
Evaluation of the Interactions between Human Serum Albumin (HSA) and Non-Steroidal Anti-Inflammatory (NSAIDs) Drugs by Multiwavelength Molecular Fluorescence, Structural and Computational Analysis

 , , , and
, , , and 
Abstract
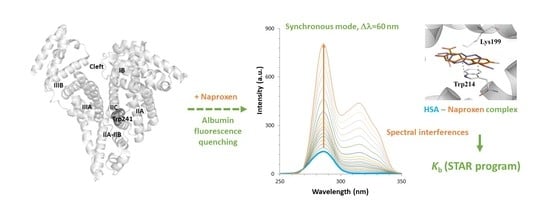
1. Introduction
2. Results and Discussion
2.1. Setup of Experimental Conditions
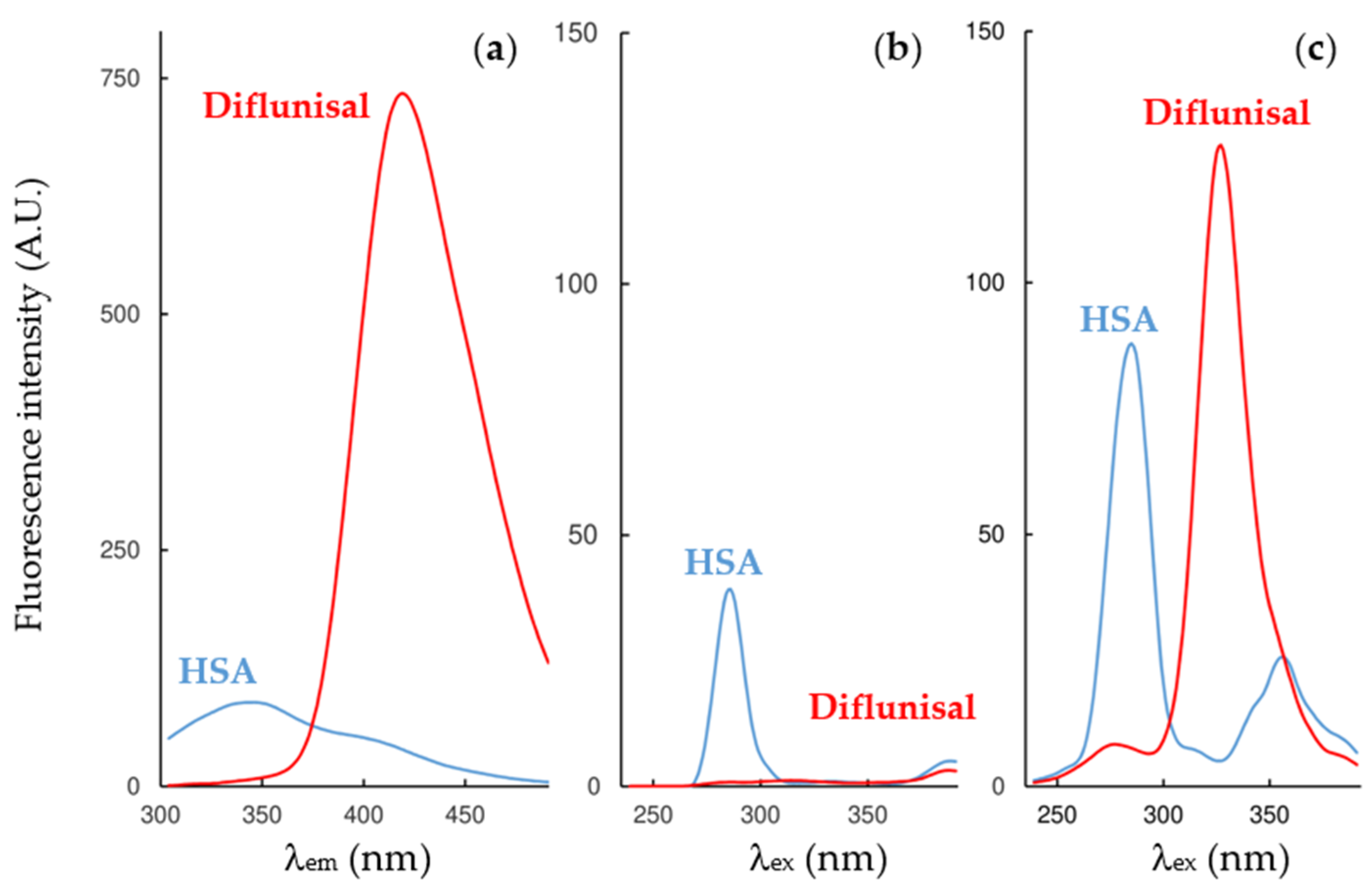
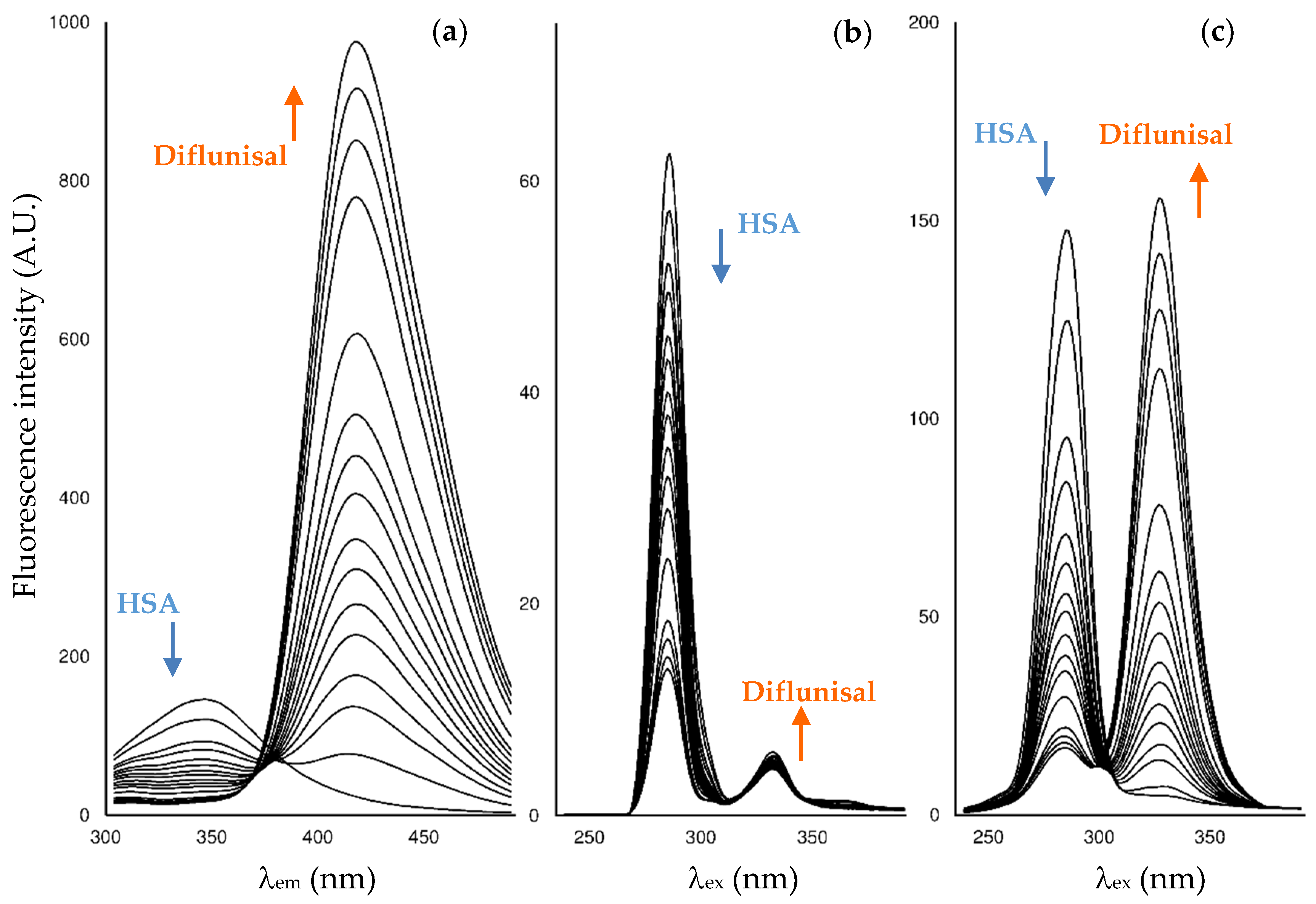
2.2. Fluorescence Measurements for Drug-Albumin Interaction
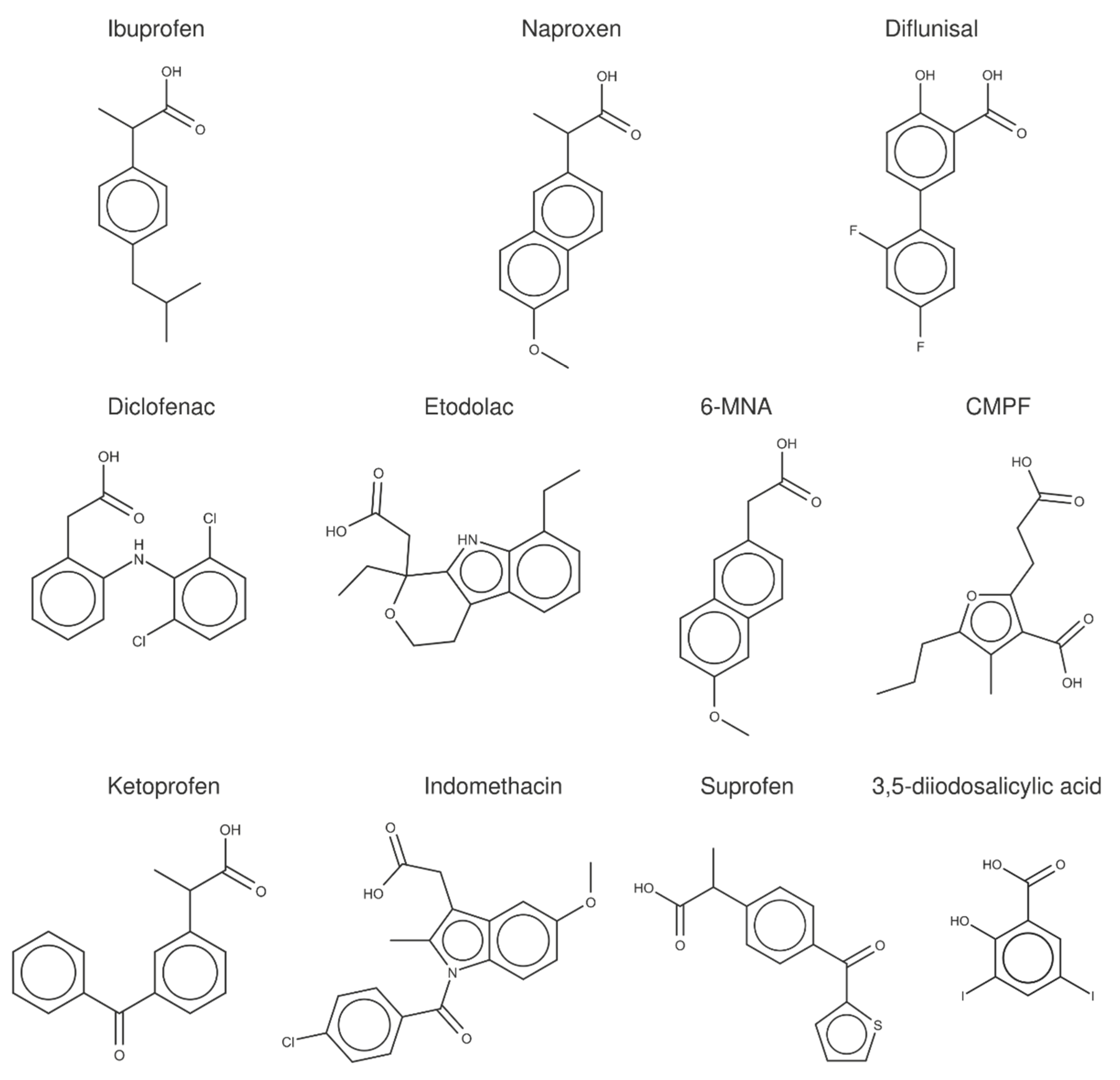
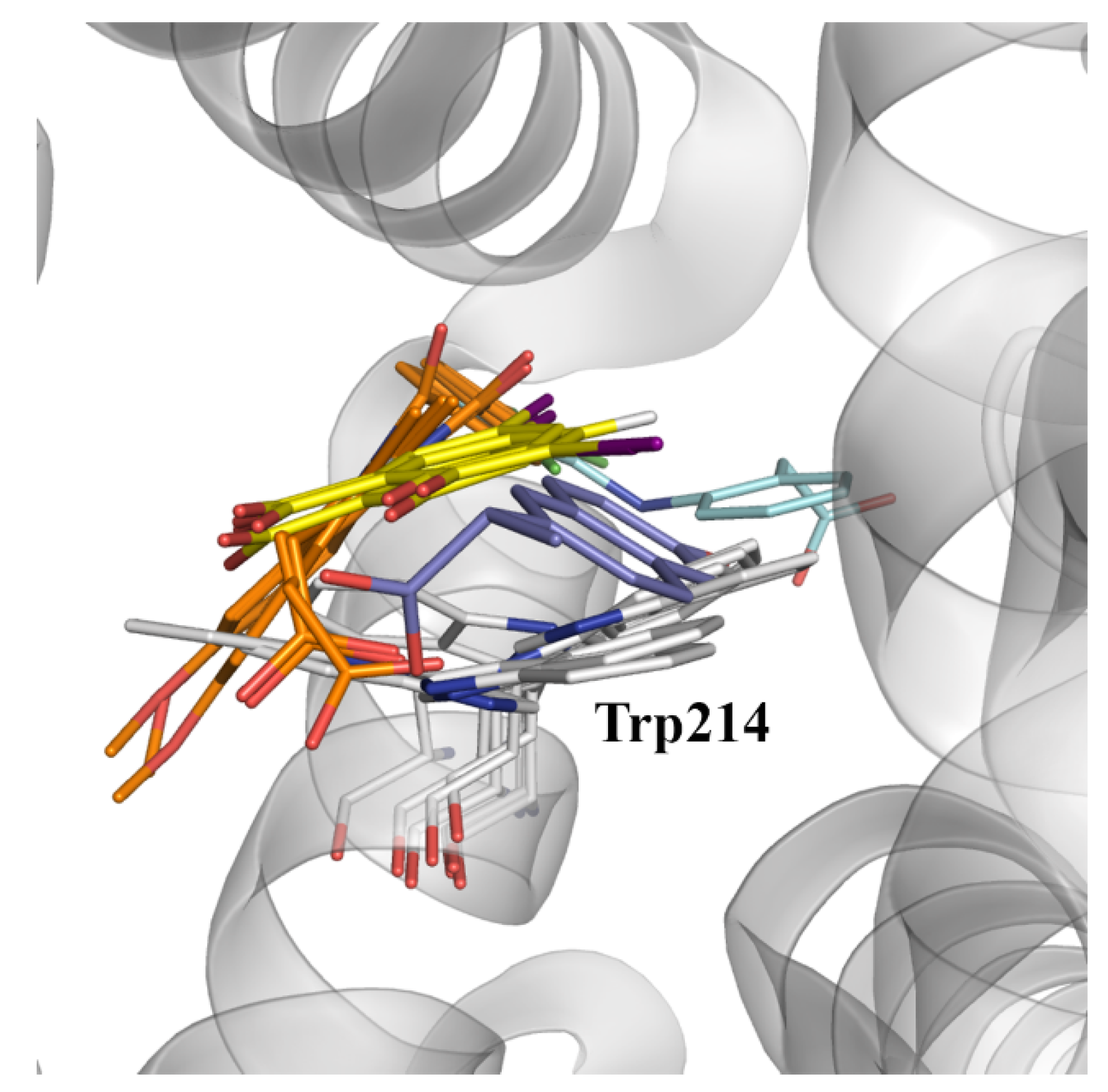
2.3. Structural Analysis of NSAID-HSA Complexes
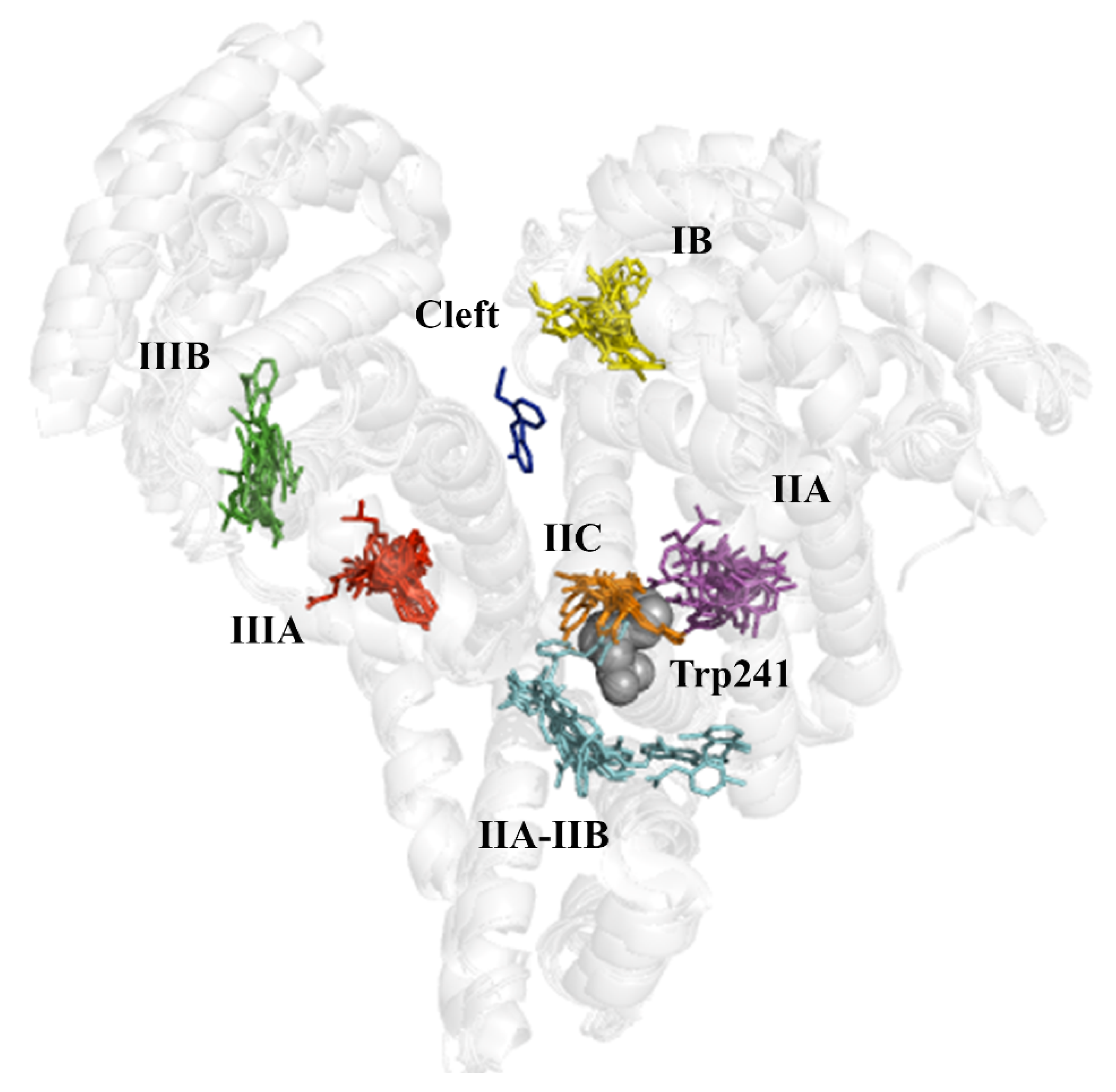
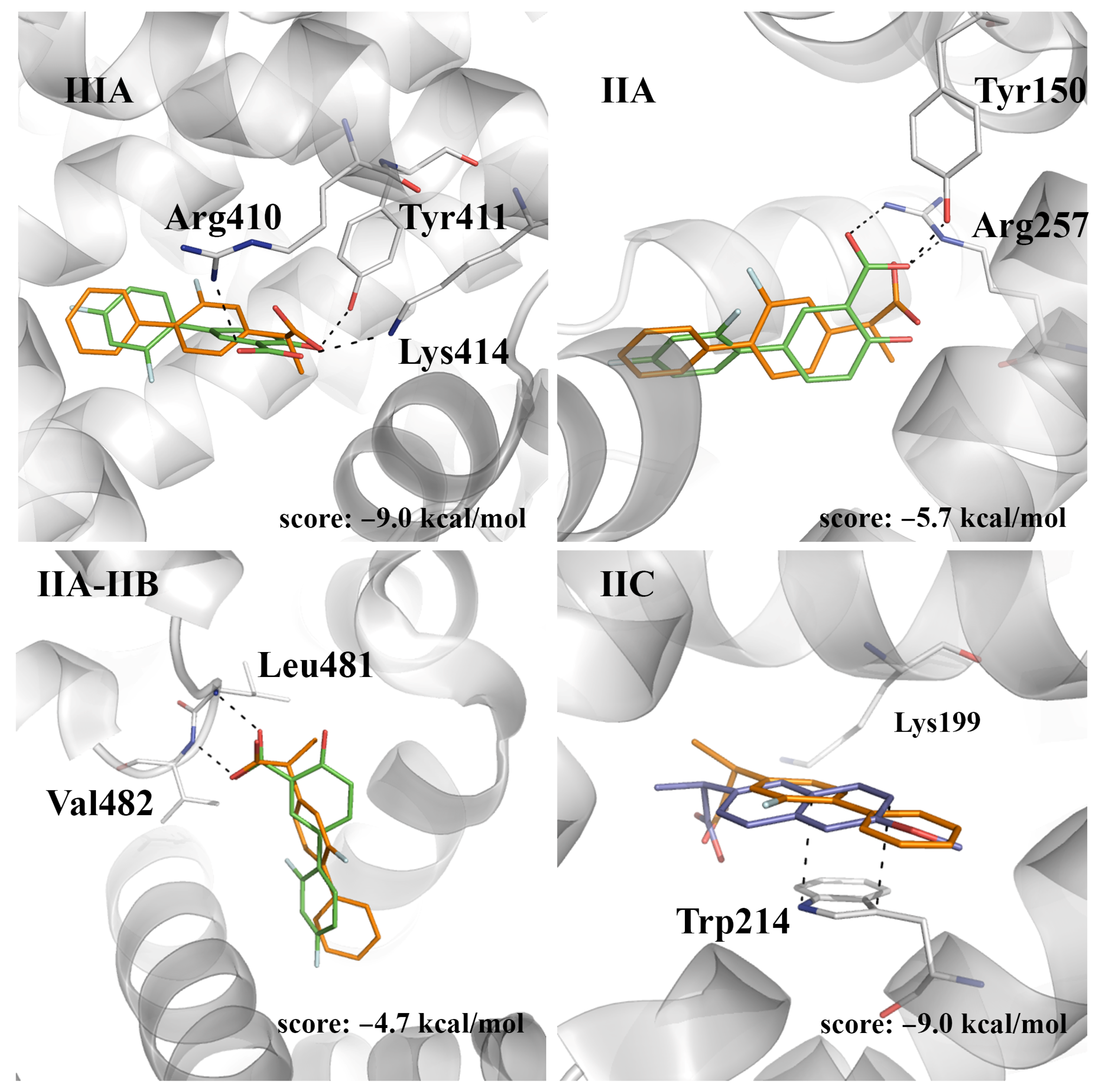
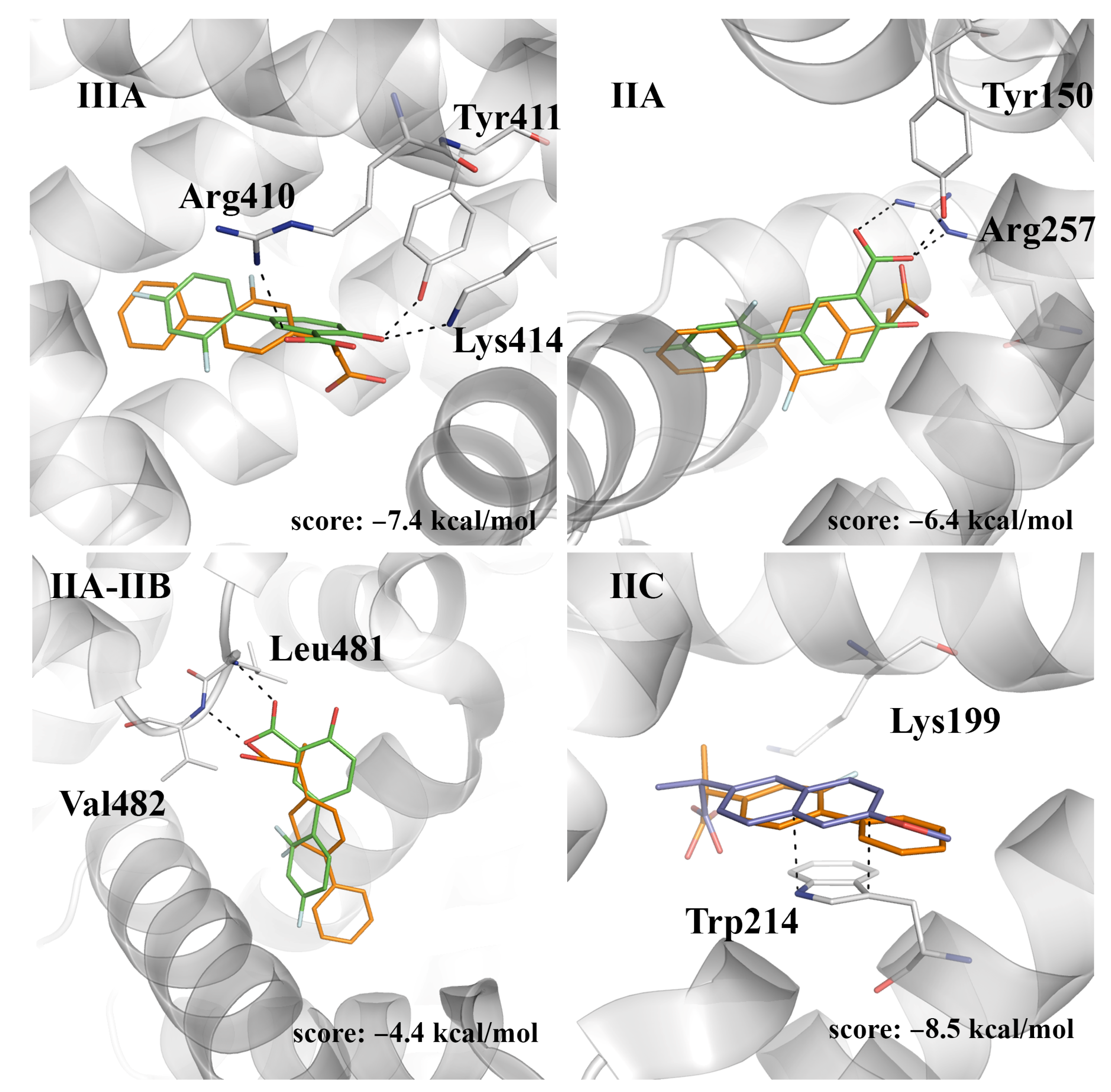
2.4. Evaluation of the Ligand Binding Events for NSAID-HSA Complexes
2.5. Prospective Study of the Flurbiprofen-HSA Complex
3. Materials and Methods
3.1. Equipment
3.2. Reagents
3.3. Fluorescence Quenching/Enhancement Measurement
3.4. Fluorescence Quenching/Enhancement Measurement
3.4.1. Double Logarithm Stern–Volmer Equation
3.4.2. STAR Program
3.4.3. Docking Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FDA. The Drug Development Process. Available online: https://www.fda.gov/ForPatients/Approvals/Drugs/default.htm (accessed on 15 January 2021).
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin-drug interaction and its clinical implication. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5435–5443. [Google Scholar] [CrossRef] [PubMed]
- Yeggoni, D.P.; Gokara, M.; Mark Manidhar, D.; Rachamallu, A.; Nakka, S.; Reddy, C.S.; Subramanyam, R. Binding and molecular dynamics studies of 7-hydroxycoumarin derivatives with human serum albumin and its pharmacological importance. Mol. Pharm. 2014, 11, 1117–1131. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fitos, I.; Hage, D.S. Chromatographic analysis of allosteric effects between ibuprofen and benzodiazepines on human serum albumin. Chirality 2006, 18, 24–36. [Google Scholar] [CrossRef]
- Yang, F.; Yue, J.; Ma, L.; Ma, Z.; Li, M.; Wu, X.; Liang, H. Interactive associations of drug-drug and drug-drug-drug with iia subdomain of human serum albumin. Mol. Pharm. 2012, 9, 3259–3265. [Google Scholar] [CrossRef]
- Faisal, Z.; Vörös, V.; Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Csepregi, R.; Koszegi, T.; Zsila, F.; Poór, M. Probing the interactions of ochratoxin b, ochratoxin c, patulin, deoxynivalenol, and t-2 toxin with human serum albumin. Toxins 2020, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Ràfols, C.; Amézqueta, S.; Fuguet, E.; Bosch, E. Molecular interactions between warfarin and human (HSA) or bovine (BSA) serum albumin evaluated by isothermal titration calorimetry (ITC), fluorescence spectrometry (FS) and frontal analysis capillary electrophoresis (FA/CE). J. Pharm. Biomed. Anal. 2018, 150, 452–459. [Google Scholar] [CrossRef]
- Van De Weert, M.; Stella, L. Fluorescence quenching and ligand binding: A critical discussion of a popular methodology. J. Mol. Struct. 2011, 998, 145–150. [Google Scholar] [CrossRef]
- Beltrán, J.L.; Codony, R.; Prats, M.D. Evaluation of stability constants from multi-wavelength absorbance data: Program STAR. Anal. Chim. Acta 1993, 276, 441–454. [Google Scholar] [CrossRef]
- Amézqueta, S.; Bolioli, A.M.; Beltrán, J.L.; Ràfols, C. Evaluation of the interactions between human serum albumin (HSA) and warfarin or diflunisal by using molecular fluorescence using two approaches. ADMET DMPK 2018, 6, 47–54. [Google Scholar] [CrossRef]
- Ràfols, C.; Zarza, S.; Bosch, E. Molecular interactions between some non-steroidal anti-inflammatory drugs (NSAID’s) and bovine (BSA) or human (HSA) serum albumin estimated by means of isothermal titration calorimetry (ITC) and frontal analysis capillary electrophoresis (FA/CE). Talanta 2014, 130, 241–250. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef]
- Greige-Gerges, H.; Khalil, R.A.; Mansour, E.A.; Magdalou, J.; Chahine, R.; Ouaini, N. Cucurbitacins from Ecballium elaterium juice increase the binding of bilirubin and ibuprofen to albumin in human plasma. Chem. Biol. Interact. 2007, 169, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lammers, I.; Lhiaubet-Vallet, V.; Ariese, F.; Miranda, M.A.; Gooijer, C. Binding of naproxen enantiomers to human serum albumin studied by fluorescence and room-temperature phosphorescence. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 105, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Ma, Z.Y.; Zhang, Y.; Li, G.Q.; Li, M.; Qin, J.K.; Lockridge, O.; Liang, H. Human serum albumin-based design of a diflunisal prodrug. Eur. J. Pharm. Biopharm. 2013, 84, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Fountoulaki, S.; Perdih, F.; Turel, I.; Kessissoglou, D.P.; Psomas, G. Non-steroidal anti-inflammatory drug diflunisal interacting with Cu(II). Structure and biological features. J. Inorg. Biochem. 2011, 105, 1645–1655. [Google Scholar] [CrossRef]
- Davilas, A.; Koupparis, M.; Macheras, P.; Valsami, G. In-vitro study on the competitive binding of diflunisal and uraemic toxins to serum albumin and human plasma using a potentiometric ion-probe technique. JPP 2006, 58, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Curry, S. Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug Metab. Pharmacokinet. 2009, 24, 342–357. [Google Scholar] [CrossRef]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef]
- Mansoor, S.E.; Dewitt, M.A.; Farrens, D.L. Distance mapping in proteins using fluorescence spectroscopy: The tryptophan-induced quenching (TrIQ) method. Biochemistry 2010, 49, 9722–9731. [Google Scholar] [CrossRef]
- Zielinski, K.; Sekula, B.; Bujacz, A.; Szymczak, I. Structural investigations of stereoselective profen binding by equine and leporine serum albumins. Chirality 2020, 32, 334–344. [Google Scholar] [CrossRef]
- Lammers, I.; Lhiaubet-Vallet, V.; Jiménez, M.C.; Ariese, F.; Miranda, M.A.; Gooijer, C. Stereoselective binding of flurbiprofen enantiomers and their methyl esters to human serum albumin studied by time-resolved phosphorescence. Chirality 2012, 24, 840–846. [Google Scholar] [CrossRef]
- Vayá, I.; Bonancía, P.; Jiménez, M.C.; Markovitsi, D.; Gustavsson, T.; Miranda, M.A. Excited state interactions between flurbiprofen and tryptophan in drug-protein complexes and in model dyads. Fluorescence studies from the femtosecond to the nanosecond time domains. Phys. Chem. Chem. Phys. 2013, 15, 4727–4734. [Google Scholar] [CrossRef]
- Pinheiro, S.; Curutchet, C. Can Förster Theory Describe Stereoselective Energy Transfer Dynamics in a Protein-Ligand Complex? J. Phys. Chem. B 2017, 121, 2265–2278. [Google Scholar] [CrossRef]
- Droge, J.H.M.; Janssen, L.H.M.; Wilting, J. The fatty-acid-induced conformational states of human serum albumin investigated by means of multiple co-binding of protons and oleic acid. Biochem. J. 1988, 250, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Chadborn, N.; Bryant, J.; Bain, A.J.; O’Shea, P. Ligand-dependent conformational equilibria of serum albumin revealed by tryptophan fluorescence quenching. Biophys. J. 1999, 76, 2198–2207. [Google Scholar] [CrossRef]
- Otosu, T.; Nishimoto, E.; Yamashita, S. Multiple conformational state of human serum albumin around single tryptophan residue at various pH revealed by time-resolved fluorescence spectroscopy. J. Biochem. 2010, 147, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Thermo-Scientific Tech Tip 6. Extinction Coefficients. Available online: http://tools.thermofisher.com/content/sfs/brochures/TR0006-Extinction-coefficients.pdf (accessed on 15 January 2021).
- Huang, C.; Gjelstad, A.; Seip, K.F.; Jensen, H.; Pedersen-Bjergaard, S. Exhaustive and stable electromembrane extraction of acidic drugs from human plasma. J. Chromatogr. A 2015, 1425, 81–87. [Google Scholar] [CrossRef]
- Patel, D.S.; Sharma, N.; Patel, M.C.; Patel, B.N.; Shrivastav, P.S.; Sanyal, M. Sensitive and Selective Determination of Diflunisal in Human Plasma by LC—MS. J. Chromatogr. Sci. 2013, 51, 872–882. [Google Scholar] [CrossRef]
- Bi, S.; Yan, L.; Pang, B.; Wang, Y. Investigation of three flavonoids binding to bovine serum albumin using molecular fluorescence technique. J. Lumin. 2012, 132, 132–140. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Li, H. Octyl gallate: An antioxidant demonstrating selective and sensitive fluorescent property. Food Chem. 2017, 219, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Dutta, S.; Maity, S.S.; Ghosh, S.; Singha Roy, A.; Ghosh, K.S.; Dasgupta, S. Spectroscopic and docking studies of the binding of two stereoisomeric antioxidant catechins to serum albumins. J. Lumin. 2012, 132, 1364–1375. [Google Scholar] [CrossRef]
- Trnková, L.; Boušová, I.; Staňková, V.; Dršata, J. Study on the interaction of catechins with human serum albumin using spectroscopic and electrophoretic techniques. J. Mol. Struct. 2011, 985, 243–250. [Google Scholar] [CrossRef]
- Li, G.; Liu, B.; Zhang, Q.; Han, R. Investigation on the effect of fluorescence quenching of bovine serum albumin by cefoxitin sodium using fluorescence spectroscopy and synchronous fluorescence spectroscopy. Luminescence 2016, 31, 1054–1062. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-2: Glide; Schrödinger LLC: New York, NY, USA, 2019.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A New approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-3: Maestro; Schrödinger LLC: New York, NY, USA, 2019.
- Schrödinger Release 2016-3: LigPrep; Schrödinger LLC: New York, NY, USA, 2016.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger LLC: New York, NY, USA, 2016.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
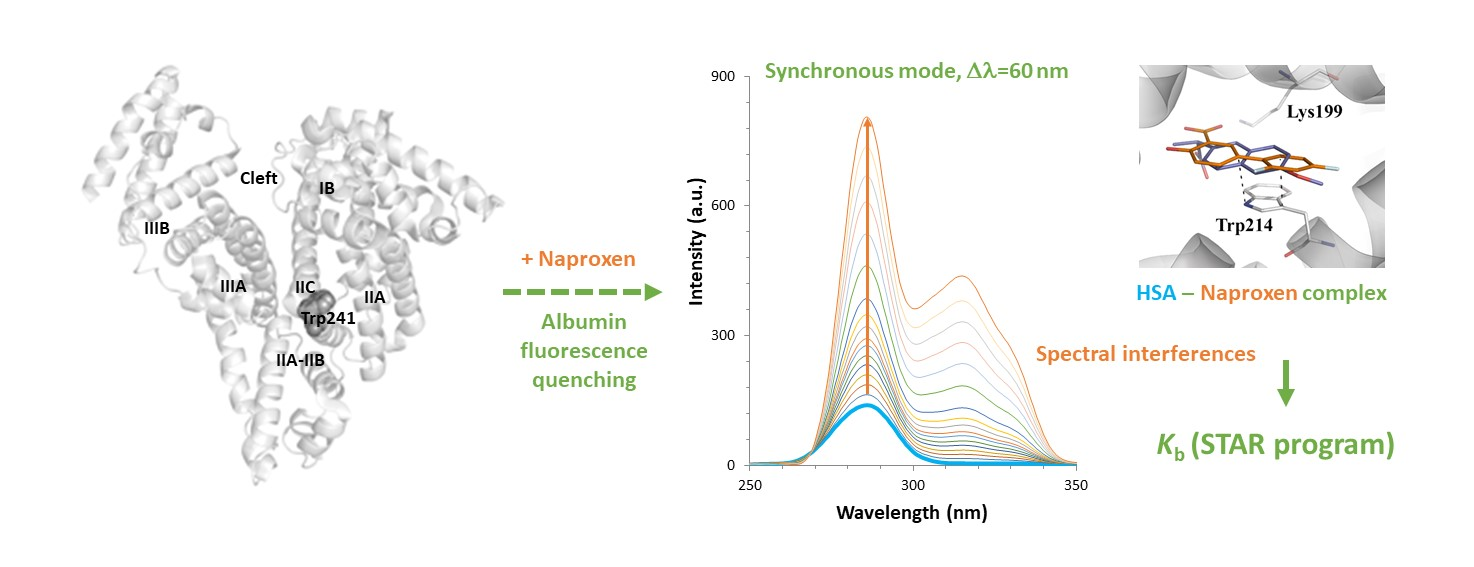
| Fluorescence Mode | Emission Wavelength | Ibuprofen | Flurbiprofen | Naproxen | Diflunisal |
|---|---|---|---|---|---|
| Emission mode, λex = 285 nm | Maximum | 346 nm (interf. 7.9%) | 346 nm (interf. 53%) | 346 nm (interf. 91%) | 346 nm (interf. 8.2%) |
| Optimum | As maximum | - 1 | 322 nm (interf. 5.3%) | As maximum | |
| Synchronous mode, ∆λ = 15 nm | Maximum | 286 nm (interf. 1.1%) | 286 nm (interf. 85%) | 286 nm (interf. 6.4%) | 285 nm (interf. 2.1%) |
| Optimum | As maximum | - | As maximum | As maximum | |
| Synchronous mode, ∆λ = 60 nm | Maximum | 286 nm (interf. 7.4%) | 286 nm (interf. 52%) | 286 nm (interf. 90%) | 285 nm (interf. 7.9%) |
| Optimum | As maximum | - | - | As maximum |
| System | Emission | Synchronous Δλ = 15 nm | Synchronous Δλ = 60 nm | |||
|---|---|---|---|---|---|---|
| nH1 | log Kb1 | nH1 | log Kb1 | nH1 | log Kb1 | |
| HSA-Ibuprofen | NQ 1 | NQ | NQ | NQ | NQ | NQ |
| HSA-Flurbiprofen | - 2 | - | - | - | - | - |
| HSA-Naproxen | 0.94 (0.01) | 4.2 (0.1) 3 | 0.89 (0.01) | 3.70 (0.08) 3 | - | - |
| HSA-Diflunisal | 1.01 (0.02) | 5.2 (0.1) 3 | 1.09 (0.02) | 5.1 (0.1) 3 | 1.04 (0.02) | 5.3 (0.1) 3 |
| System | Temperature | log Kb1 | log Kb2 |
|---|---|---|---|
| HSA-Ibuprofen | 20–37 °C | ND 1 | |
| HSA-Flurbiprofen 2 | 20 °C | 4.91 (0.01) | 5.53 (0.01) |
| 25 °C | 4.96 (0.02) | 5.52 (0.01) | |
| 37 °C | 4.98 (0.02) | 5.22 (0.01) | |
| HSA-Naproxen | 20 °C | 4.88 (0.01) | |
| 25 °C | 4.80 (0.01) | ||
| 37 °C | 4.66 (0.01) | ||
| HSA-Diflunisal | 20 °C | 5.74 (0.01) | 4.57 (0.01) |
| 25 °C | 5.86 (0.01) | 4.70 (0.01) | |
| 37 °C | 5.62 (0.01) | 4.49 (0.01) |
| Drug | n1 | log Kb1 | n2 | log Kb2 | n3 | log Kb3 |
|---|---|---|---|---|---|---|
| Ibuprofen | ||||||
| HSA ITC | 0.84 | 5.95 | - | - | - | - |
| HSA CE/FA | - | - | 5.2 | 4.9 | - | - |
| BSA ITC | 0.8 | 5.90 | - | - | - | - |
| BSA CE/FA | - | - | 7.2 | 4.2 | - | - |
| Naproxen | ||||||
| HSA ITC | 1.00 | 5.95 | 2.5 | 4.85 | - | - |
| HSA CE/FA | - | - | 3.5 | 4.9 | - | - |
| BSA ITC | 0.81 | 7.17 | - | - | - | - |
| BSA CE/FA | - | - | 4.0 | 4.2 | - | - |
| Flurbiprofen | ||||||
| HSA ITC | 0.71 | 6.70 | 4.8 | 4.78 | - | - |
| HSA CE/FA | - | - | 5.0 | 4.5 | - | - |
| BSA ITC | 0.80 | 6.3 | - | - | - | - |
| BSA CE/FA | - | - | 6.4 | 4.3 | 8.6 | 3.9 |
| Drug | n1 | log Kb1 | pH | T (°C) | Buffer (Concentration) | Ref. |
|---|---|---|---|---|---|---|
| Ibuprofen | 1 | 3.92 1 | 7.4 | 25 | PBS (50 mM) | [14] |
| Ibuprofen | 1.18 | 6.39 1 | 7.4 | 25 | PBS (67 mM) | [15] |
| Naproxen | 1 | 5.59 | 7.4 | NA 2 | PBS (10 mM) | [16] |
| Diflunisal | 1 | 5.16 | NA | NA | NA | [17] |
| Diflunisal | 1 | 5.93 3 | 7.0 | 25 | 15 mM trisodium citrate (I = 150 mM) | [18] |
| PDB Code | Organism | Resolution (Å) | Ligand | Binding Sites |
|---|---|---|---|---|
| 2BXA | Homo sapiens | 2.35 | CMPF | IIIA, IIA |
| 2BXE | Homo sapiens | 2.95 | Diflunisal | IIIA, IIA-IIB, IIA |
| 2BXG | Homo sapiens | 2.70 | (S)-Ibuprofen | IIIA, IIA-IIB |
| 2BXK | Homo sapiens | 2.40 | Indomethacin | IIC |
| 2BXL | Homo sapiens | 2.60 | 3,5-diiodosalicylic acid | IIA |
| 2BXM | Homo sapiens | 2.50 | Indomethacin | IB, IIC |
| 2BXQ | Homo sapiens | 2.60 | Indomethacin | IB, IIC |
| 2VDB | Homo sapiens | 2.52 | (S)-Naproxen | IB |
| 4Z69 | Homo sapiens | 2.19 | Diclofenac | IIA-IIB, IIA, IB |
| 4J2V | Equus caballus | 2.12 | 3,5-diiodosalicylic acid | IIIA, IIA, IB, IIIB |
| 4ZBQ | Equus caballus | 1.92 | Diclofenac | IIIA, IIIB |
| 4ZBR | Equus caballus | 2.19 | Diclofenac(S)-Naproxen | IIIBIIIA, IIA-IIB |
| 4OT2 | Bos taurus | 2.42 | (S)-Naproxen | IIIA, IIA-IIB |
| 5DBY | Equus caballus | 2.35 | Diclofenac(S)-Naproxen | IIIBIIIA |
| 5V0V | Equus caballus | 2.45 | Etodolac | IIA-IIB, IIA, IB |
| 6OCI | Equus caballus | 2.54 | (S)-Ibuprofen | IIIA, IIA-IIB |
| 6OCJ | Equus caballus | 2.50 | Suprofen | IIIA |
| 6U4R | Equus caballus | 2.45 | Ketoprofen | IIIB |
| 6U4X | Equus caballus | 2.25 | (S)-Ibuprofen | IIIA |
| 6U5A | Equus caballus | 2.65 | 6-MNA | IIIA, IIA-IIB, IIIB |
| 4JK4 | Bos taurus | 2.65 | 3,5-diiodosalicylic acid | IIIA, IIA, IB, IIC |
| 4OR0 | Bos taurus | 2.58 | (S)-Naproxen | IIIA, IIA-IIB, IIC |
| 6QS9 | Bos taurus | 2.80 | Ketoprofen | IIA |
| 6OCK | Oryctolagus cuniculus | 1.90 | Ketoprofen | IIIA, IIIB |
| 6OCL | Oryctolagus cuniculus | 2.35 | Suprofen | IIIA |
| 4LUH | Ovis aries | 2.20 | 3,5-diiodosalicylic acid | IIA, IIC |
| 6HN0 | Ovis aries | 2.12 | Diclofenac | IIIA, IIA-IIB, IB, IIIB, Cleft |
| 5OSW | Capra haircus | 1.78 | 3,5-diiodosalicylic acid | IIIA, IIA, IB, IIIB, IIC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amézqueta, S.; Beltrán, J.L.; Bolioli, A.M.; Campos-Vicens, L.; Luque, F.J.; Ràfols, C. Evaluation of the Interactions between Human Serum Albumin (HSA) and Non-Steroidal Anti-Inflammatory (NSAIDs) Drugs by Multiwavelength Molecular Fluorescence, Structural and Computational Analysis. Pharmaceuticals 2021, 14, 214. https://doi.org/10.3390/ph14030214
Amézqueta S, Beltrán JL, Bolioli AM, Campos-Vicens L, Luque FJ, Ràfols C. Evaluation of the Interactions between Human Serum Albumin (HSA) and Non-Steroidal Anti-Inflammatory (NSAIDs) Drugs by Multiwavelength Molecular Fluorescence, Structural and Computational Analysis. Pharmaceuticals. 2021; 14(3):214. https://doi.org/10.3390/ph14030214
Chicago/Turabian StyleAmézqueta, Susana, José Luís Beltrán, Anna Maria Bolioli, Lluís Campos-Vicens, Francisco Javier Luque, and Clara Ràfols. 2021. "Evaluation of the Interactions between Human Serum Albumin (HSA) and Non-Steroidal Anti-Inflammatory (NSAIDs) Drugs by Multiwavelength Molecular Fluorescence, Structural and Computational Analysis" Pharmaceuticals 14, no. 3: 214. https://doi.org/10.3390/ph14030214
APA StyleAmézqueta, S., Beltrán, J. L., Bolioli, A. M., Campos-Vicens, L., Luque, F. J., & Ràfols, C. (2021). Evaluation of the Interactions between Human Serum Albumin (HSA) and Non-Steroidal Anti-Inflammatory (NSAIDs) Drugs by Multiwavelength Molecular Fluorescence, Structural and Computational Analysis. Pharmaceuticals, 14(3), 214. https://doi.org/10.3390/ph14030214