
Acquired Drug Resistance Enhances Imidazoquinoline Efflux by P-Glycoprotein



Abstract
:1. Introduction
2. Results
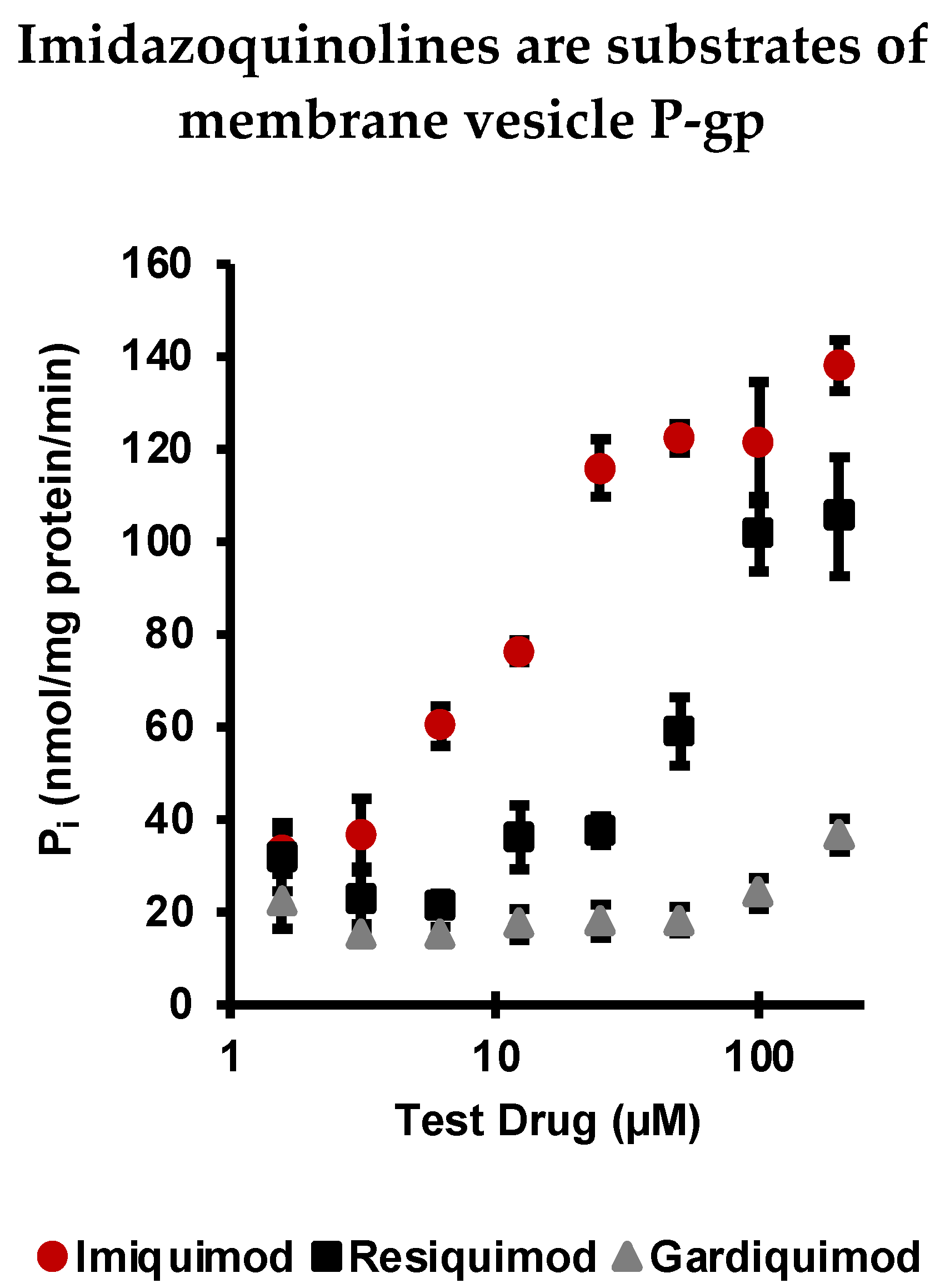
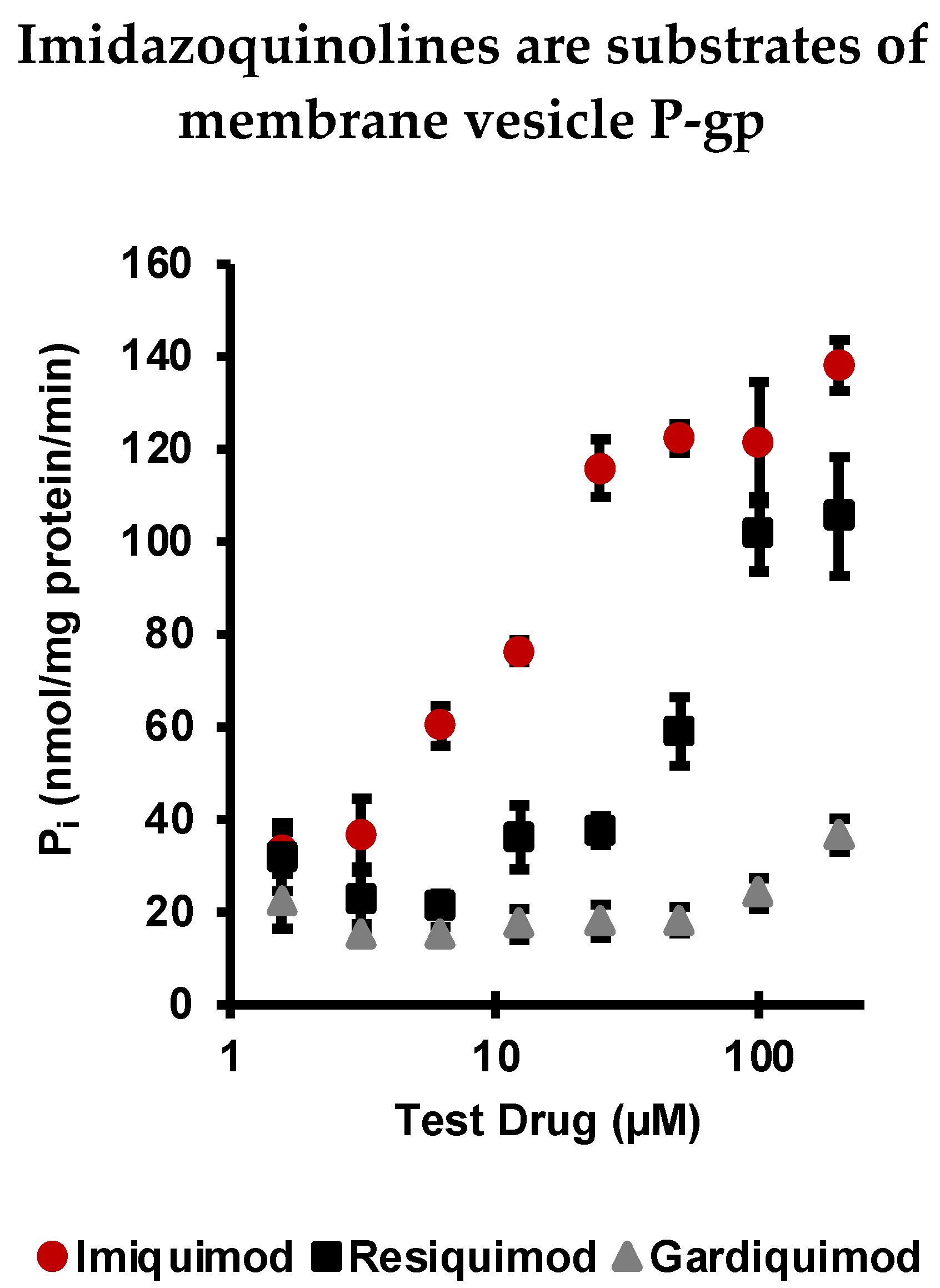
2.1. Imiquimod, Resiquimod, and Gardiquimod Are Substrates of P-gp
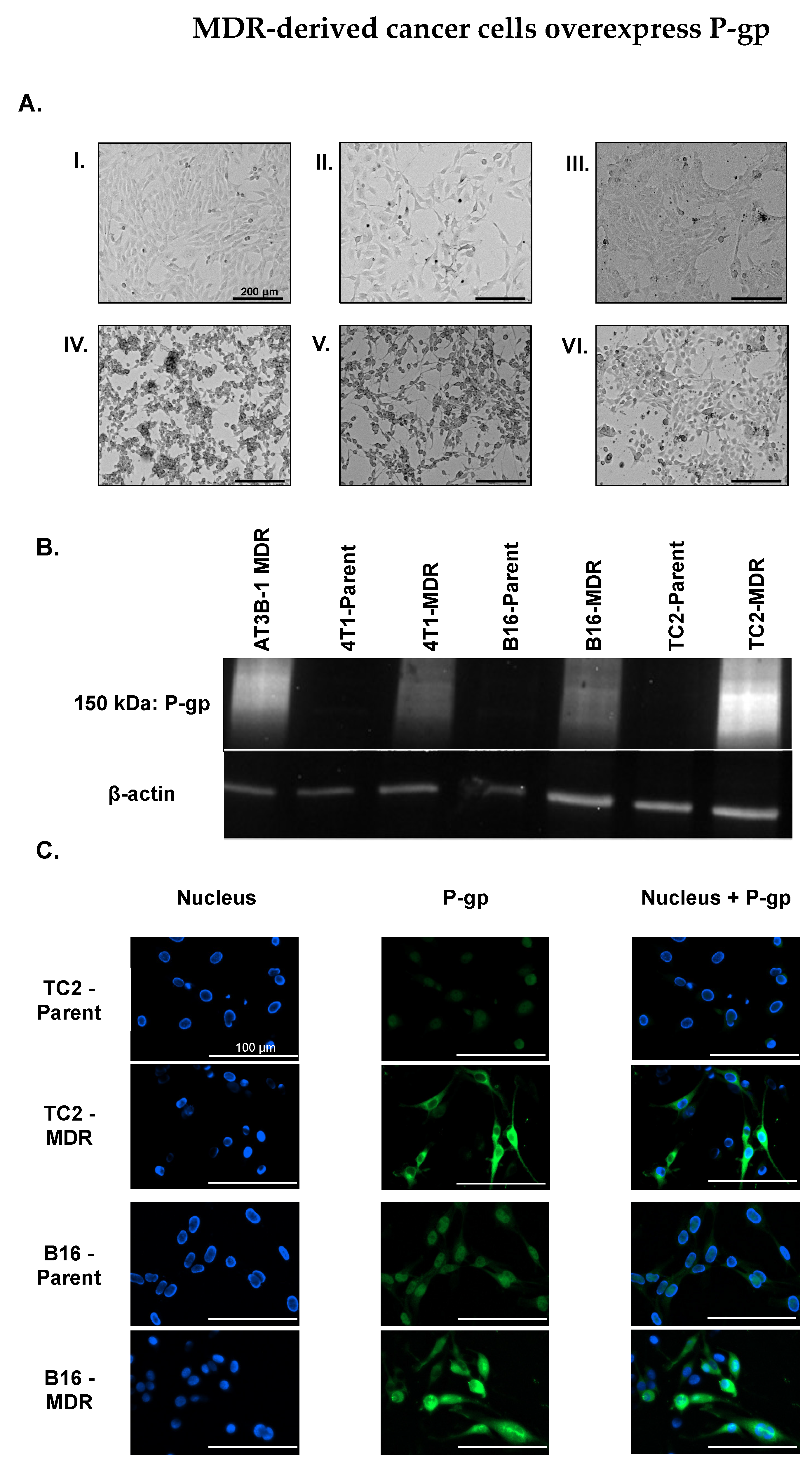
2.2. MDR Cancer Cell Lines Enhance P-gp Expression
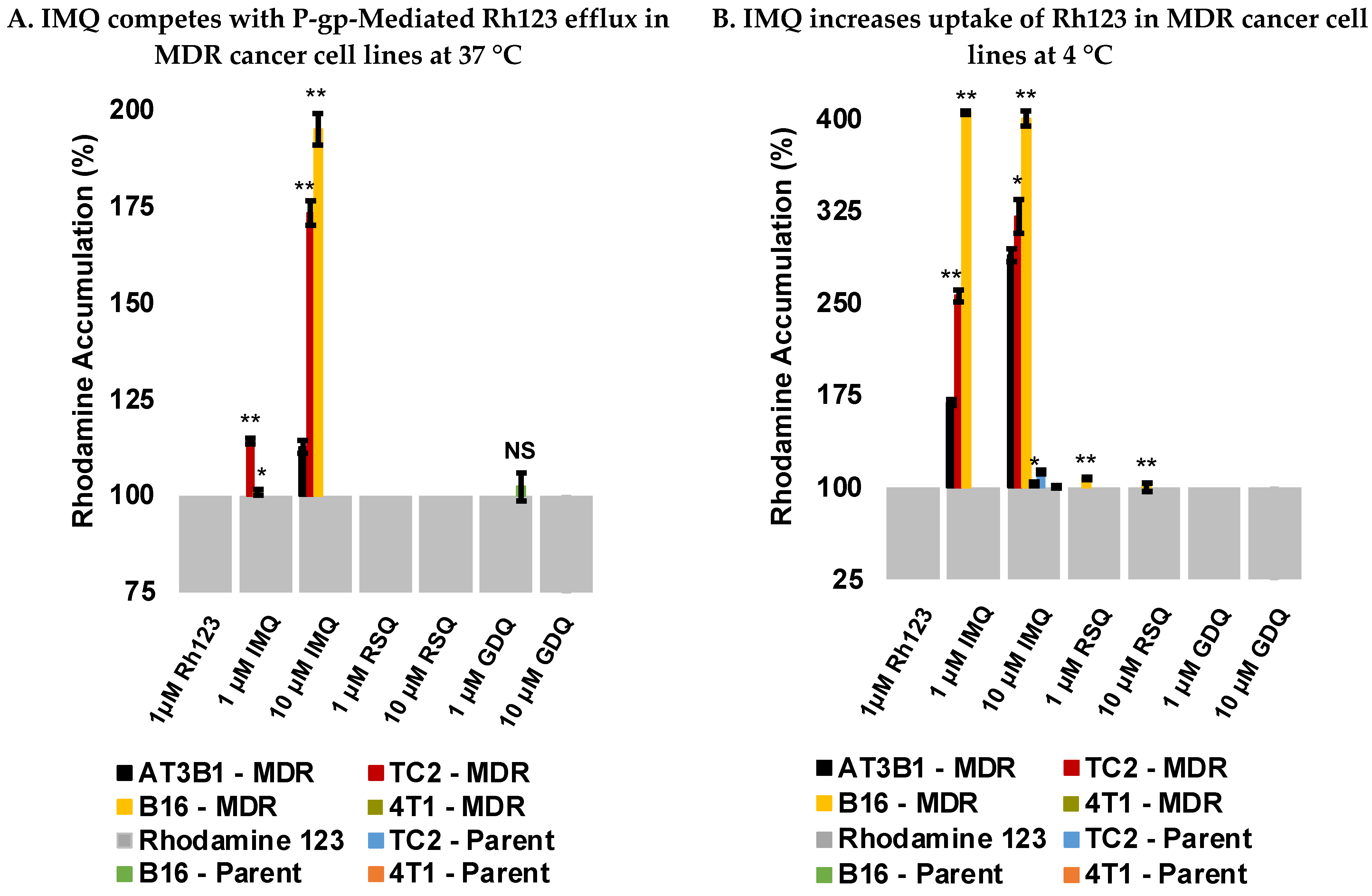
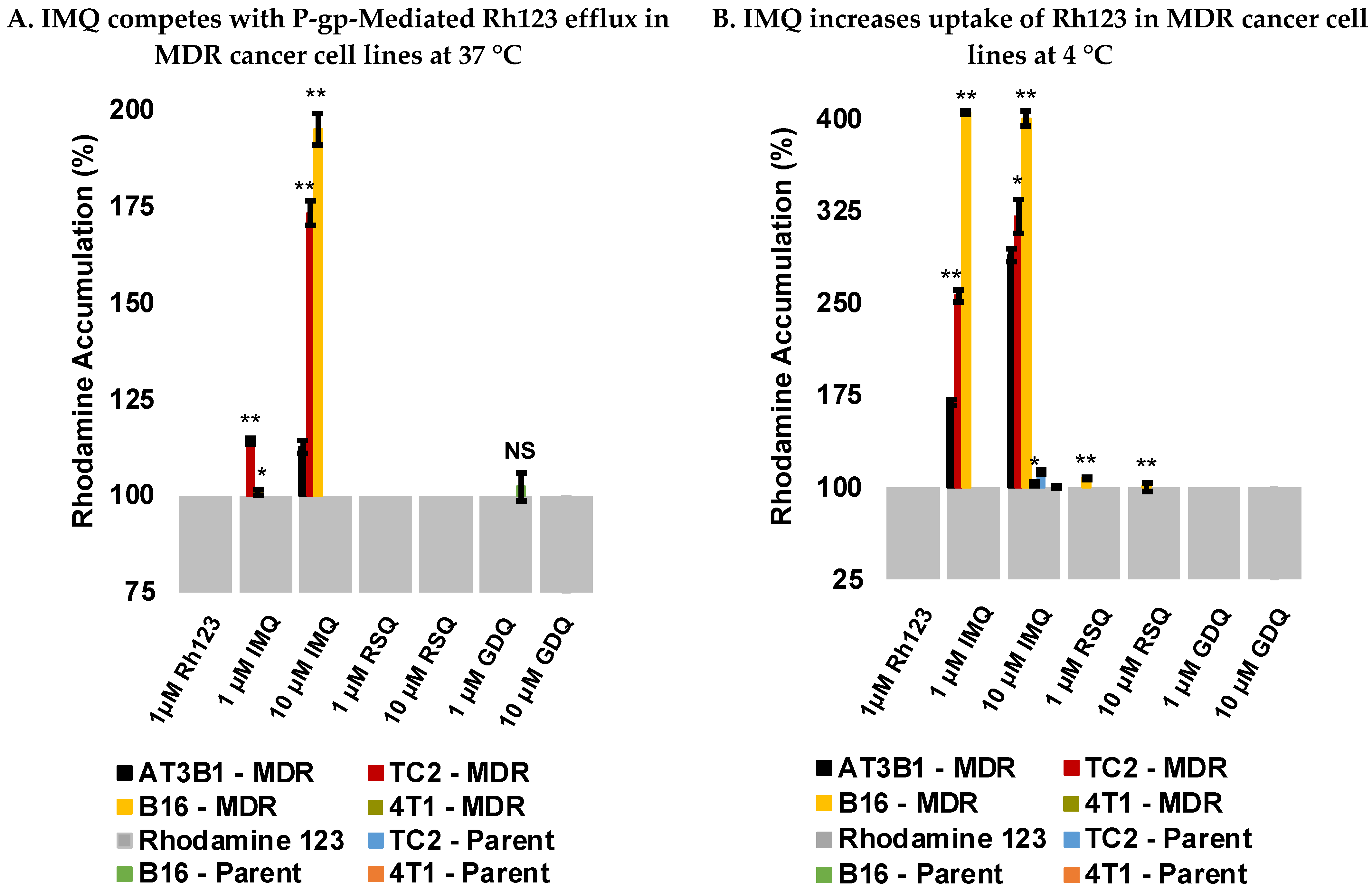
2.3. Imiquimod Competes with Rhodamine 123 Efflux in MDR-Derived Cancer Cell Lines
2.4. Imiquimod Increases Rhodamine 123 Uptake under Passive Diffusion Conditions
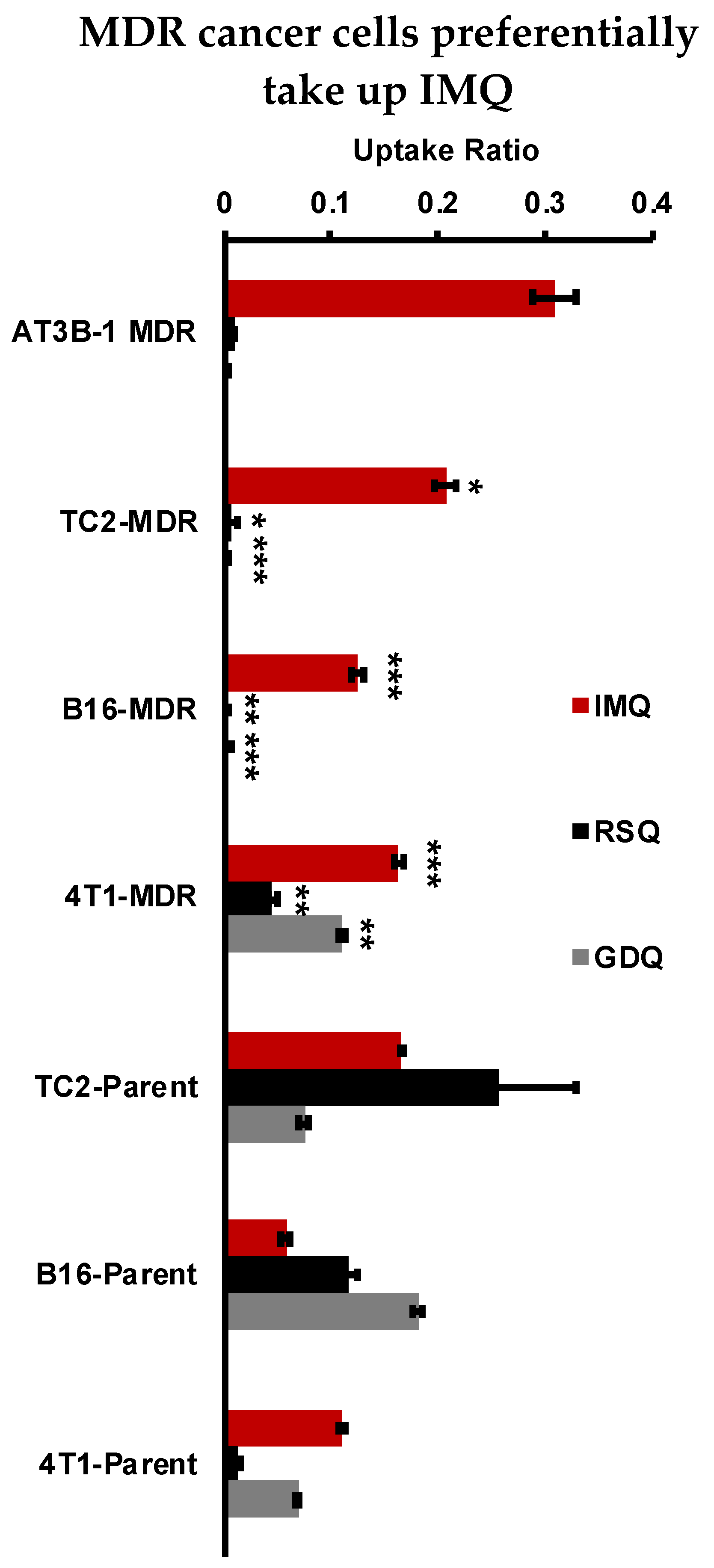
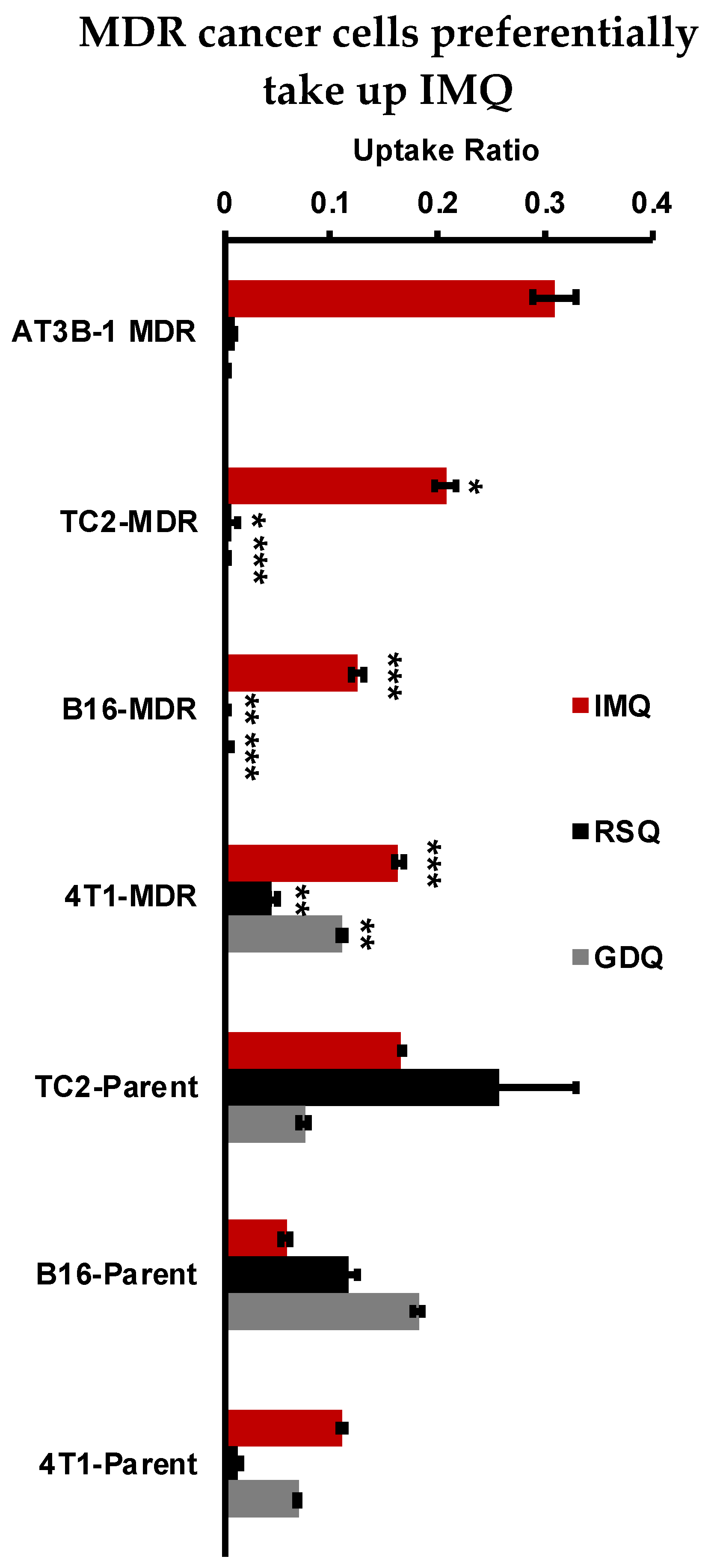
2.5. Resiquimod and Gardiquimod Are Not Passively Taken up by MDR Cancer Cells
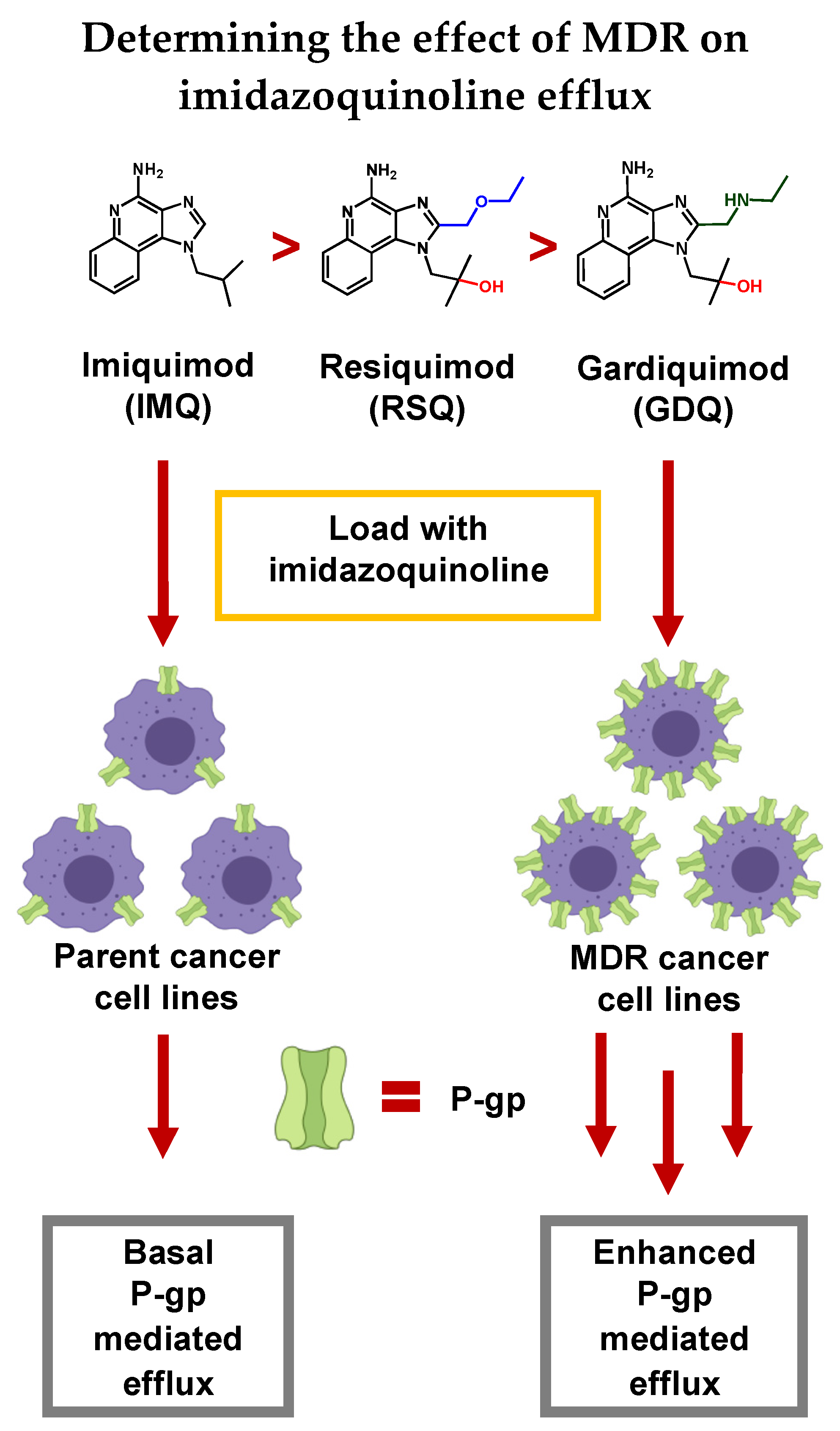
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. ATPase Assay
4.3. Western Blot
4.4. Immunofluorescence Assay
4.5. Competitive Efflux Studies with Rhodamine 123
4.6. Uptake Studies with Rhodamine 123
4.7. Cellular Uptake Studies
4.8. Cytotoxicity Assay
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Longley, D.B.; Johnston, P.G. Molecular Mechanisms of Drug Resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting Multidrug Resistance in Cancer. Nat. Rev. Drug. Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The Role of ABC Transporters in Drug Resistance, Metabolism and Toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef]
- Borst, P.; Elferink, R.O. Mammalian ABC Transporters in Health and Disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [Green Version]
- Juliano, R.L.; Ling, V. A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. Biochim. Biophys. Acta (BBA)-Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Riordan, J.R.; Ling, V. Purification of P-Glycoprotein from Plasma Membrane Vesicles of Chinese Hamster Ovary Cell Mutants with Reduced Colchicine Permeability. J. Biol. Chem. 1979, 254, 12701–12705. [Google Scholar] [CrossRef]
- Juranka, P.F.; Zastawny, R.L.; Ling, V. P-Glycoprotein: Multidrug-Resistance and a Superfamily of Membrane-Associated Transport Proteins. FASEB J. 1989, 3, 2583–2592. [Google Scholar] [CrossRef]
- Ferreira, R.J.; dos Santos, D.J.; Ferreira, M.-J.U. P-Glycoprotein and Membrane Roles in Multidrug Resistance. Future Med. Chem. 2015, 7, 929–946. [Google Scholar] [CrossRef]
- Ueda, K.; Cardarelli, C.; Gottesman, M.M.; Pastan, I. Expression of a Full-Length CDNA for the Human “MDR1” Gene Confers Resistance to Colchicine, Doxorubicin, and Vinblastine. Proc. Natl. Acad. Sci. USA 1987, 84, 3004–3008. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Gatlik-Landwojtowicz, E.; Äänismaa, P.; Seelig, A. Quantification and Characterization of P-Glycoprotein−Substrate Interactions. Biochemistry 2006, 45, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, M.; Ambudkar, S.V.; Chen, D.; Hrycyna, C.A.; Dey, S.; Gottesman, M.M.; Pastan, I. Human P-Glycoprotein Exhibits Reduced Affinity for Substrates during a Catalytic Transition State. Biochemistry 1998, 37, 5010–5019. [Google Scholar] [CrossRef]
- Pawagi, A.B.; Wang, J.; Silverman, M.; Reithmeier, R.A.F.; Deber, C.M. Transmembrane Aromatic Amino Acid Distribution in P-Glycoprotein. J. Mol. Biol. 1994, 235, 554–564. [Google Scholar] [CrossRef]
- Matsson, P.; Pedersen, J.M.; Norinder, U.; Bergström, C.A.S.; Artursson, P. Identification of Novel Specific and General Inhibitors of the Three Major Human ATP-Binding Cassette Transporters P-Gp, BCRP and MRP2 Among Registered Drugs. Pharm. Res. 2009, 26, 1816–1831. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A. Structural Modifications That Alter the P-Glycoprotein Efflux Properties of Compounds. J. Med. Chem. 2012, 55, 4877–4895. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Ling, V. The Molecular Basis of Multidrug Resistance in Cancer: The Early Years of P-Glycoprotein Research. FEBS Lett. 2006, 580, 998–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coley, W.B., II. Contribution to the Knowledge of Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef]
- Coley, W.B. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus Erysipelas and the Bacillus Prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48. [Google Scholar] [CrossRef] [Green Version]
- Smits, E.L.J.M.; Ponsaerts, P.; Berneman, Z.N.; Van Tendeloo, V.F.I. The Use of TLR7 and TLR8 Ligands for the Enhancement of Cancer Immunotherapy. Oncologist 2008, 13, 859–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, R.M.; Craft, N.; Bruhn, K.W.; Khan-Farooqi, H.; Koya, R.C.; Stripecke, R.; Miller, J.F.; Liau, L.M. The TLR-7 Agonist, Imiquimod, Enhances Dendritic Cell Survival and Promotes Tumor Antigen-Specific T Cell Priming: Relation to Central Nervous System Antitumor Immunity. J. Immunol. 2006, 176, 157–164. [Google Scholar] [CrossRef]
- Caron, G.; Duluc, D.; Frémaux, I.; Jeannin, P.; David, C.; Gascan, H.; Delneste, Y. Direct Stimulation of Human T Cells via TLR5 and TLR7/8: Flagellin and R-848 Up-Regulate Proliferation and IFN-γ Production by Memory CD4+ T Cells. J. Immunol. 2005, 175, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.J.; Hijnen, D.; Murphy, G.F.; Kupper, T.S.; Calarese, A.W.; Mollet, I.G.; Schanbacher, C.F.; Miller, D.M.; Schmults, C.D.; Clark, R.A. Imiquimod Enhances IFN-γ Production and Effector Function of T Cells Infiltrating Human Squamous Cell Carcinomas of the Skin. J. Investig. Dermatol. 2009, 129, 2676–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, O.M.; Athie-Morales, V.; O’Connor, G.M.; Gardiner, C.M. TLR7/8-Mediated Activation of Human NK Cells Results in Accessory Cell-Dependent IFN-γ Production. J. Immunol. 2005, 175, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Peng, G. Toll-Like Receptor 8-Mediated Reversal of CD4+ Regulatory T Cell Function. Science 2005, 309, 1380–1384. [Google Scholar] [CrossRef]
- Yin, T.; He, S.; Wang, Y. Toll-like Receptor 7/8 Agonist, R848, Exhibits Antitumoral Effects in a Breast Cancer Model. Mol. Med. Rep. 2015, 12, 3515–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.-H.; Lee, J.; Jeon, S.-J.; Choi, E.-S.; Cho, S.-D.; Kim, B.-Y.; Kim, D.-J.; Park, J.-H.; Park, J.-H. In Vitro and in Vivo Growth Inhibition of Prostate Cancer by the Small Molecule Imiquimod. Int. J. Oncol. 2013, 42, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Mullins, S.R.; Vasilakos, J.P.; Deschler, K.; Grigsby, I.; Gillis, P.; John, J.; Elder, M.J.; Swales, J.; Timosenko, E.; Cooper, Z.; et al. Intratumoral Immunotherapy with TLR7/8 Agonist MEDI9197 Modulates the Tumor Microenvironment Leading to Enhanced Activity When Combined with Other Immunotherapies. J. Immunother. Cancer 2019, 7, 244. [Google Scholar] [CrossRef]
- Hantho, J.D.; Strayer, T.A.; Nielsen, A.E.; Mancini, R.J. An Enzyme-Directed Imidazoquinoline for Cancer Immunotherapy. ChemMedChem 2016, 11, 2496–2500. [Google Scholar] [CrossRef]
- Burt, A.J.; Hantho, J.D.; Nielsen, A.E.; Mancini, R.J. An Enzyme-Directed Imidazoquinoline Activated by Drug Resistance. Biochemistry 2018, 57, 2184–2188. [Google Scholar] [CrossRef]
- Ryan, A.T.; Pulukuri, A.J.; Davaritouchaee, M.; Abbasi, A.; Hendricksen, A.T.; Opp, L.K.; Burt, A.J.; Nielsen, A.E.; Mancini, R.J. Comparing the Immunogenicity of Glycosidase-Directed Resiquimod Prodrugs Mediated by Cancer Cell Metabolism. Acta Pharm. Sin. 2020, 41, 995–1004. [Google Scholar] [CrossRef]
- Li, H.; Van Herck, S.; Liu, Y.; Hao, Y.; Ding, X.; Nuhn, L.; Zhong, Z.; Combes, F.; Sanders, N.N.; Lienenklaus, S.; et al. Imidazoquinoline-Conjugated Degradable Coacervate Conjugate for Local Cancer Immunotherapy. ACS Biomater. Sci. Eng. 2020, 6, 4993–5000. [Google Scholar] [CrossRef] [PubMed]
- Mottas, I.; Bekdemir, A.; Cereghetti, A.; Spagnuolo, L.; Yang, Y.-S.S.; Müller, M.; Irvine, D.J.; Stellacci, F.; Bourquin, C. Amphiphilic Nanoparticle Delivery Enhances the Anticancer Efficacy of a TLR7 Ligand via Local Immune Activation. Biomaterials 2019, 190–191, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuhn, L.; De Koker, S.; Van Lint, S.; Zhong, Z.; Catani, J.P.; Combes, F.; Deswarte, K.; Li, Y.; Lambrecht, B.N.; Lienenklaus, S.; et al. Nanoparticle-Conjugate TLR7/8 Agonist Localized Immunotherapy Provokes Safe Antitumoral Responses. Adv. Mater. 2018, 30, 1803397. [Google Scholar] [CrossRef]
- Yoo, E.; Salyer, A.C.D.; Brush, M.J.H.; Li, Y.; Trautman, K.L.; Shukla, N.M.; De Beuckelaer, A.; Lienenklaus, S.; Deswarte, K.; Lambrecht, B.N.; et al. Hyaluronic Acid Conjugates of TLR7/8 Agonists for Targeted Delivery to Secondary Lymphoid Tissue. Bioconjugate Chem. 2018, 29, 2741–2754. [Google Scholar] [CrossRef]
- Theile, D.; Wagner, L.; Haefeli, W.E.; Weiss, J. In Vitro Evidence Suggesting That the Toll-like Receptor 7 and 8 Agonist Resiquimod (R-848) Unlikely Affects Drug Levels of Co-Administered Compounds. Eur. J. Pharm. Sci. 2021, 162, 105826. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Weinman, S. Mechanisms of Doxorubicin Resistance in Hepatocellular Carcinoma. Hepatic Oncol. 2016, 3, 57–59. [Google Scholar] [CrossRef]
- Ledwitch, K.V.; Gibbs, M.E.; Barnes, R.W.; Roberts, A.G. Cooperativity between Verapamil and ATP Bound to the Efflux Transporter P-Glycoprotein. Biochem. Pharmacol. 2016, 118, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Durie, B.G.M.; Dalton, W.S. Reversal of Drug-Resistance in Multiple Myeloma with Verapamil. Br. J. Haematol. 1988, 68, 203–206. [Google Scholar] [CrossRef]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M.D. Tariquidar Is an Inhibitor and Not a Substrate of Human and Mouse P-Glycoprotein. Drug Metab. Dispos. 2016, 44, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Urbatsch, I.L.; Sankaran, B.; Weber, J.; Senior, A.E. P-Glycoprotein Is Stably Inhibited by Vanadate-Induced Trapping of Nucleotide at a Single Catalytic Site. J. Biol. Chem. 1995, 270, 19383–19390. [Google Scholar] [CrossRef] [Green Version]
- Schön, M.P.; Schön, M. Immune Modulation and Apoptosis Induction: Two Sides of the Antitumoral Activity of Imiquimod. Apoptosis 2004, 9, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachalaki, S.; Ebrahimi, M.; Mohamed Khosroshahi, L.; Mohammadinejad, S.; Baradaran, B. Cancer Chemoresistance; Biochemical and Molecular Aspects: A Brief Overview. Eur. J. Pharm. Sci. 2016, 89, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Salehan, M.R.; Morse, H.R. DNA Damage Repair and Tolerance: A Role in Chemotherapeutic Drug Resistance. Br. J. Biomed. Sci. 2013, 70, 31–40. [Google Scholar] [CrossRef]
- Mariani, M.; Supino, R. Morphological Alterations Induced by Doxorubicin in B16 Melanoma Cells. Cancer Lett. 1990, 51, 209–212. [Google Scholar] [CrossRef]
- Foster, B.A.; Gingrich, J.R.; Kwon, E.D.; Madias, C.; Greenberg, N.M. Characterization of Prostatic Epithelial Cell Lines Derived from Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) Model. Cancer Res. 1997, 57, 3325. [Google Scholar]
- Bao, L.; Haque, A.; Jackson, K.; Hazari, S.; Moroz, K.; Jetly, R.; Dash, S. Increased Expression of P-Glycoprotein Is Associated with Doxorubicin Chemoresistance in the Metastatic 4T1 Breast Cancer Model. Am. J. Pathol. 2011, 178, 838–852. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Park, C.-S.; Lee, Y.-R.; Im, S.-A.; Song, S.; Lee, C.-K. Resiquimod, a TLR7/8 Agonist, Promotes Differentiation of Myeloid-Derived Suppressor Cells into Macrophages and Dendritic Cells. Arch. Pharm. Res. 2014, 37, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Toll-like Receptor 9 (TLR9) Agonists in the Treatment of Cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Khong, H.; Dai, Z.; Huang, X.-F.; Wargo, J.A.; Cooper, Z.A.; Vasilakos, J.P.; Hwu, P.; Overwijk, W.W. Effective Innate and Adaptive Antimelanoma Immunity through Localized TLR7/8 Activation. J. Immunol. 2014, 193, 4722–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youhanna, S.; Lauschke, V.M. The Past, Present and Future of Intestinal In Vitro Cell Systems for Drug Absorption Studies. J. Pharm. Sci. 2021, 110, 50–65. [Google Scholar] [CrossRef]
- Replogle-Schwab, T.S.; Schwab, E.D.; Pienta, K.J. Development of Doxorubicin Resistant Rat Prostate Cancer Cell Lines. Anticancer Res. 1997, 17, 4535–4538. [Google Scholar] [PubMed]
- Liang, J.F.; Yang, V.C. Synthesis of Doxorubicin–Peptide Conjugate with Multidrug Resistant Tumor Cell Killing Activity. Bioorganic Med. Chem. Lett. 2005, 15, 5071–5075. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Lee, H.-J.; Ko, H.-J.; Yoon, B.-I.; Choe, J.; Kim, K.-C.; Hahn, T.-W.; Han, J.A.; Choi, S.S.; Jung, Y.M.; et al. The TLR7 Agonist Imiquimod Induces Anti-Cancer Effects via Autophagic Cell Death and Enhances Anti-Tumoral and Systemic Immunity during Radiotherapy for Melanoma. Oncotarget 2017, 8, 24932–24948. [Google Scholar] [CrossRef] [Green Version]
- Jurk, M.; Heil, F.; Vollmer, J.; Schetter, C.; Krieg, A.M.; Wagner, H.; Lipford, G.; Bauer, S. Human TLR7 or TLR8 Independently Confer Responsiveness to the Antiviral Compound R-848. Nat. Immunol. 2002, 3, 499. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative Expression of Toll-Like Receptor 1–10 MRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crozat, K.; Vivier, E.; Dalod, M. Crosstalk between Components of the Innate Immune System: Promoting Anti-Microbial Defenses and Avoiding Immunopathologies. Immunol. Rev. 2009, 227, 129–149. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, J.; Zhang, J.; Zhang, C. The TLR7 Agonists Imiquimod and Gardiquimod Improve DC-Based Immunotherapy for Melanoma in Mice. Cell Mol. Immunol. 2010, 7, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Brouty-Boyé, D.; Kolonias, D.; Wu, C.J.; Savaraj, N.; Lampidis, T.J. Relationship of Multidrug Resistance to Rhodamine-123 Selectivity between Carcinoma and Normal Epithelial Cells: Taxol and Vinblastine Modulate Drug Efflux. Cancer Res. 1995, 55, 1633. [Google Scholar]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-Binding Cassette (ABC) Transporter Family. Hum. Genom. 2008, 3, 281. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, B.J.; Liao, M. ABCG2 Transports Anticancer Drugs via a Closed-to-Open Switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.F.; Bikadi, Z.; Hazai, E.; Starborg, T.; Kelley, L.; Chayen, N.E.; Ford, R.C.; Mao, Q. Three-Dimensional Structure of the Human Breast Cancer Resistance Protein (BCRP/ABCG2) in an Inward-Facing Conformation. Acta Cryst. D Biol. Cryst. 2015, 71, 1725–1735. [Google Scholar] [CrossRef] [Green Version]
- Greer, D.A.; Ivey, S. Distinct N-Glycan Glycosylation of P-Glycoprotein Isolated from the Human Uterine Sarcoma Cell Line MES-SA/Dx5. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2007, 1770, 1275–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Hirai, K.; Nishida, K.; Taguchi, H. 6-(4-Amino-2-Butyl-Imidazoquinolyl)-Norleucine: Toll-like Receptor 7 and 8 Agonist Amino Acid for Self-Adjuvanting Peptide Vaccine. Amino Acids 2016, 48, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Gerster, J.F.; Lindstrom, K.J.; Miller, R.L.; Tomai, M.A.; Birmachu, W.; Bomersine, S.N.; Gibson, S.J.; Imbertson, L.M.; Jacobson, J.R.; Knafla, R.T.; et al. Synthesis and Structure−Activity-Relationships of 1H-Imidazo[4,5-c]Quinolines That Induce Interferon Production. J. Med. Chem. 2005, 48, 3481–3491. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.M.; Malladi, S.S.; Mutz, C.A.; Balakrishna, R.; David, S.A. Structure−Activity Relationships in Human Toll-Like Receptor 7-Active Imidazoquinoline Analogues. J. Med. Chem. 2010, 53, 4450–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, N.M.; Kimbrell, M.R.; Malladi, S.S.; David, S.A. Regioisomerism-Dependent TLR7 Agonism and Antagonism in an Imidazoquinoline. Bioorg. Med. Chem. Lett. 2009, 19, 2211–2214. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Imidazoquinoline | cLogP | KD (μM) |
|---|---|---|
| Imiquimod | 1.428 | 7.66 |
| Resiquimod | 0.036 | 24.37 |
| Gardiquimod | −0.254 | N.D. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulukuri, A.J.; Burt, A.J.; Opp, L.K.; McDowell, C.M.; Davaritouchaee, M.; Nielsen, A.E.; Mancini, R.J. Acquired Drug Resistance Enhances Imidazoquinoline Efflux by P-Glycoprotein. Pharmaceuticals 2021, 14, 1292. https://doi.org/10.3390/ph14121292
Pulukuri AJ, Burt AJ, Opp LK, McDowell CM, Davaritouchaee M, Nielsen AE, Mancini RJ. Acquired Drug Resistance Enhances Imidazoquinoline Efflux by P-Glycoprotein. Pharmaceuticals. 2021; 14(12):1292. https://doi.org/10.3390/ph14121292
Chicago/Turabian StylePulukuri, Anunay J., Anthony J. Burt, Larissa K. Opp, Colin M. McDowell, Maryam Davaritouchaee, Amy E. Nielsen, and Rock J. Mancini. 2021. "Acquired Drug Resistance Enhances Imidazoquinoline Efflux by P-Glycoprotein" Pharmaceuticals 14, no. 12: 1292. https://doi.org/10.3390/ph14121292
APA StylePulukuri, A. J., Burt, A. J., Opp, L. K., McDowell, C. M., Davaritouchaee, M., Nielsen, A. E., & Mancini, R. J. (2021). Acquired Drug Resistance Enhances Imidazoquinoline Efflux by P-Glycoprotein. Pharmaceuticals, 14(12), 1292. https://doi.org/10.3390/ph14121292