
Discovery of a Novel Template, 7-Substituted 7-Deaza-4′-Thioadenosine Derivatives as Multi-Kinase Inhibitors

 , , , , , , , , , and
, , , , , , , , , and
Abstract
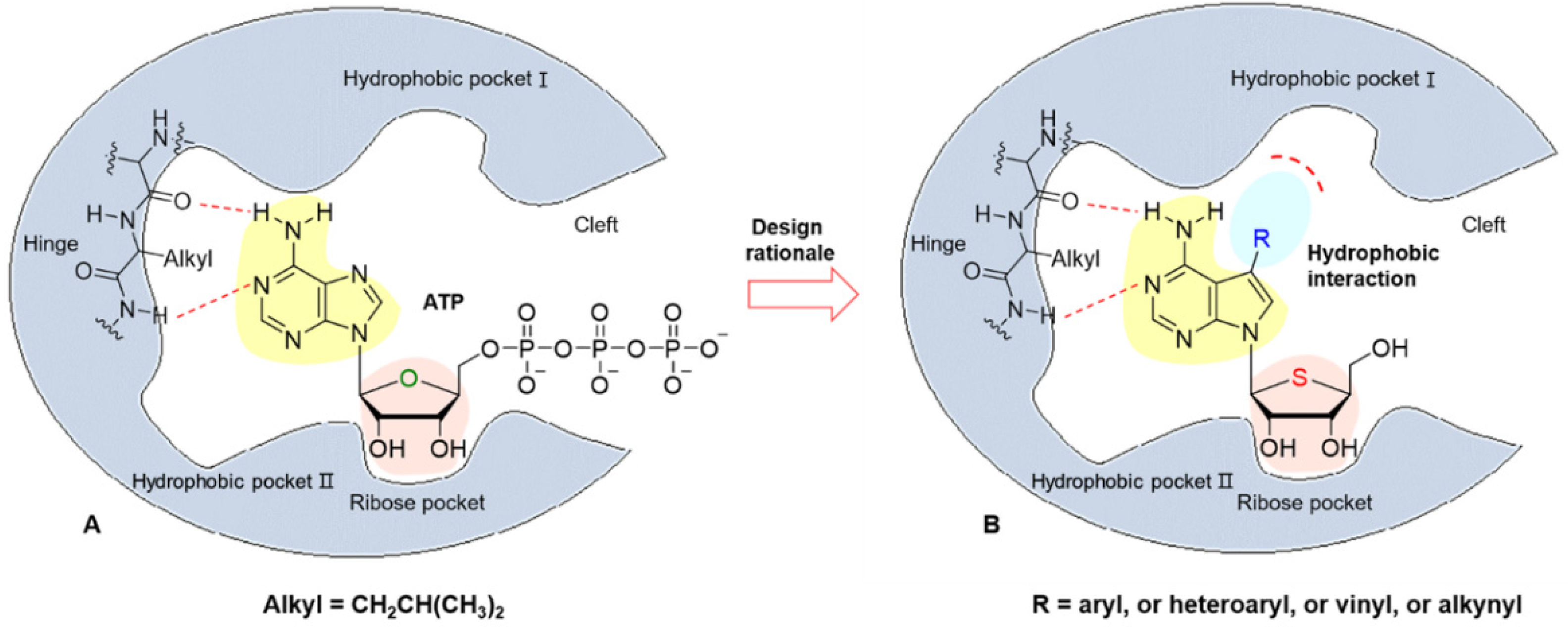
1. Introduction
2. Results and Discussion
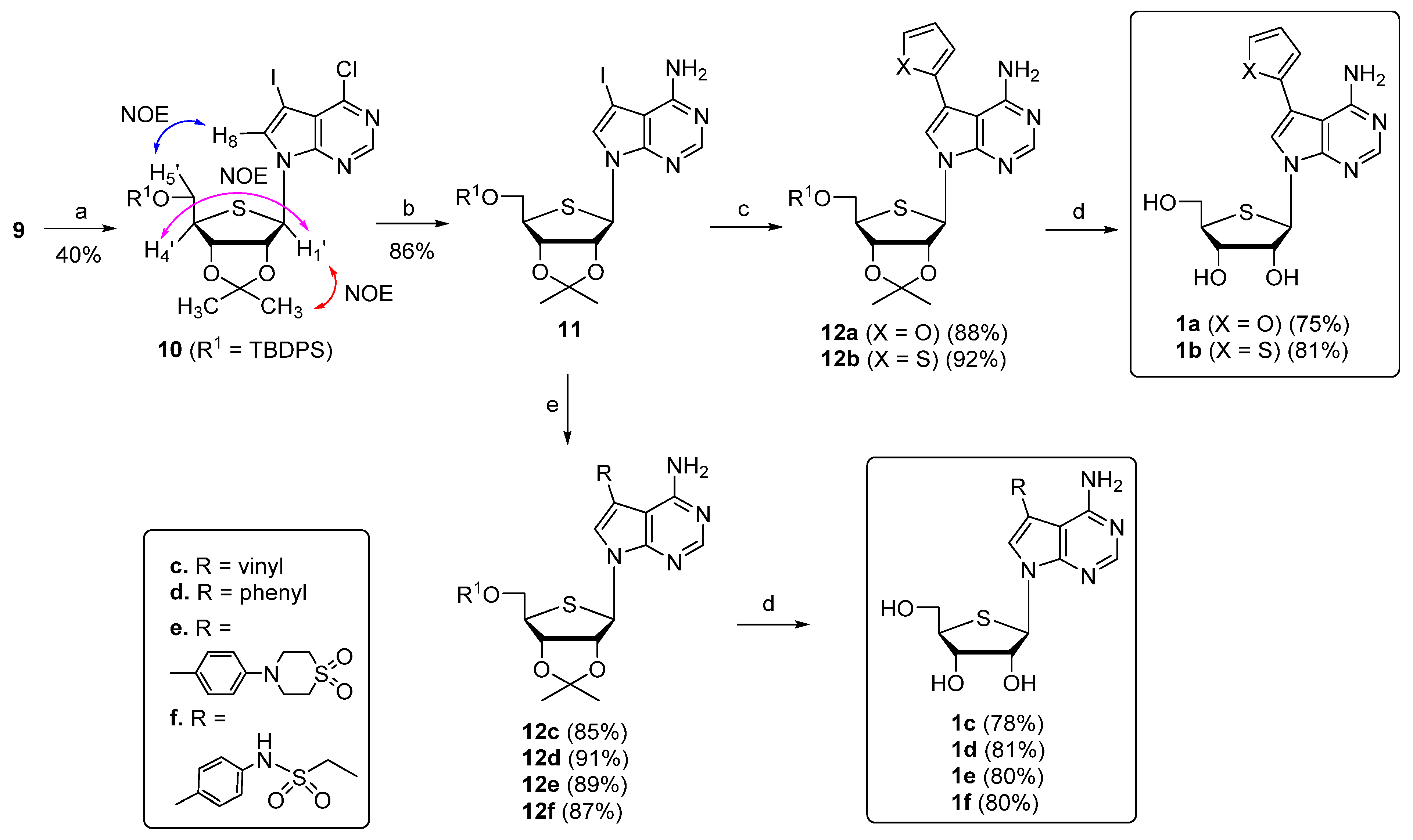
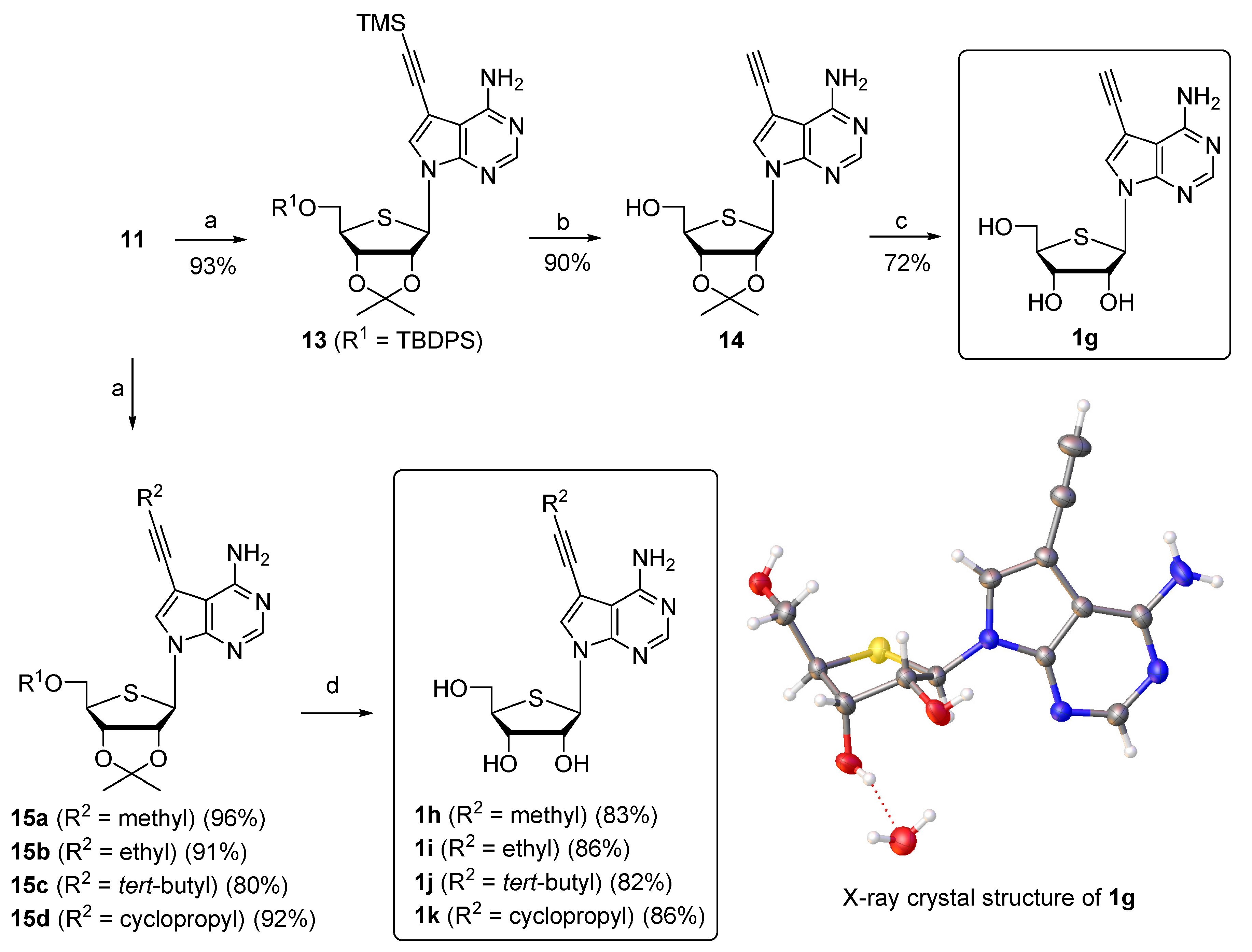
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Antiproliferative Activity
2.2.2. Kinome Scan Profile
2.2.3. Antiproliferative Activity against KM12 and ACHN Cell Lines
2.2.4. Metabolic Stability and CYP Inhibition
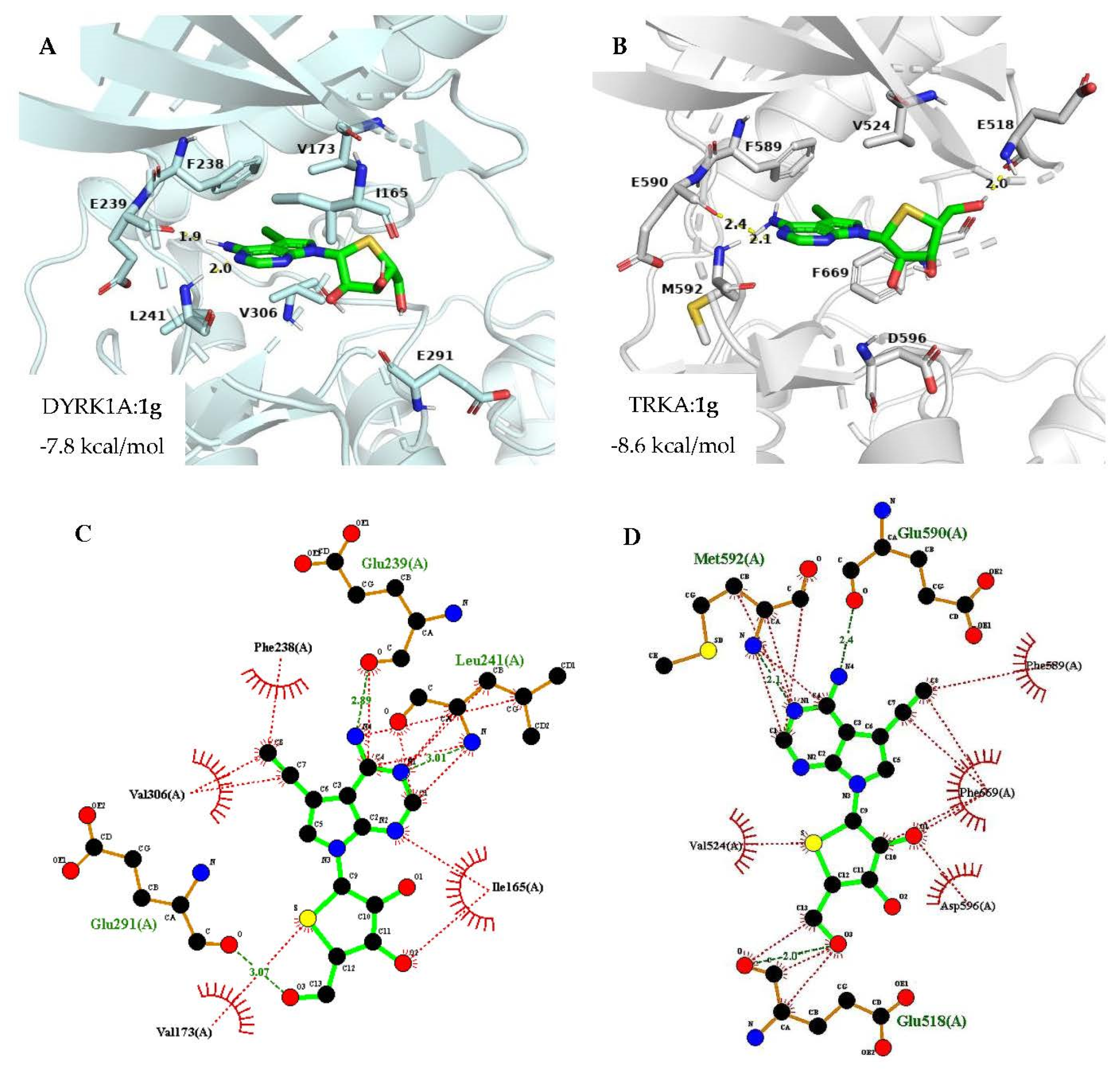
2.3. Docking Analysis
3. Materials and Methods
3.1. General Methods
3.2. Chemical Synthesis
3.2.1. (3aR,6S,6aR)-6-(Hydroxymethyl)-2,2-dimethyldihydrofuro[3,4-d][1,3]dioxol-4(3aH)-one (3)
3.2.2. (3aR,6S,6aR)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyldihydrofuro[3,4-d][1,3]dioxol-4(3aH)-one (4)
3.2.3. (S)-2-((tert-butyldiphenylsilyl)oxy)-1-((4S,5R)-5-(hydroxymethyl)-2,2-dimethyl-1,3-dioxolan-4-yl)ethan-1-ol (5)
3.2.4. (S)-2-((tert-butyldiphenylsilyl)oxy)-1-((4R,5R)-2,2-dimethyl-5-(((methylsulfonyl)oxy)methyl)-1,3-dioxolan-4-yl)ethyl Methanesulfonate (6)
3.2.5. tert-Butyl(((3aS,4R,6aR)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4 yl)methoxy)diphenylsilane (7)
3.2.6. (3aS,4R,5S,6aR)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxole 5-oxide (8)
3.2.7. (3aR,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl acetate (9)
3.2.8. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-4-chloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine (10)
3.2.9. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-iodo-7H-pyrrolo[2,3-d]pyrimidin-4-amine (11)
3.2.10. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-(furan-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (12a)
3.2.11. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-(thiophen-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (12b)
3.2.12. (2R,3R,4S,5R)-2-(4-Amino-5-(furan-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1a)
3.2.13. (2R,3R,4S,5R)-2-(4-Amino-5-(thiophen-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1b)
3.2.14. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-vinyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (12c)
3.2.15. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (12d)
3.2.16. 4-(4-(4-Amino-7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)thiomorpholine 1,1-dioxide (12e)
3.2.17. N-(4-(4-Amino-7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)ethanesulfonamide (12f)
3.2.18. (2R,3R,4S,5R)-2-(4-Amino-5-vinyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1c)
3.2.19. (2R,3R,4S,5R)-2-(4-Amino-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1d)
3.2.20. 4-(4-(4-Amino-7-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrothiophen-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)thiomorpholine 1,1-dioxide (1e)
3.2.21. N-(4-(4-Amino-7-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrothiophen-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)ethanesulfonamide (1f)
3.2.22. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-((trimethylsilyl)ethynyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (13)
3.2.23. ((3aS,4R,6R,6aR)-6-(4-Amino-5-ethynyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)methanol (14)
3.2.24. (2R,3R,4S,5R)-2-(4-Amino-5-ethynyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1g)
3.2.25. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-(prop-1-yn-1-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (15a)
3.2.26. 5-(But-1-yn-1-yl)-7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (15b)
3.2.27. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-(3,3-dimethylbut-1-yn-1-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (15c)
3.2.28. 7-((3aR,4R,6R,6aS)-6-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-5-(cyclopropylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (15d)
3.2.29. (2R,3R,4S,5R)-2-(4-Amino-5-(prop-1-yn-1-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1h)
3.2.30. (2R,3R,4S,5R)-2-(4-Amino-5-(but-1-yn-1-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1i)
3.2.31. (2R,3R,4S,5R)-2-(4-Amino-5-(3,3-dimethylbut-1-yn-1-yl)-7H-pyrrolo[2, 3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1j)
3.2.32. (2R,3R,4S,5R)-2-(4-Amino-5-(cyclopropylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)tetrahydrothiophene-3,4-diol (1k)
3.3. Cell Proliferation Inhibition Assay (SRB Assay)
Cell Culture
3.4. Kinome Scan Assays
3.5. Metabolic Stability
3.6. CYP Inhibition Assay
3.7. Computational Docking Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Khamisipour, G.; Jadidi-Niaragh, F.; Jahromi, A.S.; Zandi, K.; Hojjat-Farsangi, M. Mechanisms of tumor cell resistance to the current targeted-therapy agents. Tumor Biol. 2016, 37, 10021–10039. [Google Scholar] [CrossRef]
- Potapova, O.; Laird, A.D.; Nannini, M.A.; Barone, A.; Li, G.; Moss, K.G.; Cherrington, J.M.; Mendel, D.B. Contribution of individual targets to the antitumor efficacy of the multitargeted receptor tyrosine kinase inhibitor SU11248. Mol. Cancer Ther. 2006, 5, 1280–1289. [Google Scholar] [CrossRef][Green Version]
- Peters, J.-U. Polypharmacology-foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A. Multitarget drug discovery and polypharmacology. Chem. Med. Chem. 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by design: A medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Chaudhari, R.; Fong, L.W.; Tan, Z.; Huang, B.; Zhang, S. An up-to-date overview of computational polypharmacology in modern drug discovery. Expert Opin. Drug Discov. 2020, 15, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Bracarda, S.; Caserta, C.; Sordini, L.; Rossi, M.; Hamzay, A.; Crino, L. Protein kinase inhibitors in the treatment of renal cell carcinoma: Sorafenib. Ann. Oncol. 2007, 18, vi22–vi25. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a pan−TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Okamoto, K.; Ikemori-Kawada, M.; Jestel, A.; von Konig, K.; Funahashi, Y.; Matsushima, T.; Tsuruoka, A.; Inoue, A.; Matsui, J. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med. Chem. Lett. 2015, 6, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Vulpetti, A.; Bosotti, R. Sequence and structural analysis of kinase ATP pocket residues. IL Farmaco 2004, 59, 759–765. [Google Scholar] [CrossRef]
- Zheng, J.; Knighton, D.R.; Ten Eyck, L.F.; Karlsson, R.; Xuong, N.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 1993, 32, 2154–2161. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Gandin, V.; Ferrarese, A.; Dalla Via, M.; Marzano, C.; Chilin, A.; Marzaro, G. Targeting kinases with anilinopyrimidines: Discovery of N-phenyl-N′-[4-(pyrimidin-4-ylamino)phenyl]urea derivatives as selective inhibitors of class III receptor tyrosine kinase subfamily. Sci. Rep. 2015, 5, 16750. [Google Scholar] [CrossRef]
- Morphy, R. Selectively nonselective kinase inhibition: Striking the right balance. J. Med. Chem. 2010, 53, 1413–1437. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H., III; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitor. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Dikic, I.; Ebner, D.; Ettmayer, P.; Fischer, C.; Frederiksen, M.; et al. The kinase chemogenomic set (KCGS): An open science resource for kinase vulnerability identification. Int. J. Mol. Sci. 2021, 22, 566. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, 1148. [Google Scholar] [CrossRef] [PubMed]
- Biabani, M.F.; Gunasekera, S.P.; Longley, R.E.; Wright, A.E.; Pomponi, S.A. Tubercidin, a cytotoxic agent from the marine sponge Caulospongia biflabellata. Pharm. Biol. 2002, 40, 302–303. [Google Scholar] [CrossRef]
- Grage, T.B.; Rochlin, D.B.; Weiss, A.J.; Wilson, W.L. Clinical studies with tubercidin administered after absorption into human erythrocytes. Cancer Res. 1970, 30, 79–81. [Google Scholar] [PubMed]
- Bourderioux, A.; Nauš, P.; Perlíková, P.; Pohl, R.; Pichová, I.; Votruba, I.; Džubák, P.; Konečný, P.; Hajdúch, M.; Stray, K.M.; et al. Synthesis and significant cytostatic activity of 7-hetaryl-7-deazaadenosines. J. Med. Chem. 2011, 54, 5498–5507. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, K.N.; Shortnacy-fowler, A.T.; Cappellacci, L.; Parker, W.B.; Waud, W.R.; Montgomery, J.A.; Secrist, J.A., III. Synthesis of 4′-thio-β-D-arabinofuranosylcytosine (4′-thio-ara-C) and comparison of its anticancer activity with that of ara-C. Nucleosides Nucleotides Nucleic Acids 2000, 19, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- Reist, E.J.; Gueffroy, D.E.; Goodman, L. Synthesis of 4-thio-D- and -L-ribofuranose and the corresponding adenine nucleosides. J. Am. Chem. Soc. 1964, 86, 5658–5663. [Google Scholar] [CrossRef]
- Gunaga, P.; Moon, H.R.; Choi, W.J.; Shin, D.H.; Park, J.G.; Jeong, L.S. Recent advances in 4′-thionucleosides as potential antiviral and antitumor agents. Curr. Med. Chem. 2004, 11, 2585–2637. [Google Scholar] [CrossRef]
- Parker, W.B.; Shaddix, S.C.; Rose, L.M.; Waud, W.R.; Shewach, D.S.; Tiwari, K.N.; Secrist, J.A., III. Metabolism of 4′-thio-β-D-arabinofuranosylcytosine in CEM cells. Biochem. Pharmacol. 2000, 60, 1925–1932. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Nanjappa, V.; Raja, R.; Sathe, G.; Puttamallesh, V.N.; Jain, A.P.; Pinto, S.M.; Balaji, S.A.; Chavan, S.; Sahasrabuddhe, N.A.; et al. A dual specificity kinase, DYRK1A, as a potential therapeutic target for head and neck squamous cell carcinoma. Sci. Rep. 2016, 6, 36132. [Google Scholar] [CrossRef]
- Friedman, E. Mirk/Dyrk1B in cancer. J. Cell. Biochem. 2007, 102, 274–279. [Google Scholar] [CrossRef]
- Knippschild, U.; Wolff, S.; Giamas, G.; Brockschmidt, C.; Wittau, M.; Wurl, P.U.; Eismann, T.; Stoter, M. The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie 2005, 28, 508–514. [Google Scholar] [CrossRef]
- Batra, H.; Moriarty, R.M.; Penmasta, R.; Sharma, V.; Stanciuc, G.; Staszewski, J.P.; Tuladhar, S.M.; Walsh, D.A.; Datla, S.; Krishnaswamy, S. A concise, efficient and production-scale synthesis of a protected L-lyxonolactone derivative: An important aldonolactone core. Org. Process Res. Dev. 2006, 10, 484–486. [Google Scholar] [CrossRef]
- Haraguchi, K.; Shimada, H.; Kimura, K.; Akutsu, G.; Tanaka, H.; Abe, H.; Hamasaki, T.; Baba, M.; Gullen, E.A.; Dutschman, G.E.; et al. Synthesis of 4′-ethynyl-2′-deoxy-4′-thioribonucleosides and discovery of a highly potent and less toxic NRTI. ACS Med. Chem. Lett. 2011, 2, 692–697. [Google Scholar] [CrossRef]
- Molloy, J.J.; Seath, C.P.; West, M.J.; McLaughlin, C.; Fazakerley, N.J.; Kennedy, A.R.; Nelson, D.J.; Watson, A.J.B. Interrogating Pd(II) anion metathesis using a bifunctional chemical probe: A transmetalation switch. J. Am. Chem. Soc. 2018, 140, 126–130. [Google Scholar] [CrossRef]
- The CIF file for X-ray crystal structure of 1g has been deposited at the Cambridge Crystallographic Data Centre (CCDC, 12 Union Road, Cambridge, CB2 1EZ (UK); Tel: (+44)1223-336-408, Fax: (+44) 1223-336-033, e-mail: Deposit@ccdc.cam.ac.uk) with deposition number: CCDC 1575257.
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Song, J.; Yu, J.; Jeong, L.S.; Lee, S.K. A novel cytarabine analog evokes synthetic lethality by targeting MK2 in p53-deficient cancer cells. Cancer Lett. 2021, 497, 54–65. [Google Scholar] [CrossRef]
- Walmsley, D.L.; Murray, J.B.; Dokurno, P.; Massey, A.J.; Benwell, K.; Fiumana, A.; Foloppe, N.; Ray, S.; Smith, J.; Surgenor, A.E.; et al. Fragment-derived selective inhibitors of dual-specificity kinases DYRK1A and DYRK1B. J. Med. Chem. 2021, 64, 8971–8991. [Google Scholar] [CrossRef]
- Skerratt, A.E.; Andrews, M.; Bagal, S.K.; Bilsland, J.; Brown, D.; Bungay, P.J.; Cole, S.; Gibson, K.R.; Jones, R.; Morao, I.; et al. The discovery of a potent, selective, and peripherally restricted pan-Trk inhibitor (PF-06273340) for the treatment of pain. J. Med. Chem. 2016, 59, 10084–10099. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Gao, N.; Li, S.Q.; Huang, Y.; Bourassa, J.L.; Huryn, D.M. Experimental design on single-time-point high-throughput microsomal stability assay. J. Pharm. Sci. 2004, 93, 1537–1544. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L. (Eds.) Chapter 29—Metabolic Stability Methods. Drug-like Properties: Concepts, Structure Design and Methods; Academic Press: San Diego, CA, USA, 2008; pp. 329–347. [Google Scholar]
- Kim, M.-J.; Kim, H.; Cha, I.-J.; Park, J.-S.; Shon, J.-H.; Liu, K.-H.; Shin, J.-G. High-throughput screening of inhibitory potential of nine cytochrome P450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2651–2658. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L. (Eds.) Chapter 32—CYP Inhibition Methods. Drug-like Properties: Concepts, Structure Design and Methods; Academic Press: San Diego, CA, USA, 2008; pp. 360–371. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System (Version 2.4.0); Schrödinger, LLC: New York, NY, USA, 2002. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound No. | IC50 (μM) a | |||||
|---|---|---|---|---|---|---|
| A549 b | HCT116 c | MDA-MB-231 d | SK-HEP-1 e | SNU638 f | PC-3 g | |
| 1a | 1.02 | 2.16 | 3.31 | 3.38 | 1.95 | 2.46 |
| 1b | 1.65 | 1.74 | 2.72 | 1.38 | 2.41 | 2.48 |
| 1c | 0.97 | 0.56 | 0.31 | 0.22 | 0.47 | 0.2 |
| 1d | 7.57 | 6.35 | 8.83 | 7.81 | 6.32 | 9.21 |
| 1e | >50 | >50 | >50 | >50 | >50 | >50 |
| 1f | 41.6 | 30.6 | 30.1 | 10.3 | 27.5 | 15.4 |
| 1g | 0.06 | 0.03 | 0.05 | 0.05 | 0.03 | 0.004 |
| 1h | >50 | >50 | 45.5 | 6.05 | >50 | >50 |
| 1i | >50 | >50 | >50 | >50 | >50 | >50 |
| 1j | >50 | >50 | 29.6 | >50 | >50 | >50 |
| 1k | >50 | >50 | 30.6 | 8.31 | >50 | >50 |
| Etoposide h | 0.36 | 1.11 | 4.9 | 0.91 | 0.41 | 23.4 |
| Gemcitabine i | 0.3 | 0.2 | 1.1 | 0.2 | 0.1 | 3.6 |
| Compound No. | Metabolic Stability (%) a | CYP Inhibition, IC50 (μM) b | ||||
|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 | ||
| 1g | 82.3 | >20 | >20 | >20 | >20 | >20 |
| Verapamil | 15.3 | - | - | - | - | - |
| Ketoconazole d | - | 95.7 c | 93.6 c | 93.6 c | 96.0 c | 27.2 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mashelkar, K.K.; Byun, W.S.; Ko, H.; Sung, K.; Tripathi, S.K.; An, S.; Yum, Y.A.; Kwon, J.Y.; Kim, M.; Kim, G.; et al. Discovery of a Novel Template, 7-Substituted 7-Deaza-4′-Thioadenosine Derivatives as Multi-Kinase Inhibitors. Pharmaceuticals 2021, 14, 1290. https://doi.org/10.3390/ph14121290
Mashelkar KK, Byun WS, Ko H, Sung K, Tripathi SK, An S, Yum YA, Kwon JY, Kim M, Kim G, et al. Discovery of a Novel Template, 7-Substituted 7-Deaza-4′-Thioadenosine Derivatives as Multi-Kinase Inhibitors. Pharmaceuticals. 2021; 14(12):1290. https://doi.org/10.3390/ph14121290
Chicago/Turabian StyleMashelkar, Karishma K., Woong Sub Byun, Hyejin Ko, Kisu Sung, Sushil K. Tripathi, Seungchan An, Yun A Yum, Jee Youn Kwon, Minjae Kim, Gibae Kim, and et al. 2021. "Discovery of a Novel Template, 7-Substituted 7-Deaza-4′-Thioadenosine Derivatives as Multi-Kinase Inhibitors" Pharmaceuticals 14, no. 12: 1290. https://doi.org/10.3390/ph14121290
APA StyleMashelkar, K. K., Byun, W. S., Ko, H., Sung, K., Tripathi, S. K., An, S., Yum, Y. A., Kwon, J. Y., Kim, M., Kim, G., Kwon, E.-J., Lee, H. W., Noh, M., Lee, S. K., & Jeong, L. S. (2021). Discovery of a Novel Template, 7-Substituted 7-Deaza-4′-Thioadenosine Derivatives as Multi-Kinase Inhibitors. Pharmaceuticals, 14(12), 1290. https://doi.org/10.3390/ph14121290