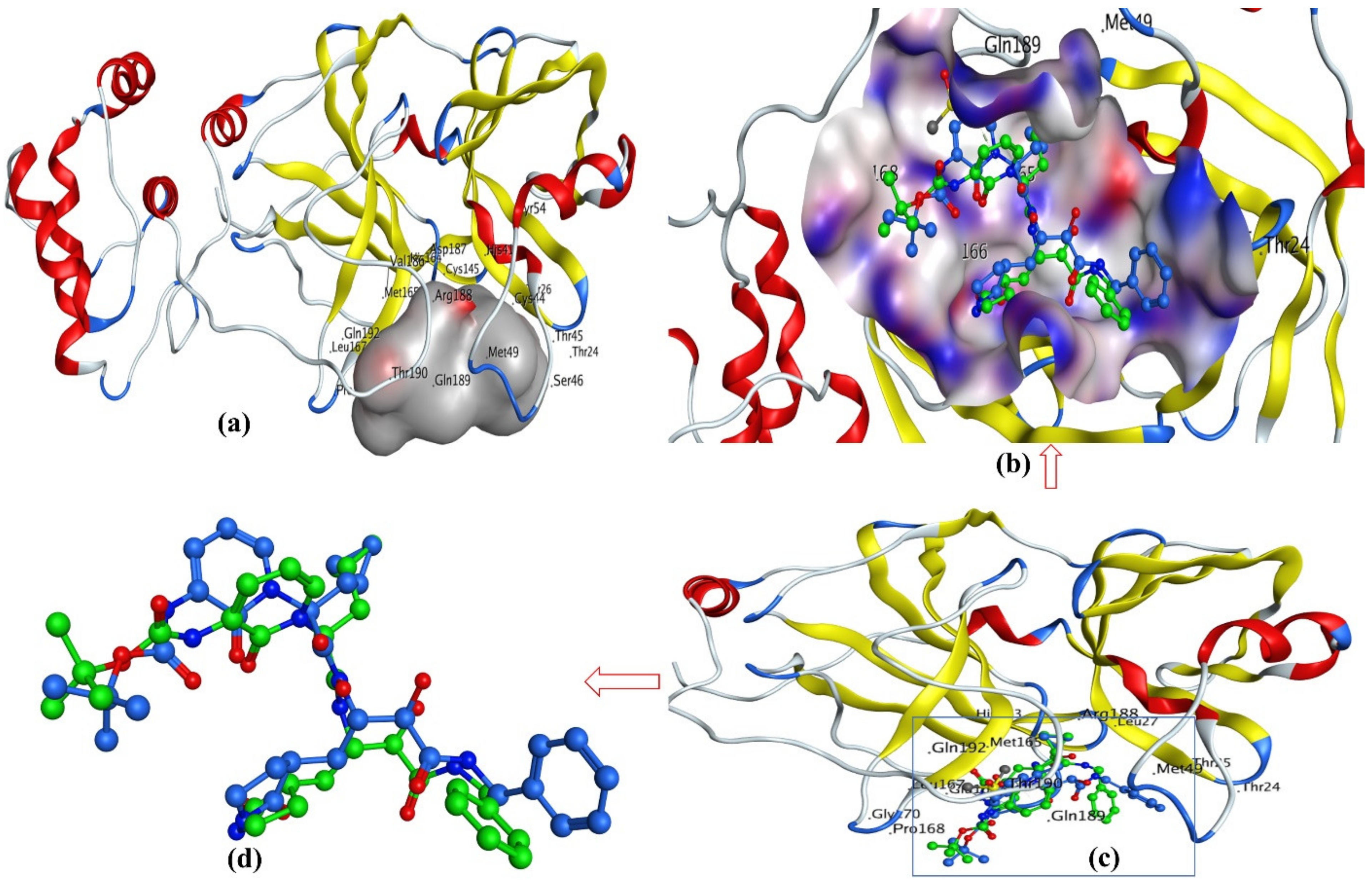
3.2. Docking Analysis
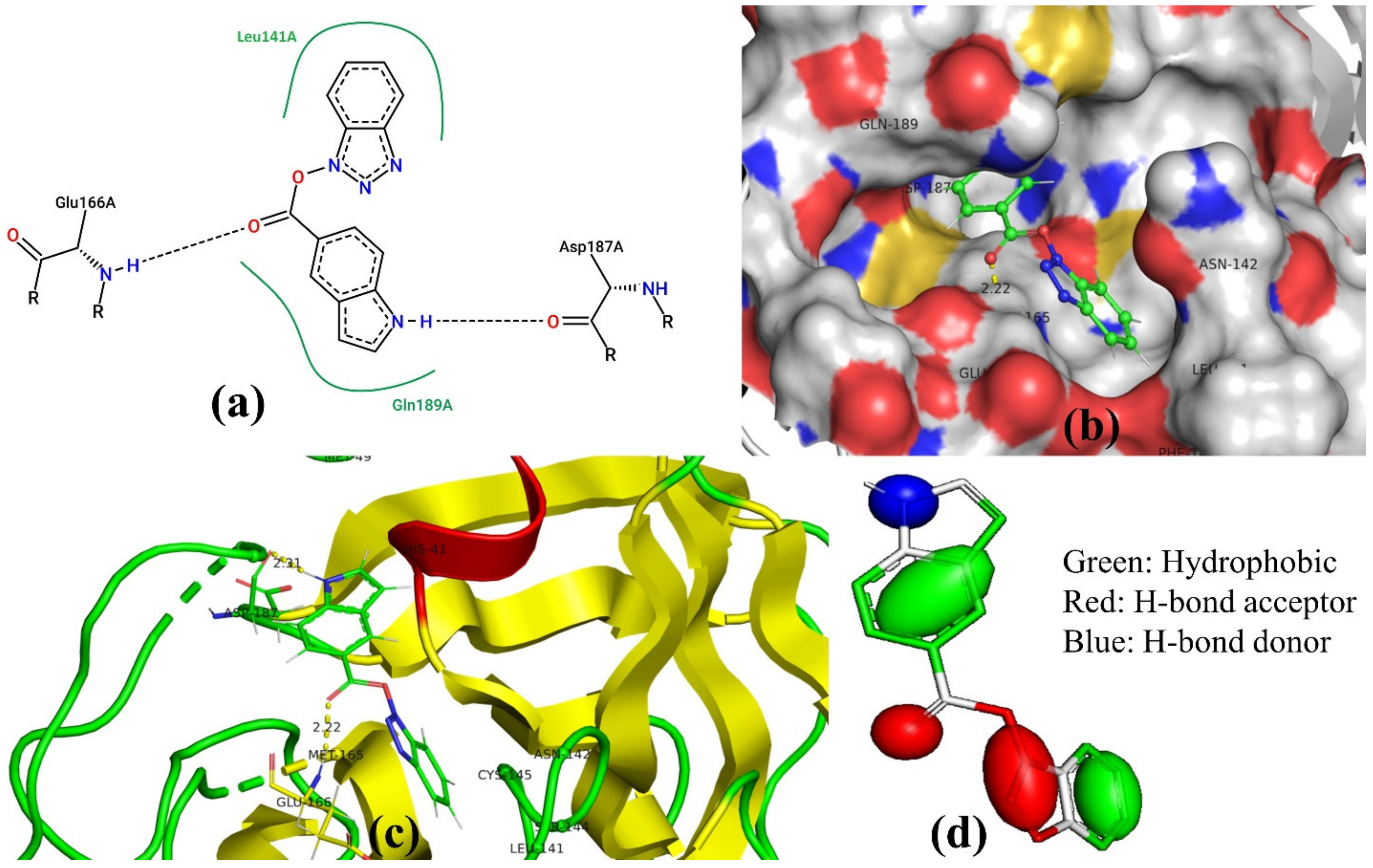
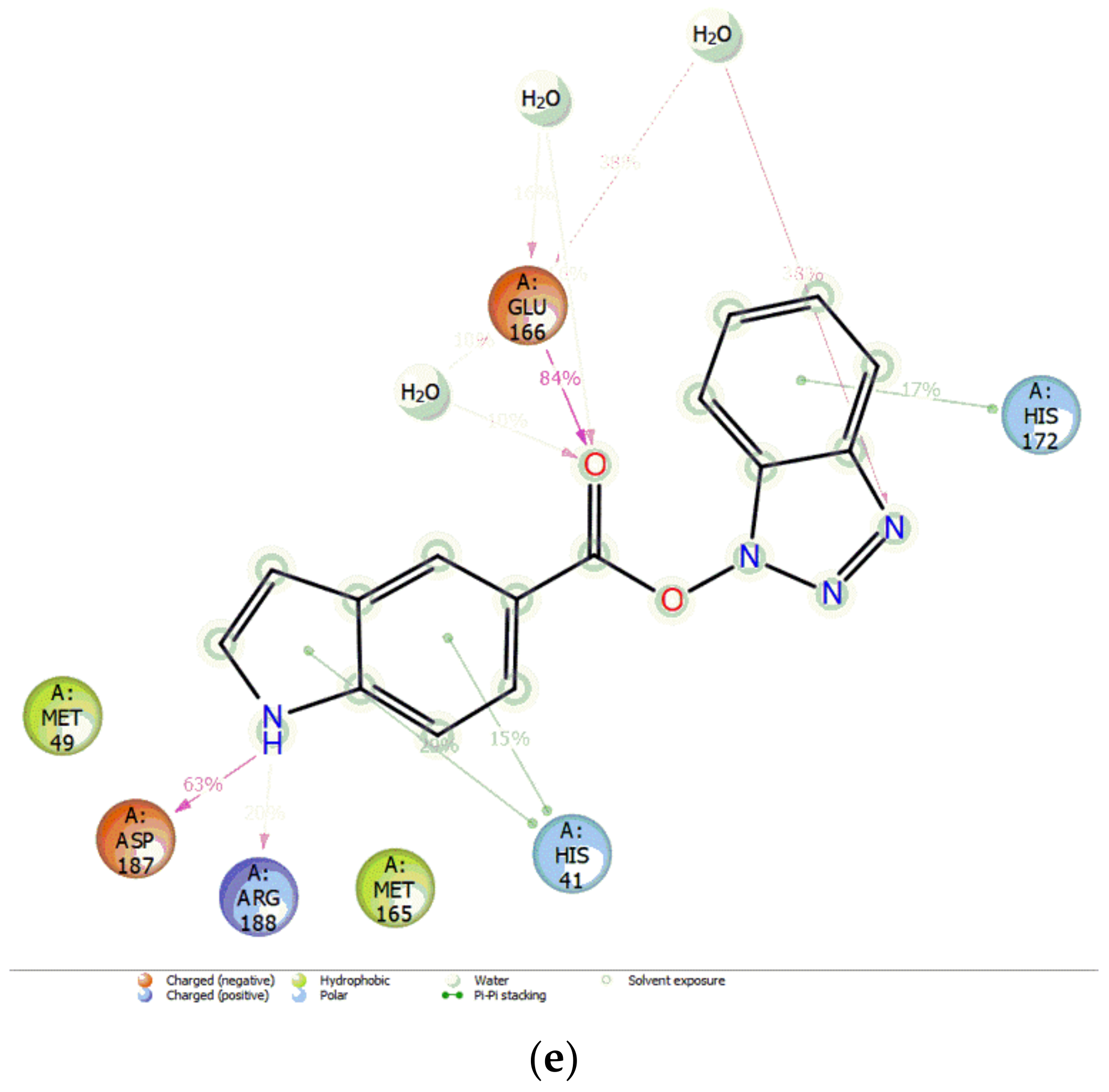
3.3. Docking Pose for Most Active Molecule
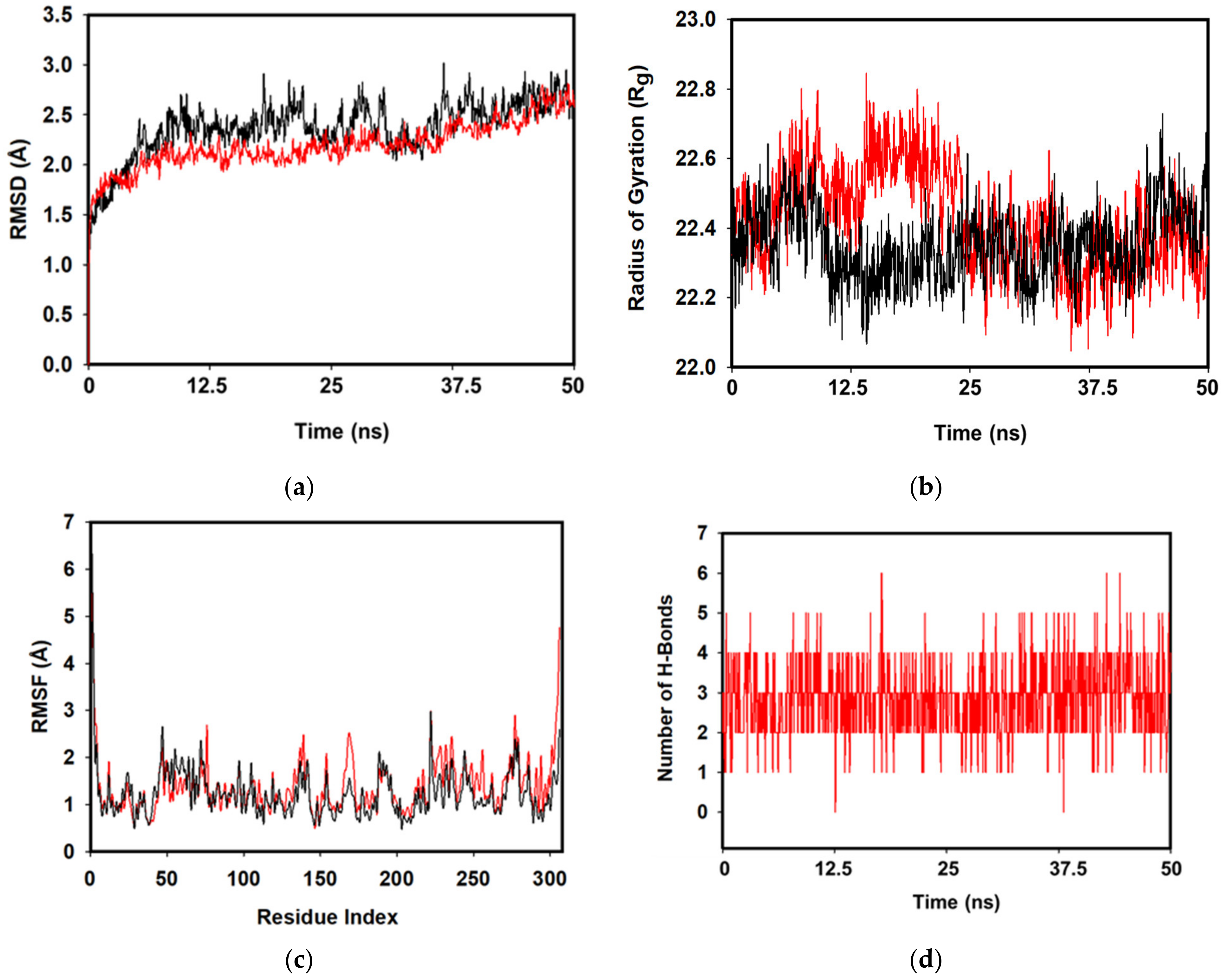
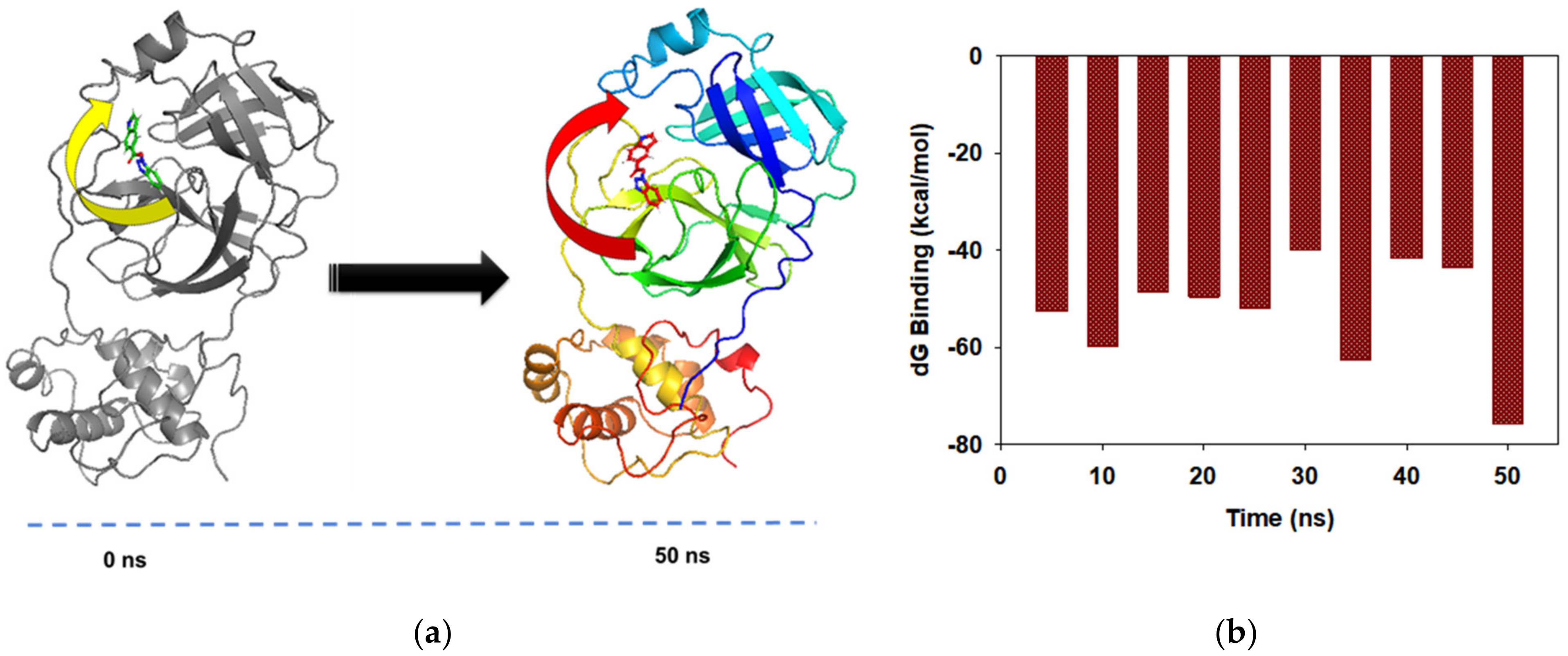
3.4. MD Simulations and MMGBSA Binding Free-Energy Calculations
4. Materials and Methods
4.1. QSAR Analysis and Model Building
4.2. QSAR-Based Virtual Screening
4.3. Molecular Docking Analysis
4.4. Molecular Dynamics and Binding Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SMILES | Simplified Molecular-Input Line-Entry System |
| GA | Genetic Algorithm |
| MLR | Multiple Linear Regression |
| QSAR | Quantitative Structure−Activity Relationship |
| WHO | World Health Organization |
| ADMET | Absorption, Distribution, Metabolism, Excretion, and Toxicity |
| OLS | Ordinary least square |
| SARS-CoV | Severe Acute Respiratory Syndrome Coronavirus |
| QSARINS | QSAR Insubria |
| HTS | High-throughput screening |
| MDS | Molecular Dynamics Simulation |
| Mpro | Main protease |
| OECD | Organisation for Economic Co-operation and Development |
References
- Gao, K.; Nguyen, D.D.; Chen, J.; Wang, R.; Wei, G.-W. Repositioning of 8565 Existing Drugs for COVID-19. J. Phys. Chem. Lett. 2020, 11, 5373–5382. [Google Scholar] [CrossRef]
- WHO. Available online: https://covid19.who.int/ or https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200414-sitrep-85-covid-19.pdf?sfvrsn=7b8629bb_4 (accessed on 4 January 2021).
- Ko, M.; Jeon, S.; Ryu, W.S.; Kim, S. Comparative analysis of antiviral efficacy of FDA-approved drugs against SARS-CoV-2 in human lung cells. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10 (Suppl. S12), S88–S97. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Leung, N.H.L.; Chu, D.K.W.; Shiu, E.Y.C.; Chan, K.-H.; McDevitt, J.J.; Hau, B.J.P.; Yen, H.-L.; Li, Y.; Ip, D.K.M.; Peiris, J.S.M.; et al. Respiratory virus shedding in exhaled breath and efficacy of face masks. Nat. Med. 2020, 26, 676–680. [Google Scholar] [CrossRef]
- Chilamakuri, R.; Agarwal, S. COVID-19: Characteristics and Therapeutics. Cells 2021, 10, 206. [Google Scholar] [CrossRef]
- Mulholland, A.J.; Amaro, R.E. COVID19—Computational Chemists Meet the Moment. J. Chem. Inf. Model. 2020, 60, 5724–5726. [Google Scholar] [CrossRef]
- Tripathi, N.; Tripathi, N.; Goshisht, M.K. COVID-19: Inflammatory responses, structure-based drug design and potential therapeutics. Mol. Divers. 2021. [Google Scholar] [CrossRef]
- Kirby, T. New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Mohamed, N.M.; Ali, E.M.H.; AboulMagd, A.M. Ligand-based design, molecular dynamics and ADMET studies of suggested SARS-CoV-2 Mpro inhibitors. RSC Adv. 2021, 11, 4523–4538. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, Z.; Wang, X.; Zhang, X.; Mu, K.; Shi, Y.; Peng, C.; Xu, Z.; Zhu, W. Ligand-based approach for predicting drug targets and for virtual screening against COVID-19. Brief. Bioinform. 2021. [Google Scholar] [CrossRef]
- Masand, V.H.; Akasapu, S.; Gandhi, A.; Rastija, V.; Patil, M.K. Structure features of peptide-type SARS-CoV main protease inhibitors: Quantitative structure activity relationship study. Chemom. Intell. Lab. Syst. 2020, 206. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Fujita, T.; Winkler, D.A. Understanding the Roles of the “Two QSARs”. J. Chem. Inf. Model. 2016, 56, 269–274. [Google Scholar] [CrossRef]
- Masand, V.H.; El-Sayed, N.N.E.; Bambole, M.U.; Patil, V.R.; Thakur, S.D. Multiple quantitative structure-activity relationships (QSARs) analysis for orally active trypanocidal N-myristoyltransferase inhibitors. J. Mol. Struct. 2019, 1175, 481–487. [Google Scholar] [CrossRef]
- Masand, V.H.; Elsayed, N.N.; Thakur, S.D.; Gawhale, N.; Rathore, M.M. Quinoxalinones Based Aldose Reductase Inhibitors: 2D and 3D-QSAR Analysis. Mol. Inform. 2019, 38, e1800149. [Google Scholar] [CrossRef]
- Masand, V.H.; El-Sayed, N.N.E.; Bambole, M.U.; Quazi, S.A. Multiple QSAR models, pharmacophore pattern and molecular docking analysis for anticancer activity of α, β-unsaturated carbonyl-based compounds, oxime and oxime ether analogues. J. Mol. Struct. 2018, 1157, 89–96. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR Modeling. Int. J. Quant. Struct. Prop. Relatsh. 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef]
- Thorne, N.; Auld, D.S.; Inglese, J. Apparent activity in high-throughput screening: Origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 2010, 14, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, A.J. Bioactive Compounds of Food: Their Role in the Prevention and Treatment of Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1–4. [Google Scholar] [CrossRef]
- Childs, C.E.; Calder, P.C.; Miles, E.A. Diet and Immune Function. Nutrients 2019, 11, 1933. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provaznik, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Masand, V.H.; El-Sayed, N.N.E.; Mahajan, D.T.; Rastija, V. QSAR analysis for 6-arylpyrazine-2-carboxamides as Trypanosoma brucei inhibitors. SAR QSAR Environ. Res. 2017, 28, 165–177. [Google Scholar] [CrossRef]
- Masand, V.H.; El-Sayed, N.N.E.; Mahajan, D.T.; Mercader, A.G.; Alafeefy, A.M.; Shibi, I.G. QSAR modeling for anti-human African trypanosomiasis activity of substituted 2-Phenylimidazopyridines. J. Mol. Struct. 2017, 1130, 711–718. [Google Scholar] [CrossRef]
- Masand, V.H.; Mahajan, D.T.; Maldhure, A.K.; Rastija, V. Quantitative structure–activity relationships (QSARs) and pharmacophore modeling for human African trypanosomiasis (HAT) activity of pyridyl benzamides and 3-(oxazolo[4,5-b]pyridin-2-yl)anilides. Med. Chem. Res. 2016, 25, 2324–2334. [Google Scholar] [CrossRef]
- Masand, V.H.; Rastija, V.; Patil, M.K.; Gandhi, A.; Chapolikar, A. Extending the identification of structural features responsible for anti-SARS-CoV activity of peptide-type compounds using QSAR modelling. SAR QSAR Environ. Res. 2020, 31, 643–654. [Google Scholar] [CrossRef]
- Konno, S.; Thanigaimalai, P.; Yamamoto, T.; Nakada, K.; Kakiuchi, R.; Takayama, K.; Yamazaki, Y.; Yakushiji, F.; Akaji, K.; Kiso, Y.; et al. Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Bioorg. Med. Chem. 2013, 21, 412–424. [Google Scholar] [CrossRef]
- Regnier, T.; Sarma, D.; Hidaka, K.; Bacha, U.; Freire, E.; Hayashi, Y.; Kiso, Y. New developments for the design, synthesis and biological evaluation of potent SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2722–2727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2019, 35, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. alpha-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.-H. An Overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Saxena, A.K.; Prathipati, P. Comparison of MLR, PLS and GA-MLR in QSAR analysis*. SAR QSAR Environ. Res. 2003, 14, 433–445. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Gaudreault, F.; Morency, L.P.; Najmanovich, R.J. NRGsuite: A PyMOL plugin to perform docking simulations in real time using FlexAID. Bioinformatics 2015, 31, 3856–3858. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, F.; Najmanovich, R.J. FlexAID: Revisiting Docking on Non-Native-Complex Structures. J. Chem. Inf. Model. 2015, 55, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Dayer, M.R.; Taleb-Gassabi, S.; Dayer, M.S. Lopinavir; A Potent Drug against Coronavirus Infection: Insight from Molecular Docking Study. Arch. Clin. Infect. Dis. 2017, 12, e13823. [Google Scholar] [CrossRef]
- Kaiser, J. SCIENCE RESOURCES: Chemists Want NIH to Curtail Database. Science 2005, 308, 774. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chowdhury, P. In silico investigation of phytoconstituents from Indian medicinal herb ‘Tinospora cordifolia (giloy)’ against SARS-CoV-2 (COVID-19) by molecular dynamics approach. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SN | Pred-pKi (M) | Pred-Ki (nM) |
|---|---|---|
| S341 | 9.226 | 0.594 |
| S337 | 9.121 | 0.757 |
| S342 | 9.121 | 0.757 |
| S338 | 9.086 | 0.82 |
| S339 | 8.842 | 1.439 |
| S340 | 8.807 | 1.56 |
| S293 | 8.499 | 3.17 |
| S161 | 8.464 | 3.436 |
| S251 | 8.464 | 3.436 |
| S317 | 8.464 | 3.436 |
| FoodS8291 | 8.46 | 3.467 |
| FoodS6189 | 8.355 | 4.416 |
| FoodS677 | 8.32 | 4.786 |
| FoodS3568 | 8.32 | 4.786 |
| FoodS4426 | 8.251 | 5.61 |
| FoodS6919 | 8.251 | 5.61 |
| FoodS4135 | 8.181 | 6.592 |
| FoodS7495 | 8.181 | 6.592 |
| FoodS1368 | 7.971 | 10.691 |
| FoodS4841 | 7.936 | 11.588 |
| SN | Structure | Docking Score (kcal/mol) |
|---|---|---|
| 1 |  | −5.997 |
| 2 |  | −7.008 |
| 3 |  | −6.42 |
| 4 |  | −5.886 |
| 5 |  | −6.041 |
| 6 |  | −6.48 |
| 7 |  | −5.866 |
| 8 |  | −6.331 |
| 9 |  | −7.774 |
| 10 |  | −9.931 |
| SN | Structure | Docking Score (kcal/mol) |
|---|---|---|
| 18 |  | −10.285 |
| 60 |  | −10.259 |
| 19 |  | −10.159 |
| 21 |  | −10.026 |
| 57 |  | −9.975 |
| 10 |  | −9.931 |
| SN | List of Interacting Amino Acids | Docking Score |
|---|---|---|
| 1 | His41, Met49, Tyr54, Phe140, Leu141, Asn142, Ser144, Cys145, His163, His164, Met165, Glu166 (strong H-bond), Asp187 (strong H-bond), Arg188, Gln189 | −10.285 |
| 62 | His41, Met49, Tyr54, Phe140, Leu141, Asn142, Ser144, Cys145, His163, His164, Met165 (weak H-bond), Glu166, Pro168, Val186, Asp187, Arg188, Gln189 (weak H-bond) | −9.358 |
| SN | SMILES | Ki (nM) | pKi (M) |
|---|---|---|---|
| 1 | c1cccc(c12)n(nn2)OC(=O)c(c3)ccc(c34)[nH]cc4 | 7.5 | 8.125 |
| 2 | c1cccc(c12)n(nn2)OC(=O)c3ccc(cc3)N(CC)CC | 11.1 | 7.955 |
| 3 | CNc(cc1)ccc1C(=O)On(nn2)c(c23)cccc3 | 12.1 | 7.917 |
| 4 | c1cccc(c12)n(nn2)OC(=O)c(c3)[nH]c(c34)cccc4 | 12.3 | 7.91 |
| 5 | c1cccc(c12)n(nn2)OC(=O)c(c3)[nH]c(c34)ccc(F)c4 | 13.8 | 7.86 |
| 58 | CCCN(CCC)C(=O)CC[C@@H](C(=O)C(F)(F)F)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)OCc1ccccc1 | 363,000 | 3.44 |
| 59 | CCN(CC)C(=O)CC[C@@H](C(=O)c1nccs1)NC(=O)[C@H](CC(C)C)NC(=O)OCc2ccccc2 | 462,000 | 3.335 |
| 60 | C1COCCN1C(=O)CC[C@@H](C(=O)c2nccs2)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)OCc3ccccc3 | 478,000 | 3.321 |
| 61 | CCCN(CCC)C(=O)CC[C@@H](C(=O)C(F)(F)F)NC(=O)[C@H](CC(C)C)NC(=O)OCc1ccccc1 | 584,000 | 3.234 |
| 62 | CCN(CC)C(=O)CC[C@@H](C(=O)c1nccs1)NC(=O)[C@H](C(C)C)NC(=O)OCc2ccccc2 | 614,000 | 3.212 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Akasapu, S.; Bajaj, S.O.; El-Sayed, N.N.E.; Ghosh, A.; Lewaa, I. Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis. Pharmaceuticals 2021, 14, 357. https://doi.org/10.3390/ph14040357
Zaki MEA, Al-Hussain SA, Masand VH, Akasapu S, Bajaj SO, El-Sayed NNE, Ghosh A, Lewaa I. Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis. Pharmaceuticals. 2021; 14(4):357. https://doi.org/10.3390/ph14040357
Chicago/Turabian StyleZaki, Magdi E. A., Sami A. Al-Hussain, Vijay H. Masand, Siddhartha Akasapu, Sumit O. Bajaj, Nahed N. E. El-Sayed, Arabinda Ghosh, and Israa Lewaa. 2021. "Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis" Pharmaceuticals 14, no. 4: 357. https://doi.org/10.3390/ph14040357
APA StyleZaki, M. E. A., Al-Hussain, S. A., Masand, V. H., Akasapu, S., Bajaj, S. O., El-Sayed, N. N. E., Ghosh, A., & Lewaa, I. (2021). Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis. Pharmaceuticals, 14(4), 357. https://doi.org/10.3390/ph14040357