
Tetrahydroimidazo[4,5-c]pyridine-Based Inhibitors of Porphyromonas gingivalis Glutaminyl Cyclase

 , and
, and
Abstract
:1. Introduction
2. Results
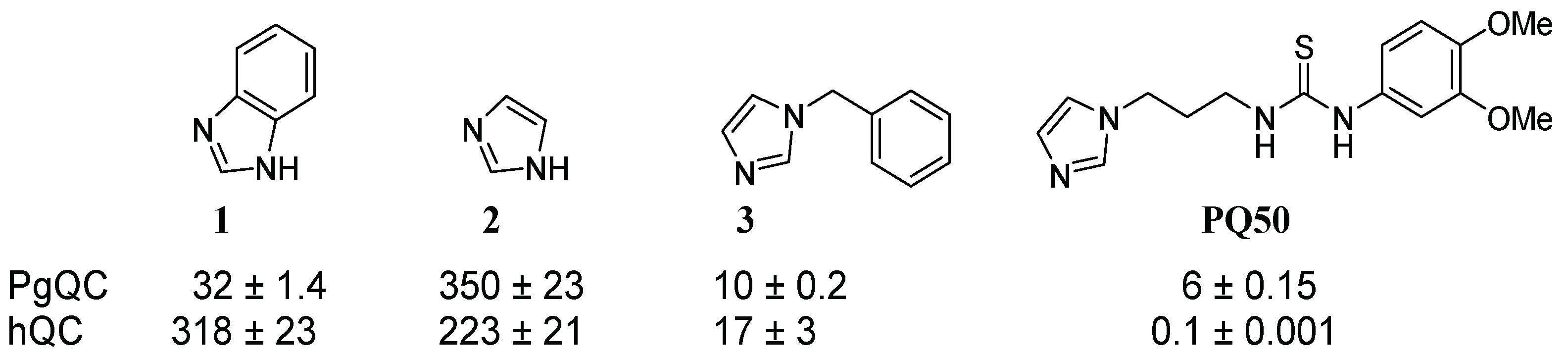
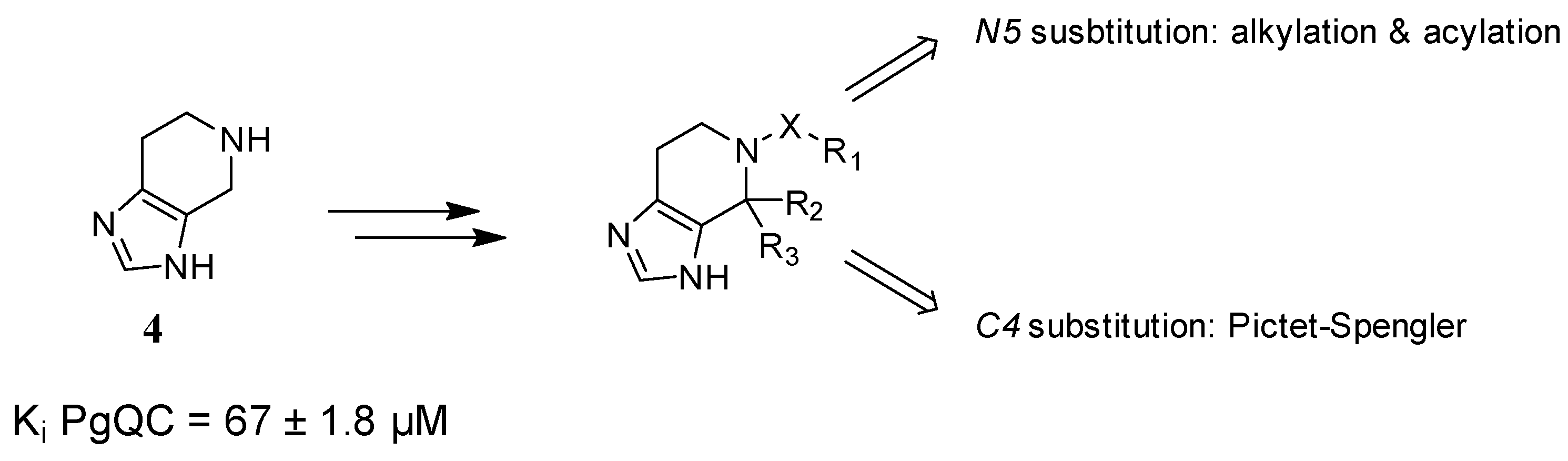
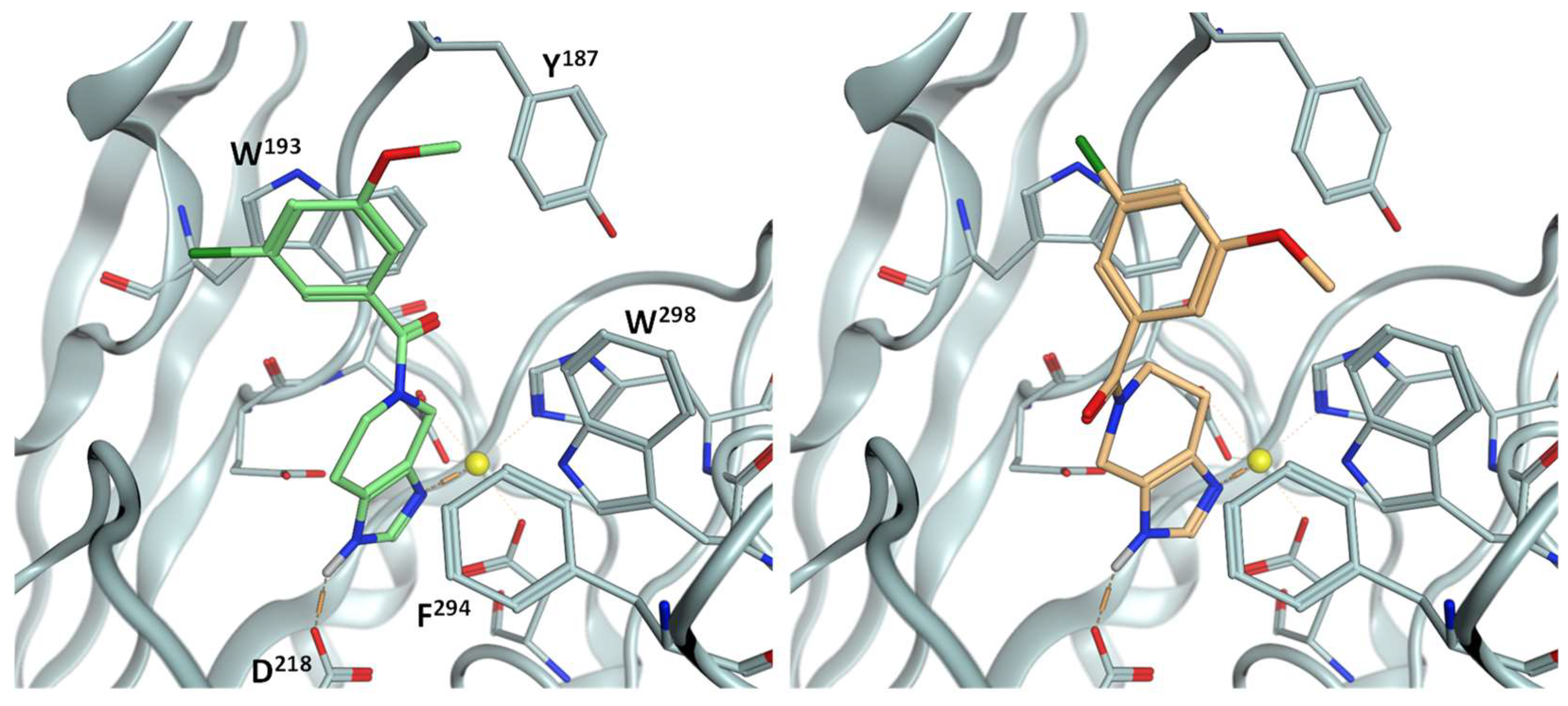
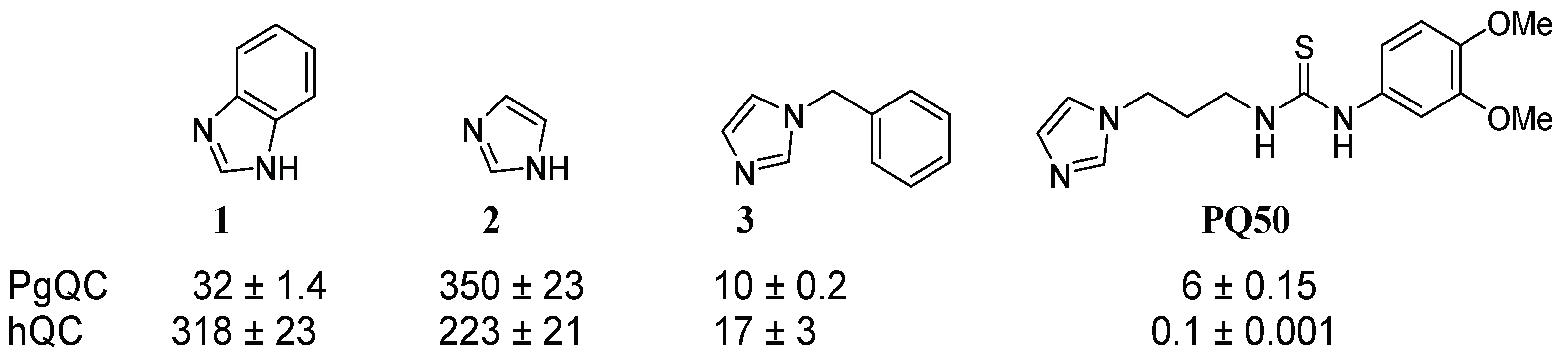
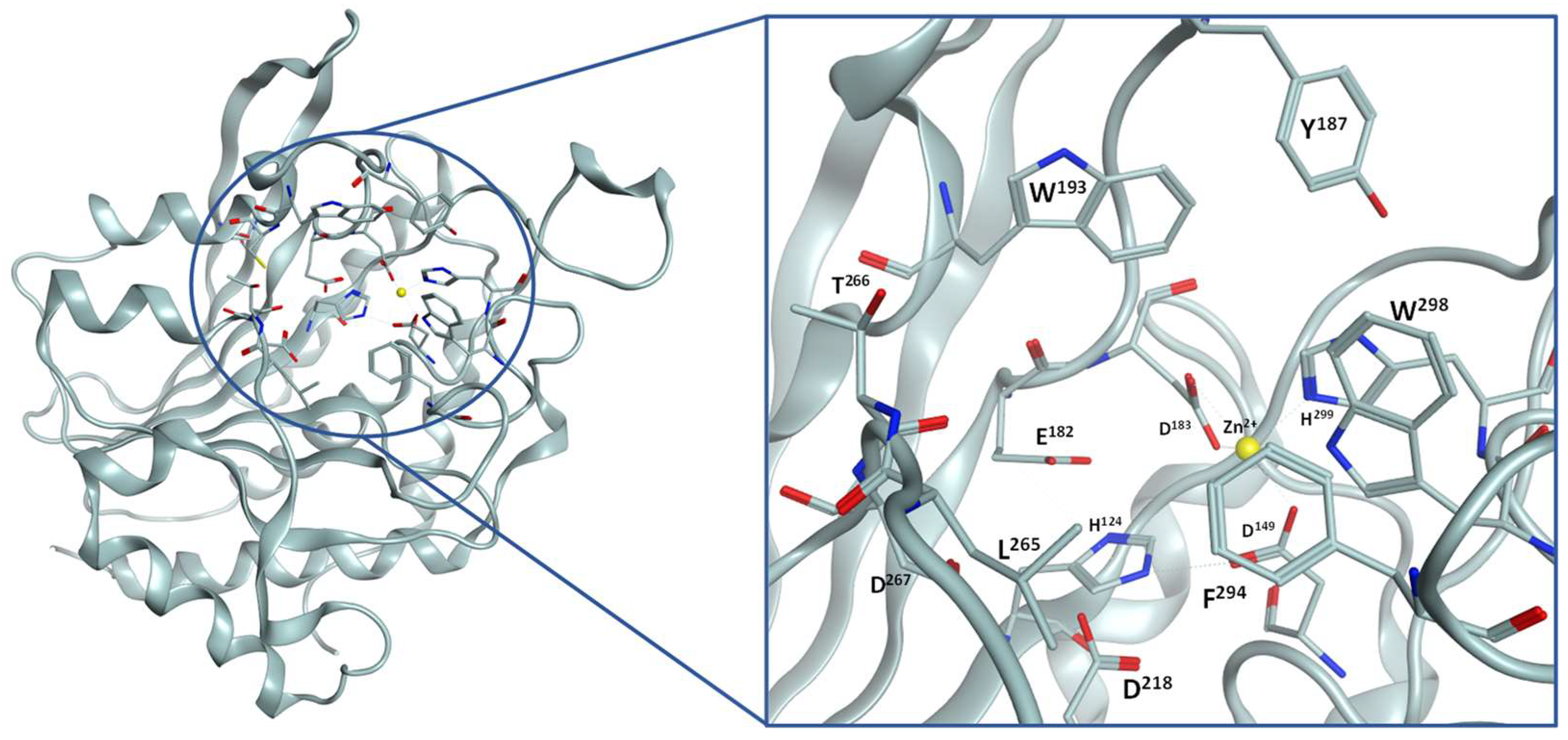
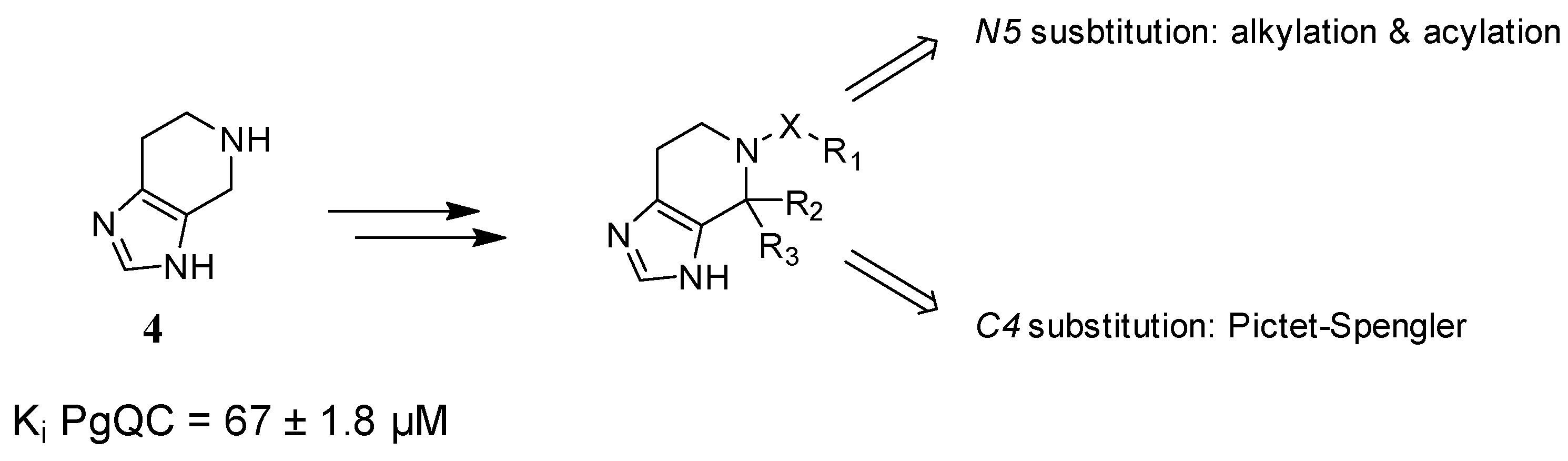
2.1. Inhibitor Design
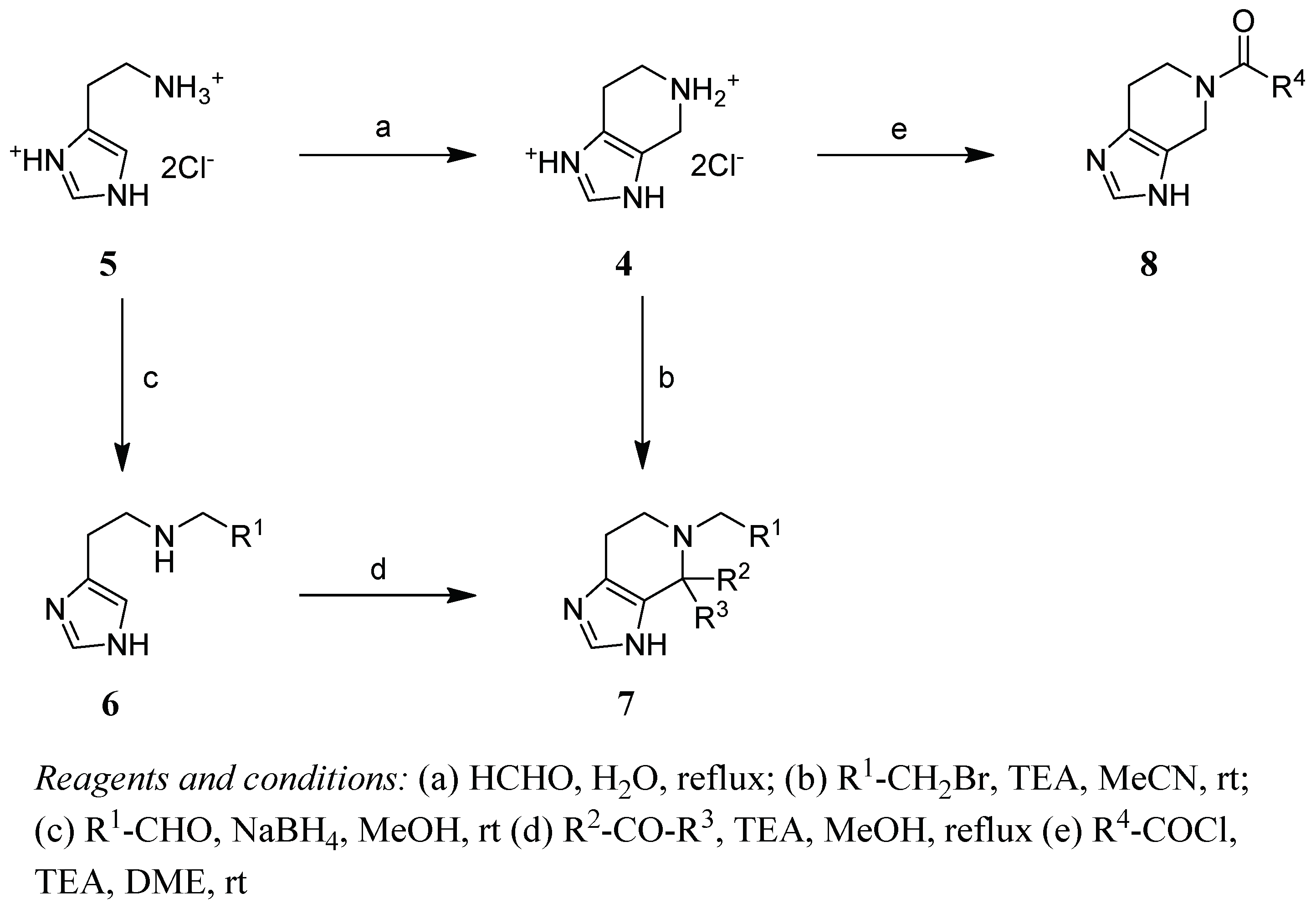
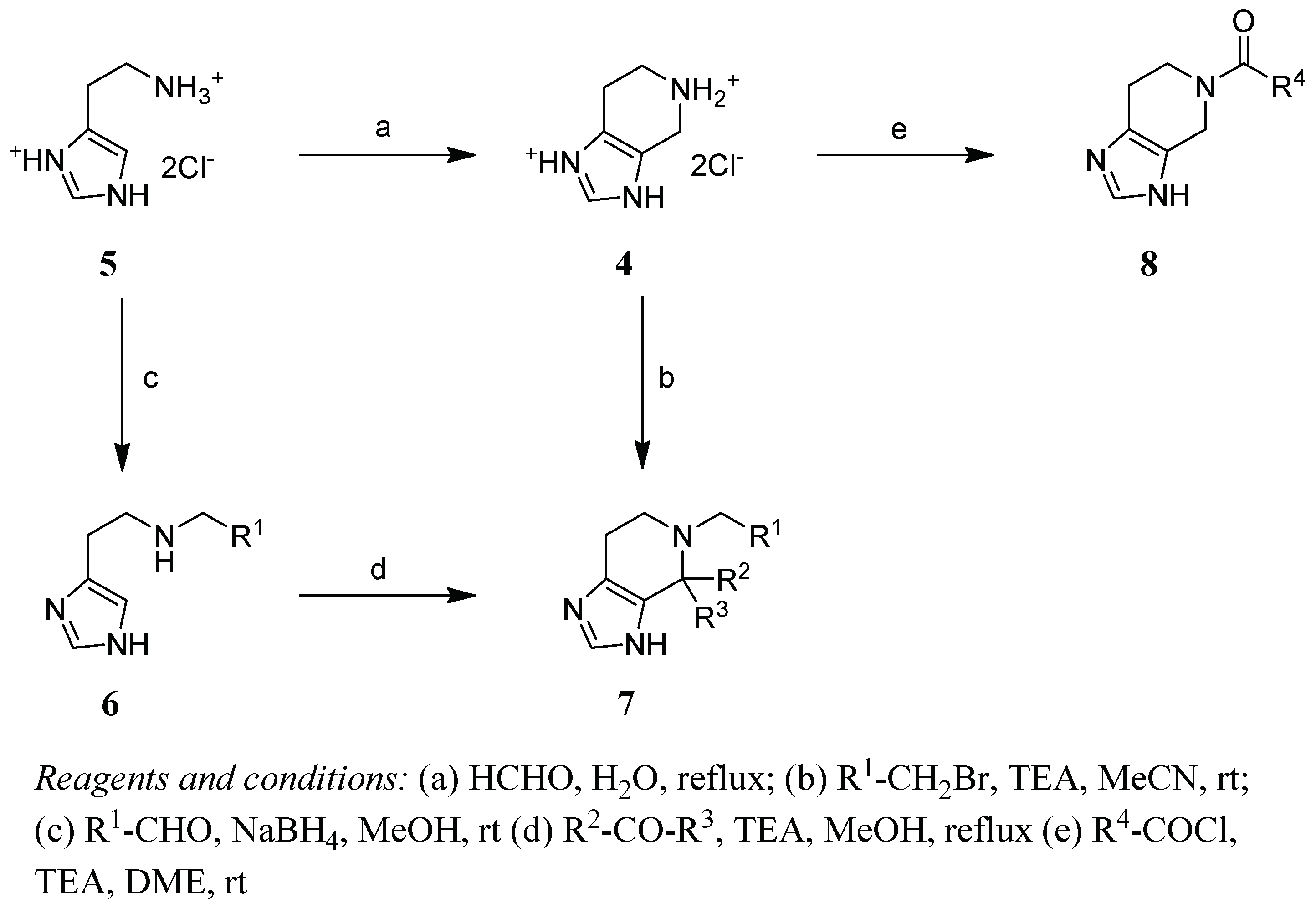
2.2. Chemistry
2.3. SAR
3. Conclusions
4. Materials and Methods
4.1. Chemistry
- 4,5,6,7-Tetrahydro-imidazo[4,5-c]pyridine (4)
- General Method N-Alkylation
- 5-Benzyl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridine (7a)
- 5-(2-Phenylethyl)-3,4,6,7-tetrahydroimidazo[4,5-c]pyridine (7b)
- Synthesis of C4 substituted derivatives
- 5-Benzylspiro[6,7-dihydro-3H-imidazo[4,5-c]pyridine-4,3′-oxetane] (7c)
- 5-Benzyl-4-methyl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridine (7d)
- 5-Benzyl-4-phenyl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridine (7e)
- General Method Acylation
- Phenyl(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8a)
- 2-Phenyl-1-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)ethanone (8b)
- 3-Phenyl-1-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)propan-1-one (8c)
- (E)-3-Phenyl-1-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)prop-2-en-1-one (8d)
- (4-Fluorophenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8e)
- (3-Fluorophenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8f)
- (3,4,6,7-Tetrahydroimidazo[4,5-c]pyridin-5-yl)(3,4,5-trifluoro-phenyl)methanone (8g)
- (4-Chlorophenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8h)
- (3-Chlorophenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8i)
- (3,4-Dichlorophenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8j)
- (3,5-Dichlorophenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8k)
- (4-Methoxyphenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8l)
- (3-Methoxyphenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8m)
- (3,4-Dimethoxyphenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8n)
- (3,5-Dimethoxyphenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8o)
- (2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)(1,4,6,7-tetrahydro-5H-imidazo-[4,5-c]pyridin-5-yl)methanone (8p)
- Benzo[d][1,3]dioxol-5-yl(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8q)
- (3-Propoxyphenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8r)
- (4-Fluoro-3-methoxyphenyl)-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8s)
- (3-Chloro-5-methoxyphenyl)(1,4,6,7-tetrahydro-5H-imidazo[4,5-c]pyridin-5-yl)methanone (8t)
- [1,1′-Biphenyl]-4-yl(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8u)
- [1,1′-Biphenyl]-3-yl-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8v)
- Naphthalen-2-yl-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)methanone (8w)
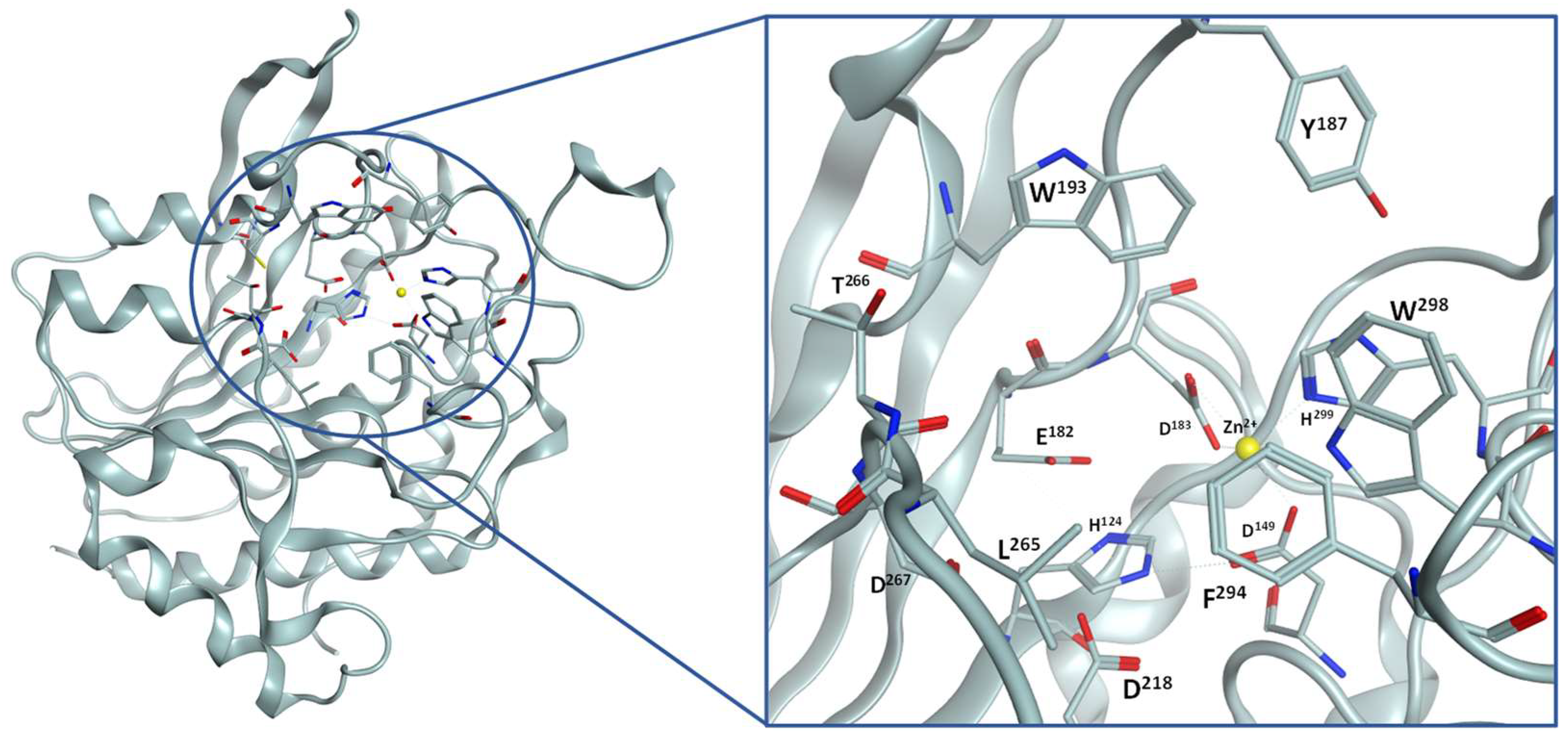
4.2. Docking
4.3. Inhibitor Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef]
- Wade, W.G. The oral microbiome in health and disease. Pharmacol. Res. 2013, 69, 137–143. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef] [PubMed]
- De Diego, I.; Veillard, F.T.; Guevara, T.; Potempa, B.; Sztukowska, M.; Potempa, J.; Gomis-Rüth, F.X. Porphyromonas gingivalis virulence factor gingipain RgpB shows a unique zymogenic mechanism for cysteine peptidases. J. Biol. Chem. 2013, 288, 14287–14296. [Google Scholar] [CrossRef] [Green Version]
- De Diego, I.; Veillard, F.; Sztukowska, M.N.; Guevara, T.; Potempa, B.; Pomowski, A.; Huntington, J.A.; Potempa, J.; Gomis-Rüth, F.X. Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 2014, 289, 32291–32302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Diaz, P.I. Porphyromonas gingivalis: Immune subversion activities and role in periodontal dysbiosis. Curr. Oral Health Rep. 2020, 7, 12–21. [Google Scholar] [CrossRef]
- Sun, Y.; Shu, R.; Li, C.-L.; Zhang, M.-Z. Gram-negative periodontal bacteria induce the activation of Toll-like receptors 2 and 4, and cytokine production in human periodontal ligament cells. J. Periodontol. 2010, 81, 1488–1496. [Google Scholar] [CrossRef]
- Veith, P.D.; Chen, Y.-Y.; Gorasia, D.G.; Chen, D.; Glew, M.D.; O’Brien-Simpson, N.M.; Cecil, J.D.; Holden, J.A.; Reynolds, E.C. Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J. Proteome Res. 2014, 13, 2420–2432. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Liu, S.; Zhang, S.; Pan, Y. The Role of Porphyromonas gingivalis Outer Membrane Vesicles in Periodontal Disease and Related Systemic Diseases. Front. Cell. Infect. Microbiol. 2020, 10, 585917. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Anan, F.; Takahashi, N.; Shinohara, T.; Nakagawa, M.; Masaki, T.; Katsuragi, I.; Tanaka, K.; Kakuma, T.; Yonemochi, H.; Eshima, N.; et al. Smoking is associated with insulin resistance and cardiovascular autonomic dysfunction in type 2 diabetic patients. Eur. J. Clin. Investig. 2006, 36, 459–465. [Google Scholar] [CrossRef]
- Bourgeois, D.; Inquimbert, C.; Ottolenghi, L.; Carrouel, F. Periodontal Pathogens as Risk Factors of Cardiovascular Diseases, Diabetes, Rheumatoid Arthritis, Cancer, and Chronic Obstructive Pulmonary Disease-Is There Cause for Consideration? Microorganisms 2019, 7, 424. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, X.; Yu, H.; Zhou, H.; Xu, S. Oral Microbiota as Promising Diagnostic Biomarkers for Gastrointestinal Cancer: A Systematic Review. OncoTargets Ther. 2019, 12, 11131–11144. [Google Scholar] [CrossRef] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugisch, O.; Wong, A.; Sroka, A.; Kantyka, T.; Koziel, J.; Neuhaus, K.; Sculean, A.; Venables, P.J.; Potempa, J.; Möller, B.; et al. Citrullination in the periodontium--a possible link between periodontitis and rheumatoid arthritis. Clin. Oral Investig. 2016, 20, 675–683. [Google Scholar] [CrossRef]
- Wada, K.; Kamisaki, Y. Roles of oral bacteria in cardiovascular diseases—From molecular mechanisms to clinical cases: Involvement of Porphyromonas gingivalis in the development of human aortic aneurysm. J. Pharmacol. Sci. 2010, 113, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonetti, M.S.; Jepsen, S.; Jin, L.; Otomo-Corgel, J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J. Clin. Periodontol. 2017, 44, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Chi, M.; Qi, M.; Lan, A.; Wang, P.; Weir, M.D.; Melo, M.A.; Sun, X.; Dong, B.; Li, C.; Wu, J.; et al. Novel Bioactive and Therapeutic Dental Polymeric Materials to Inhibit Periodontal Pathogens and Biofilms. Int. J. Mol. Sci. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smiley, C.J.; Tracy, S.L.; Abt, E.; Michalowicz, B.S.; John, M.T.; Gunsolley, J.; Cobb, C.M.; Rossmann, J.; Harrel, S.K.; Forrest, J.L.; et al. Systematic review and meta-analysis on the nonsurgical treatment of chronic periodontitis by means of scaling and root planing with or without adjuncts. J. Am. Dent. Assoc. 2015, 146, 508–524.e5. [Google Scholar] [CrossRef]
- Kajikawa, T.; Briones, R.A.; Resuello, R.R.G.; Tuplano, J.V.; Reis, E.S.; Hajishengallis, E.; Garcia, C.A.G.; Yancopoulou, D.; Lambris, J.D.; Hajishengallis, G. Safety and Efficacy of the Complement Inhibitor AMY-101 in a Natural Model of Periodontitis in Non-human Primates. Mol. Ther. Methods Clin. Dev. 2017, 6, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, T.; Abe, T.; Hajishengallis, E.; Hosur, K.B.; DeAngelis, R.A.; Ricklin, D.; Lambris, J.D.; Hajishengallis, G. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J. Immunol. 2014, 192, 6020–6027. [Google Scholar] [CrossRef] [Green Version]
- Balta, M.G.; Loos, B.G.; Nicu, E.A. Emerging Concepts in the Resolution of Periodontal Inflammation: A Role for Resolvin E1. Front. Immunol. 2017, 8, 1682. [Google Scholar] [CrossRef] [Green Version]
- Bochtler, M.; Mizgalska, D.; Veillard, F.; Nowak, M.L.; Houston, J.; Veith, P.; Reynolds, E.C.; Potempa, J. The Bacteroidetes Q-Rule: Pyroglutamate in Signal Peptidase I Substrates. Front. Microbiol. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taudte, N.; Linnert, M.; Rahfeld, J.-U.; Piechotta, A.; Ramsbeck, D.; Buchholz, M.; Kolenko, P.; Parthier, C.; Houston, J.A.; Veillard, F.; et al. Mammalian-like type II glutaminyl cyclases in Porphyromonas gingivalis and other oral pathogenic bacteria as targets for treatment of periodontitis. J. Biol. Chem. 2021, 296, 100263. [Google Scholar] [CrossRef]
- Hutcherson, J.A.; Gogeneni, H.; Yoder-Himes, D.; Hendrickson, E.L.; Hackett, M.; Whiteley, M.; Lamont, R.J.; Scott, D.A. Comparison of inherently essential genes of Porphyromonas gingivalis identified in two transposon-sequencing libraries. Mol. Oral Microbiol. 2016, 31, 354–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, B.A.; Tenorio, E.L.; Lazinski, D.W.; Camilli, A.; Duncan, M.J.; Hu, L.T. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genom. 2012, 13, 578. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, M.; Heiser, U.; Schilling, S.; Niestroj, A.J.; Zunkel, K.; Demuth, H.-U. The first potent inhibitors for human glutaminyl cyclase: Synthesis and structure-activity relationship. J. Med. Chem. 2006, 49, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Hamann, A.; Aust, S.; Brandt, W.; Böhme, L.; Hoffmann, T.; Schilling, S.; Demuth, H.-U.; Heiser, U. Inhibitors for human glutaminyl cyclase by structure based design and bioisosteric replacement. J. Med. Chem. 2009, 52, 7069–7080. [Google Scholar] [CrossRef]
- Ramsbeck, D.; Buchholz, M.; Koch, B.; Böhme, L.; Hoffmann, T.; Demuth, H.-U.; Heiser, U. Structure–Activity Relationships of Benzimidazole-Based Glutaminyl Cyclase Inhibitors Featuring a Heteroaryl Scaffold. J. Med. Chem. 2013, 56, 6613–6625. [Google Scholar] [CrossRef]
- Hoang, V.-H.; Tran, P.-T.; Cui, M.; van Ngo, T.H.; Ann, J.; Park, J.; Lee, J.; Choi, K.; Cho, H.; Kim, H.; et al. Discovery of Potent Human Glutaminyl Cyclase Inhibitors as Anti-Alzheimer’s Agents Based on Rational Design. J. Med. Chem. 2017, 60, 2573–2590. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Dong, Y.; Yu, X.; Li, Y.; Zou, Y.; Zheng, Y.; He, Z.; Liu, Z.; Quan, J.; Bu, X.; et al. Synthesis and Evaluation of Diphenyl Conjugated Imidazole Derivatives as Potential Glutaminyl Cyclase Inhibitors for Treatment of Alzheimer’s Disease. J. Med. Chem. 2017, 60, 6664–6677. [Google Scholar] [CrossRef]
- van Ngo, T.H.; Hoang, V.-H.; Tran, P.-T.; Ann, J.; Cui, M.; Park, G.; Choi, S.; Lee, J.; Kim, H.; Ha, H.-J.; et al. Potent human glutaminyl cyclase inhibitors as potential anti-Alzheimer’s agents: Structure-activity relationship study of Arg-mimetic region. Bioorg. Med. Chem. 2018, 26, 1035–1049. [Google Scholar] [CrossRef]
- van Ngo, T.H.; Hoang, V.-H.; Tran, P.-T.; van Manh, N.; Ann, J.; Kim, E.; Cui, M.; Choi, S.; Lee, J.; Kim, H.; et al. Structure-activity relationship investigation of Phe-Arg mimetic region of human glutaminyl cyclase inhibitors. Bioorg. Med. Chem. 2018, 26, 3133–3144. [Google Scholar] [CrossRef]
- Hoang, V.-H.; van Ngo, T.H.; Cui, M.; van Manh, N.; Tran, P.-T.; Ann, J.; Ha, H.-J.; Kim, H.; Choi, K.; Kim, Y.-H.; et al. Discovery of Conformationally Restricted Human Glutaminyl Cyclase Inhibitors as Potent Anti-Alzheimer’s Agents by Structure-Based Design. J. Med. Chem. 2019, 62, 8011–8027. [Google Scholar] [CrossRef]
- Kupski, O.; Funk, L.-M.; Sautner, V.; Seifert, F.; Worbs, B.; Ramsbeck, D.; Meyer, F.; Diederichsen, U.; Buchholz, M.; Schilling, S.; et al. Hydrazides Are Potent Transition-State Analogues for Glutaminyl Cyclase Implicated in the Pathogenesis of Alzheimer’s Disease. Biochemistry 2020, 59, 2585–2591. [Google Scholar] [CrossRef]
- Coimbra, J.R.M.; Salvador, J.A.R. A patent review of glutaminyl cyclase inhibitors (2004-present). Expert Opin. Ther. Pat. 2021, 31, 809–836. [Google Scholar] [CrossRef] [PubMed]
- Dileep, K.V.; Sakai, N.; Ihara, K.; Kato-Murayama, M.; Nakata, A.; Ito, A.; Sivaraman, D.M.; Shin, J.W.; Yoshida, M.; Shirouzu, M.; et al. Piperidine-4-carboxamide as a new scaffold for designing secretory glutaminyl cyclase inhibitors. Int. J. Biol. Macromol. 2021, 170, 415–423. [Google Scholar] [CrossRef]
- Xu, C.; Zou, H.; Yu, X.; Xie, Y.; Cai, J.; Shang, Q.; Ouyang, N.; Wang, Y.; Xu, P.; He, Z.; et al. Repurposing FDA-Approved Compounds for the Discovery of Glutaminyl Cyclase Inhibitors as Drugs Against Alzheimer’s Disease. ChemistryOpen 2021, 10, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Hielscher-Michael, S.; Griehl, C.; Buchholz, M.; Demuth, H.-U.; Arnold, N.; Wessjohann, L.A. Natural Products from Microalgae with Potential against Alzheimer’s Disease: Sulfolipids Are Potent Glutaminyl Cyclase Inhibitors. Mar. Drugs 2016, 14, 203. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
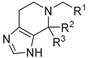 | R1 | R2 | R3 | Ki [µM] |
|---|---|---|---|---|
| 6a |  | - | no inhibition @ 10 µM | |
| 7a | | H | H | 0.909 ± 0.005 |
| 7b |  | H | H | 6.470 ± 0.400 |
| 7c | | -CH2OCH2- | 1.755 ± 0.145 | |
| 7d | | CH3 | - | 3.080 ± 0.040 |
| 7e | | | - | no inhibition @ 10 µM |
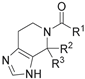 | ||||
| 8a | | H | H | 0.435 ± 0.015 |
| 8b | | H | H | 1.680 ± 0.080 |
| 8c | 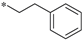 | H | H | 1.750 ± 0.060 |
| 8d |  | H | H | 1.575 ± 0.025 |
 | R1 | R2 | R3 | Ki (µM) |
|---|---|---|---|---|
| 8e | H | F | H | 0.879 ± 0.008 |
| 8f | F | H | H | 0.322 ± 0.006 |
| 8g | F | F | F | 0.904 ± 0.007 |
| 8h | H | Cl | H | 0.682 ± 0.025 |
| 8i | Cl | H | H | 0.174 ± 0.014 |
| 8j | Cl | Cl | H | 0.257 ± 0.016 |
| 8k | Cl | H | Cl | 0.100 ± 0.006 |
| 8l | H | OCH3 | H | 0.833 ± 0.049 |
| 8m | OCH3 | H | H | 0.335 ± 0.006 |
| 8n | OCH3 | OCH3 | H | 1.465 ± 0.304 |
| 8o | OCH3 | H | OCH3 | 0.928 ± 0.021 |
| 8p | -OCH2CH2O- | H | 1.335 ± 0.219 | |
| 8q | -OCH2O- | H | 0.560 ± 0.004 | |
| 8r | OnPr | H | H | 0.780 ± 0.024 |
| 8s | OCH3 | F | H | 1.240 ± 0.113 |
| 8t | OCH3 | H | Cl | 0.087 ± 0.006 |
| 8u | H | Ph | H | 1.900 ± 0.071 |
| 8v | Ph | H | H | 0.547 ± 0.041 |
| 8w | Ph | H | 0.126 ± 0.009 | |
| 8k | 8t | 8w | |
|---|---|---|---|
| Ki (µM) | |||
| PiQC | 0.063 ± 0.007 | 0.096 ± 0.012 | 0.117 ± 0.002 |
| TfQC | 0.176 ± 0.003 | 0.221 ± 0.019 | 0.185 ± 0.010 |
| hQC | 0.387 ± 0.064 | 0.385 ± 0.096 | 0.132 ± 0.011 |
| Cell viability @ 30 µM (%) | |||
| SHSY-5Y | 119 | 128 | 127 |
| Hep-G2 | 92 | 96 | 94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsbeck, D.; Taudte, N.; Jänckel, N.; Strich, S.; Rahfeld, J.-U.; Buchholz, M. Tetrahydroimidazo[4,5-c]pyridine-Based Inhibitors of Porphyromonas gingivalis Glutaminyl Cyclase. Pharmaceuticals 2021, 14, 1206. https://doi.org/10.3390/ph14121206
Ramsbeck D, Taudte N, Jänckel N, Strich S, Rahfeld J-U, Buchholz M. Tetrahydroimidazo[4,5-c]pyridine-Based Inhibitors of Porphyromonas gingivalis Glutaminyl Cyclase. Pharmaceuticals. 2021; 14(12):1206. https://doi.org/10.3390/ph14121206
Chicago/Turabian StyleRamsbeck, Daniel, Nadine Taudte, Nadine Jänckel, Stefanie Strich, Jens-Ulrich Rahfeld, and Mirko Buchholz. 2021. "Tetrahydroimidazo[4,5-c]pyridine-Based Inhibitors of Porphyromonas gingivalis Glutaminyl Cyclase" Pharmaceuticals 14, no. 12: 1206. https://doi.org/10.3390/ph14121206
APA StyleRamsbeck, D., Taudte, N., Jänckel, N., Strich, S., Rahfeld, J.-U., & Buchholz, M. (2021). Tetrahydroimidazo[4,5-c]pyridine-Based Inhibitors of Porphyromonas gingivalis Glutaminyl Cyclase. Pharmaceuticals, 14(12), 1206. https://doi.org/10.3390/ph14121206