
Heterocyclic Cathinones as Inhibitors of Kynurenine Aminotransferase II—Design, Synthesis, and Evaluation

 , , , , ,
, , , , ,  and
and
Abstract
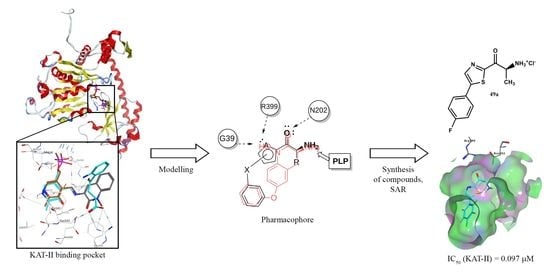
:
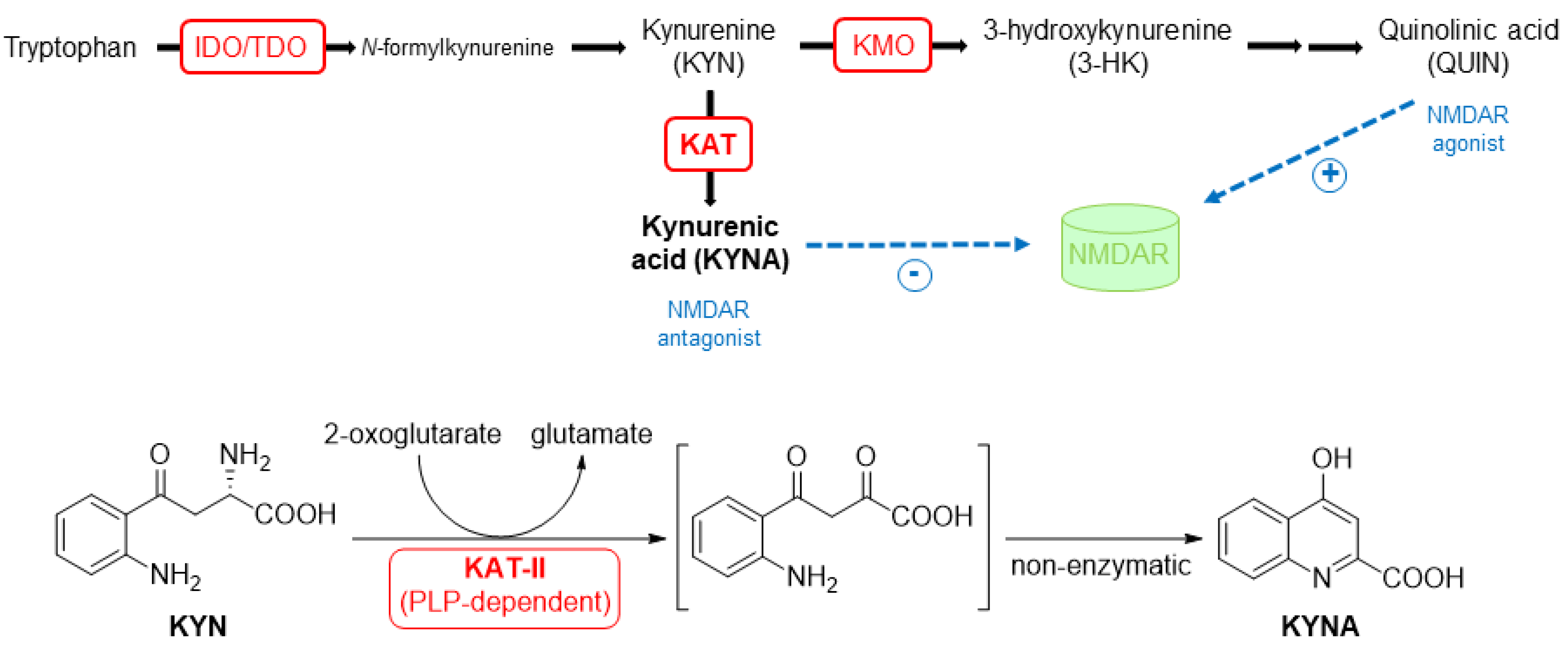
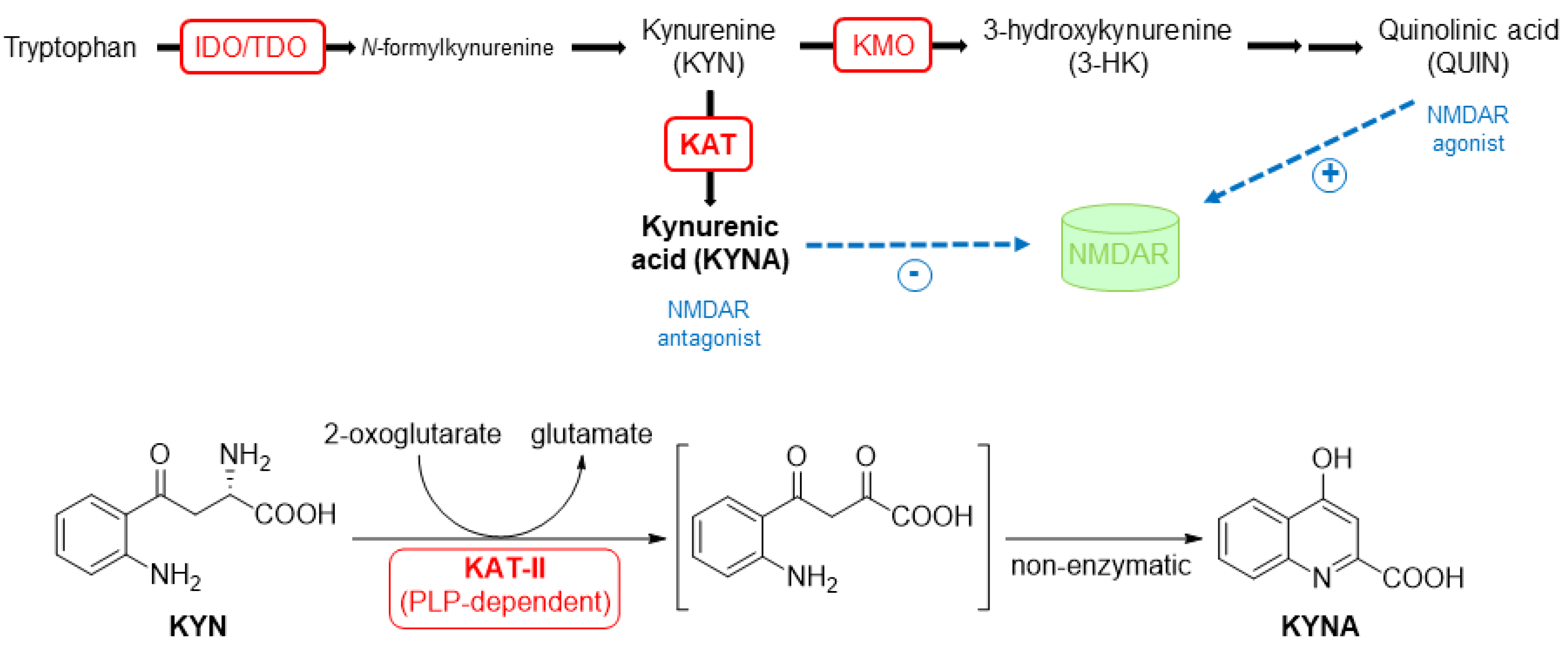
1. Introduction
2. Results and Discussion
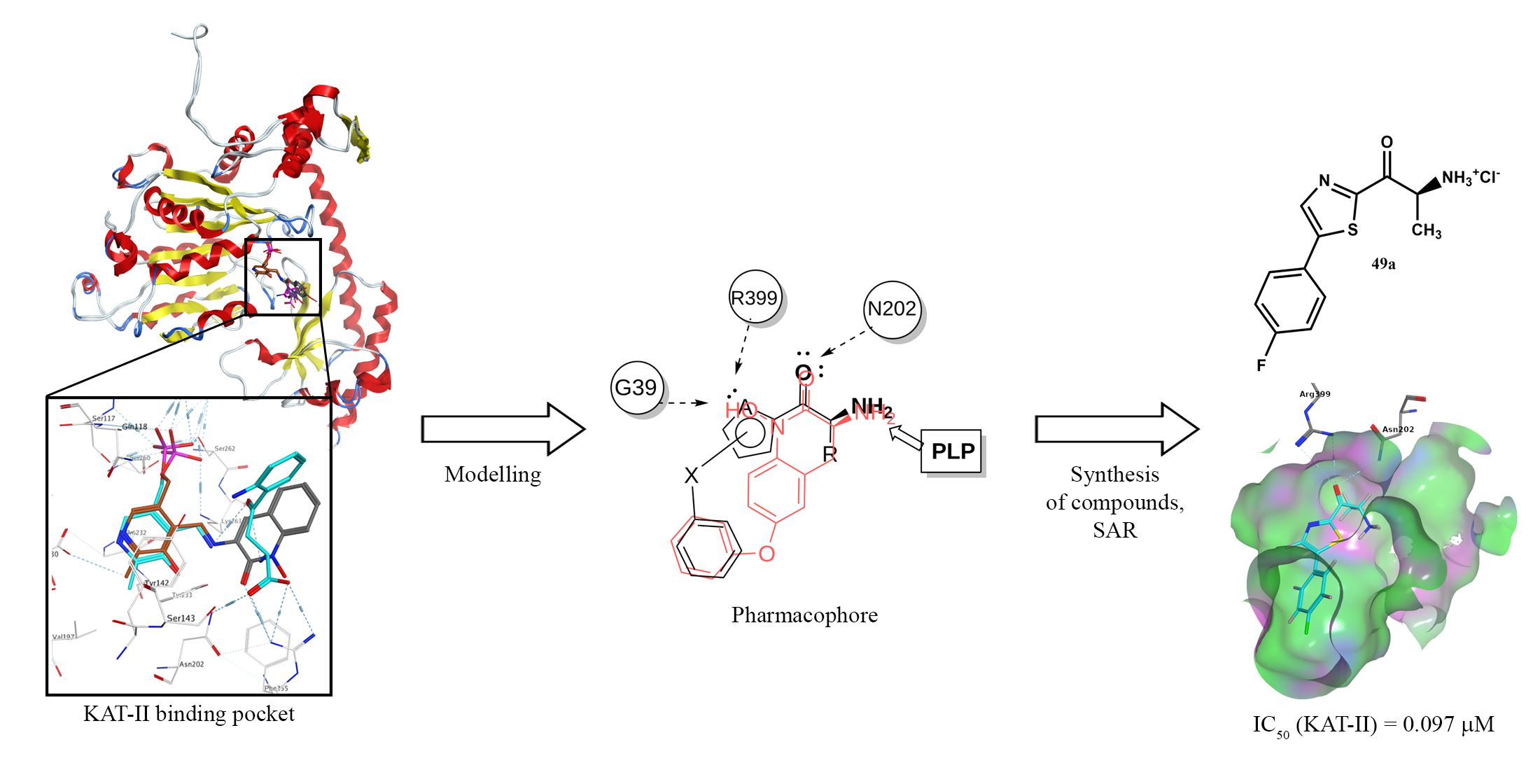
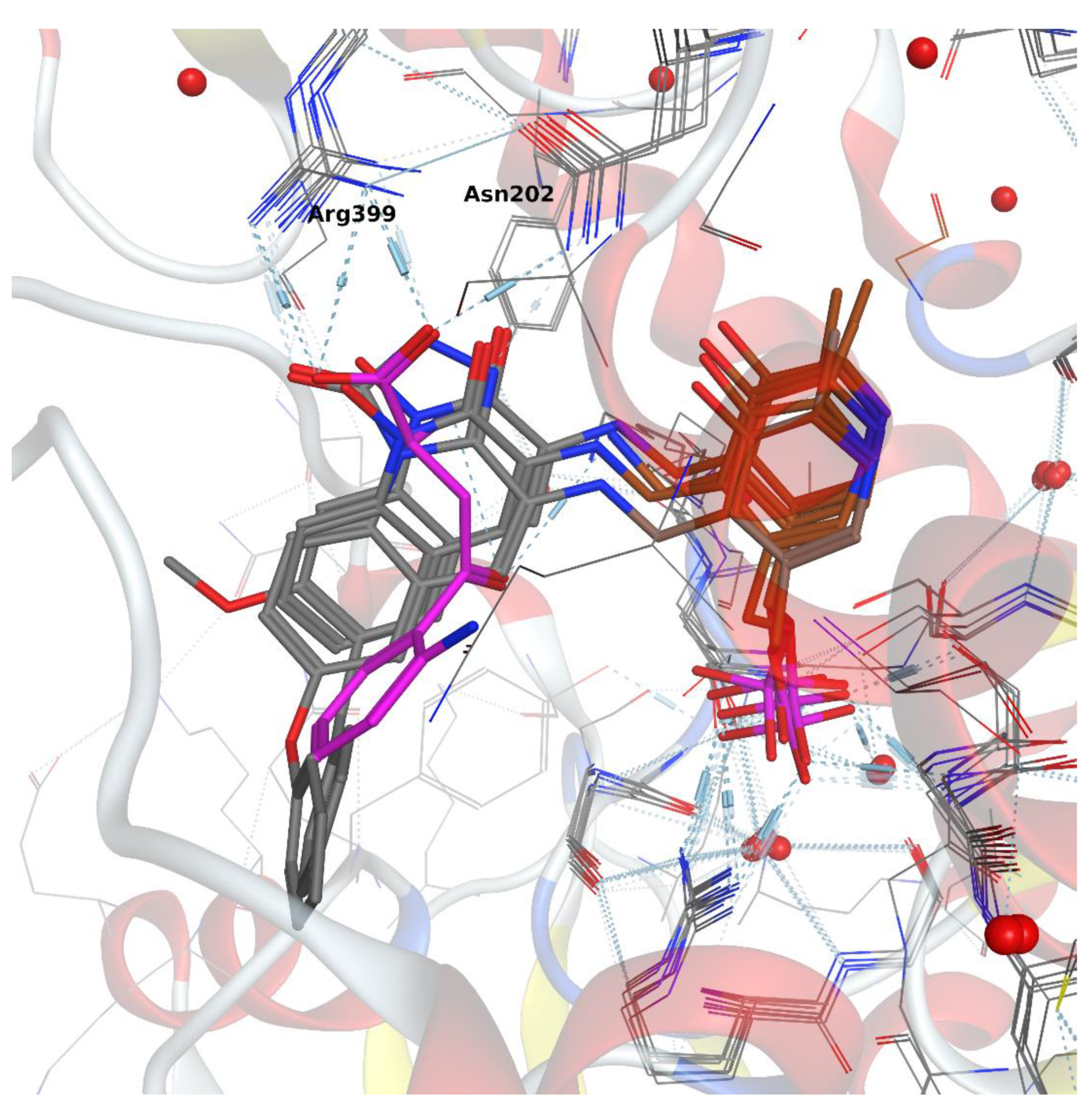
2.1. Pharmacophore Modeling and Design
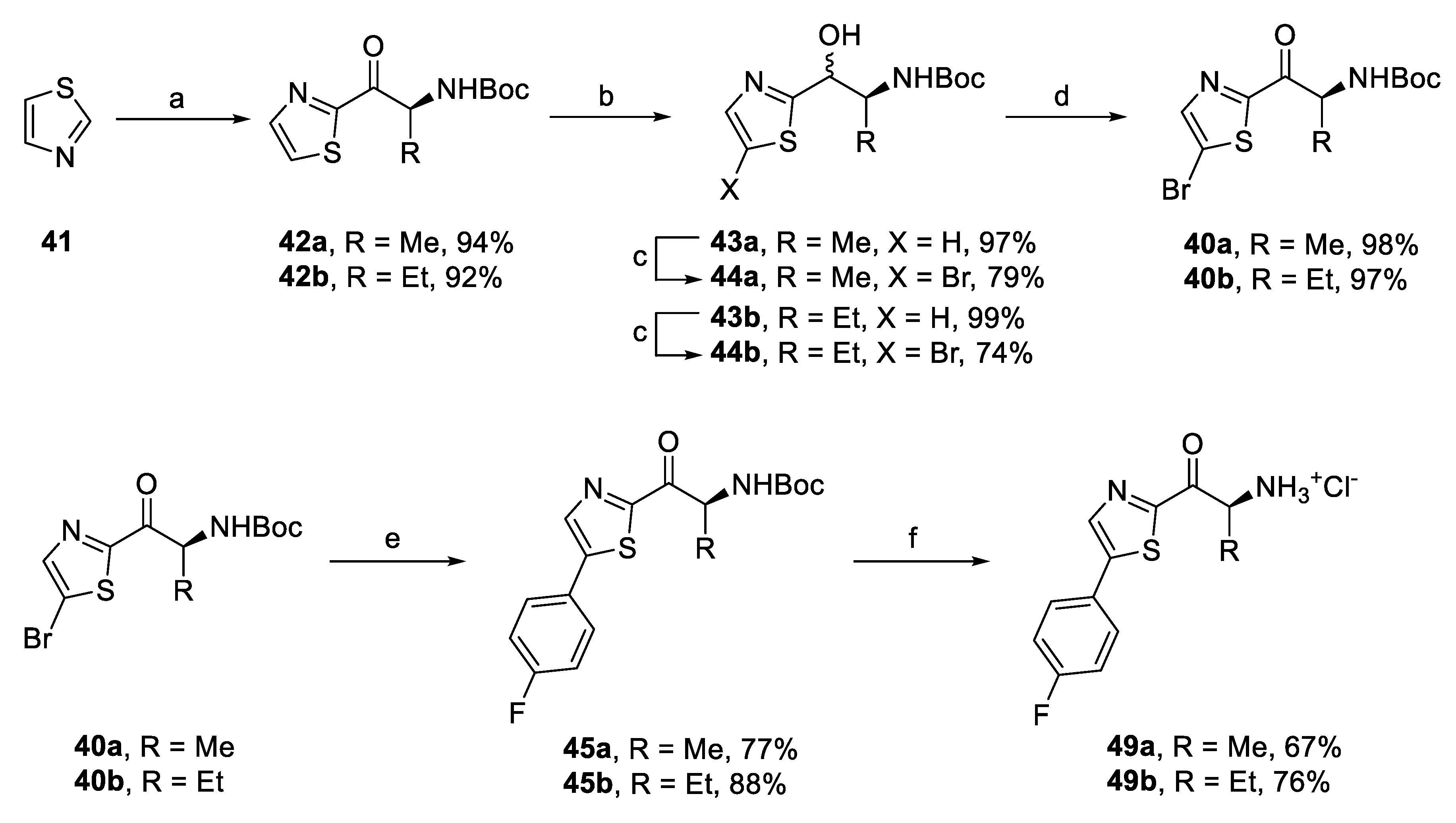
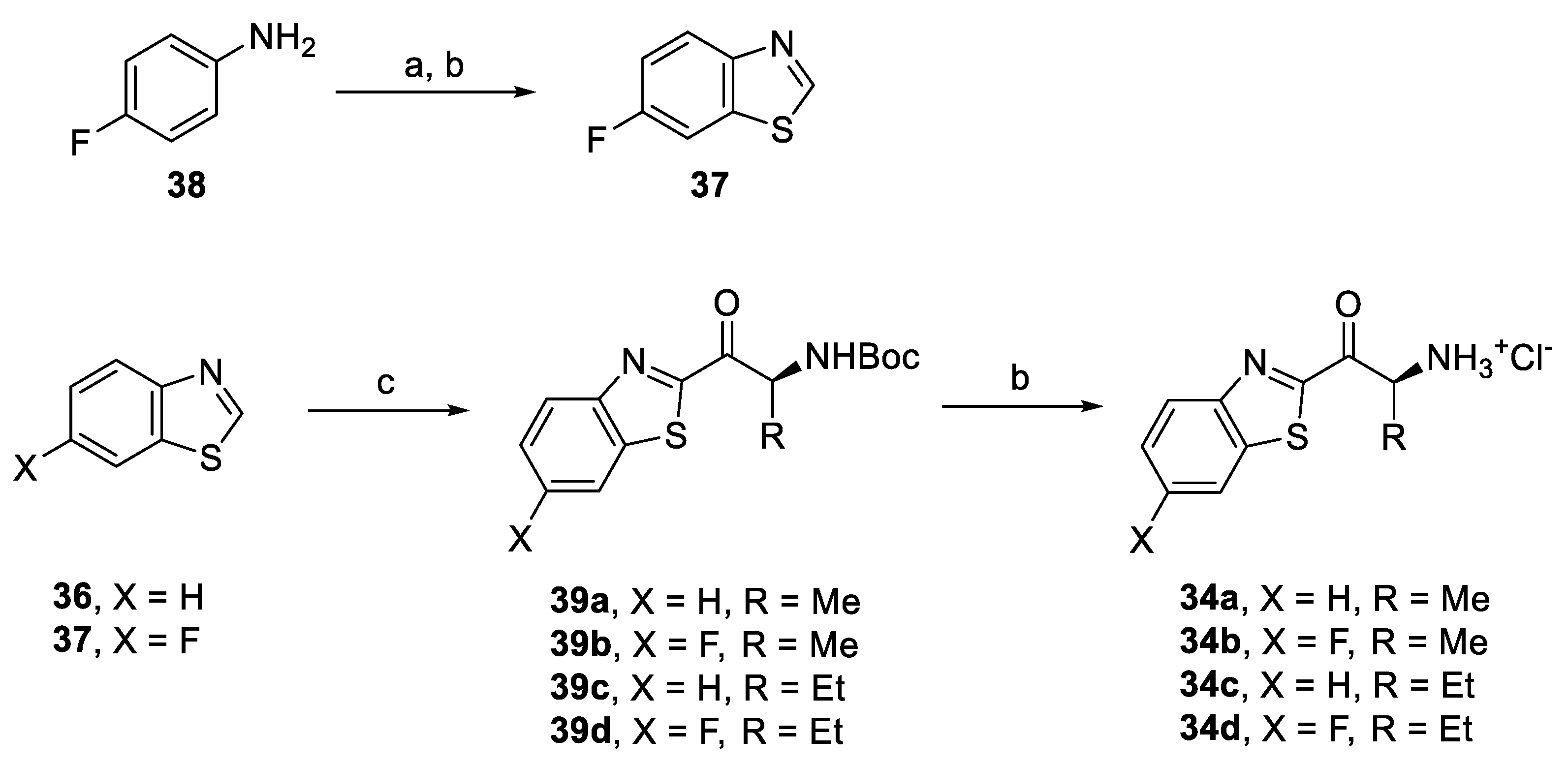
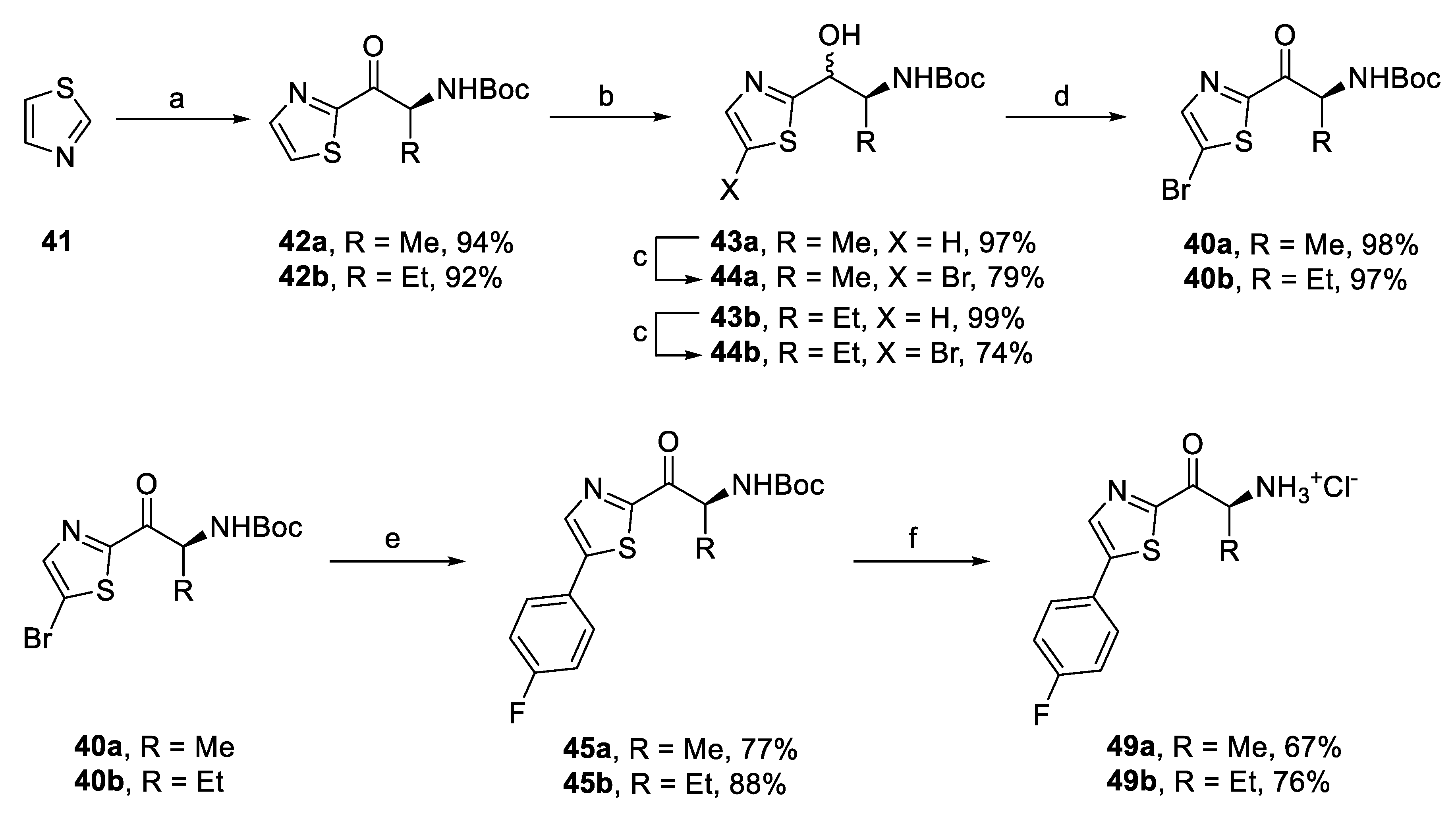
2.2. Synthesis of In Silico Designed Amino Ketones
2.3. Inhibitory Activity of the Designed Lead Compounds
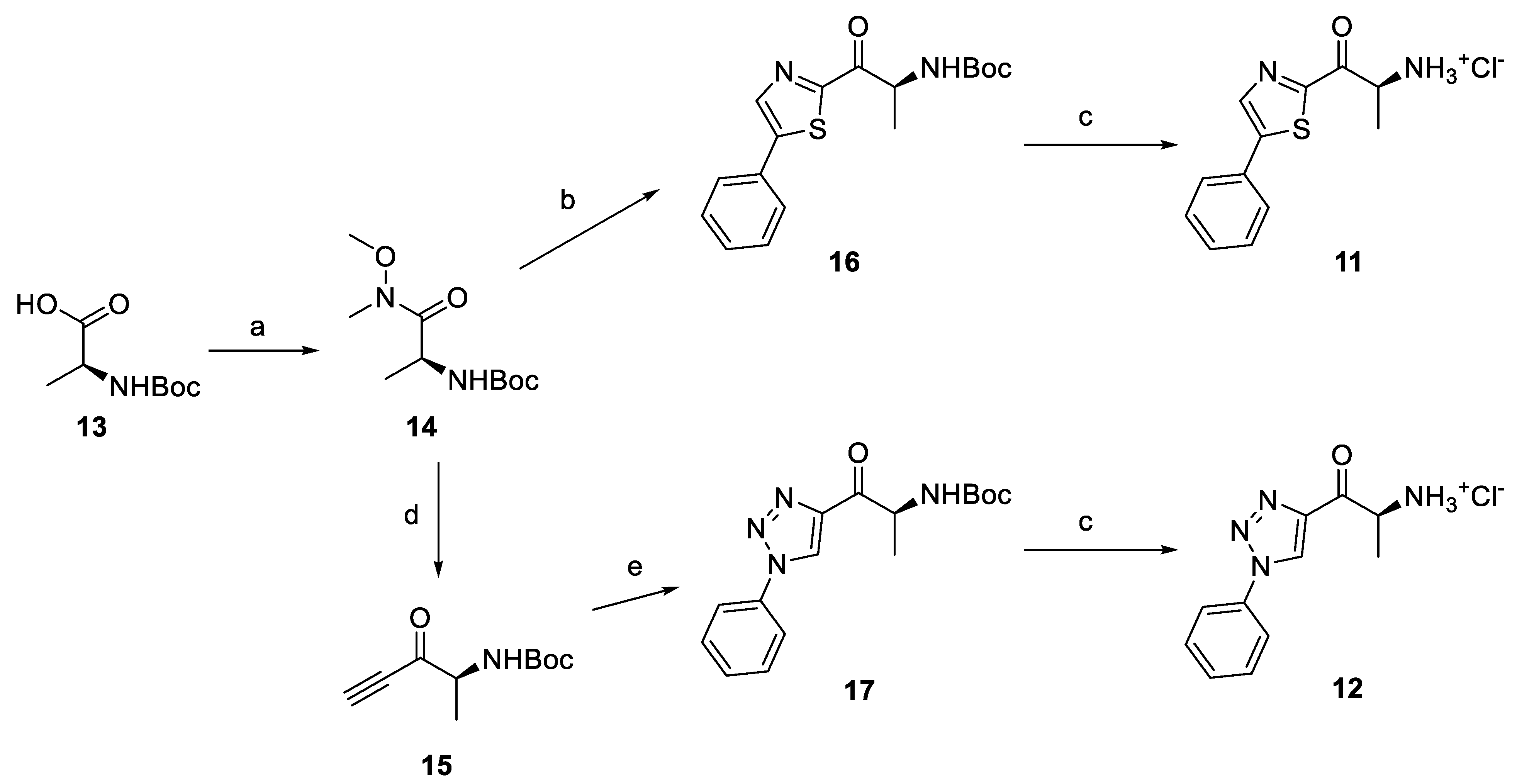
2.4. Synthesis of Derivatives for SAR Study
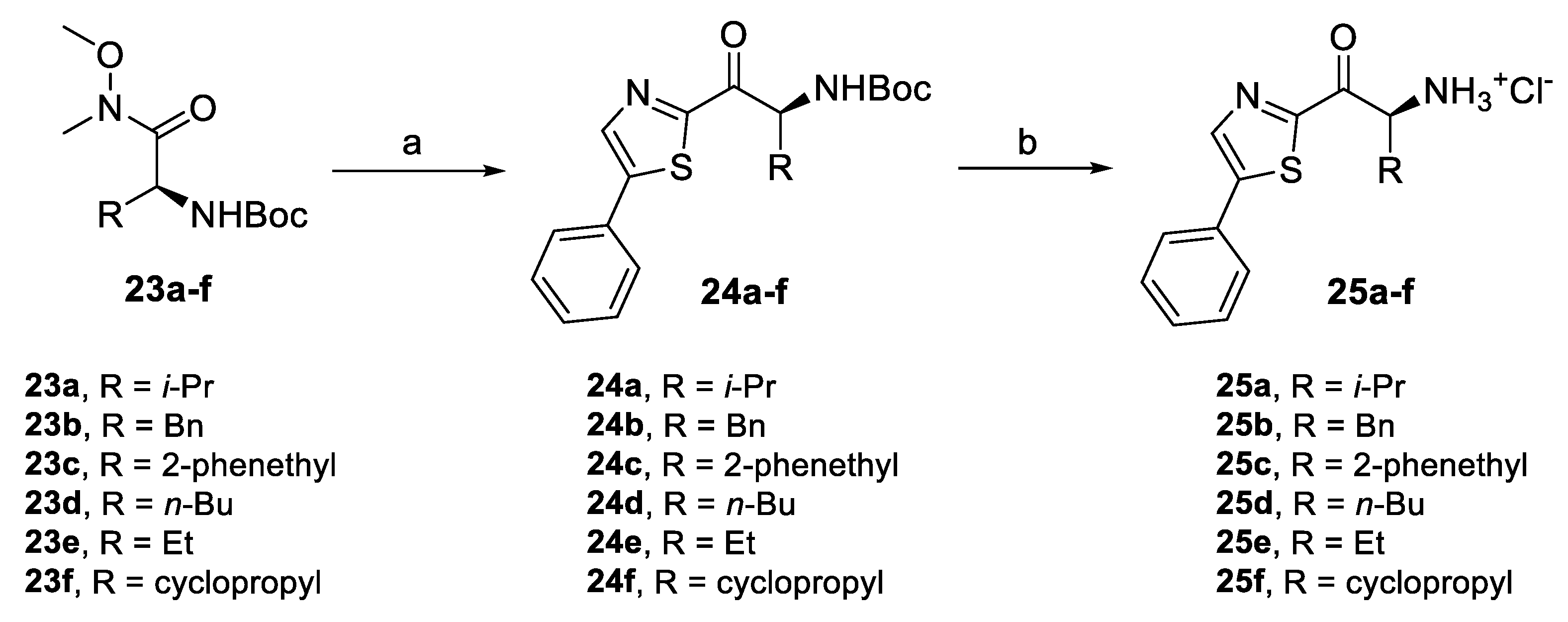
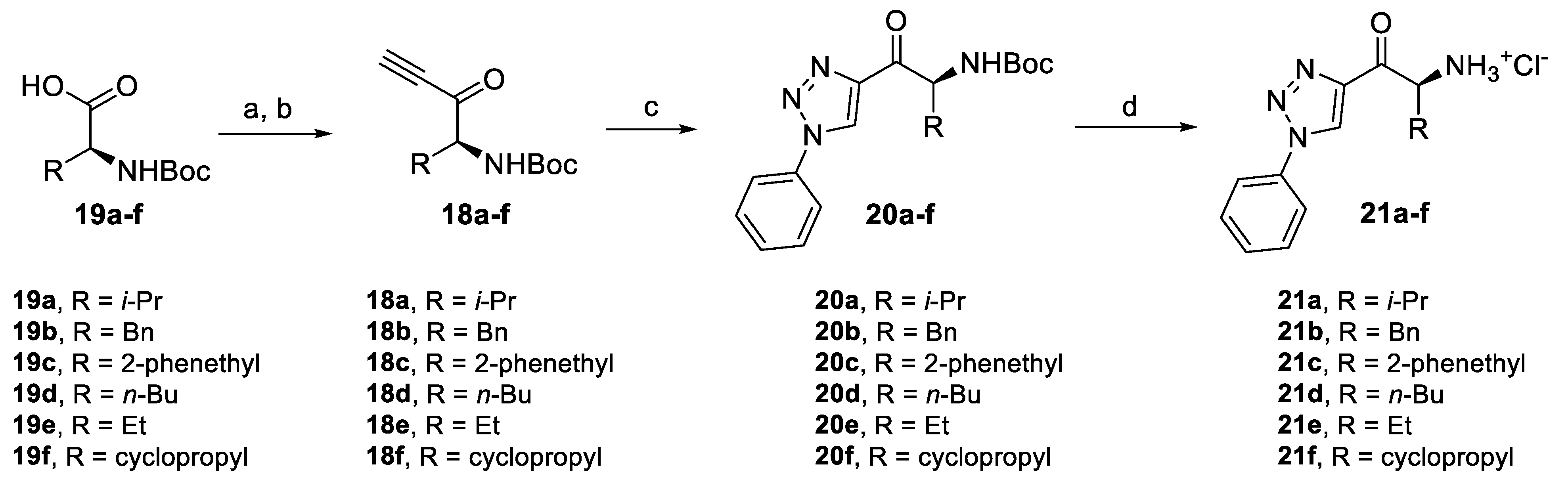
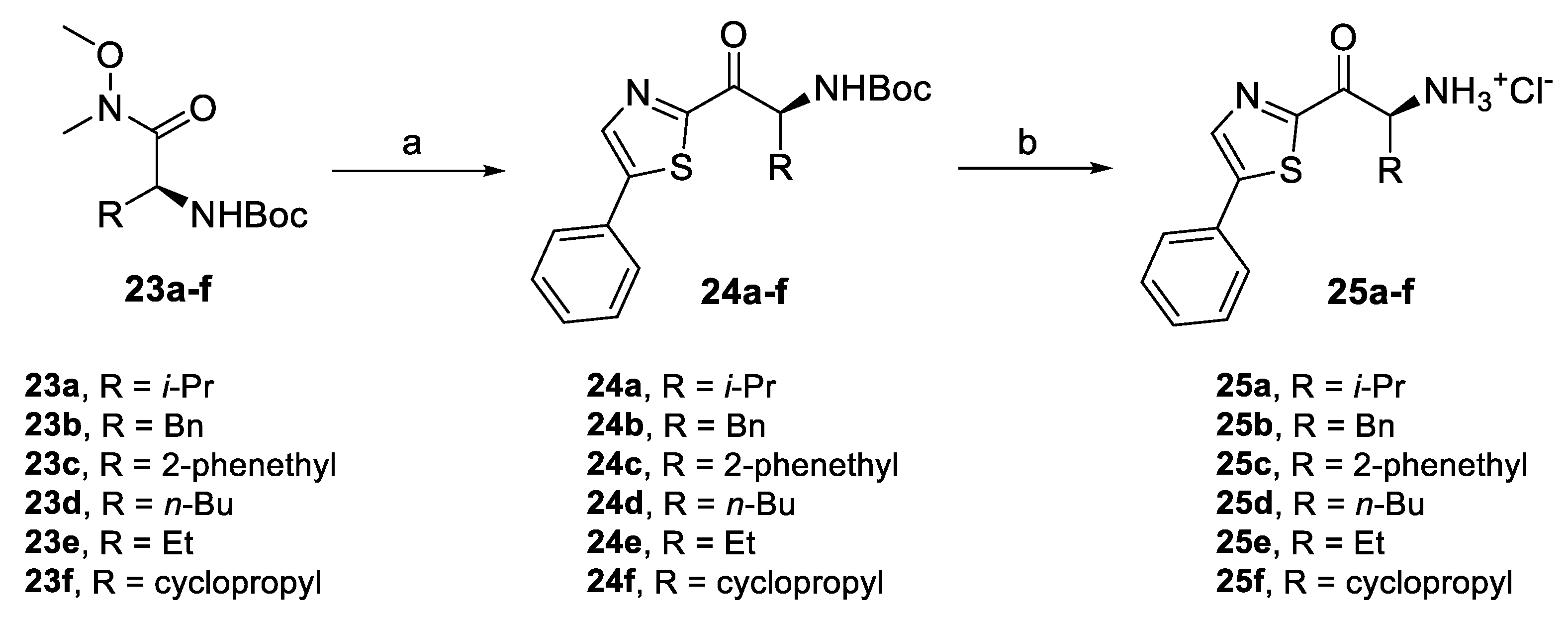
2.4.1. Amino Ketone Derivatives with Variation of the Alkyl Chain
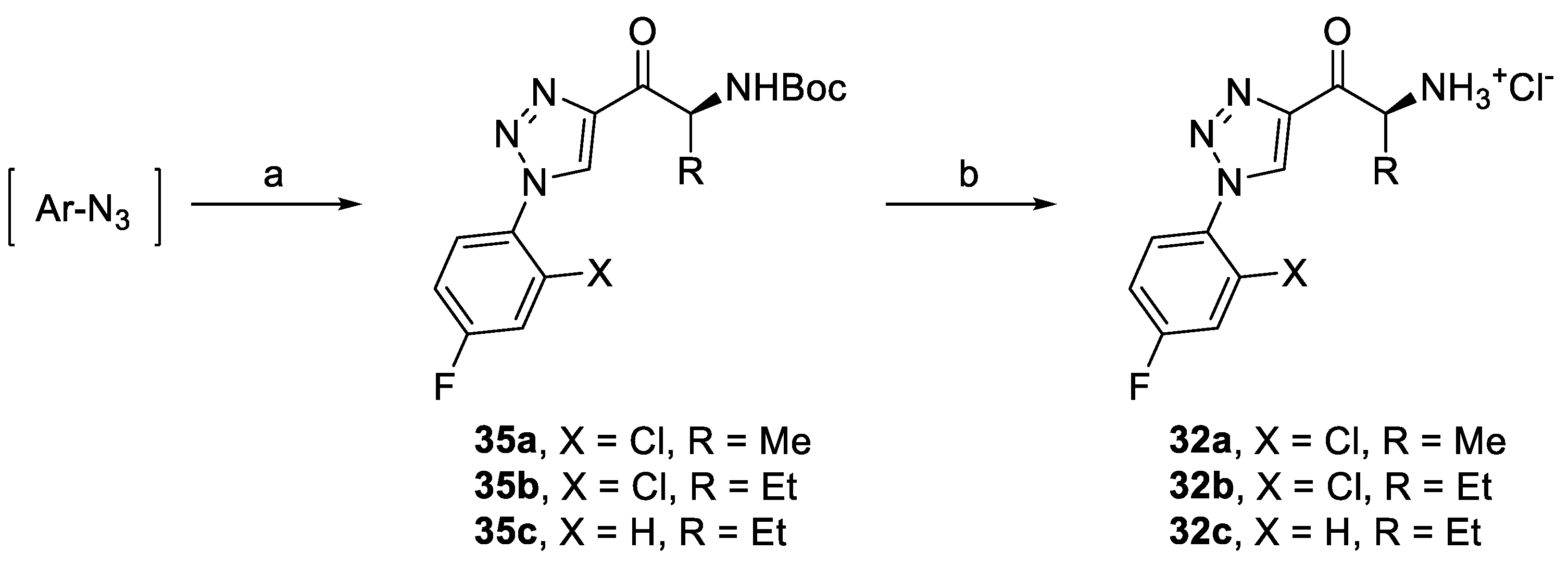
2.4.2. Triazole Derivatives with Variation of Aryl Substituent
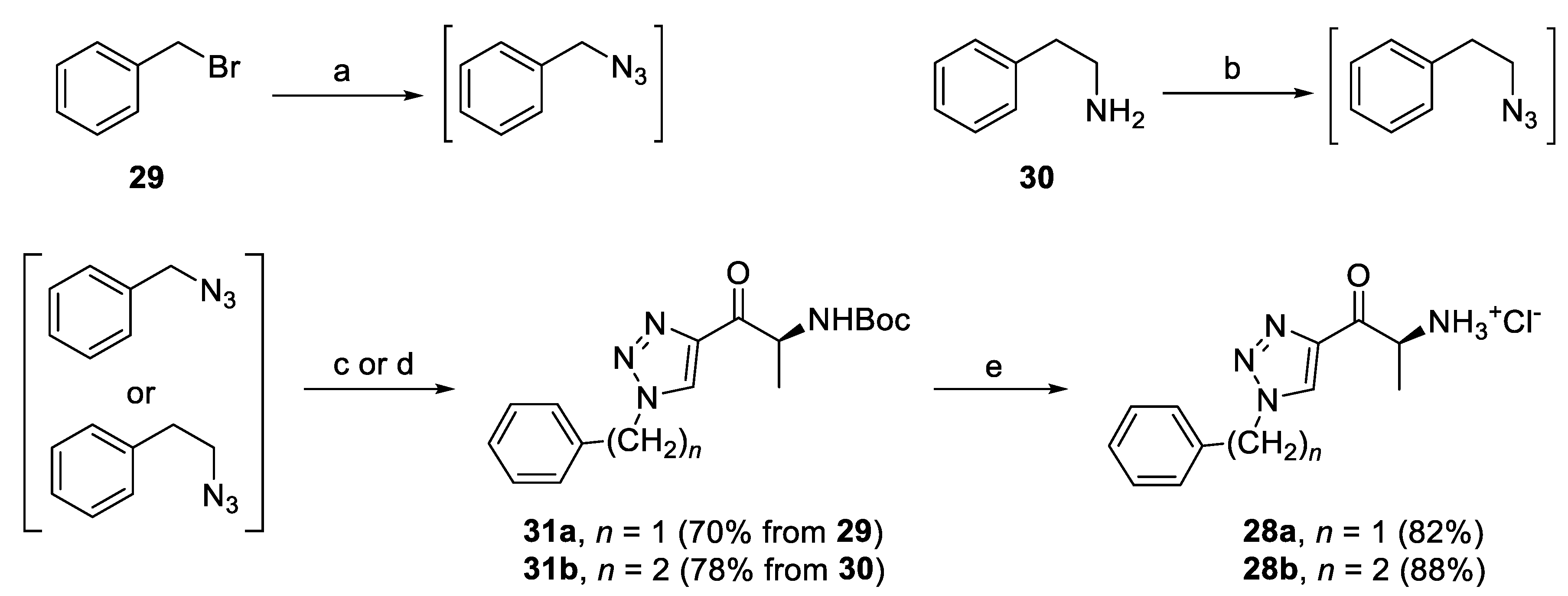
2.4.3. Triazole Derivatives with Alkyl Linker
2.5. Evaluation of Inhibitory Activity of SAR Derivatives
2.6. Synthesis of Optimized Inhibitors Based on SAR Evaluation
2.7. Inhibitory Activity of Structurally Optimized Derivatives
3. Materials and Methods
3.1. Computational Methods
3.1.1. Pharmacophore Modeling
3.1.2. Docking
3.2. Enzyme Production and Purification
3.3. Activity Assay of hKAT-II
3.4. Coupled Fluorescence In Vitro Inhibition Assay
3.5. Chemistry
3.5.1. General Procedure 1 (GP1): Synthesis of Weinreb Amides from Boc-l-Amino Acids
3.5.2. General Procedure 2 (GP2): Synthesis of Alkynones from Weinreb Amides
3.5.3. General Procedure 3 (GP3): Acylation of Thiazoles and Benzo[d]thiazoles with Weinreb Amides
3.5.4. General Procedure 4 (GP4): Synthesis of Aryl Azides
3.5.5. General Procedure 5 (GP5): Synthesis of Triazole Derivatives by CuAAC
3.5.6. General Procedure 6 (GP6): Suzuki Coupling of 2-acyl-5-bromothiazoles
3.5.7. General Procedure 7 (GP7): Synthesis of Heterocyclic Cathinones by Deprotection of the Boc Protecting Group
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Foster, A.C.; Vezzani, A.; French, E.D.; Schwarcz, R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci. Lett. 1984, 48, 273–278. [Google Scholar] [CrossRef]
- Winn, P.; Stone, T.W.; Latimer, M.; Hastings, M.H.; Clark, A.J.M. A comparison of excitotoxic lesions of the basal forebrain by kainate, quinolinate, ibotenate, N-methyl-d-aspartate or quisqualate, and the effects on toxicity of 2-amino-5-phosphonovaleric acid and kynurenic acid in the rat. Br. J. Pharmacol. 1991, 102, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Modoux, M.; Rolhion, N.; Mani, S.; Sokol, H. Tryptophan Metabolism as a Pharmacological Target. Trends Pharmacol. Sci. 2021, 42, 60–73. [Google Scholar] [CrossRef]
- Stone, J.M.; Morrison, P.D.; Pilowsky, L.S. Review: Glutamate and dopamine dysregulation in schizophrenia—A synthesis and selective review. J. Psychopharmacol. 2007, 21, 440–452. [Google Scholar] [CrossRef]
- Plitman, E.; Iwata, Y.; Caravaggio, F.; Nakajima, S.; Chung, J.K.; Gerretsen, P.; Kim, J.; Takeuchi, H.; Chakravarty, M.M.; Remington, G.; et al. Kynurenic Acid in Schizophrenia: A Systematic Review and Meta-analysis. Schizophr. Bull. 2017, 43, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Blennow, K.; Nordin, C.; Skogh, E.; Lindström, L.H.; Engberg, G. Kynurenic acid levels ae elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci. Lett. 2001, 313, 96–98. [Google Scholar] [CrossRef]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef]
- Pocivavsek, A.; Elmer, G.I.; Schwarcz, R. Inhibition of kynurenine aminotransferase II attenuates hippocampus-dependent memory deficit in adult rats treated prenatally with kynurenine. Hippocampus 2019, 29, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Tutakhail, A.; Boulet, L.; Khabil, S.; Nazari, Q.A.; Hamid, H.; Coudoré, F. Neuropathology of Kynurenine Pathway of Tryptophan Metabolism. Curr. Pharmacol. Rep. 2020, 6, 8–23. [Google Scholar] [CrossRef]
- Kindler, J.; Lim, C.K.; Weickert, C.S.; Boerrigter, D.; Galletly, C.; Liu, D.; Jacobs, K.R.; Balzan, R.; Bruggemann, J.; O’Donnell, M.; et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol. Psychiatry 2020, 25, 2860–2872. [Google Scholar] [CrossRef] [Green Version]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocivavsek, A.; Notarangelo, F.M.; Wu, H.-Q.; Bruno, J.P.; Schwarcz, R. Astrocytes as Pharmacological Targets in the Treatment of Schizophrenia: Focus on Kynurenic Acid. In Modeling the Psychopathological Dimensions of Schizophrenia; Pletnikov, M.V., Waddington, J.L., Eds.; Handbook of Behavioral Neuroscience; Elsevier: Amsterdam, The Netherlands, 2016; Volume 23, pp. 423–443. [Google Scholar]
- Fukuwatari, T. Possibility of Amino Acid Treatment to Prevent the Psychiatric Disorders via Modulation of the Production of Tryptophan Metabolite Kynurenic Acid. Nutrients 2020, 12, 1403. [Google Scholar] [CrossRef]
- Potter, M.C.; Elmer, G.I.; Bergeron, R.; Albuquerque, E.X.; Guidetti, P.; Wu, H.-Q.; Schwarcz, R. Reduction of Endogenous Kynurenic Acid Formation Enhances Extracellular Glutamate, Hippocampal Plasticity, and Cognitive Behavior. Neuropsychopharmacology 2010, 35, 1734–1742. [Google Scholar] [CrossRef]
- Rossi, F.; Valentina, C.; Garavaglia, S.; Sathyasaikumar, K.V.; Schwarcz, R.; Kojima, S.-I.; Okuwaki, K.; Ono, S.-I.; Kajii, Y.; Rizzi, M. Crystal structure-based selective targeting of the pyridoxal 5’-phosphate dependent enzyme kynurenine aminotransferase II for cognitive enhancement. J. Med. Chem. 2010, 53, 5684–5689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicciari, R.; Venturoni, F.; Bellocchi, D.; Carotti, A.; Marinozzi, M.; Macchiarulo, A.; Amori, L.; Schwarcz, R. Sequence Variants in Kynurenine Aminotransferase II (KAT II) Orthologs Determine Different Potencies of the Inhibitor S-ESBA. ChemMedChem 2008, 3, 1199–1202. [Google Scholar] [CrossRef]
- Pellicciari, R.; Rizzo, R.C.; Costantino, G.; Marinozzi, M.; Amori, L.; Guidetti, P.; Wu, H.-Q.; Schwarcz, R. Modulators of the Kynurenine Pathway of Tryptophan Metabolism: Synthesis and Preliminary Biological Evaluation of (S)-4-(Ethylsulfonyl)benzoylalanine, a Potent and Selective Kynurenine Aminotransferase II (KAT II) Inhibitor. ChemMedChem 2006, 1, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Dounay, A.B.; Anderson, M.; Bechle, B.M.; Campbell, B.M.; Claffey, M.M.; Evdokimov, A.; Evrard, E.; Fonseca, K.R.; Gan, X.; Ghosh, S.; et al. Discovery of Brain-Penetrant, Irreversible Kynurenine Aminotransferase II Inhibitors for Schizophrenia. ACS Med. Chem. Lett. 2012, 3, 187–192. [Google Scholar] [CrossRef]
- Imbeault, S.; Olivé, M.G.; Jungholm, O.; Erhardt, S.; Wigström, H.; Engberg, G.; Jardemark, K. Blockade of KAT II Facilitates LTP in Kynurenine 3-Monooxygenase Depleted Mice. Int. J. Tryptophan Res. 2021, 14, 1–8. [Google Scholar] [CrossRef]
- Koshy Cherian, A.; Gritton, H.; Johnson, D.E.; Young, D.; Kozak, R.; Sarter, M. A systemically-available kynurenine aminotransferase II (KAT II) inhibitor restores nicotine-evoked glutamatergic activity in the cortex of rats. Neuropharmacology 2014, 82, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Linderholm, K.R.; Alm, M.T.; Larsson, M.K.; Olsson, S.K.; Goiny, M.; Hajos, M.; Erhardt, S.; Engberg, G. Inhibition of kynurenine aminotransferase II reduces activity of midbrain dopamine neurons. Neuropharmacology 2016, 102, 42–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herédi, J.; Cseh, E.K.; Berkó, A.M.; Veres, G.; Zádori, D.; Toldi, J.; Kis, Z.; Vécsei, L.; Ono, E.; Gellért, L. Investigating KYNA production and kynurenergic manipulation on acute mouse brain slice preparations. Brain Res. Bull. 2019, 146, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Zakhary, G.; Sherchan, P.; Li, Q.; Tang, J.; Zhang, J.H. Modification of kynurenine pathway via inhibition of kynurenine hydroxylase attenuates surgical brain injury complications in a male rat model. J. Neurosci. Res. 2020, 98, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Yamasaki, S.; Kaneko, O.; Taoka, N.; Tomimoto, Y.; Namatame, I.; Yahata, T.; Kuromitsu, S.; Cantley, L.C.; Lyssiotis, C.A. A covalent small molecule inhibitor of glutamate-oxaloacetate transaminase 1 impairs pancreatic cancer growth. Biochem. Biophys. Res. Commun. 2020, 522, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Klausing, A.D.; Fukuwatari, T.; Bucci, D.J.; Schwarcz, R. Stress-induced impairment in fear discrimination is causally related to increased kynurenic acid formation in the prefrontal cortex. Psychopharmacology 2020, 237, 1931–1941. [Google Scholar] [CrossRef]
- Kozak, R.; Campbell, B.M.; Strick, C.A.; Horner, W.; Hoffmann, W.E.; Kiss, T.; Chapin, D.S.; McGinnis, D.; Abbott, A.L.; Roberts, B.M.; et al. Reduction of Brain Kynurenic Acid Improves Cognitive Function. J. Neurosci. 2014, 34, 10592–10602. [Google Scholar] [CrossRef]
- Henderson, J.L.; Sawant-Basak, A.; Tuttle, J.B.; Dounay, A.B.; McAllister, L.A.; Pandit, J.; Rong, S.; Hou, X.; Bechle, B.M.; Kim, J.-Y.; et al. Discovery of hydroxamate bioisosteres as KAT II inhibitors with improved oral bioavailability and pharmacokinetics. Med. Chem. Commun. 2013, 4, 125–129. [Google Scholar] [CrossRef]
- Tuttle, J.B.; Anderson, M.; Bechle, B.M.; Campbell, B.M.; Chang, C.; Dounay, A.B.; Evrard, E.; Fonseca, K.R.; Gan, X.; Ghosh, S.; et al. Structure-Based Design of Irreversible Human KAT II Inhibitors: Discovery of New Potency-Enhancing Interactions. ACS Med. Chem. Lett. 2013, 4, 37–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claffey, M.M.; Dounay, A.B.; Gan, X.; Hayward, M.M.; Rong, S.; Tuttle, J.B.; Verhoest, P.R. Bicyclic and Tricyclic Compounds as KAT II Inhibitors. WO Patent 2010/146488A1, 23 December 2010. [Google Scholar]
- Dounay, A.B.; McAllister, L.A.; Parikh, V.D.; Rong, S.; Verhoest, P.R. KAT II Inhibitors. WO Patent 2012/073143A1, 7 June 2012. [Google Scholar]
- Dounay, A.B.; Anderson, M.; Bechle, B.M.; Evrard, E.; Gan, X.; Kim, J.-Y.; McAllister, L.A.; Pandit, J.; Rong, S.; Salafia, M.A.; et al. PF-04859989 as a template for structure-based drug design: Identification of new pyrazole series of irreversible KAT II inhibitors with improved lipophilic efficiency. Bioorg. Med. Chem. Lett. 2013, 23, 1961–1966. [Google Scholar] [CrossRef]
- Dounay, A.B.; Tuttle, J.B.; Verhoest, P.R. Preparation of Tricyclic Compounds as KAT II Inhibitors. WO Patent 2013/186666A1, 19 December 2013. [Google Scholar]
- Wu, H.-Q.; Okuyama, M.; Kajii, Y.; Pocivavsek, A.; Bruno, J.P.; Schwarcz, R. Targeting Kynurenine Aminotransferase II in Psychiatric Diseases: Promising Effects of an Orally Active Enzyme Inhibitor. Schizophr. Bull. 2014, 40, S152–S158. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Fujigaki, H.; Kato, K.; Yamazaki, K.; Fujigaki, S.; Kunisawa, K.; Yamamoto, Y.; Mouri, A.; Oda, A.; Nabeshima, T.; et al. Selective and competitive inhibition of kynurenine aminotransferase 2 by glycyrrhizic acid and its analogues. Sci. Rep. 2019, 9, 10243. [Google Scholar] [CrossRef] [Green Version]
- Jayawickrama, G.S.; Nematollahi, A.; Sun, G.; Church, W.B. Improvement of kynurenine aminotransferase-II inhibitors guided by mimicking sulfate esters. PLoS ONE 2018, 13, e0196404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuyama, M.; Fukunaga, K.; Usui, K.; Hayashi, N.; Iijima, D.; Horiuchi, H.; Itagaki, N. Novel Bicyclic or Tricyclic Heterocyclic Compounds and Their Pharmaceutical Compositions for Prophylactic and Therapeutic Treatment of KAT (Kynurenine Aminotransferase) II-Associated Disorders. WO Patent 2015/163339A1, 29 October 2015. [Google Scholar]
- Kalliokoski, T.; Rummakko, P.; Rantanen, M.; Blaesse, M.; Augustin, M.; Ummenthala, G.R.; Choudhary, S.; Venäläinen, J. Discovery of sulfonamides and 9-oxo-2,8-diazaspiro[5,5]undecane-2-carboxamides as human kynurenine aminotransferase 2 (KAT2) inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 127060. [Google Scholar] [CrossRef] [PubMed]
- Morwick, T.; Hrapchak, M.; DeTuri, M.; Campbell, S. A practical approach to the synthesis of 2,4-disubstituted oxazoles from amino acids. Org. Lett. 2002, 4, 2665–2668. [Google Scholar] [CrossRef] [PubMed]
- Pirc, S.; Bevk, D.; Golobič, A.; Stanovnik, B.; Svete, J. Transformation of amino acids into nonracemic 1-(heteroaryl)ethanamines by the enamino ketone methodology. Helv. Chim. Acta 2006, 89, 30–44. [Google Scholar] [CrossRef]
- Liu, J.; Ikemoto, N.; Petrillo, D.; Armstrong, J.D. Improved syntheses of α-BOC-aminoketones from α-BOC-amino-Weinreb amides using a pre-deprotonation protocol. Tetrahedron Lett. 2002, 43, 8223–8226. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Madsen, U.; Björkling, F.; Liang, X. Rapid Synthesis of Aryl Azides from Aryl Halides under Mild Conditions. Synlett 2005, 2209–2213. [Google Scholar] [CrossRef]
- Berry, M.T.; Castrejon, D.; Hein, J.E. Oxidative Esterification of Aldehydes Using Mesoionic 1,2,3-Triazolyl Carbene Organocatalysts. Org. Lett. 2014, 16, 3676–3679. [Google Scholar] [CrossRef]
- Ran, R.Q.; He, J.; Xiu, S.D.; Wang, K.B.; Li, C.Y. Synthesis of 3-pyrrolin-2-ones by rhodium-catalyzed transannulation of 1-sulfonyl-1,2,3-triazole with ketene silyl acetal. Org. Lett. 2014, 16, 3704–3707. [Google Scholar] [CrossRef]
- Kitamura, M.; Kato, S.; Yano, M.; Tashiro, N.; Shiratake, Y.; Sando, M.; Okauchi, T. A reagent for safe and efficient diazo-transfer to primary amines: 2-azido-1,3-dimethylimidazolinium hexafluorophosphate. Org. Biomol. Chem. 2014, 12, 4397–4406. [Google Scholar] [CrossRef]
- Sharma, D.K.; Spencer, T.; Adams, J.; Liebmann, K.L.; Miller, S.C. Rapid Access to a Broad Range of 6′-Substituted Firefly Luciferin Analogues Reveals Surprising Emitters and Inhibitors. Org. Lett. 2017, 19, 5836–5839. [Google Scholar] [CrossRef]
- Tsuruoka, A.; Kaku, Y.; Kakinuma, H.; Tsukada, I.; Yanagisawa, M.; Nara, K.; Naito, T. Synthesis and Antifungal Activity of Novel Thiazole-Containing Triazole Antifungals. II. Optically Active ER-30346 and Its Derivatives. Chem. Pharm. Bull. 1998, 46, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skácel, J.; Dračínský, M.; Janeba, Z. Synthesis of Tetrasubstituted Thiophenes via Direct Metalation. J. Org. Chem. 2020, 85, 788–797. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021.
- Lu, H.; Kopcho, L.; Ghosh, K.; Witmer, M.; Parker, M.; Gupta, S.; Paul, M.; Krishnamurthy, P.; Laksmaiah, B.; Xie, D.; et al. Development of a RapidFire mass spectrometry assay and a fluorescence assay for the discovery of kynurenine aminotransferase II inhibitors to treat central nervous system disorders. Anal. Biochem. 2016, 501, 56–65. [Google Scholar] [CrossRef]
- García-Urricelqui, A.; de Cózar, A.; Mielgo, A.; Palomo, C. Probing α-Amino Aldehydes as Weakly Acidic Pronucleophiles: Direct Access to Quaternary α-Amino Aldehydes by an Enantioselective Michael Addition Catalyzed by Brønsted Bases. Chem. – A Eur. J. 2021, 27, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Stephenson, C.R.J. Total Synthesis of Syringolin A. Org. Lett. 2010, 12, 3453–3455. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, H.Y.; Lei, M.; Li, Z.Y.; Zhu, Y.Q. An Efficient Preparation of Novel Epoxyketone Intermediates for the Synthesis of Carfilzomib and Its Derivatives. J. Chem. Res. 2016, 40, 82–86. [Google Scholar] [CrossRef]
- Ripa, L.; Edman, K.; Dearman, M.; Edenro, G.; Hendrickx, R.; Ullah, V.; Chang, H.-F.; Lepistö, M.; Chapman, D.; Geschwindner, S.; et al. Discovery of a Novel Oral Glucocorticoid Receptor Modulator (AZD9567) with Improved Side Effect Profile. J. Med. Chem. 2018, 61, 1785–1799. [Google Scholar] [CrossRef] [Green Version]
- Audin, P.; Pothion, C.; Fehrentz, J.-A.; Loffet, A.; Martinez, J.; Paris, J. Diastereoselective Synthesis of N-Protected β-Amino-α-Hydroxyacids (Norstatines) from Urethane N-Carboxyanhydrides (UNCAs). J. Chem. Res. Synopses 1999, 4, 282–283. [Google Scholar] [CrossRef]
- Gouault, N.; Le Roch, M.; Cornée, C.; David, M.; Uriac, P. Synthesis of Substituted Pyrrolin-4-Ones from Amino Acids in Mild Conditions via a Gold-Catalyzed Approach. J. Org. Chem. 2009, 74, 5614–5617. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Kim, E.; Kim, M.; Hwang, Y.J.; Padhi, B.; Choi, J.; Lee, Y.; Joo, J.M. Divergent Strategies for the π-Extension of Heteroaryl Halides Using Norbornadiene as an Acetylene Synthon. Org. Lett. 2020, 22, 9670–9676. [Google Scholar] [CrossRef] [PubMed]
- Steert, K.; Berg, M.; Mottram, J.C.; Westrop, G.D.; Coombs, G.H.; Cos, P.; Maes, L.; Joossens, J.; Van der Veken, P.; Haemers, A.; et al. α-Ketoheterocycles as Inhibitors of Leishmania Mexicana Cysteine Protease CPB. ChemMedChem 2010, 5, 1734–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Wu, X.; Yu, Y.; An, L.; Yin, X. A Convenient Synthesis of 2-Acyl Benzothiazoles/Thiazoles from Benzothiazole/Thiazole and N,N’-Carbonyldiimidazole Activated Carboxylic Acids. Tetrahedron Lett. 2019, 60, 1667–1670. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | Inhibitory Activity [%I ± SEM] c = 1 µM | IC50 ± SEM [µM] |
|---|---|---|---|
| 1 | 11 | 41.41 ± 4.15 | 1.62 ± 1.43 |
| 2 | 12 | 43.19 ± 2.30 | 3.21 ± 0.26 |
 Reagents and conditions: (a) CuI, N,N’-dimethylethylenediamine (DMEDA), NaN3, sodium ascorbate, EtOH/H2O, reflux (X = Br, I); (b) (i) NaNO2, HCl, 0 °C, (ii) NaHCO3, NaN3, 0 °C–r.t. (X = NH2); (c) 15, CuSO4.5H2O, sodium ascorbate, t-BuOH/H2O 1:1, r.t.; (d) 15, CuTC, toluene, r.t.; and (e) AcCl, MeOH, r.t. Reagents and conditions: (a) CuI, N,N’-dimethylethylenediamine (DMEDA), NaN3, sodium ascorbate, EtOH/H2O, reflux (X = Br, I); (b) (i) NaNO2, HCl, 0 °C, (ii) NaHCO3, NaN3, 0 °C–r.t. (X = NH2); (c) 15, CuSO4.5H2O, sodium ascorbate, t-BuOH/H2O 1:1, r.t.; (d) 15, CuTC, toluene, r.t.; and (e) AcCl, MeOH, r.t. | |||||||||
| Entry | Ar | X | Boc Derivative | Amino Ketone | Entry | Ar | X | Boc Derivative | Amino Ketone |
| 1 |  | NH2 | 27a, 65% | 26a, 82% | 12 |  | Br | 27l, 67% | 26l, 88% a |
| 2 | 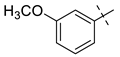 | Br | 27b, 67% | 26b, 65% | 13 |  | Br | 27m, 66% | 26m, 93% a |
| 3 |  | NH2 | 27c, 88% | 26c, 79% | 14 | 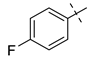 | I | 27n, 72% | 26n, 95% |
| 4 |  | I | 27d, 83% | 26d, 89% | 15 |  | NH2 | 27o, 66% | 26o, 71% |
| 5 |  | Br | 27e, 76% | 26e, 85% | 16 |  | NH2 | 27p, 69% | 26p, 70% |
| 6 |  | NH2 | 27f, 67% | 26f, 72% | 17 |  | NH2 | 27q, 72% | 26q, 75% |
| 7 |  | NH2 | 27g, 73% | 26g, 71% | 18 | 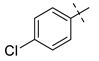 | Br | 27r, 44% | 26r, 79% |
| 8 |  | NH2 | 27h, 72% | 26h, 70% | 19 |  | NH2 | 27s, 78% | 26s, 67% |
| 9 |  | Br | 27i, 58% | 26i, 90% | 20 |  | NH2 | 27t, 69% | 26t, 72% |
| 10 |  | Br | 27j, 86% | 26j, 91% | 21 |  | I | 27u, 82% | 26u, 76% |
| 11 |  | Br | 27k, 47% | 26k, 79% | 22 |  | NH2 | 27v, 77% | 26v, 79% |
| Entry | Compound | Inhibitory Activity [%I ± SEM] c = 1 µM |
|---|---|---|
| Triazole derivatives with the alkyl chain variation | ||
| 1 | 21a | 10.32 ± 4.81 |
| 2 | 21b | 14.52 ± 3.71 |
| 3 | 21c | 16.50 ± 3.71 |
| 4 | 21d | 13.46 ± 5.46 |
| 5 | 21e | 21.82 ± 4.80 |
| 6 | 21f | 7.44 ± 3.75 |
| Thiazole derivatives with the alkyl chain variation | ||
| 7 | 25a | 31.24 ± 3.64 |
| 8 | 25b | 7.22 ± 2.70 |
| 9 | 25c | 31.32 ± 0.88 |
| 10 | 25d | 37.22 ± 2.53 |
| 11 | 25e | 44.18 ± 3.42 |
| 12 | 25f | 6.12 ± 2.66 |
| Triazole derivatives with alkyl linker | ||
| 13 | 28a | 14.54 ± 3.19 |
| 14 | 28b | 11.84 ± 5.76 |
| Triazole derivatives with the aryl variation | ||
| 15 | 26a | 38.50 ± 2.21 |
| 16 | 26b | 18.20 ± 2.27 |
| 17 | 26c | 23.41 ± 3.97 |
| 18 | 26d | 0.98 ± 1.98 |
| 19 | 26e | 18.56 ± 2.33 |
| 20 | 26f | 9.10 ± 2.07 |
| 21 | 26g | 41.60 ± 6.39 |
| 22 | 26h | 25.38 ± 2.22 |
| 23 | 26i | 4.32 ± 1.32 |
| 24 | 26j | 27.81 ± 3.33 |
| 25 | 26k | 20.73 ± 1.83 |
| 26 | 26l | 23.63 ± 2.30 |
| 27 | 26m | 33.48 ± 4.88 |
| 28 | 26n | 41.77 ± 3.01 |
| 29 | 26o | 9.70 ± 1.67 |
| 30 | 26p | 41.43 ± 2.67 |
| 31 | 26q | 41.73 ± 2.66 |
| 32 | 26r | 23.86 ± 2.95 |
| 33 | 26s | 20.63 ± 3.78 |
| 34 | 26t | 35.52 ± 3.63 |
| 35 | 26u | 19.33 ± 1.06 |
| 36 | 26v | 23.62 ± 4.30 |
| Entry | Compound | Inhibitory Activity [%I ± SEM] c = 1 µM | IC50 ± SEM [µM] |
|---|---|---|---|
| 1 | 32a | 25.30 ± 1.10 | N.D. |
| 2 | 32b | 21.25 ± 0.67 | N.D. |
| 3 | 32c | 35.94 ± 2.87 | N.D. |
| 4 | 34a | 52.49 ± 1.31 | N.D. |
| 5 | 34b | 56.60 ± 3.68 | 0.987 ± 0.088 |
| 6 | 34c | 36.16 ± 3.81 | N.D. |
| 7 | 34d | 9.73 ± 1.41 | N.D. |
| 8 | 49a | 61.54 ± 7.16 | 0.097 ± 0.014 |
| 9 | 49b | 57.07 ± 0.31 | N.D. |
| 10 | 49c | 47.65 ± 1.71 | N.D. |
| 11 | 49d | 59.71 ± 0.97 | 0.304 ± 0.039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maryška, M.; Svobodová, L.; Dehaen, W.; Hrabinová, M.; Rumlová, M.; Soukup, O.; Kuchař, M. Heterocyclic Cathinones as Inhibitors of Kynurenine Aminotransferase II—Design, Synthesis, and Evaluation. Pharmaceuticals 2021, 14, 1291. https://doi.org/10.3390/ph14121291
Maryška M, Svobodová L, Dehaen W, Hrabinová M, Rumlová M, Soukup O, Kuchař M. Heterocyclic Cathinones as Inhibitors of Kynurenine Aminotransferase II—Design, Synthesis, and Evaluation. Pharmaceuticals. 2021; 14(12):1291. https://doi.org/10.3390/ph14121291
Chicago/Turabian StyleMaryška, Michal, Lucie Svobodová, Wim Dehaen, Martina Hrabinová, Michaela Rumlová, Ondřej Soukup, and Martin Kuchař. 2021. "Heterocyclic Cathinones as Inhibitors of Kynurenine Aminotransferase II—Design, Synthesis, and Evaluation" Pharmaceuticals 14, no. 12: 1291. https://doi.org/10.3390/ph14121291
APA StyleMaryška, M., Svobodová, L., Dehaen, W., Hrabinová, M., Rumlová, M., Soukup, O., & Kuchař, M. (2021). Heterocyclic Cathinones as Inhibitors of Kynurenine Aminotransferase II—Design, Synthesis, and Evaluation. Pharmaceuticals, 14(12), 1291. https://doi.org/10.3390/ph14121291