
From Far West to East: Joining the Molecular Architecture of Imidazole-like Ligands in HO-1 Complexes

 ,
,  , , ,
, , ,  ,
,  and
and 
Abstract
:
1. Introduction
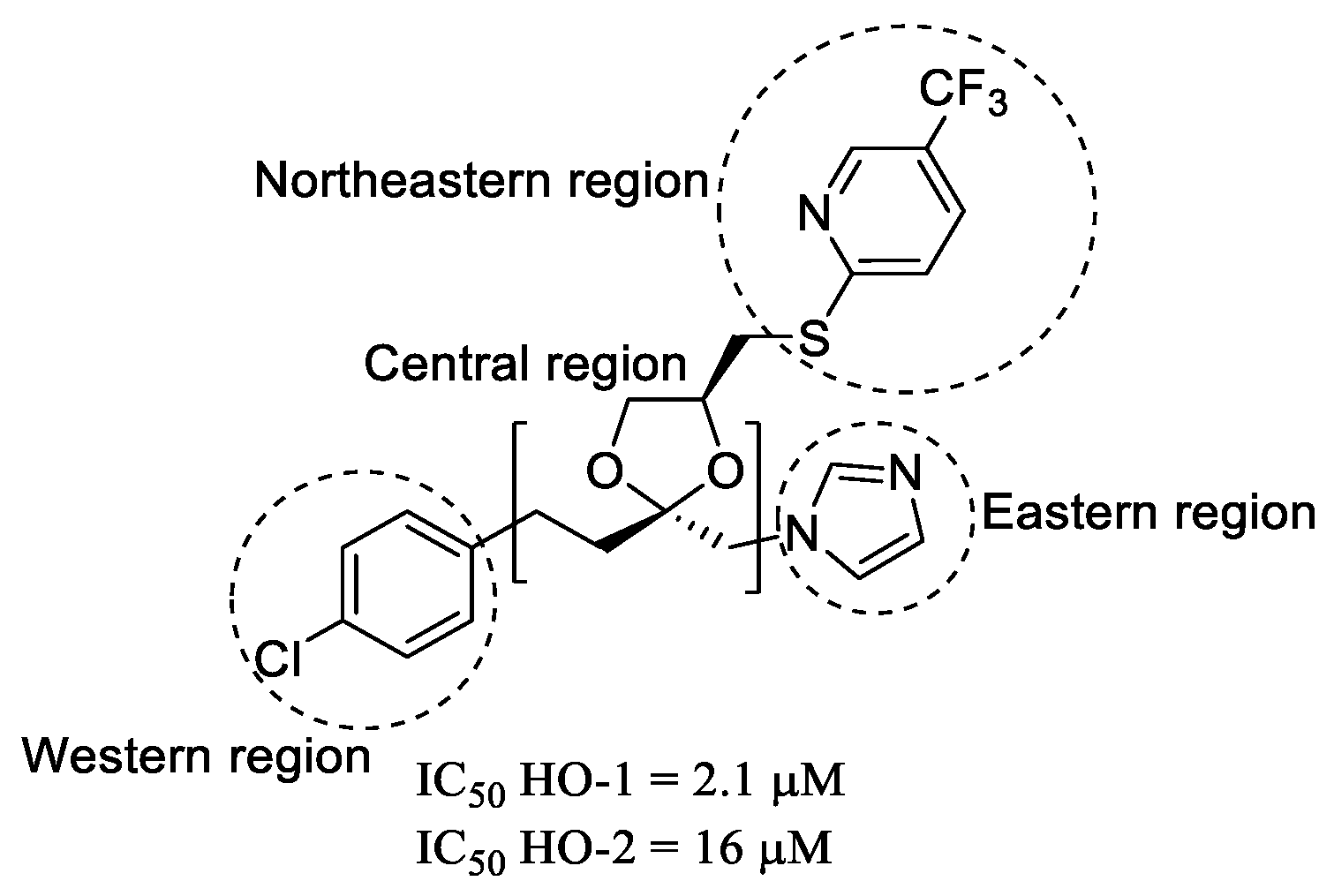
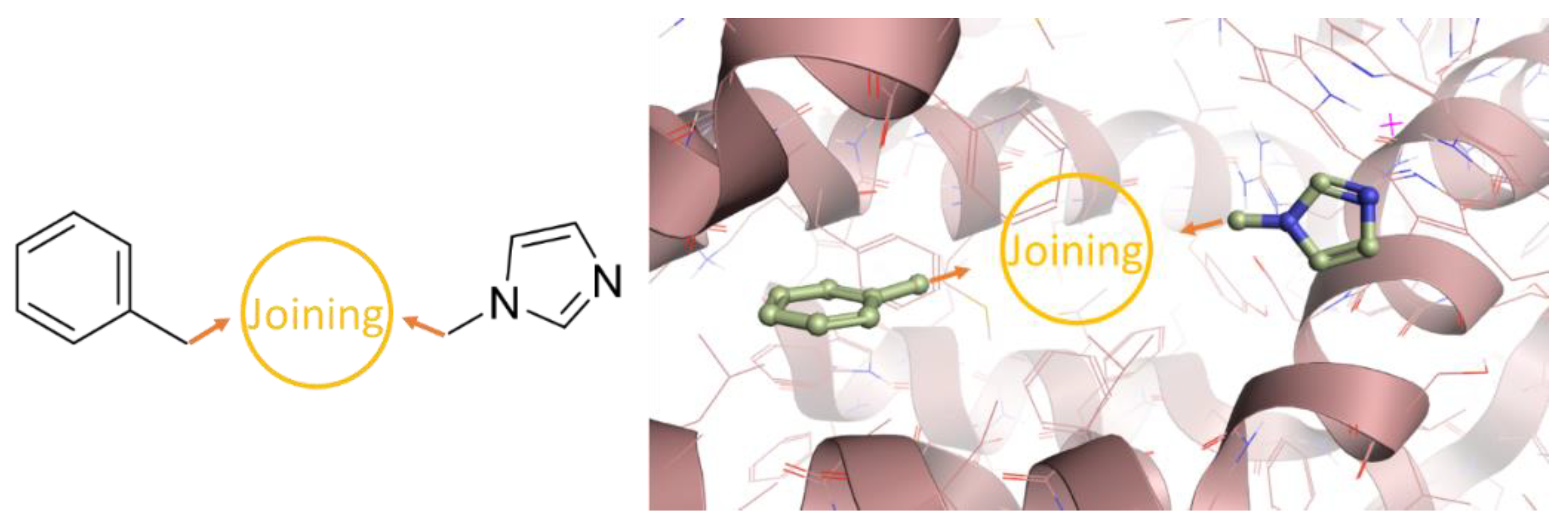
2. Results and Discussion
2.1. Molecular Modeling
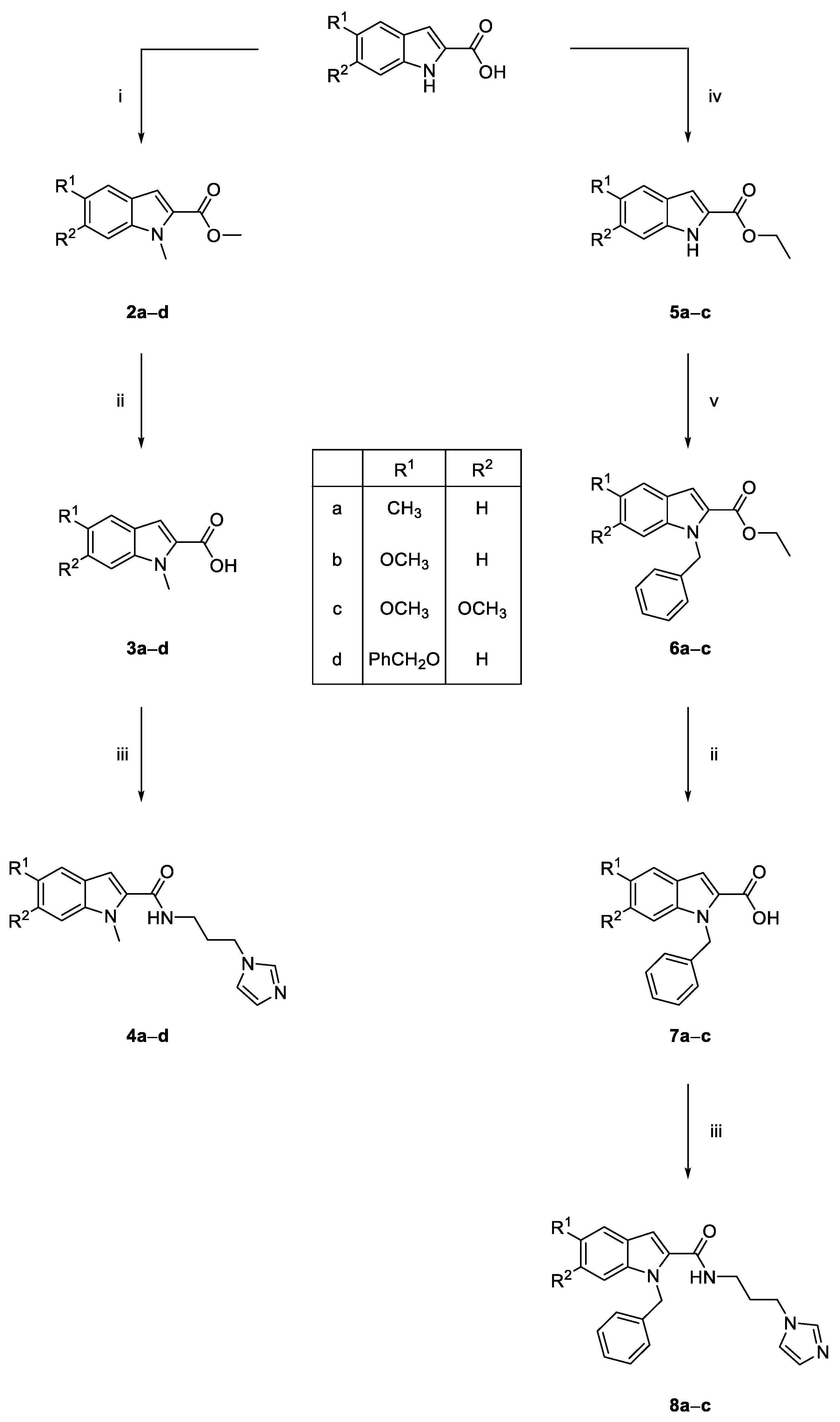
2.2. Chemistry
2.3. In Vitro HO-1 Inhibition and Structure–Activity Relationships Studies
3. Materials and Methods
3.1. Chemistry
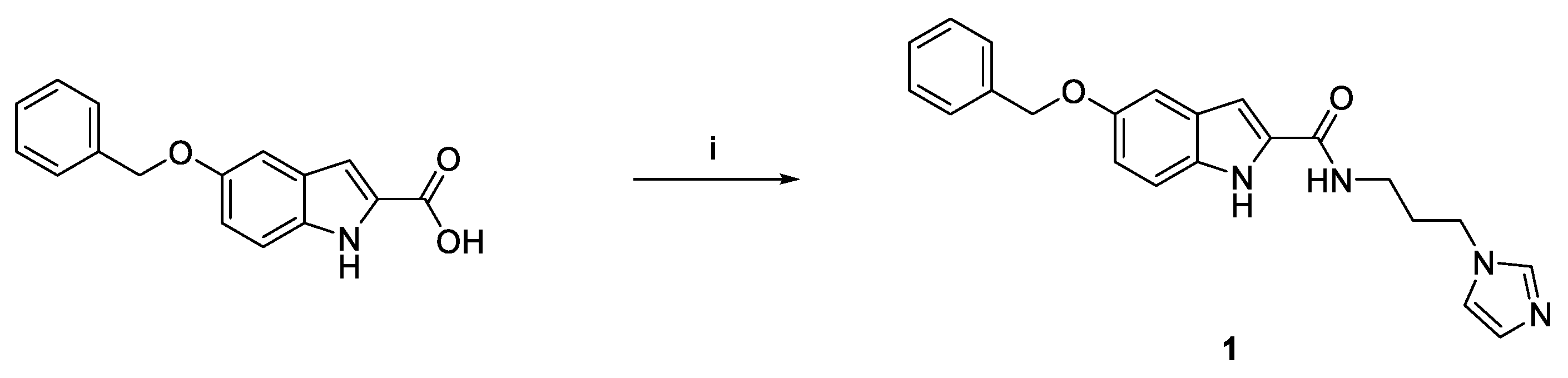
3.1.1. Synthesis of N-(3-(1H-Imidazol-1-yl)propyl)-5-(benzyloxy)-1H-indole-2-carboxamide (1)
3.1.2. General Procedure for the Synthesis of Substituted 1-Methyl-1H-Indole Methyl Esters (2a–d)
Methyl 1,5-Dimethyl-1H-indole-2-carboxylate (2a)
Methyl 5-Methoxy-1-methyl-1H-indole-2-carboxylate (2b)
Methyl 5,6-Dimethoxy-1-methyl-1H-indole-2-carboxylate (2c)
Methyl 5-(Benzyloxy)-1-methyl-1H-indole-2-carboxylate (2d)
3.1.3. General Procedure for the Synthesis of N-Substituted 1H-Indole-2-carboxylic Acids (3a–d, 7a–c)
1,5-Dimethyl-1H-indole-2-carboxylic Acid (3a)
5-Methoxy-1-methyl-1H-indole-2-carboxylic Acid (3b)
5,6-Dimethoxy-1-methyl-1H-indole-2-carboxylic Acid (3c)
5-(Benzyloxy)-1-methyl-1H-indole-2-carboxylic Acid (3d)
1-Benzyl-5-methyl-1H-indole-2-carboxylic Acid (7a)
1-Benzyl-5-methoxy-1H-indole-2-carboxylic Acid (7b)
1-Benzyl-5,6-dimethoxy-1H-indole-2-carboxylic Acid (7c)
3.1.4. General Procedure for the Synthesis of N-Substituted 1H-Indole-2-carboxamides (4a–d, 8a–c)
N-(3-(1H-Imidazol-1-yl)propyl)-1,5-dimethyl-1H-indole-2-carboxamide (4a)
N-(3-(1H-Imidazol-1-yl)propyl)-5-methoxy-1-methyl-1H-indole-2-carboxamide (4b)
N-(3-(1H-Imidazol-1-yl)propyl)-5,6-dimethoxy-1-methyl-1H-indole-2-carboxamide (4c)
N-(3-(1H-Imidazol-1-yl)propyl)-5-(benzyloxy)-1-methyl-1H-indole-2-carboxamide (4d)
N-(3-(1H-Imidazol-1-yl)propyl)-1-benzyl-5-methyl-1H-indole-2-carboxamide (8a)
N-(3-(1H-Imidazol-1-yl)propyl)-1-benzyl-5-methoxy-1H-indole-2-carboxamide (8b)
N-(3-(1H-Imidazol-1-yl)propyl)-1-benzyl-5,6-dimethoxy-1H-indole-2-carboxamide (8c)
3.1.5. General Procedure for the Synthesis of Substituted 1H-Indole Ethyl Esters (5a–c)
Ethyl 5-Methyl-1H-indole-2-carboxylate (5a)
Ethyl 5-Methoxy-1H-indole-2-carboxylate (5b)
Ethyl 5,6-Dimethoxy-1H-indole-2-carboxylate (5c)
3.1.6. General Procedure for the Synthesis of Substituted 1-Benzyl-1H-indole Ethyl Esters (6a–c)
Ethyl 1-Benzyl-5-methyl-1H-indole-2-carboxylate (6a)
Ethyl 1-Benzyl-5-methoxy-1H-indole-2-carboxylate (6)
Ethyl 1-Benzyl-5,6-dimethoxy-1H-indole-2-carboxylate (6c)
3.2. Molecular Modeling
3.3. Biology
3.3.1. Preparation of Spleen Microsomal Fractions
3.3.2. Preparation of Biliverdin Reductase
3.3.3. Measurement of HO-1 Enzymatic Activity in the Microsomal Fraction of Rat Spleen
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Chau, L.Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Was, H.; Dulak, J.; Jozkowicz, A. Heme oxygenase-1 in tumor biology and therapy. Curr. Drug Targets 2010, 11, 1551–1570. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aztatzi-Santillán, E.; Nares-López, F.E.; Márquez-Valadez, B.; Aguilera, P.; Chánez-Cárdenas, M.E. The protective role of heme oxygenase-1 in cerebral ischemia. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 310–316. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfenova, H.; Leffler, C.W.; Basuroy, S.; Liu, J.; Fedinec, A.L. Antioxidant roles of heme oxygenase, carbon monoxide, and bilirubin in cerebral circulation during seizures. J. Cereb. Blood Flow Metab. 2012, 32, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186. [Google Scholar] [CrossRef]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Kutty, R.K.; Kutty, G.; Rodriguez, I.R.; Chader, G.J.; Wiggert, B. Chromosomal localization of the human heme oxygenase genes: Heme oxygenase-1 (HMOX1) maps to chromosome 22q12 and heme oxygenase-2 (HMOX2) maps to chromosome 16p13.3. Genomics 1994, 20, 513–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intagliata, S.; Salerno, L.; Ciaffaglione, V.; Leonardi, C.; Fallica, A.N.; Carota, G.; Amata, E.; Marrazzo, A.; Pittalà, V.; Romeo, G. Heme Oxygenase-2 (HO-2) as a therapeutic target: Activators and inhibitors. Eur. J. Med. Chem. 2019, 183, 111703. [Google Scholar] [CrossRef]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittalà, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef]
- Hemmati, M.; Yousefi, B.; Bahar, A.; Eslami, M. Importance of Heme Oxygenase-1 in Gastrointestinal Cancers: Functions, Inductions, Regulations, and Signaling. J. Gastrointest. Cancer 2021, 52, 454–461. [Google Scholar] [CrossRef]
- Li Volti, G.; Tibullo, D.; Vanella, L.; Giallongo, C.; Di Raimondo, F.; Forte, S.; Di Rosa, M.; Signorelli, S.S.; Barbagallo, I. The Heme Oxygenase System in Hematological Malignancies. Antioxid. Redox Signal. 2017, 27, 363–377. [Google Scholar] [CrossRef]
- Raval, C.M.; Lee, P.J. Heme oxygenase-1 in lung disease. Curr. Drug Targets 2010, 11, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Romeo, G.; Modica, M.N.; Amata, E.; Sorrenti, V.; Barbagallo, I.; Pittalà, V. Heme oxygenase-1: A new druggable target in the management of chronic and acute myeloid leukemia. Eur. J. Med. Chem. 2017, 142, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.S.; Jiang, W.Y.; Chi, J.H.; Jin, H.; Park, W.C.; Sohn, D.H.; Park, P.H.; Lee, S.H. Heme oxygenase-1 promotes tumor progression and metastasis of colorectal carcinoma cells by inhibiting antitumor immunity. Oncotarget 2015, 6, 19792–19806. [Google Scholar] [CrossRef] [Green Version]
- Salerno, L.; Vanella, L.; Sorrenti, V.; Consoli, V.; Ciaffaglione, V.; Fallica, A.N.; Canale, V.; Zajdel, P.; Pignatello, R.; Intagliata, S. Novel mutual prodrug of 5-fluorouracil and heme oxygenase-1 inhibitor (5-FU/HO-1 hybrid): Design and preliminary in vitro evaluation. J. Enzyme Inhib. Med. Chem. 2021, 36, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Ciaffaglione, V.; Modica, M.N.; Pittalà, V.; Romeo, G.; Salerno, L.; Intagliata, S. Mutual Prodrugs of 5-Fluorouracil: From a Classic Chemotherapeutic Agent to Novel Potential Anticancer Drugs. ChemMedChem 2021, 16, 3496–3512. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of TET-dependent DNA demethylation. Cell Death Dis. 2014, 5, 1183. [Google Scholar] [CrossRef]
- Podkalicka, P.; Mucha, O.; Józkowicz, A.; Dulak, J.; Łoboda, A. Heme oxygenase inhibition in cancers: Possible tools and targets. Contemp. Oncol. (Pozn) 2018, 22, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Siracusa, M.A.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Tibullo, D.; Sorrenti, V. Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorg. Med. Chem. 2013, 21, 5145–5153. [Google Scholar] [CrossRef]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Marrazzo, A.; Siracusa, M.A.; Sorrenti, V.; Di Giacomo, C.; Vanella, L.; Parayath, N.N.; et al. Novel imidazole derivatives as heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2) inhibitors and their cytotoxic activity in human-derived cancer cell lines. Eur. J. Med. Chem. 2015, 96, 162–172. [Google Scholar] [PubMed]
- Sorrenti, V.; Pittalà, V.; Romeo, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Turnaturi, R.; Prezzavento, O.; Barbagallo, I.; Vanella, L.; et al. Targeting heme Oxygenase-1 with hybrid compounds to overcome Imatinib resistance in chronic myeloid leukemia cell lines. Eur. J. Med. Chem. 2018, 158, 937–950. [Google Scholar]
- Greish, K.F.; Salerno, L.; Al Zahrani, R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef] [Green Version]
- Ciaffaglione, V.; Intagliata, S.; Pittalà, V.; Marrazzo, A.; Sorrenti, V.; Vanella, L.; Rescifina, A.; Floresta, G.; Sultan, A.; Greish, K.; et al. New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 1923. [Google Scholar]
- Floresta, G.; Carotti, A.; Ianni, F.; Sorrenti, V.; Intagliata, S.; Rescifina, A.; Salerno, L.; Di Michele, A.; Sardella, R.; Pittalà, V. Chromatograpic resolution of phenylethanolic-azole racemic compounds highlighted stereoselective inhibition of heme oxygenase-1 by (R)-enantiomers. Bioorg. Chem. 2020, 99, 103777. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Fallica, A.N.; Romeo, G.; Sorrenti, V.; Salerno, L.; Rescifina, A.; Pittalà, V. Identification of a potent heme oxygenase-2 (HO-2) inhibitor by targeting the secondary hydrophobic pocket of the HO-2 western region. Bioorg. Chem. 2020, 104, 104310. [Google Scholar] [CrossRef]
- Salerno, L.; Amata, E.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Floresta, G.; Sorrenti, V.; Barbagallo, I.; Rescifina, A.; Pittalà, V. Potholing of the hydrophobic heme oxygenase-1 western region for the search of potent and selective imidazole-based inhibitors. Eur. J. Med. Chem. 2018, 148, 54–62. [Google Scholar] [CrossRef]
- Fallica, A.N.; Sorrenti, V.; D’Amico, A.G.; Salerno, L.; Romeo, G.; Intagliata, S.; Consoli, V.; Floresta, G.; Rescifina, A.; D’Agata, V.; et al. Discovery of Novel Acetamide-Based Heme Oxygenase-1 Inhibitors with Potent In Vitro Antiproliferative Activity. J. Med. Chem. 2021, 64, 13373–13393. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Heme Oxygenase Database (HemeOxDB) and QSAR Analysis of Isoform 1 Inhibitors. ChemMedChem 2017, 12, 1873–1881. [Google Scholar] [CrossRef]
- Floresta, G.; Pittalà, V.; Sorrenti, V.; Romeo, G.; Salerno, L.; Rescifina, A. Development of new HO-1 inhibitors by a thorough scaffold-hopping analysis. Bioorg. Chem. 2018, 81, 334–339. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Salerno, L.; Romeo, G.; Prezzavento, O.; Pittalà, V.; Rescifina, A. Identification of Potentially Potent Heme Oxygenase 1 Inhibitors through 3D-QSAR Coupled to Scaffold-Hopping Analysis. ChemMedChem 2018, 13, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Boström, J. Where Do Recent Small Molecule Clinical Development Candidates Come from? J. Med. Chem. 2018, 61, 9442–9468. [Google Scholar] [CrossRef]
- Bienstock, R.J. Computational methods for fragment-based ligand design: Growing and linking. Methods Mol. Biol. 2015, 1289, 119–135. [Google Scholar]
- Floresta, G.; Amata, E.; Gentile, D.; Romeo, G.; Marrazzo, A.; Pittalà, V.; Salerno, L.; Rescifina, A. Fourfold Filtered Statistical/Computational Approach for the Identification of Imidazole Compounds as HO-1 Inhibitors from Natural Products. Mar. Drugs 2019, 17, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floresta, G.; Fallica, A.N.; Salerno, L.; Sorrenti, V.; Pittalà, V.; Rescifina, A. Growing the molecular architecture of imidazole-like ligands in HO-1 complexes. Bioorg. Chem. 2021, 117, 105428. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Floresta, G.; Patamia, V.; Chiaramonte, R.; Mauro, G.L.; Rescifina, A.; Vecchio, M. An Integrated Pharmacophore/Docking/3D-QSAR Approach to Screening a Large Library of Products in Search of Future Botulinum Neurotoxin A Inhibitors. Int. J. Mol. Sci. 2020, 21, 9470. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Alemán, C.; Luque, F.J.; Orozco, M. Suitability of the PM3-derived molecular electrostatic potentials. J. Comput. Chem. 1993, 14, 799–808. [Google Scholar] [CrossRef]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View–Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistarà, V.; Pittalà, V.; Prezzavento, O.; Amata, E. Hyphenated 3D-QSAR statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017, 139, 884–891. [Google Scholar] [CrossRef]
- Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules 2018, 23, 2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Vlahakis, J.Z.; Lazar, C.; Roman, G.; Vukomanovic, D.; Nakatsu, K.; Szarek, W.A. Heme oxygenase inhibition by alpha-(1H-imidazol-1-yl)-omega-phenylalkanes: Effect of introduction of heteroatoms in the alkyl linker. ChemMedChem 2012, 7, 897–902. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
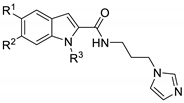 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | HO-1 In Silico (μM) a | HO-1 IC50 (μM) b |
| 1 | PhCH2O | H | H | 1.19 | 89.60 ± 6.10 |
| 4a | CH3 | H | CH3 | — | 367.31 ± 34.50 |
| 4b | OCH3 | H | CH3 | — | 113.33 ± 18.50 |
| 4c | OCH3 | OCH3 | CH3 | — | 55.48 ± 3.82 |
| 4d | PhCH2O | H | CH3 | 1.03 | 1.03 ± 0.13 |
| 8a | CH3 | H | PhCH2 | 1.49 | 2063 ± 412 |
| 8b | OCH3 | H | PhCH2 | 1.29 | 349.24 ± 37.00 |
| 8c | OCH3 | OCH3 | PhCH2 | 1.30 | 81.03 ± 4.65 |
| Azalanstat c | — | — | — | — | 5.30 ± 0.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Floresta, G.; Fallica, A.N.; Patamia, V.; Sorrenti, V.; Greish, K.; Rescifina, A.; Pittalà, V. From Far West to East: Joining the Molecular Architecture of Imidazole-like Ligands in HO-1 Complexes. Pharmaceuticals 2021, 14, 1289. https://doi.org/10.3390/ph14121289
Floresta G, Fallica AN, Patamia V, Sorrenti V, Greish K, Rescifina A, Pittalà V. From Far West to East: Joining the Molecular Architecture of Imidazole-like Ligands in HO-1 Complexes. Pharmaceuticals. 2021; 14(12):1289. https://doi.org/10.3390/ph14121289
Chicago/Turabian StyleFloresta, Giuseppe, Antonino Nicolò Fallica, Vincenzo Patamia, Valeria Sorrenti, Khaled Greish, Antonio Rescifina, and Valeria Pittalà. 2021. "From Far West to East: Joining the Molecular Architecture of Imidazole-like Ligands in HO-1 Complexes" Pharmaceuticals 14, no. 12: 1289. https://doi.org/10.3390/ph14121289
APA StyleFloresta, G., Fallica, A. N., Patamia, V., Sorrenti, V., Greish, K., Rescifina, A., & Pittalà, V. (2021). From Far West to East: Joining the Molecular Architecture of Imidazole-like Ligands in HO-1 Complexes. Pharmaceuticals, 14(12), 1289. https://doi.org/10.3390/ph14121289