
Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches

 ,
,  ,
,  ,
, 
 , , ,
, , , 
Abstract
1. Introduction
2. Results and Discussion
2.1. High-Throughput Virtual Screening of Compound Databases
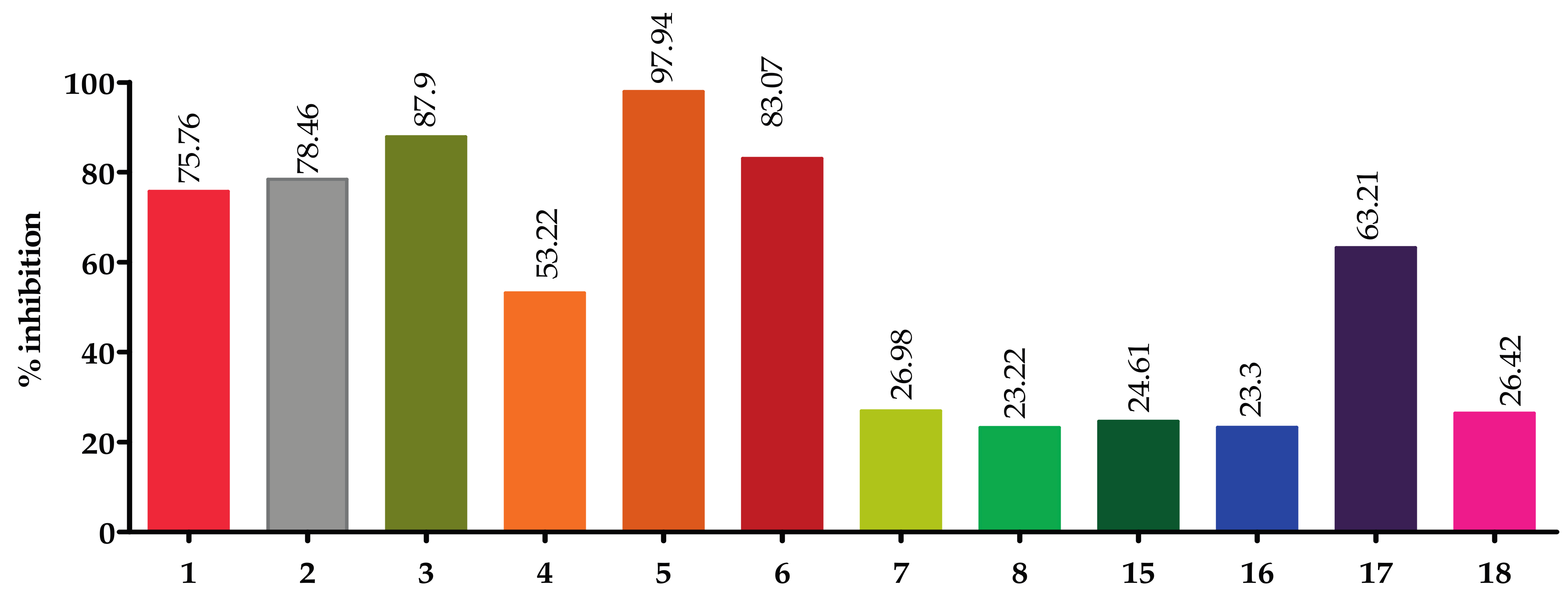
2.2. Results of In-Vitro Inhibition of SARS-CoV-2 MPRO by Selected Natural Product Isolates
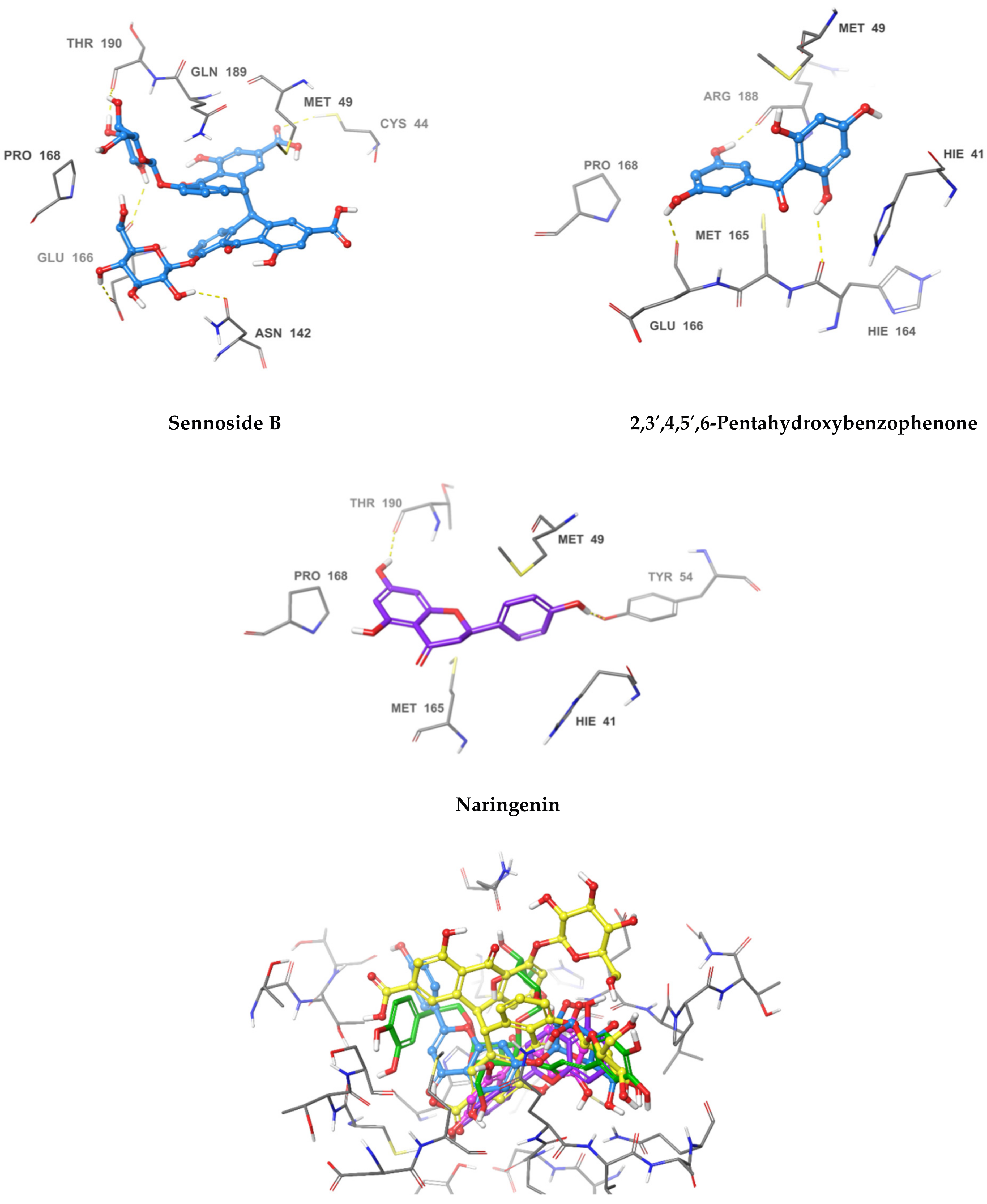
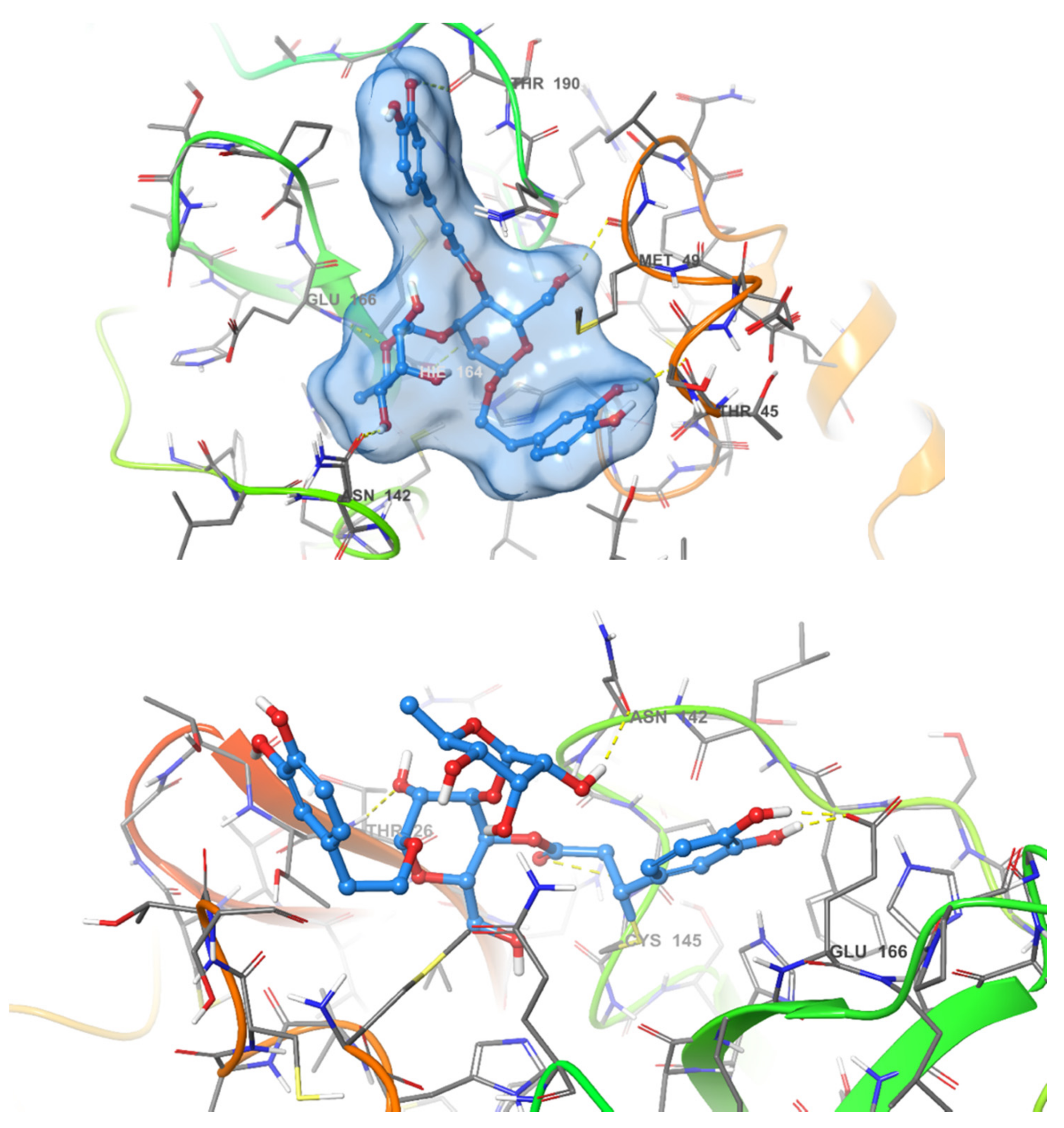
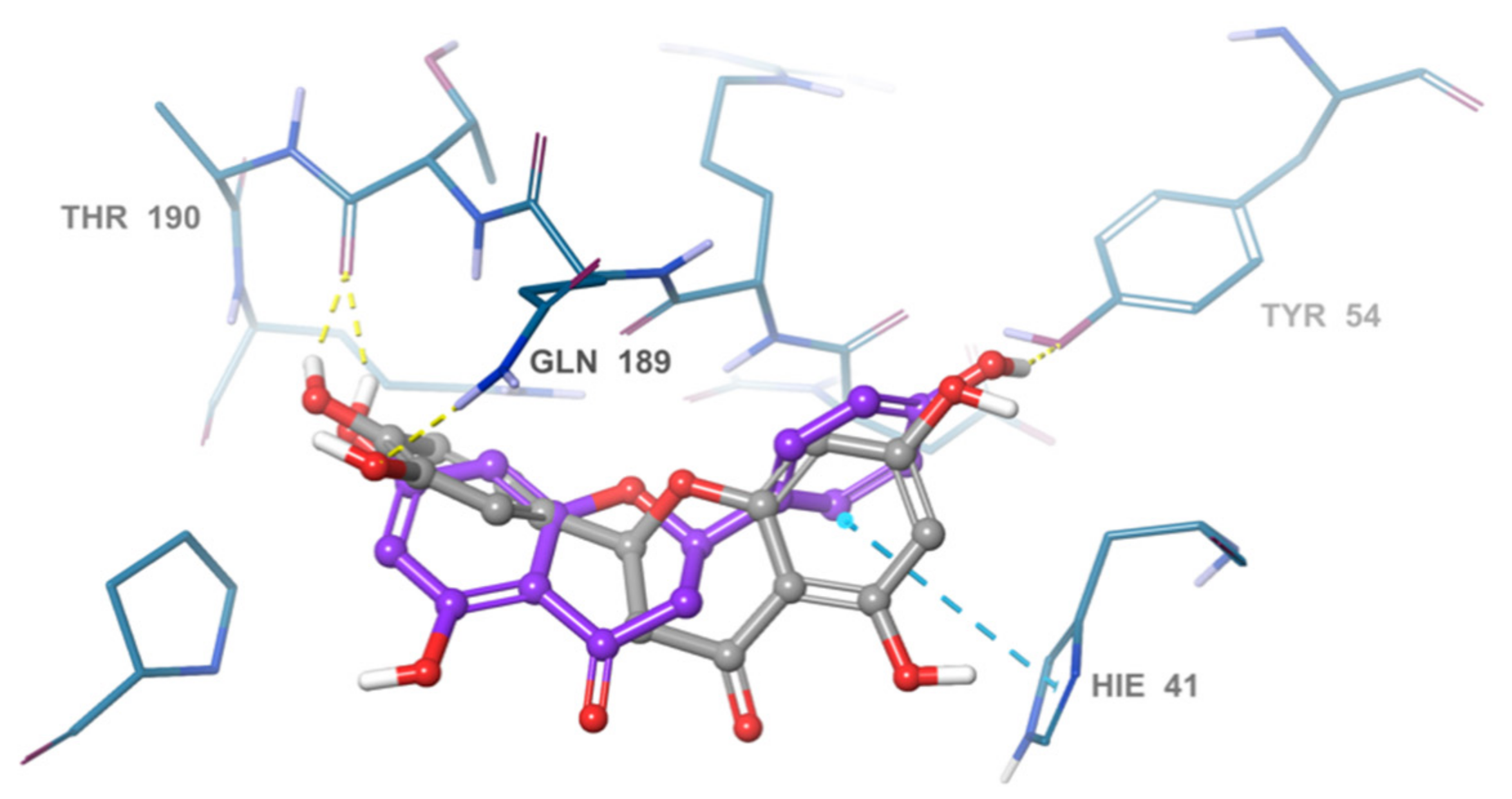
2.3. Pose Prediction of the In Vitro-Active Compounds
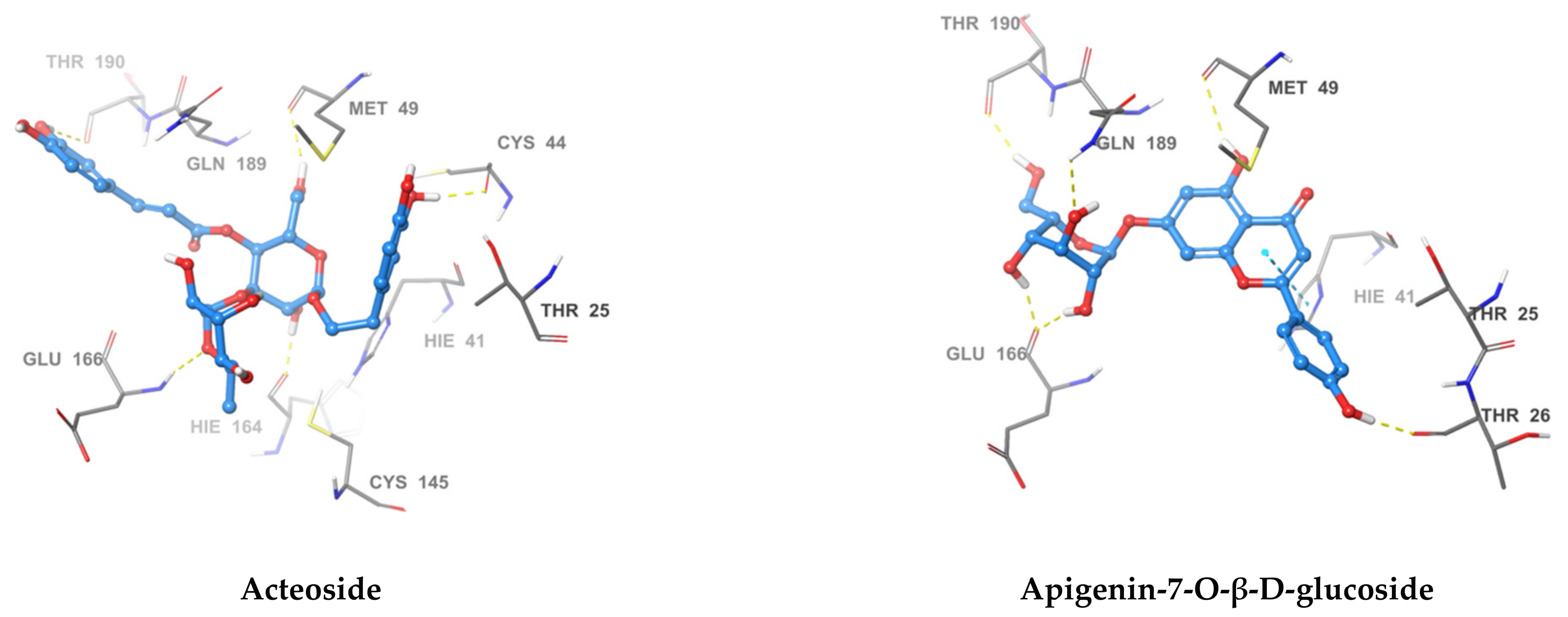
2.4. Acteoside Binds to SARS-CoV-2 MPRO in a Non-Covalent Manner
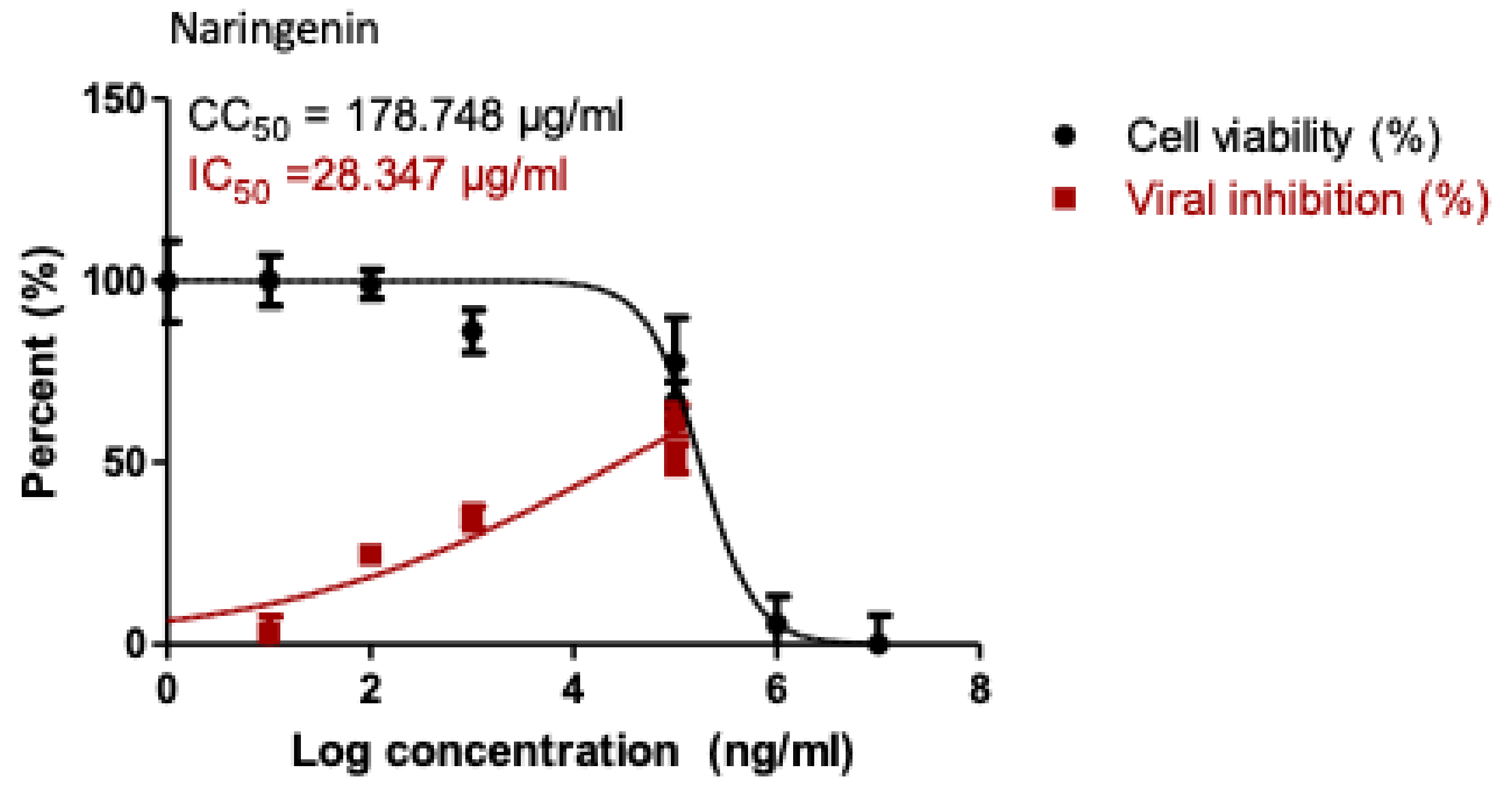
2.5. Antiviral Activity of the Most Potent SARS COV-2 Viral Main Protease Inhibitors
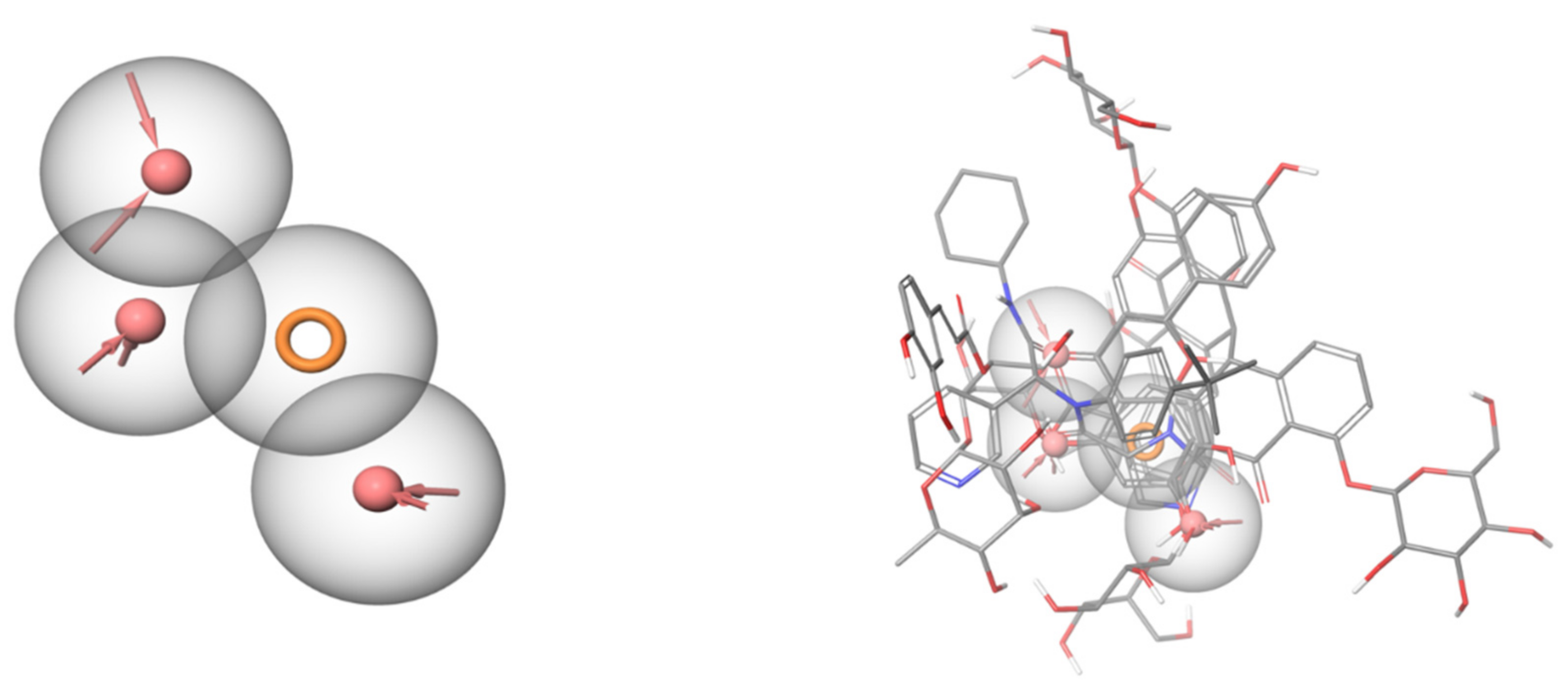
2.6. Pharmacophore Identification
2.7. Drug Likeness
3. Materials and Methods
3.1. Molecular Modeling
3.2. Source of the Tested Isolates
3.3. In Vitro Screening
3.4. Drug Likeness
3.5. Cytotoxicity Assay
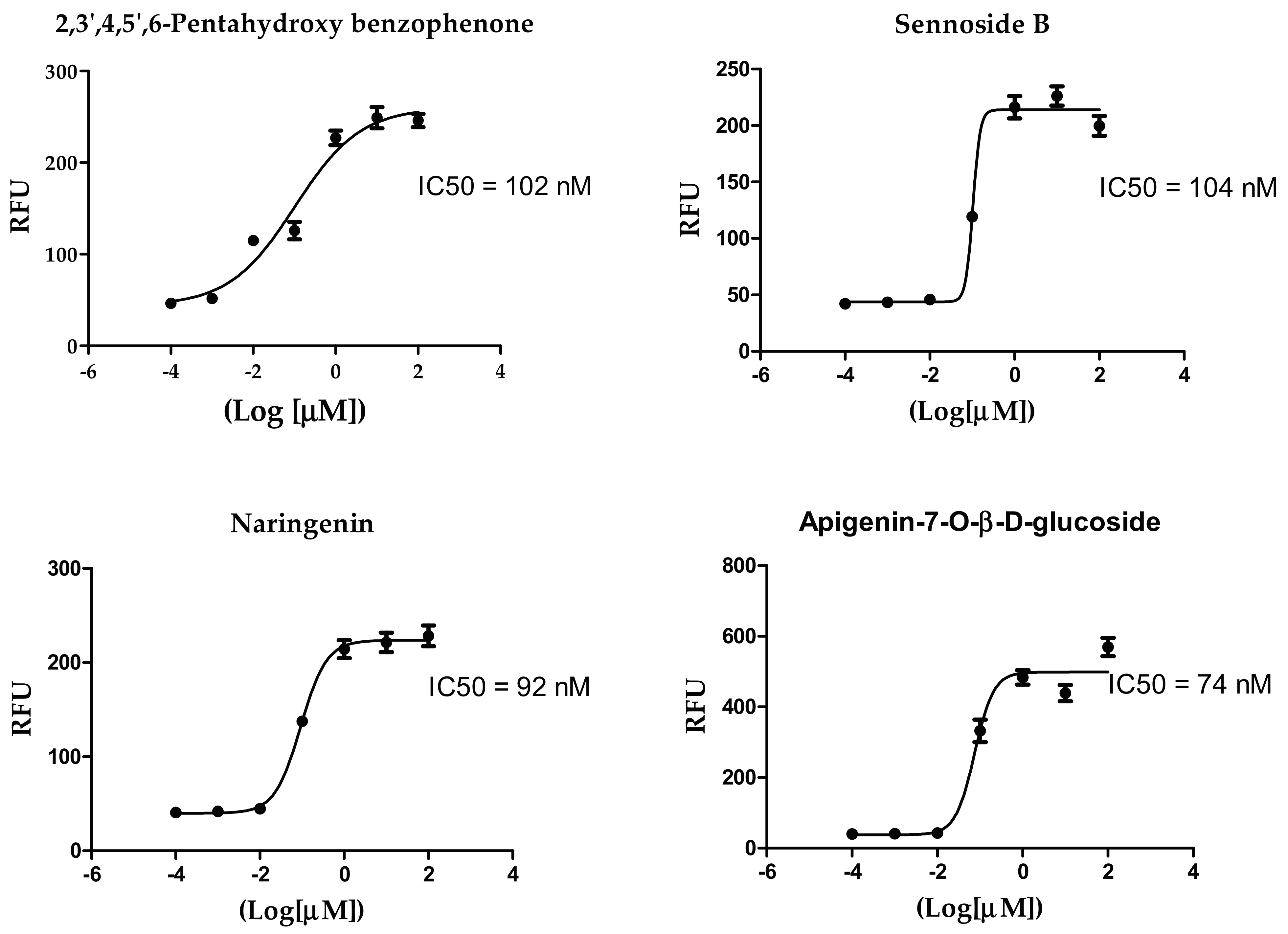
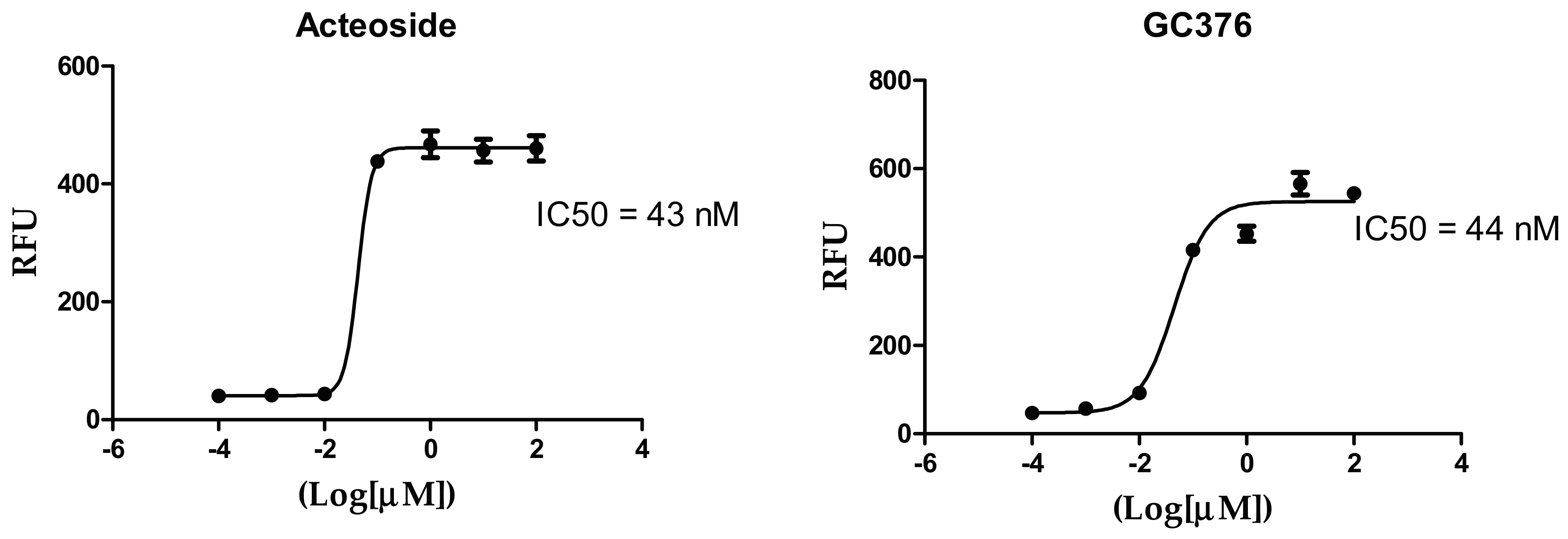
3.6. Inhibitory Concentration 50 (IC50) Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, P.; Lou, Y.X.; Wang, X.; Hu, B.; Zhang, L.; Zhang, W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Fan, W.; Su, Z.; Bin, Y.; Yan-Mei, C.; Wen, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar]
- Gorbalenya, A.; Baker, S.; Baric, R.; de Groot, R.; Drosten, C.; Gulyaeva, A.; Haagmans, B.; Lauber, C.; Leontovich, A.; Neuman, B. CSG of the IC on T. of Viruses. 2020. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Thiel, V.; Herold, J.; Schelle, B.; Siddell, S.G. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 2001, 75, 6676–6681. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Denaro, M.; Smeriglio, A.; Barreca, D.; De Francesco, C.; Occhiuto, C.; Milano, G.; Trombetta, D. Antiviral activity of plants and their isolated bioactive compounds: An update. Phytother. Res. 2020, 34, 742–768. [Google Scholar] [CrossRef] [PubMed]
- Mirzaie, A.; Halaji, M.; Dehkordi, F.S.; Ranjbar, R.; Noorbazargan, H. A narrative literature review on traditional medicine options for treatment of corona virus disease 2019 (COVID-19). Complementary Ther. Clin. Pract. 2020, 40, 101214. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Huang, J.; Yeung, A.W.K.; Tzvetkov, N.T.; Horbańczuk, J.O.; Willschke, H.; Gai, Z.; Atanasov, A.G. The significance of natural product derivatives and traditional medicine for COVID-19. Processes 2020, 8, 937. [Google Scholar] [CrossRef]
- Hong-Zhi, D.; Xiao-Ying, H.; Yu-Huan, M.; Huang, B.-S.; Da-Hui, L. Traditional Chinese Medicine: An effective treatment for 2019 novel coronavirus pneumonia (NCP). Chin. J. Nat. Med. 2020, 18, 206–210. [Google Scholar]
- Koshak, A.E.; Koshak, E.A. Nigella sativa l. as a potential phytotherapy for covid-19: A mini-review of in-silico studies. Curr. Ther. Res. 2020, 93, 100602. [Google Scholar] [CrossRef]
- Lung, J.; Lin, Y.S.; Yang, Y.H.; Chou, Y.L.; Shu, L.H.; Cheng, Y.C.; Liu, H.T.; Wu, C.Y. The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J. Med. Virol. 2020, 92, 693–697. [Google Scholar] [CrossRef]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.; Xie, H.; Ke, C.; Gao, M.; Yu, K. Discovery of baicalin and baicalein as novel, natural product inhibitors of SARS-CoV-2 3CL protease in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Quimque, M.T.J.; Notarte, K.I.R.; Fernandez, R.A.T.; Mendoza, M.A.O.; Liman, R.A.D.; Lim, J.A.K.; Pilapil, L.A.E.; Ong, J.K.H.; Pastrana, A.M.; Khan, A. Virtual Screening-Driven Drug Discovery of SARS-CoV2 Enzyme Inhibitors Targeting Viral Attachment, Replication, Post-Translational Modification and Host Immunity Evasion Infection Mechanisms. J. Biomol. Struct. Dyn. 2020, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC−A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Tahir Ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Grum-Tokars, V.; Ratia, K.; Begaye, A.; Baker, S.C.; Mesecar, A.D. Evaluating the 3C-like protease activity of SARS-Coronavirus: Recommendations for standardized assays for drug discovery. Virus Res. 2008, 133, 63–73. [Google Scholar] [CrossRef]
- Du, Q.S.; Wang, S.Q.; Zhu, Y.; Wei, D.Q.; Guo, H.; Sirois, S.; Chou, K.C. Polyprotein cleavage mechanism of SARS CoV Mpro and chemical modification of the octapeptide. Peptides 2004, 25, 1857–1864. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Tyagi, A.K.; Deshmukh-Taskar, P.; Hinojosa, M.; Prasad, S.; Aggarwal, B.B. Downregulation of tumor necrosis factor and other proinflammatory biomarkers by polyphenols. Arch. Biochem. Biophys. 2014, 559, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zeng, W.; Zhang, F.; Zhang, C.; Liang, W. Naringenin ameliorates acute inflammation by regulating intracellular cytokine degradation. J. Immunol. 2017, 199, 3466–3477. [Google Scholar] [CrossRef]
- Alberca, R.W.; Teixeira, F.M.E.; Beserra, D.R.; de Oliveira, E.A.; de Souza Andrade, M.M.; Pietrobon, A.J.; Sato, M.N. Perspective: The potential effects of naringenin in COVID-19. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Cheng, L.; Zheng, W.; Li, M.; Huang, J.; Bao, S.; Xu, Q.; Ma, Z. Citrus Fruits Are Rich in Flavonoids for Immunoregulation and Potential Targeting ACE2. 2020. Available online: https://www.preprints.org/manuscript/202002.0313/v1 (accessed on 3 March 2021).
- Diaz, J.H. Hypothesis: Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J. Travel Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Clementi, N.; Scagnolari, C.; D’Amore, A.; Palombi, F.; Criscuolo, E.; Frasca, F.; Pierangeli, A.; Mancini, N.; Antonelli, G.; Clementi, M. Naringenin is a powerful inhibitor of SARS-CoV-2 infection in vitro. Pharmacol. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Khaerunnisa, S.; Kurniawan, H.; Awaluddin, R.; Suhartati, S.; Soetjipto, S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints 2020. [Google Scholar] [CrossRef]
- Bai, Y.; Peng, W.; Yang, C.; Zou, W.; Liu, M.; Wu, H.; Fan, L.; Li, P.; Zeng, X.; Su, W. Pharmacokinetics and metabolism of naringin and active metabolite naringenin in rats, dogs, humans, and the differences between species. Front. Pharmacol. 2020, 11, 364. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Beyl, R.A.; Lertora, J.J.; Greenway, F.L.; Ravussin, E.; Ribnicky, D.M.; Poulev, A.; Kennedy, B.J.; Castro, H.F.; Campagna, S.R. Safety and pharmacokinetics of naringenin: A randomized, controlled, single-ascending-dose clinical trial. Diabetes Obes. Metab. 2020, 22, 91–98. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Zakeri, M.P.; Tajerzadeh, H.; Eslamboulchilar, Z.; Barzegar, S.; Valizadeh, H. The relation between molecular properties of drugs and their transport across the intestinal membrane. DARU J. Pharm. Sci. 2006, 14, 164–171. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, G.A.; Al-Abd, A.M.; El-Halawany, A.M.; Abdallah, H.M.; Ibrahim, S.R. New xanthones and cytotoxic constituents from Garcinia mangostana fruit hulls against human hepatocellular, breast, and colorectal cancer cell lines. J. Ethnopharmacol. 2017, 198, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.; Abdallah, H.M.; El-Halawany, A.M.; Nafady, A.M.; Mohamed, G.A. Mangostanaxanthone VIII, a new xanthone from Garcinia mangostana and its cytotoxic activity. Nat. Prod. Res. 2019, 33, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Elghamdi, A.A.; Abdallah, H.M.; Shehata, I.A.; Mohamed, G.A.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Koshak, A.E.; Ibrahim, S.R. Cyclocuneatol and Cuneatannin, New Cycloartane Triterpenoid and Ellagitannin Glycoside from Euphorbia Cuneata. Chem. Select 2019, 4, 12375–12379. [Google Scholar] [CrossRef]
- El-Halawany, A.M.; Abdallah, H.M.; Hamed, A.R.; Khalil, H.E.; Almohammadi, A.M. Phenolics from Barleria cristata var. Alba as carcinogenesis blockers against menadione cytotoxicity through induction and protection of quinone reductase. BMC Complementary Altern. Med. 2018, 18, 163. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.; Mohamed, G.A.; Elkhayat, E.S.; Fouad, M.A.; Proksch, P. Sagitol C, a new cytotoxic pyridoacridine alkaloid from the sponge Oceanapia sp. Bull. Fac. Pharm. Cairo Univ. 2013, 51, 229–232. [Google Scholar] [CrossRef]
- Ibrahim, S.R.; Mohamed, G.A.; Ross, S.A. Aspernolides L and M, new butyrolactones from the endophytic fungus Aspergillus versicolor. Z. Naturforsch C. 2017, 72, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Elkhayat, E.S.; Ibrahim, S.R.; Mohamed, G.A.; Ross, S.A. Terrenolide S, a new antileishmanial butenolide from the endophytic fungus Aspergillus terreus. Nat. Prod. Res. 2016, 30, 814–820. [Google Scholar] [CrossRef]
- Ibrahim, S.R.; Mohamed, G.A. Ingenines C and D, new cytotoxic pyrimidine-β-carboline alkaloids from the Indonesian sponge Acanthostrongylophora ingens. Phytochem. Lett. 2016, 18, 168–171. [Google Scholar] [CrossRef]
- Zhu, L.; George, S.; Schmidt, M.F.; Al-Gharabli, S.I.; Rademann, J.; Hilgenfeld, R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antivir. Res. 2011, 92, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound Name | Structure | Glide G-Score |
|---|---|---|---|
| 1 | Acteoside |  | −10.127 |
| 2 | Sennoside B |  | −9.009 |
| 3 | 2,3′,4,5′,6-Pentahydroxy benzophenone |  | −8.341 |
| 4 | Ochnaflavone-4′-methyl ether |  | −8.310 |
| 5 | Naringenin |  | −7.824 |
| 6 | Apigenin-7-O-β-d-glucoside |  | −7.562 |
| 7 | Sagitol C |  | −7.505 |
| 8 | Maclurin-6-O-β-d-glucopyranoside (Rhodanthenone) |  | −7.441 |
| 15 | Iso-Acteoside |  | −7.724 |
| 16 | Kaempferol-7-O-β-d-glucoside |  | −7.206 |
| 17 | Thymoquinon |  | −6.693 |
| 18 | Oleuropein |  | −5.509 |
| Compound Name | IC50 (nM) |
|---|---|
| 2,3′,4,5′,6-Pentahydroxybenzophenone | 102 |
| Sennoside B | 104 |
| Naringenin | 92 |
| Apigenin-7-O-β-d-glucoside | 74 |
| Acteoside | 43 |
| GC376 (positive control) | 44 |
| Compound Name | Hydrogen Bond | Pi-Pi Stacking | Van der Waal Interactions | XP GScore |
|---|---|---|---|---|
| Acteoside | Cys44, Met49, Asn142, Hie164, Glu166, Thr190 | Pro168, Glu189, Thr25, Hie41, Cys145, Met49 | −12.341 | |
| Apigenin-7-O-β-d-glucoside | Thr26, Met49, Glu166, Gln189, Thr190 | Hie41 | Met49, Thr25, Hie41 | −10.834 |
| Sennoside B | Thr190, Glu166, Asn142, Cys44 | Pro168, Gln189, Met49 | −10.336 | |
| 2,3′,4,5′,6-pentahydroxybenzophenone | Hie164, Glu166, Arg188, Tyr54 | Hie41, Met49, Met165, Pro168 | −8.105 | |
| Naringenin | Tyr54 Thr190 | Hie41, Met49, Met165, Pro168 | −7.083 |
| Compound | miLogP | TPSA | natoms | MW | nON | nOHNH | nrotb | % ABS |
|---|---|---|---|---|---|---|---|---|
| Acetoside | 0.4 | 245.2 | 44 | 624.5 | 15 | 9 | 11 | 24.4% |
| Sennoside B | 0.8 | 347.9 | 62 | 862.7 | 20 | 12 | 9 | - |
| Naringenin | 2.1 | 86.9 | 20 | 272.2 | 5 | 3 | 1 | 78.9% |
| Apigenin-7-glucoside | 0.6 | 170.0 | 31 | 432.3 | 10 | 6 | 4 | 50.35 |
| 2,3′,4,5′,6-Pentahydroxybenzophenone | 1.7 | 118.2 | 19 | 262.2 | 6 | 5 | 2 | 68.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdallah, H.M.; El-Halawany, A.M.; Sirwi, A.; El-Araby, A.M.; Mohamed, G.A.; Ibrahim, S.R.M.; Koshak, A.E.; Asfour, H.Z.; Awan, Z.A.; A. Elfaky, M. Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches. Pharmaceuticals 2021, 14, 213. https://doi.org/10.3390/ph14030213
Abdallah HM, El-Halawany AM, Sirwi A, El-Araby AM, Mohamed GA, Ibrahim SRM, Koshak AE, Asfour HZ, Awan ZA, A. Elfaky M. Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches. Pharmaceuticals. 2021; 14(3):213. https://doi.org/10.3390/ph14030213
Chicago/Turabian StyleAbdallah, Hossam M., Ali M. El-Halawany, Alaa Sirwi, Amr M. El-Araby, Gamal A. Mohamed, Sabrin R. M. Ibrahim, Abdulrahman E. Koshak, Hani Z. Asfour, Zuhier A. Awan, and Mahmoud A. Elfaky. 2021. "Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches" Pharmaceuticals 14, no. 3: 213. https://doi.org/10.3390/ph14030213
APA StyleAbdallah, H. M., El-Halawany, A. M., Sirwi, A., El-Araby, A. M., Mohamed, G. A., Ibrahim, S. R. M., Koshak, A. E., Asfour, H. Z., Awan, Z. A., & A. Elfaky, M. (2021). Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches. Pharmaceuticals, 14(3), 213. https://doi.org/10.3390/ph14030213