
Obesity as a Condition Determined by Food Addiction: Should Brain Endocannabinoid System Alterations Be the Cause and Its Modulation the Solution?




Abstract
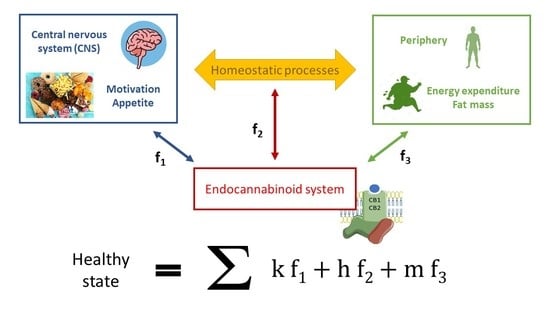
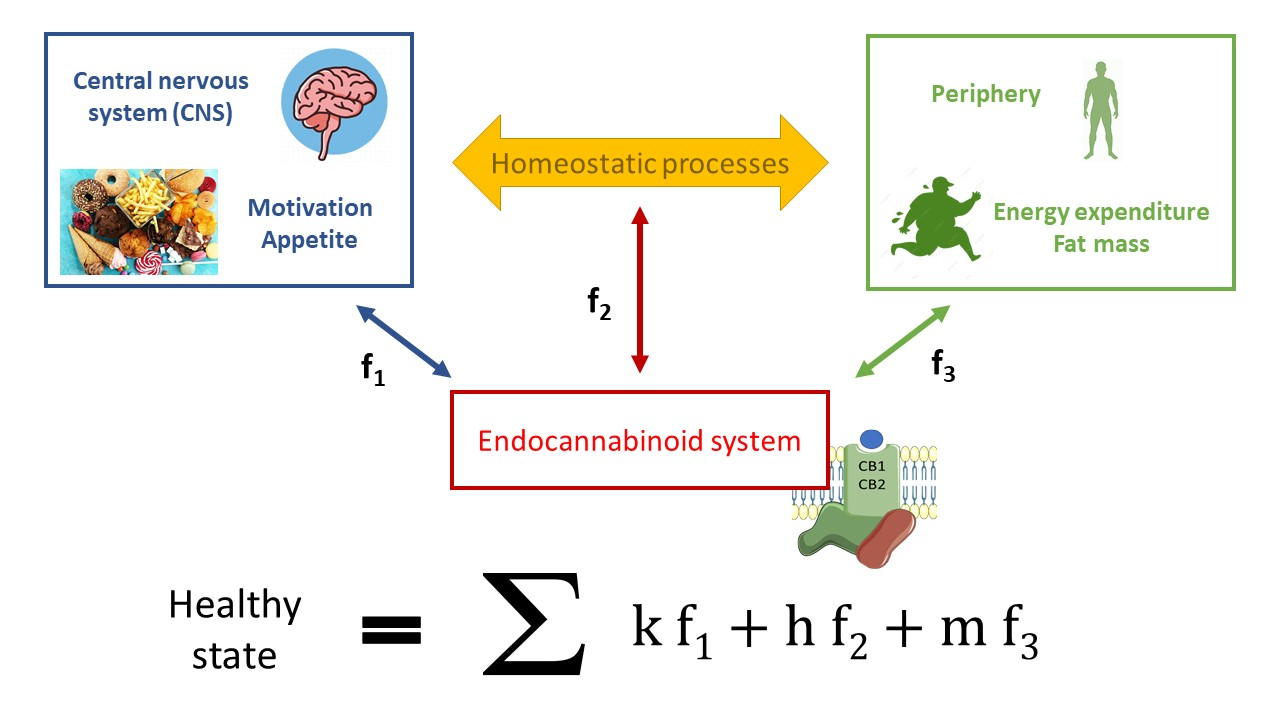
1. Introduction
2. Food Addiction
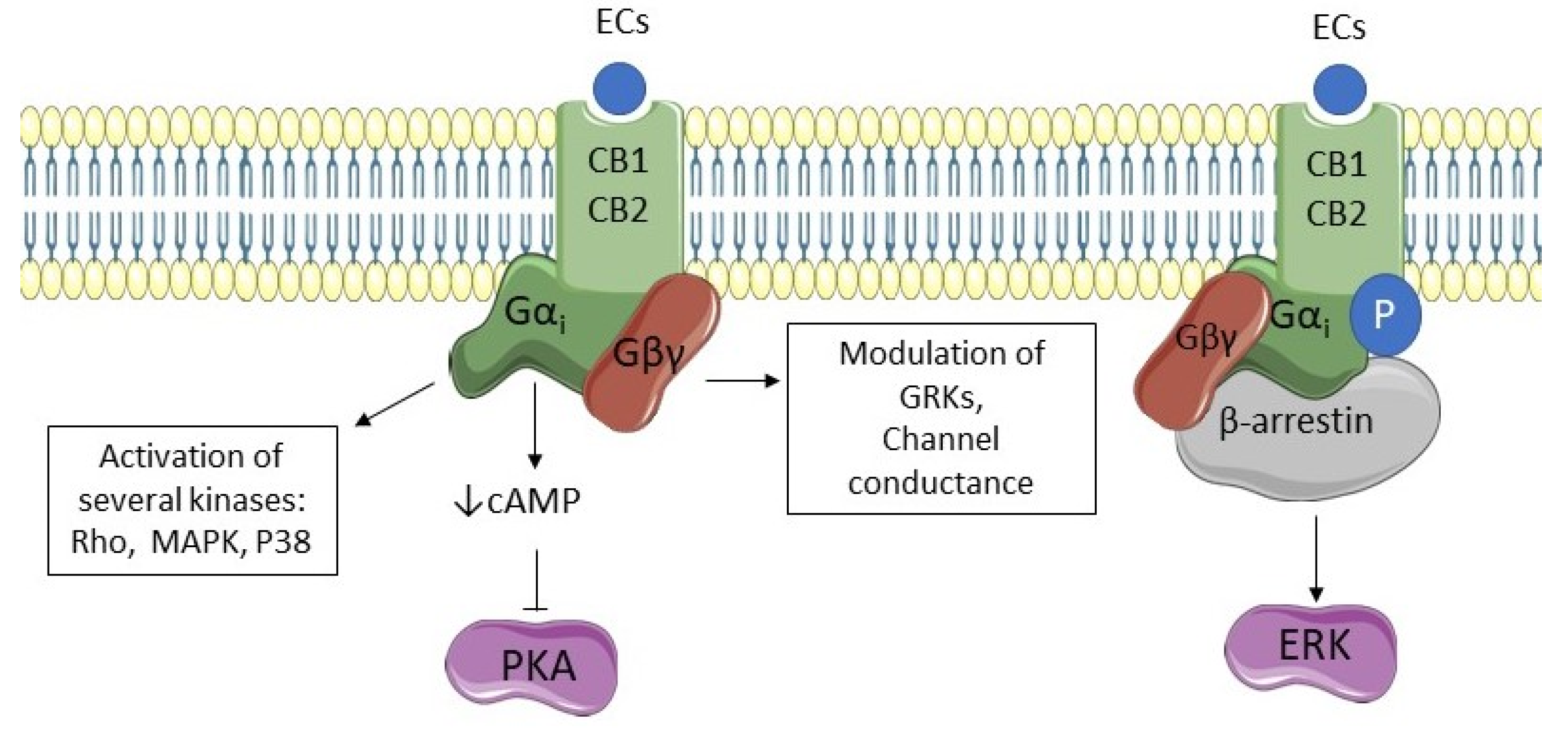
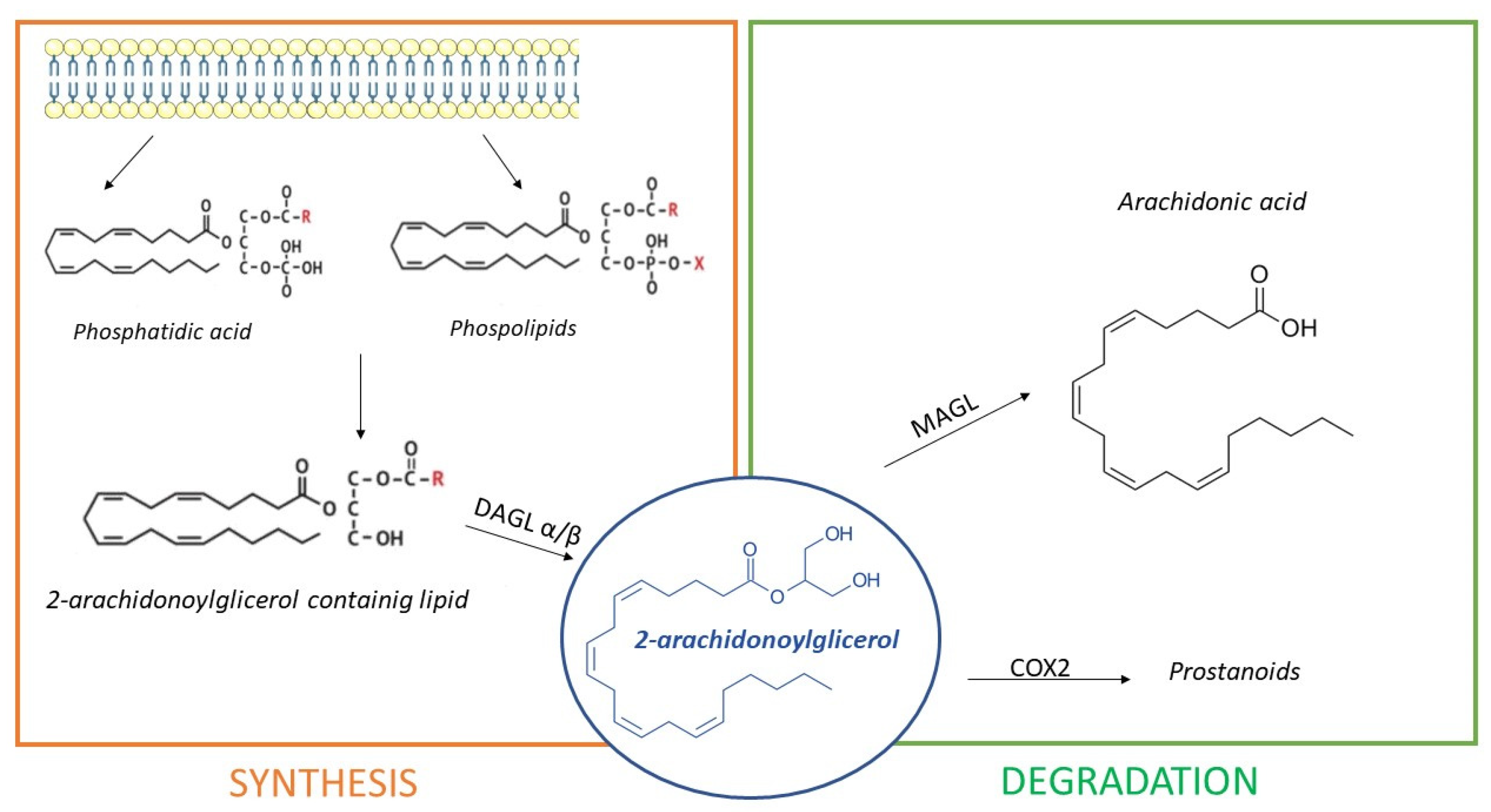
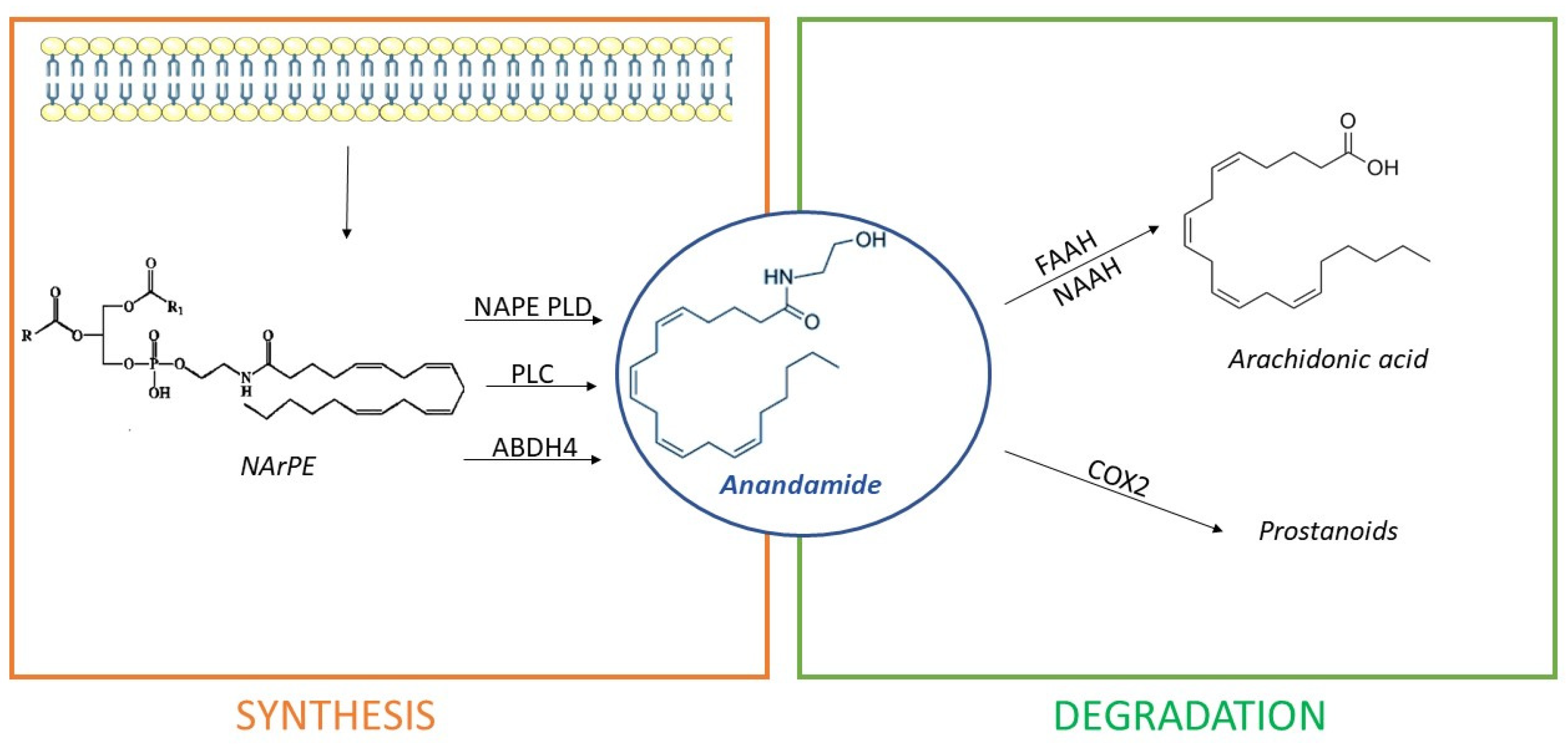
3. The Endocannabinoid System
- Directly, from the hydrolysis of N-arachidonoyl-phosphatidyl ethanolamine (NArPE), a phospholipid precursor belonging to a complex family of lipids, the N-acylethanolamides (NAEs), through the enzymatic action of N-acyl phosphatidylethanolamine phospholipase D or NAPE-PLD;
- Through NArPE deacetylation by α/β-hydrolase domain type-4 (ABHD4) and the hydrolysis of glycerophosphoethanolamine by glycerophosphodiesterase GDE1;
- Via the PLC-mediated hydrolysis of NArPE to yield phosphoanandamide, which is dephosphorylated to AEA by a phosphatase.
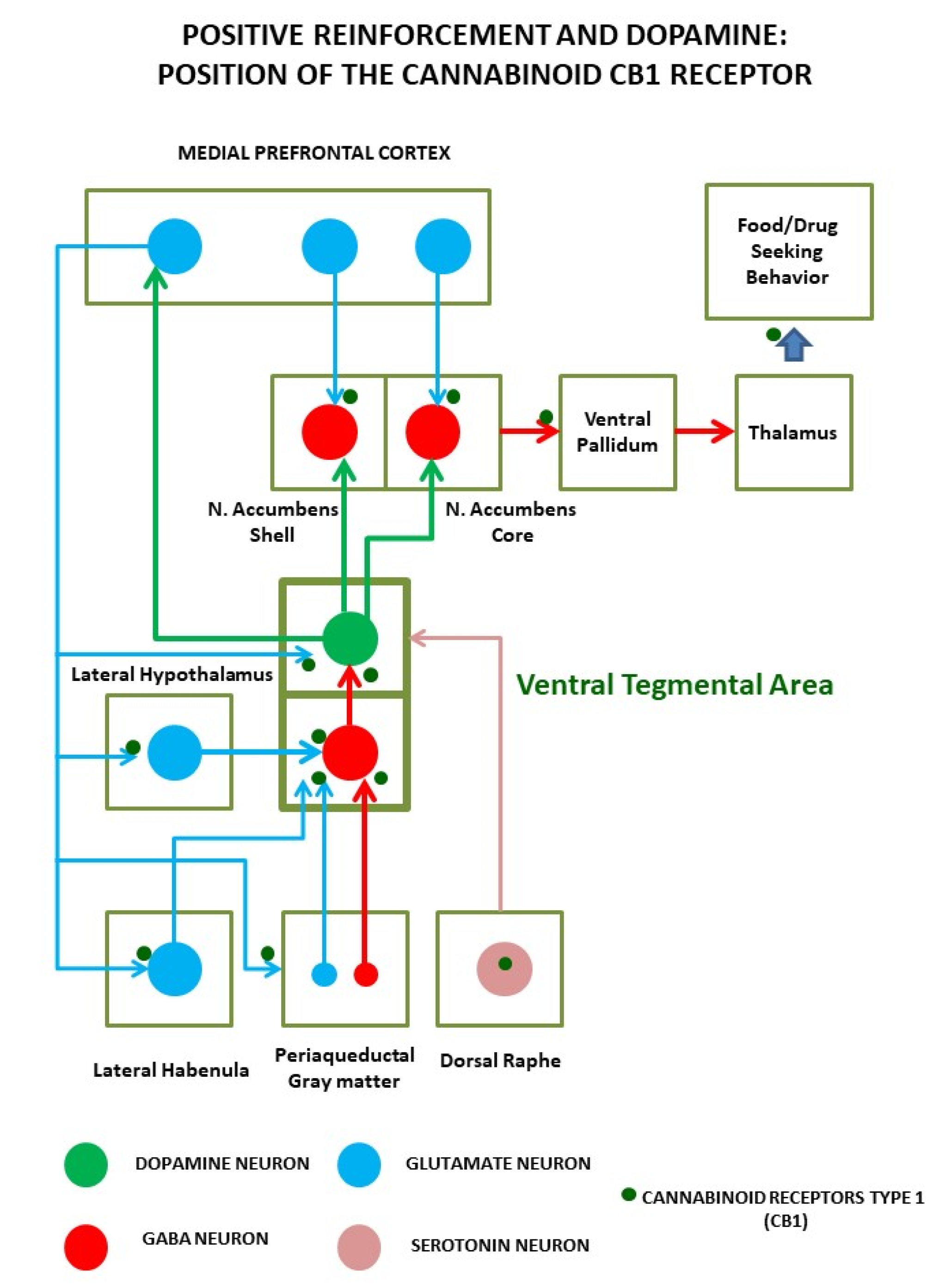
4. Endocannabinoid System Physiopathology in Obesity and Food Addiction
5. Endocannabinoid System as a Pharmacological Target for Obesity and Food Addiction
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Most Obese Countries. 2020. Available online: https://worldpopulationreview.com/countries/most-obese-countries/ (accessed on 29 April 2020).
- WHO. Controlling the Global Obesity Epidemic; WHO: Geneva, Switzerland, 2013. [Google Scholar]
- Rippe, J.M.; Crossley, S.; Ringer, R. Obesity as a chronic disease: Modern medical and lifestyle management. J. Am. Diet. Assoc. 1998, 98, S9–S15. [Google Scholar] [CrossRef]
- Meldrum, D.R.; Morris, M.A.; Gambone, J.C. Obesity pandemic: Causes, consequences, and solutions—But do we have the will? Fertil. Steril. 2017, 107, 833–839. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, Z.; Heshka, S.; Heo, M.; Faith, M.S.; Heymsfield, S.B. Waist circumference and obesity-associated risk factors among whites in the third National Health and Nutrition Examination Survey: Clinical action thresholds. Am. J. Clin. Nutr. 2002, 76, 743. [Google Scholar] [CrossRef]
- Skelton, J.A.; Irby, M.B.; Grzywacz, J.G.; Miller, G. Etiologies of obesity in children: Nature and nurture. Pediatr. Clin. N. Am. 2011, 58, 1333–1354. [Google Scholar] [CrossRef]
- McAllister, E.J.; Dhurandhar, N.V.; Keith, S.W.; Aronne, L.J.; Barger, J.; Baskin, M.; Benca, R.M.; Biggio, J.; Boggiano, M.M.; Eisenmann, J.C.; et al. Ten putative contributors to the obesity epidemic. Crit. Rev. Food Sci. Nutr. 2009, 49, 868–913. [Google Scholar] [CrossRef]
- Webber, J. Energy balance in obesity. Proc. Nutr. Soc. 2003, 62, 539–543. [Google Scholar] [CrossRef]
- Barsh, G.S.; Farooqi, I.S.; O’rahilly, S. Genetics of body-weight regulation. Nature 2000, 404, 644–651. [Google Scholar] [CrossRef]
- Leigh, S.J.; Morris, M.J. The role of reward circuitry and food addiction in the obesity epidemic: An update. Biol. Psychol. 2018, 131, 31–42. [Google Scholar] [CrossRef]
- Berthoud, H.R. The neurobiology of food intake in an obesogenic environment. Proc. Nutr. Soc. 2012, 71, 478–487. [Google Scholar] [CrossRef]
- Morris, M.J.; Beilharz, J.E.; Maniam, J.; Reichelt, A.C.; Westbrook, R.F. Why is obesity such a problem in the 21st century? The intersection of palatable food, cues and reward pathways, stress, and cognition. Neurosci. Biobehav. Rev. 2015, 58, 36–45. [Google Scholar] [CrossRef]
- Dallman, M.F.; Pecoraro, N.; Akana, S.F.; la Fleur, S.E.; Gomez, F.; Houshyar, H.; Bell, M.E.; Bhatnagar, S.; Laugero, K.D.; Manalo, S. Chronic stress and obesity: A new view of comfort food. Proc. Natl. Acad. Sci. USA 2003, 100, 11696–11701. [Google Scholar] [CrossRef]
- Roth, J.; Qiang, X.; Marbán, S.L.; Redelt, H.; Lowell, B.C. The Obesity Pandemic: Where Have We Been and Where Are We Going? Obes. Res. 2004, 12, 88S–101S. [Google Scholar] [CrossRef]
- Stamler, R. Weight and blood pressure. Findings in hypertension screening of 1 million Americans. JAMA J. Am. Med. Assoc. 1978, 240, 1607–1610. [Google Scholar] [CrossRef]
- Piegas, L.S.; Avezum, Á.; Pereira, J.C.R.; Neto, J.M.R.; Hoepfner, C.; Farran, J.A.; Ramos, R.F.; Timerman, A.; Esteves, J.P. Risk factors for myocardial infarction in Brazil. Am. Heart J. 2003, 146, 331–338. [Google Scholar] [CrossRef]
- Walker, S.P.; Rimm, E.B.; Ascherio, A.; Kawachi, I.; Stampfer, M.J.; Willett, W.C. Body size and fat distribution as predictors of stroke among US men. Am. J. Epidemiol. 1996, 144, 1143–1150. [Google Scholar] [CrossRef]
- Harris, M.I.; Flegal, K.M.; Cowie, C.C.; Eberhardt, M.S.; Goldstein, D.E.; Little, R.R.; Wiedmeyer, H.M.; Byrd-Holt, D.D. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults: The Third National Health and Nutrition Examination Survey, 1988–1994. Diabetes Care 1998, 21, 518–524. [Google Scholar] [CrossRef]
- Combe, H.; Vol, S.; Thévenot, A.; Lasfargues, G.; Cacès, E.; Tichet, J.; Lecomte, P. Comparison of men with impaired fasting glycaemia to controls and to diabetic subjects with fasting glycaemia from 7.0 to 7.7 mmol/L: Clinical, nutritional and biological status. Diabetes Metab. 2004, 30, 167–174. [Google Scholar] [CrossRef]
- Kanaya, A.M.; Barrett-Connor, E.; Gildengorin, G.; Yaffe, K. Change in cognitive function by glucose tolerance status in older adults: A 4-year prospective study of the Rancho Bernardo Study cohort. Arch. Intern. Med. 2004, 164, 1327–1333. [Google Scholar] [CrossRef]
- Gustafson, D.; Rothenberg, E.; Blennow, K.; Steen, B.; Skoog, I. An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 2003, 163, 1524–1528. [Google Scholar] [CrossRef]
- Lew, E.A.; Garfinkel, L. Variations in mortality by weight among 750,000 men and women. J. Chronic Dis. 1979, 32, 563–576. [Google Scholar] [CrossRef]
- Luder, E.; Ehrlich, R.I.; Lou, W.Y.W.; Melnik, T.A.; Kattan, M. Body mass index and the risk of asthma in adults. Respir. Med. 2004, 98, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Anderson, J.J.; Naimark, A.; Walker, A.M.; Meenan, R.F. Obesity and knee osteoarthritis. The Framingham Study. Ann. Intern. Med. 1988, 109, 18–24. [Google Scholar] [CrossRef]
- Pasquali, R.; Pelusi, C.; Genghini, S.; Cacciari, M.; Gambineri, A. Obesity and reproductive disorders in women. Hum. Reprod. Update 2003, 9, 359–372. [Google Scholar] [CrossRef]
- Dong, C.; Sanchez, L.E.; Price, R.A. Relationship of obesity to depression: A family-based study. Int. J. Obes. 2004, 28, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Butler, A.A.; Kozak, L.P. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes 2010, 59, 323–329. [Google Scholar] [CrossRef]
- Donath, M.Y.; Böni-Schnetzler, M.; Ellingsgaard, H.; Halban, P.A.; Ehses, J.A. Cytokine production by islets in health and diabetes: Cellular origin, regulation and function. Trends Endocrinol. Metab. 2010, 21, 261–267. [Google Scholar] [CrossRef]
- Li, Z.; Soloski, M.J.; Diehl, A.M. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology 2005, 42, 880–885. [Google Scholar] [CrossRef]
- Saghizadeh, M.; Ong, J.M.; Garvey, W.T.; Henry, R.R.; Kern, P.A. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J. Clin. Investig. 1996, 97, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.H. Making sense of metabolic obesity and hedonic obesity. J. Diabetes 2017, 9, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Mittendorfer, B.; Klein, S. Metabolically healthy obesity: Facts and fantasies. J. Clin. Investig. 2019, 129, 3978–3989. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.H.; Vasselli, J.R.; Zhang, Y.; Mechanick, J.I.; Korner, J.; Peterli, R. Metabolic vs. hedonic obesity: A conceptual distinction and its clinical implications. Obes. Rev. 2015, 16, 234–247. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.-J.; Fowler, J.S.; Telang, F. Overlapping neuronal circuits in addiction and obesity: Evidence of systems pathology. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3191–3200. [Google Scholar] [CrossRef]
- Randolph, T.G. The descriptive features of food addiction; addictive eating and drinking. Q. J. Stud. Alcohol 1956, 17, 198–224. [Google Scholar] [CrossRef] [PubMed]
- Gearhardt, A.N.; Corbin, W.R.; Brownell, K.D. Preliminary validation of the Yale Food Addiction Scale. Appetite 2009, 52, 430–436. [Google Scholar] [CrossRef]
- Pursey, K.; Stanwell, P.; Gearhardt, A.; Collins, C.; Burrows, T. The Prevalence of Food Addiction as Assessed by the Yale Food Addiction Scale: A Systematic Review. Nutrients 2014, 6, 4552–4590. [Google Scholar] [CrossRef]
- Epel, E.S.; Tomiyama, A.J.; Mason, A.E.; Laraia, B.A.; Hartman, W.; Ready, K.; Acree, M.; Adam, T.C.; Jeor, S.S.; Kessler, D. The Reward-Based Eating Drive Scale: A Self-Report Index of Reward-Based Eating. PLoS ONE 2014, 9, e101350. [Google Scholar] [CrossRef]
- Mason, A.E.; Lustig, R.H.; Brown, R.R.; Acree, M.; Bacchetti, P.; Moran, P.J.; Dallman, M.; Laraia, B.; Adler, N.; Hecht, F.M.; et al. Acute responses to opioidergic blockade as a biomarker of hedonic eating among obese women enrolled in a mindfulness-based weight loss intervention trial. Appetite 2015, 91, 311–320. [Google Scholar] [CrossRef]
- Bechara, A. Decision making, impulse control and loss of willpower to resist drugs: A neurocognitive perspective. Nat. Neurosci. 2005, 8, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.; Clarke, M.; Petry, N.M. Obesity and its relationship to addictions: Is overeating a form of Addictive Behavior? Am. J. Addict. 2009, 18, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.; Hendrikse, J.; Lee, N.; Yücel, M.; Verdejo-Garcia, A.; Andrews, Z.; Hall, W. The Neurobiology of ‘Food Addiction’ and Its Implications for Obesity Treatment and Policy. Annu. Rev. Nutr. 2016, 36, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Sutin, A.R.; Ferrucci, L.; Zonderman, A.B.; Terracciano, A. Personality and obesity across the adult life span. J. Pers. Soc. Psychol. 2011, 101, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L. What nutritional physiology tells us about diet, sugar and obesity. Int. J. Obes. 2016, 40, S28–S29. [Google Scholar] [CrossRef]
- Meule, A.; Gearhardt, A. Food Addiction in the Light of DSM-5. Nutrients 2014, 6, 3653–3671. [Google Scholar] [CrossRef] [PubMed]
- Gearhardt, A.N.; Grilo, C.M.; Dileone, R.J.; Brownell, K.D.; Potenza, M.N. Can food be addictive? Public health and policy implications. Addiction 2011, 106, 1208–1212. [Google Scholar] [CrossRef]
- Schulte, E.M.; Avena, N.M.; Gearhardt, A.N. Which Foods May Be Addictive? The Roles of Processing, Fat Content, and Glycemic Load. PLoS ONE 2015, 10, e0117959. [Google Scholar] [CrossRef]
- Münzberg, H.; Qualls-Creekmore, E.; Yu, S.; Morrison, C.D.; Berthoud, H.R. Hedonics Act in Unison with the Homeostatic System to Unconsciously Control Body Weight. Front. Nutr. 2016, 3. [Google Scholar] [CrossRef]
- Carlier, N.; Marshe, V.; Cmorejova, J.; Davis, C.; Müller, D. Genetic Similarities between Compulsive Overeating and Addiction Phenotypes: A Case for ‘Food Addiction’? Curr. Psychiatry Rep. 2015, 17, 96. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.J.; Tomasi, D.; Baler, R.D. Obesity and addiction: Neurobiological overlaps. Obes. Rev. 2013, 14, 2–18. [Google Scholar] [CrossRef]
- Davis, C.; Curtis, C.; Levitan, R.D.; Carter, J.C.; Kaplan, A.S.; Kennedy, J.L. Evidence that food addiction is a valid phenotype of obesity. Appetite 2011, 57, 711–717. [Google Scholar] [CrossRef]
- Lindgren, E.; Gray, K.; Miller, G.; Tyler, R.; Wiers, C.E.; Volkow, N.D.; Wang, G.J. Food addiction: A common neurobiological mechanism with drug abuse. Front. Biosci. Landmark 2018, 23, 811–836. [Google Scholar] [CrossRef]
- Smith, D.; Robbins, T. The neurobiological underpinnings of obesity and binge eating: A rationale for adopting the food addiction model. Biol. Psychiatry 2013, 73, 804–810. [Google Scholar] [CrossRef]
- Barbano, M.F.; Cador, M. Opioids for hedonic experience and dopamine to get ready for it. Psychopharmacology 2007, 191, 497–506. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Nicoll, R. Endocannabinoid signaling in the brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB₁ receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Zottola, A.C.P.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Piomelli, D.; Giuffrida, A.; Calignano, A.; de Fonseca, F.R. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol. Sci. 2000, 21, 218–224. [Google Scholar] [CrossRef]
- Mechoulam, R.; Parker, L.A. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef]
- Howlett, A.C.; Abood, M.E. CB1 and CB2 Receptor Pharmacology. Adv. Pharmacol. 2017, 80, 169–206. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef]
- Lu, H.C.; MacKie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Baggelaar, M.P.; Maccarrone, M.; van der Stelt, M. 2-Arachidonoylglycerol: A signaling lipid with manifold actions in the brain. Prog. Lipid Res. 2018, 71, 1–17. [Google Scholar] [CrossRef]
- Scherma, M.; Masia, P.; Satta, V.; Fratta, W.; Fadda, P.; Tanda, G. Brain activity of anandamide: A rewarding bliss? Acta Pharmacol. Sin. 2019, 40, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Smita, K.; Kumar, V.S.; Premendran, J.S. Anandamide: An update. Fundam. Clin. Pharmacol. 2007, 21, 1–8. [Google Scholar] [CrossRef]
- De Fonseca, F.R.; Navarro, M.; Gómez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodríguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S.; et al. An anorexic lipid mediator regulated by feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, S.; Dipasquale, P.; Romano, A.; Righetti, L.; Cassano, T.; Piomelli, D.; Cuomo, V. Chapter 5 The Endocannabinoid System as A Target for Novel Anxiolytic and Antidepressant Drugs. Int. Rev. Neurobiol. 2009, 85, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Tempesta, B.; Provensi, G.; Passani, M.B.; Gaetani, S. Central mechanisms mediating the hypophagic effects of oleoylethanolamide and N-acylphosphatidylethanolamines: Different lipid signals? Front. Pharmacol. 2015, 6, 137. [Google Scholar] [CrossRef]
- Orio, L.; Alen, F.; Pavón, F.J.; Serrano, A.; García-Bueno, B. Oleoylethanolamide, neuroinflammation, and alcohol abuse. Front. Mol. Neurosci. 2019, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Tellez, L.A.; Medina, S.; Han, W.; Ferreira, J.G.; Licona-Limón, P.; Ren, X.; Lam, T.T.; Schwartz, G.J.; de Araujo, I.E. A gut lipid messenger links excess dietary fat to dopamine deficiency. Science 2013, 341, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Gómez, R.; Navarro, M.; Ferrer, B.; Trigo, J.M.; Bilbao, A.; del Arco, I.; Cippitelli, A.; Nava, F.; Piomelli, D.; de Fonseca, F.R. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J. Neurosci. 2002, 22, 9612–9617. [Google Scholar] [CrossRef] [PubMed]
- De Azua, I.R.; Lutz, B. Multiple endocannabinoid-mediated mechanisms in the regulation of energy homeostasis in brain and peripheral tissues. Cell. Mol. Life Sci. 2019, 76, 1341–1363. [Google Scholar] [CrossRef]
- Kirkham, T. Cannabinoids and appetite: Food craving and food pleasure. Int. Rev. Psychiatry 2009, 21, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, L.; Lafentre, P.; Cannich, A.; Cota, D.; Puente, N.; Grandes, P.; Chaouloff, F.; Piazza, P.V.; Marsicano, G. Bimodal control of stimulated food intake by the endocannabinoid system. Nat. Neurosci. 2010, 13, 281–283. [Google Scholar] [CrossRef]
- Mahler, S.V.; Smith, K.S.; Berridge, K.C. Endocannabinoid hedonic hotspot for sensory pleasure: Anandamide in nucleus accumbens shell enhances liking of a sweet reward. Neuropsychopharmacology 2007, 32, 2267–2278. [Google Scholar] [CrossRef]
- Lau, B.K.; Cota, D.; Cristino, L.; Borgland, S.L. Endocannabinoid modulation of homeostatic and non-homeostatic feeding circuits. Neuropharmacology 2017, 124, 38–51. [Google Scholar] [CrossRef]
- Cruz-Martínez, A.M.; Tejas-Juárez, J.G.; Mancilla-Díaz, J.M.; Florán-Garduño, B.; López-Alonso, V.E.; Escartín-Pérez, R.E. CB1 receptors in the paraventricular nucleus of the hypothalamus modulate the release of 5-HT and GABA to stimulate food intake in rats. Eur. Neuropsychopharmacol. 2018, 28, 1247–1259. [Google Scholar] [CrossRef]
- Koch, M.; Varela, L.; Kim, J.G.; Kim, J.D.; Hernández-Nuño, F.; Simonds, S.E.; Castorena, C.M.; Vianna, C.R.; Elmquist, J.K.; Morozov, Y.M.; et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 2015, 519, 45–50. [Google Scholar] [CrossRef]
- Coccurello, R.; Maccarrone, M. Hedonic eating and the delicious circle: From lipid-derived mediators to brain dopamine and back. Front. Neurosci. 2018, 12, 271. [Google Scholar] [CrossRef] [PubMed]
- Mazier, W.; Saucisse, N.; Gatta-Cherifi, B.; Cota, D. The Endocannabinoid System: Pivotal Orchestrator of Obesity and Metabolic Disease. Trends Endocrinol. Metab. 2015, 26, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, C.; Di Marzo, V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef]
- DiPatrizio, N.V.; Piomelli, D. The thrifty lipids: Endocannabinoids and the neural control of energy conservation. Trends Neurosci. 2012, 35, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.C.; Scott, T.M.; Murray, S.; Harismendy, O.; Simon, G.M.; Cravatt, B.F.; Waalen, J. Biomarkers of endocannabinoid system activation in severe obesity. PLoS ONE 2010, 5, e8792. [Google Scholar] [CrossRef] [PubMed]
- Engeli, S. Dysregulation of the Endocannabinoid System in Obesity. J. Neuroendocrinol. 2008, 20, 110–115. [Google Scholar] [CrossRef]
- Forte, N.; Fernández-Rilo, A.; Palomba, L.; di Marzo, V.; Cristino, L. Obesity Affects the Microbiota-Gut-Brain Axis and the Regulation Thereof by Endocannabinoids and Related Mediators. Int. J. Mol. Sci. 2020, 21, 1554. [Google Scholar] [CrossRef]
- Rossi, F.; Punzo, F.; Umano, G.R.; Argenziano, M.; del Giudice, E.M. Role of cannabinoids in obesity. Int. J. Mol. Sci. 2018, 19, 2690. [Google Scholar] [CrossRef]
- Vaitheesvaran, B.; Yang, L.; Hartil, K.; Glaser, S.; Yazulla, S.; Bruce, J.E.; Kurland, I.J. Peripheral effects of FAAH deficiency on fuel and energy homeostasis: Role of dysregulated lysine acetylation. PLoS ONE 2012, 7, e33717. [Google Scholar] [CrossRef]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta 2008, 1781, 200–212. [Google Scholar] [CrossRef]
- Banni, S.; di Marzo, V. Effect of dietary fat on endocannabinoids and related mediators: Consequences on energy homeostasis, inflammation and mood. Mol. Nutr. Food Res. 2010, 54, 82–92. [Google Scholar] [CrossRef]
- Melis, M.; Carta, G.; Pintus, S.; Pintus, P.; Piras, C.A.; Murru, E.; Manca, C.; di Marzo, V.; Banni, S.; Barbarossa, I.T. Polymorphism rs1761667 in the CD36 Gene Is Associated to Changes in Fatty Acid Metabolism and Circulating Endocannabinoid Levels Distinctively in Normal Weight and Obese Subjects. Front. Physiol. 2017, 8, 1006. [Google Scholar] [CrossRef] [PubMed]
- Barbarossa, I.T.; Carta, G.; Murru, E.; Melis, M.; Zonza, A.; Vacca, C.; Muroni, P.; di Marzo, V.; Banni, S. Taste sensitivity to 6-n-propylthiouracil is associated with endocannabinoid plasma levels in normal-weight individuals. Nutrition 2013, 29, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Melis, M.; Pintus, S.; Pintus, P.; Piras, C.A.; Muredda, L.; Demurtas, D.; di Marzo, V.; Banni, S.; Barbarossa, I.T. Participants with Normal Weight or with Obesity Show Different Relationships of 6-n-Propylthiouracil (PROP) Taster Status with BMI and Plasma Endocannabinoids. Sci. Rep. 2017, 7, 1361. [Google Scholar] [CrossRef] [PubMed]
- Kunos, G. Understanding metabolic homeostasis and imbalance: What is the role of the endocannabinoid system? Am. J. Med. 2007, 120 (Suppl. S1), S18–S24. [Google Scholar] [CrossRef] [PubMed]
- Freitas, H.R.; Isaac, A.R.; Malcher-Lopes, R.; Diaz, B.L.; Trevenzoli, I.H.; De Melo Reis, R.A. Polyunsaturated fatty acids and endocannabinoids in health and disease. Nutr. Neurosci. 2018, 21, 695–714. [Google Scholar] [CrossRef]
- Miralpeix, C.; Fosch, A.; Casas, J.; Baena, M.; Herrero, L.; Serra, D.; Rodríguez-Rodríguez, R.; Casals, N. Hypothalamic endocannabinoids inversely correlate with the development of diet-induced obesity in male and female mice. J. Lipid Res. 2019, 60, 1260–1269. [Google Scholar] [CrossRef]
- Pucci, M.; Zaplatic, E.; di Bonaventura, M.V.M.; di Bonaventura, E.M.; de Cristofaro, P.; Maccarrone, M.; Cifani, C.; D’Addario, C. On the Role of Central Type-1 Cannabinoid Receptor Gene Regulation in Food Intake and Eating Behaviors. Int. J. Mol. Sci. 2021, 22, 398. [Google Scholar] [CrossRef]
- Martin, G.G.; Landrock, D.; Chung, S.; Dangott, L.J.; Seeger, D.R.; Murphy, E.J.; Golovko, M.Y.; Kier, A.B.; Schroeder, F. Fabp1 gene ablation inhibits high-fat diet-induced increase in brain endocannabinoids. J. Neurochem. 2017, 140, 294–306. [Google Scholar] [CrossRef]
- Yang, D.; Xu, L.; Guo, F.; Sun, X.; Zhang, D.; Wang, M. Orexin-A and endocannabinoid signaling regulate glucose-responsive arcuate nucleus neurons and feeding behavior in obese rats. Neuropeptides 2018, 69, 26–38. [Google Scholar] [CrossRef] [PubMed]
- DiPatrizio, N.V. Endocannabinoids and the Gut-Brain Control of Food Intake and Obesity. Nutrients 2021, 13, 1214. [Google Scholar] [CrossRef] [PubMed]
- Harrold, J.; Elliott, J.; King, J.; Widdowson, P.; Williams, G. Down-regulation of cannabinoid-1 (CB-1) receptors in specific extrahypothalamic regions of rats with dietary obesity: A role for endogenous cannabinoids in driving appetite for palatable food? Brain Res. 2002, 952, 232–238. [Google Scholar] [CrossRef]
- Bourdy, R.; Hertz, A.; Filliol, D.; Andry, V.; Goumon, Y.; Mendoza, J.; Olmstead, M.C.; Befort, K. The endocannabinoid system is modulated in reward and homeostatic brain regions following diet-induced obesity in rats: A cluster analysis approach. Eur. J. Nutr. 2021, 1–13. [Google Scholar] [CrossRef]
- Labouèbe, G.; Liu, S.; Dias, C.; Zou, S.; Wong, J.; Karunakaran, S.; Clee, S.; Phillips, A.; Boutrel, B.; Borgland, S. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat. Neurosci. 2013, 16, 300–308. [Google Scholar] [CrossRef]
- Salaya-Velazquez, N.F.; López-Muciño, L.A.; Mejía-Chávez, S.; Sánchez-Aparicio, P.; Domínguez-Guadarrama, A.A.; Venebra-Muñoz, A. Anandamide and sucralose change ΔFosB expression in the reward system. Neuroreport 2020, 31, 240–244. [Google Scholar] [CrossRef]
- Yoshida, R.; Ohkuri, T.; Jyotaki, M.; Yasuo, T.; Horio, N.; Yasumatsu, K.; Sanematsu, K.; Shigemura, N.; Yamamoto, T.; Margolskee, R.; et al. Endocannabinoids selectively enhance sweet taste. Proc. Natl. Acad. Sci. USA 2010, 107, 935–939. [Google Scholar] [CrossRef]
- Cooper, S. Endocannabinoids and food consumption: Comparisons with benzodiazepine and opioid palatability-dependent appetite. Eur. J. Pharmacol. 2004, 500, 37–49. [Google Scholar] [CrossRef]
- Berridge, K.; Ho, C.; Richard, J.; DiFeliceantonio, A. The tempted brain eats: Pleasure and desire circuits in obesity and eating disorders. Brain Res. 2010, 1350, 43–64. [Google Scholar] [CrossRef]
- Cota, D.; Barrera, J.; Seeley, R. Leptin in energy balance and reward: Two faces of the same coin? Neuron 2006, 51, 678–680. [Google Scholar] [CrossRef]
- Berry, E.; Mechoulam, R. Tetrahydrocannabinol and endocannabinoids in feeding and appetite. Pharmacol. Ther. 2002, 95, 185–190. [Google Scholar] [CrossRef]
- Laksmidewi, P.; Soejitno, A. Endocannabinoid and dopaminergic system: The pas de deux underlying human motivation and behaviors. J. Neural Transm. 2021, 128, 615. [Google Scholar] [CrossRef] [PubMed]
- Oleson, E.B.; Beckert, M.V.; Morra, J.T.; Lansink, C.S.; Cachope, R.; Abdullah, R.A.; Loriaux, A.L.; Schetters, D.; Pattij, T.; Roitman, M.F.; et al. Endocannabinoids shape accumbal encoding of cue-motivated behavior via CB1 receptor activation in the ventral tegmentum. Neuron 2012, 73, 360–373. [Google Scholar] [CrossRef] [PubMed]
- D’Addario, C.; di Bonaventura, M.V.M.; Pucci, M.; Romano, A.; Gaetani, S.; Ciccocioppo, R.; Cifani, C.; Maccarrone, M. Endocannabinoid signaling and food addiction. Neurosci. Biobehav. Rev. 2014, 47, 203–224. [Google Scholar] [CrossRef]
- Tung, L.; Lu, G.; Lee, Y.; Yu, L.; Lee, H.; Leishman, E.; Bradshaw, H.; Hwang, L.; Hung, M.; Mackie, K.; et al. Orexins contribute to restraint stress-induced cocaine relapse by endocannabinoid-mediated disinhibition of dopaminergic neurons. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Parsons, L.; Hurd, Y. Endocannabinoid signalling in reward and addiction. Nat. Rev. Neurosci. 2015, 16, 579–594. [Google Scholar] [CrossRef]
- Cottone, P.; Sabino, V.; Roberto, M.; Bajo, M.; Pockros, L.; Frihauf, J.B.; Fekete, E.M.; Steardo, L.; Rice, K.C.; Grigoriadis, D.E.; et al. CRF system recruitment mediates dark side of compulsive eating. Proc. Natl. Acad. Sci. USA 2009, 106, 20016–20020. [Google Scholar] [CrossRef]
- Domingo-Rodriguez, L.; de Azua, I.R.; Dominguez, E.; Senabre, E.; Serra, I.; Kummer, S.; Navandar, M.; Baddenhausen, S.; Hofmann, C.; Andero, R.; et al. A specific prelimbic-nucleus accumbens pathway controls resilience versus vulnerability to food addiction. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- DiPatrizio, N.V. Is fat taste ready for primetime? Physiol. Behav. 2014, 136, 145–154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chaperon, F.; Soubrié, P.; Puech, A.; Thiébot, M. Involvement of central cannabinoid (CB1) receptors in the establishment of place conditioning in rats. Psychopharmacology 1998, 135, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Novelle, M.; Diéguez, C. Unravelling the role and mechanism of adipokine and gastrointestinal signals in animal models in the nonhomeostatic control of energy homeostasis: Implications for binge eating disorder. Eur. Eat. Disord. Rev. 2018, 26, 551–568. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Melis, M.; Fadda, P.; Pistis, M.; Fratta, W. The endocannabinoid system and nondrug rewarding behaviours. Exp. Neurol. 2010, 224, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.; Stark, J.; McKie, S.; Williams, S.; Luckman, S. Central cannabinoid signaling mediating food intake: A pharmacological-challenge magnetic resonance imaging and functional histology study in rat. Neuroscience 2009, 163, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.; Solinas, M.; Bimpisidis, Z.; Goldberg, S.; di Chiara, G. Cannabinoid facilitation of behavioral and biochemical hedonic taste responses. Neuropharmacology 2012, 63, 161–168. [Google Scholar] [CrossRef]
- Sanchis-Segura, C.; Cline, B.; Marsicano, G.; Lutz, B.; Spanagel, R. Reduced sensitivity to reward in CB1 knockout mice. Psychopharmacology 2004, 176, 223–232. [Google Scholar] [CrossRef]
- Mancino, S.; Burokas, A.; Gutiérrez-Cuesta, J.; Gutiérrez-Martos, M.; Martín-García, E.; Pucci, M.; Falconi, A.; D’Addario, C.; Maccarrone, M.; Maldonado, R. Epigenetic and Proteomic Expression Changes Promoted by Eating Addictive-Like Behavior. Neuropsychopharmacology 2015, 40, 2788–2800. [Google Scholar] [CrossRef]
- Soria-Gómez, E.; Bellocchio, L.; Reguero, L.; Lepousez, G.; Martin, C.; Bendahmane, M.; Ruehle, S.; Remmers, F.; Desprez, T.; Matias, I.; et al. The endocannabinoid system controls food intake via olfactory processes. Nat. Neurosci. 2014, 17, 407–415. [Google Scholar] [CrossRef]
- Han, J.E.; Frasnelli, J.; Zeighami, Y.; Larcher, K.; Boyle, J.; McConnell, T.; Malik, S.; Jones-Gotman, M.; Dagher, A. Ghrelin Enhances Food Odor Conditioning in Healthy Humans: An fMRI Study. Cell Rep. 2018, 25, 2643–2652. [Google Scholar] [CrossRef]
- Argueta, D.A.; DiPatrizio, N.V. Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol. Behav. 2017, 171, 32. [Google Scholar] [CrossRef]
- Mansouri, E.; Nobrega, J.; Hill, M.N.; Tyndale, R.; Lee, F.; Hendershot, C.; Best, L.; di Ciano, P.; Balsevich, G.; Sloan, M.; et al. D3 dopamine receptors and a missense mutation of fatty acid amide hydrolase linked in mouse and men: Implication for addiction. Neuropsychopharmacology 2020, 45, 745–752. [Google Scholar] [CrossRef]
- Maccarrone, M.; Gasperi, V.; Catani, M.; Diep, T.; Dainese, E.; Hansen, H.; Avigliano, L. The endocannabinoid system and its relevance for nutrition. Annu. Rev. Nutr. 2010, 30, 423–440. [Google Scholar] [CrossRef]
- Dipatrizio, N.V.; Simansky, K.J. Inhibiting parabrachial fatty acid amide hydrolase activity selectively increases the intake of palatable food via cannabinoid CB1 receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1409–R1414. [Google Scholar] [CrossRef]
- Monteleone, P.; Matias, I.; Martiadis, V.; de Petrocellis, L.; Maj, M.; di Marzo, V. Blood levels of the endocannabinoid anandamide are increased in anorexia nervosa and in binge-eating disorder, but not in bulimia nervosa. Neuropsychopharmacology 2005, 30, 1216–1221. [Google Scholar] [CrossRef]
- Monteleone, P.; Piscitelli, F.; Scognamiglio, P.; Monteleone, A.; Canestrelli, B.; di Marzo, V.; Maj, M. Hedonic eating is associated with increased peripheral levels of ghrelin and the endocannabinoid 2-arachidonoyl-glycerol in healthy humans: A pilot study. J. Clin. Endocrinol. Metab. 2012, 97, E917–E924. [Google Scholar] [CrossRef]
- Monteleone, A.; di Marzo, V.; Monteleone, P.; Grave, R.D.; Aveta, T.; Ghoch, M.; Piscitelli, F.; Volpe, U.; Calugi, S.; Maj, M. Responses of peripheral endocannabinoids and endocannabinoid-related compounds to hedonic eating in obesity. Eur. J. Nutr. 2016, 55, 1799–1805. [Google Scholar] [CrossRef]
- Higuchi, S.; Irie, K.; Yamaguchi, R.; Katsuki, M.; Araki, M.; Ohji, M.; Hayakawa, K.; Mishima, S.; Akitake, Y.; Matsuyama, K.; et al. Hypothalamic 2-arachidonoylglycerol regulates multistage process of high-fat diet preferences. PLoS ONE 2012, 7, e38609. [Google Scholar] [CrossRef]
- Moreira, F.; Crippa, J. The psychiatric side-effects of rimonabant. Rev. Bras. Psiquiatr. 2009, 31, 145–153. [Google Scholar] [CrossRef]
- Navarro, M.; Hernández, E.; Muñoz, R.M.; del Arco, I.; Villanúa, M.A.; Carrera, M.R.; de Fonseca, F.R. Acute administration of the CB1 cannabinoid receptor antagonist SR 141716A induces anxiety-like responses in the rat. Neuroreport 1997, 8, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Blasio, A.; Iemolo, A.; Sabino, V.; Petrosino, S.; Steardo, L.; Rice, K.; Orlando, P.; Iannotti, F.; di Marzo, V.; Zorrilla, E.; et al. Rimonabant precipitates anxiety in rats withdrawn from palatable food: Role of the central amygdala. Neuropsychopharmacology 2013, 38, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Jackson, V.M.; Price, D.A.; Carpino, P.A. Investigational drugs in Phase II clinical trials for the treatment of obesity: Implications for future development of novel therapies. Expert Opin. Investig. Drugs 2014, 23, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Jackson, V.M.; Breen, D.M.; Fortin, J.P.; Liou, A.; Kuzmiski, J.B.; Loomis, A.K.; Rives, M.L.; Shah, B.; Carpino, P.A. Latest approaches for the treatment of obesity. Expert Opin. Drug Discov. 2015, 10, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.; Lang, S.; Truby, H.; Brennan, L.; Gibson, S. Tackling the challenge of treating obesity using design research methods: A scoping review. Obes. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Ho, M.-S.; Huang, W.-T.; Chou, Y.-T.; King, K. Modulation of Glucagon-like Peptide-1 (GLP-1) Potency by Endocannabinoid-like Lipids Represents a Novel Mode of Regulating GLP-1 Receptor Signaling. J. Biol. Chem. 2015, 290, 14302–14313. [Google Scholar] [CrossRef] [PubMed]
- Radziszewska, E.; Bojanowska, E. Effects of glucagon-like peptide-1 receptor stimulation and blockade on food consumption and body weight in rats treated with a cannabinoid CB1 receptor agonist WIN 55,212-2 BCDEF Elżbieta Radziszewska ACDEFG Ewa Bojanowska. Med. Sci. Monit. Basic Res. 2013, 19, 6. [Google Scholar] [CrossRef]
- González-Mariscal, I.; Krzysik-Walker, S.M.; Kim, W.; Rouse, M.; Egan, J.M. Blockade of cannabinoid 1 receptor improves GLP-1R mediated insulin secretion in mice. Mol. Cell. Endocrinol. 2016, 423, 1–10. [Google Scholar] [CrossRef]
- Murphy, T.; Le Foll, B. Targeting the Endocannabinoid CB1 Receptor to Treat Body Weight Disorders: A Preclinical and Clinical Review of the Therapeutic Potential of Past and Present CB1 Drugs. Biomolecules 2020, 10, 855. [Google Scholar] [CrossRef]
- Addy, C.; Wright, H.; van Laere, K.; Gantz, I.; Erondu, N.; Musser, B.J.; Lu, K.; Yuan, J.; Sanabria-Bohórquez, S.M.; Stoch, A.; et al. The acyclic CB1R inverse agonist taranabant mediates weight loss by increasing energy expenditure and decreasing caloric intake. Cell Metab. 2008, 7, 68–78. [Google Scholar] [CrossRef]
- Salamone, J.D.; McLaughlin, P.J.; Sink, K.; Makriyannis, A.; Parker, L.A. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: Effects on food intake, food-reinforced behavior and food aversions. Physiol. Behav. 2007, 91, 383–388. [Google Scholar] [CrossRef]
- Merroun, I.; Sánchez-González, C.; Martínez, R.; López-Chaves, C.; Porres, J.; Aranda, P.; Llopis, J.; Galisteo, M.; Zarzuelo, A.; Errami, M.; et al. Novel effects of the cannabinoid inverse agonist AM 251 on parameters related to metabolic syndrome in obese Zucker rats. Metabolism 2013, 62, 1641–1650. [Google Scholar] [CrossRef]
- Cifuentes, L.; Acosta, A. Homeostatic regulation of food intake. Clin. Res. Hepatol. Gastroenterol. 2021, 101794. [Google Scholar] [CrossRef]
- Suárez, J.; Rivera, P.; Rey, A.A.; Pérez-Martín, M.; Arrabal, S.; de Fonseca, F.R.; de Azua, I.R.; Lutz, B. Adipocyte cannabinoid CB1 receptor deficiency alleviates high fat diet-induced memory deficit, depressive-like behavior, neuroinflammation and impairment in adult neurogenesis. Psychoneuroendocrinology 2019, 110, 104418. [Google Scholar] [CrossRef]
- Pavon, F.; Bilbao, A.; Hernández-Folgado, L.; Cippitelli, A.; Jagerovic, N.; Abellán, G.; Rodríguez-Franco, M.; Serrano, A.; Macias, M.; Gómez, R.; et al. Antiobesity effects of the novel in vivo neutral cannabinoid receptor antagonist 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-3-hexyl-1H-1,2,4-triazole—LH 21. Neuropharmacology 2006, 51, 358–366. [Google Scholar] [CrossRef]
- Alonso, M.; Serrano, A.; Vida, M.; Crespillo, A.; Hernandez-Folgado, L.; Jagerovic, N.; Goya, P.; Reyes-Cabello, C.; Perez-Valero, V.; Decara, J.; et al. Anti-obesity efficacy of LH-21, a cannabinoid CB (1) receptor antagonist with poor brain penetration, in diet-induced obese rats. Br. J. Pharmacol. 2012, 165, 2274–2291. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Gong, H.; Chen, Y.; Wu, H.; Wu, J.; Deng, Y.; Song, X. LH-21, A Peripheral Cannabinoid Receptor 1 Antagonist, Exerts Favorable Metabolic Modulation Including Antihypertensive Effect in KKAy Mice by Regulating Inflammatory Cytokines and Adipokines on Adipose Tissue. Front. Endocrinol. 2018, 9, 167. [Google Scholar] [CrossRef]
- Noerregaard, P.K.; Fridberg, M.; Elling, C.E. TM38837–A novel second generation peripheral selective CB1 receptor antagonist with efficacy and potency in rodent obesity models equal to brain-penetrant CB1 antagonist rimonabant. In Proceedings of the 20th Annual Symposium of the International Cannabinoid Research Society, Lund, Sweden; 2010; p. 39. [Google Scholar]
- Han, J.H.; Shin, H.; Park, J.-Y.; Rho, J.G.; Son, D.H.; Kim, K.W.; Seong, J.K.; Yoon, S.-H.; Kim, W. A novel peripheral cannabinoid 1 receptor antagonist, AJ5012, improves metabolic outcomes and suppresses adipose tissue inflammation in obese mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 4314–4326. [Google Scholar] [CrossRef]
- Han, J.H.; Shin, H.; Rho, J.G.; Kim, J.-E.; Son, D.H.; Yoon, J.; Lee, Y.J.; Park, J.-H.; Song, B.J.; Choi, C.-S.; et al. Peripheral cannabinoid 1 receptor blockade mitigates adipose tissue inflammation via NLRP3 inflammasome in mouse models of obesity. Diabetes Obes. Metab. 2018, 20, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.-S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.-C.; Shia, K.-S.; Wang, Y.-T.; Yeh, Y.-N.; Chang, C.-P.; Lin, Y.; Chen, P.-H.; Wu, C.-H.; Chao, Y.-S.; Hung, M.-S. A novel peripheral cannabinoid receptor 1 antagonist, BPR0912, reduces weight independently of food intake and modulates thermogenesis. Diabetes Obes. Metab. 2015, 17, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, R.S.; Lin, Y.; Shia, K.-S.; Yeh, Y.-N.; Hsieh, W.-P.; Hsiao, W.-C.; Chang, C.-P.; Chao, Y.-S.; Hung, M.-S. Induction of fatty acid oxidation resists weight gain, ameliorates hepatic steatosis and reduces cardiometabolic risk factors. Int. J. Obes. 2012, 36, 999–1006. [Google Scholar] [CrossRef]
- Chen, W.; Shui, F.; Liu, C.; Zhou, X.; Li, W.; Zheng, Z.; Fu, W.; Wang, L. Novel Peripherally Restricted Cannabinoid 1 Receptor Selective Antagonist TXX-522 with Prominent Weight-Loss Efficacy in Diet Induced Obese Mice. Front. Pharmacol. 2017, 8, 707. [Google Scholar] [CrossRef]
- Méndez-Díaz, M.; Amancio-Belmont, O.; Hernández-Vázquez, E.; Ruiz-Contreras, A.E.; Hernández-Luis, F.; Prospéro-García, O. ENP11, a potential CB1R antagonist, induces anorexia in rats. Pharmacol. Biochem. Behav. 2015, 135, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Cinar, R.; Iyer, M.R.; Kunos, G. The therapeutic potential of second and third generation CB 1 R antagonists. Pharmacol. Ther. 2020, 208, 107477. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.B.; Maxwell, B.D.; Burrell, R.; Bonacorsi, S.J.J. The syntheses of isotopically labelled CB-1 antagonists for the treatment of obesity. J. Label. Comp. Radiopharm. 2016, 59, 665–672. [Google Scholar] [CrossRef]
- Klumpers, L.; Fridberg, M.; de Kam, M.; Little, P.; Jensen, N.; Kleinloog, H.; Elling, C.; van Gerven, J. Peripheral selectivity of the novel cannabinoid receptor antagonist TM38837 in healthy subjects. Br. J. Clin. Pharmacol. 2013, 76, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Knani, I.; Earley, B.J.; Udi, S.; Nemirovski, A.; Hadar, R.; Gammal, A.; Cinar, R.; Hirsch, H.J.; Pollak, Y.; Gross, I.; et al. Targeting the endocannabinoid/CB1 receptor system for treating obesity in Prader-Willi syndrome. Mol. Metab. 2016, 5, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Kale, V.; Gibbs, S.; Taylor, J.; Novak, A.; Patton, K.; Sparrow, B.; Gorospe, J.; Anand, S.; Cinar, R.; Kunos, G.; et al. Preclinical toxicity evaluation of JD5037, a peripherally restricted CB 1 receptor inverse agonist, in rats and dogs for treatment of nonalcoholic steatohepatitis. Regul. Toxicol. Pharmacol. 2019, 109, 104483. [Google Scholar] [CrossRef]
- Matthews, J.; McNally, J.; Connolly, P.; Xia, M.; Zhu, B.; Black, S.; Chen, C.; Hou, C.; Liang, Y.; Tang, Y.; et al. Tetrahydroindazole derivatives as potent and peripherally selective cannabinoid-1 (CB1) receptor inverse agonists. Bioorg. Med. Chem. Lett. 2016, 26, 5346–5349. [Google Scholar] [CrossRef]
- Le Foll, B.; Trigo, J.; Sharkey, K.; Le Strat, Y. Cannabis and Δ9-tetrahydrocannabinol (THC) for weight loss? Med. Hypotheses 2013, 80, 564–567. [Google Scholar] [CrossRef]
- Plescia, F.; Plescia, F.; Raffa, D.; Cavallaro, A.; Lavanco, G.; Maggio, B.; Raimondi, M.; Daidone, G.; Brancato, A.; Cannizzaro, C. The role of (E)-6-chloro-3-(3-methyl-1-phenyl-1H-pyrazol-5-yl)-2-styrylquinazolin-4(3H)-one in the modulation of cannabinoidergic system. A pilot study. Pharmacol. Rep. 2018, 70, 1124–1132. [Google Scholar] [CrossRef]
- Takeda, S.; Usami, N.; Yamamoto, I.; Watanabe, K. Cannabidiol-2, 6-dimethyl ether, a cannabidiol derivative, is a highly potent and selective 15-lipoxygenase inhibitor. Drug Metab. Dispos. 2009, 37, 1733–1737. [Google Scholar] [CrossRef]
- Behl, T.; Chadha, S.; Sachdeva, M.; Sehgal, A.; Kumar, A.; Venkatachalam, T.; Hafeez, A.; Aleya, L.; Arora, S.; Batiha, G.; et al. Understanding the possible role of endocannabinoid system in obesity. Prostaglandins Other Lipid Mediat. 2021, 152. [Google Scholar] [CrossRef]
- Cluny, N.; Keenan, C.; Reimer, R.; Le Foll, B.; Sharkey, K. Prevention of Diet-Induced Obesity Effects on Body Weight and Gut Microbiota in Mice Treated Chronically with Δ9-Tetrahydrocannabinol. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Cluny, N.; Vemuri, V.K.; Chambers, A.; Limebeer, C.; Bedard, H.; Wood, J.; Lutz, B.; Zimmer, A.; Parker, L.A.; Makriyannis, A.; et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br. J. Pharmacol. 2010, 161, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Vemuri, V.; Liu, J.; Bátkai, S.; Mukhopadhyay, B.; Godlewski, G.; Osei-Hyiaman, D.; Ohnuma, S.; Ambudkar, S.; Pickel, J.; et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Investig. 2010, 120, 2953–2966. [Google Scholar] [CrossRef]
- Wargent, E.; Zaibi, M.; Silvestri, C.; Hislop, D.; Stocker, C.; Stott, C.; Guy, G.; Duncan, M.; di Marzo, V.; Ma, C. The cannabinoid Δ(9)-tetrahydrocannabivarin (THCV) ameliorates insulin sensitivity in two mouse models of obesity. Nutr. Diabetes 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Bielawiec, P.; Harasim-Symbor, E.; Chabowski, A. Phytocannabinoids: Useful Drugs for the Treatment of Obesity? Special Focus on Cannabidiol. Front. Endocrinol. 2020, 11, 114. [Google Scholar] [CrossRef]
- Rzepa, E.; Tudge, L.; McCabe, C. The CB1 Neutral Antagonist Tetrahydrocannabivarin Reduces Default Mode Network and Increases Executive Control Network Resting State Functional Connectivity in Healthy Volunteers. Int. J. Neuropsychopharmacol. 2015, 19, 1–7. [Google Scholar] [CrossRef]
- Mastinu, A.; Pira, M.; Pinna, G.A.; Pisu, C.; Casu, M.A.; Reali, R.; Marcello, S.; Murineddu, G.; Lazzari, P. NESS06SM reduces body weight with an improved profile relative to SR141716A. Pharmacol. Res. 2013, 74, 94–108. [Google Scholar] [CrossRef]
- Fois, G.R.; Fattore, L.; Murineddu, G.; Salis, A.; Pintore, G.; Asproni, B.; Pinna, G.A.; Diana, M. The novel cannabinoid antagonist SM-11 reduces hedonic aspect of food intake through a dopamine-dependent mechanism. Pharmacol. Res. 2016, 113, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Seltzman, H.H.; Maitra, R.; Bortoff, K.; Henson, J.; Reggio, P.H.; Wesley, D.; Tam, J. Metabolic Profiling of CB1 Neutral Antagonists. Methods Enzymol. 2017, 593, 199–215. [Google Scholar] [CrossRef]
- Nava-Molina, L.; Uchida-Fuentes, T.; Ramos-Tovar, H.; Fregoso-Padilla, M.; Rodríguez-Monroy, M.; Vega, A.; Navarrete-Vázquez, G.; Andrade-Jorge, E.; Villalobos-Molina, R.; Ortiz-Ortega, R.; et al. Novel CB1 receptor antagonist BAR-1 modifies pancreatic islet function and clinical parameters in prediabetic and diabetic mice. Nutr. Diabetes 2020, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, P.; Serra, V.; Marcello, S.; Pira, M.; Mastinu, A. Metabolic side effects induced by olanzapine treatment are neutralized by CB1 receptor antagonist compounds co-administration in female rats. Eur. Neuropsychopharmacol. 2017, 27, 667–678. [Google Scholar] [CrossRef]
- Boström, J.; Olsson, R.; Tholander, J.; Greasley, P.; Ryberg, E.; Nordberg, H.; Hjorth, S.; Cheng, L. Novel thioamide derivatives as neutral CB1 receptor antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 479–482. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Greco, M.N.; Macielag, M.J.; Teleha, C.A.; DesJarlais, R.L.; Tang, Y.; Ho, G.; Hou, C.; Chen, C.; Zhao, S.; et al. 6-Benzhydryl-4-amino-quinolin-2-ones as Potent Cannabinoid Type 1 (CB1) Receptor Inverse Agonists and Chemical Modifications for Peripheral Selectivity. J. Med. Chem. 2018, 61, 10276–10298. [Google Scholar] [CrossRef]
- Hirsch, S.; Tam, J. Cannabis: From a Plant That Modulates Feeding Behaviors toward Developing Selective Inhibitors of the Peripheral Endocannabinoid System for the Treatment of Obesity and Metabolic Syndrome. Toxins 2019, 11, 275. [Google Scholar] [CrossRef] [PubMed]
- Black, M.D.; Stevens, R.J.; Rogacki, N.; Featherstone, R.E.; Senyah, Y.; Giardino, O.; Borowsky, B.; Stemmelin, J.; Cohen, C.; Pichat, P.; et al. AVE1625, a cannabinoid CB1 receptor antagonist, as a co-treatment with antipsychotics for schizophrenia: Improvement in cognitive function and reduction of antipsychotic-side effects in rodents. Psychopharmacol. 2010, 215, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Amato, G.; Khan, N.; Maitra, R. A patent update on cannabinoid receptor 1 antagonists (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 261–269. [Google Scholar] [CrossRef]
- Udi, S.; Hinden, L.; Ahmad, M.; Drori, A.; Iyer, M.; Cinar, R.; Herman-Edelstein, M.; Tam, J. Dual inhibition of cannabinoid CB 1 receptor and inducible NOS attenuates obesity-induced chronic kidney disease. Br. J. Pharmacol. 2020, 177, 110–127. [Google Scholar] [CrossRef]
- Santos-Molina, L.; Herrerias, A.; Zawatsky, C.N.; Gunduz-Cinar, O.; Cinar, R.; Iyer, M.R.; Wood, C.M.; Lin, Y.; Gao, B.; Kunos, G.; et al. Effects of a Peripherally Restricted Hybrid Inhibitor of CB1 Receptors and iNOS on Alcohol Drinking Behavior and Alcohol-Induced Endotoxemia. Molecules 2021, 26, 5089. [Google Scholar] [CrossRef]
- Cotrim, B.A.; Joglar, J.; Rojas, M.J.L.; Del Olmo, J.M.D.; Macias-González, M.; Cuevas, M.R.; Fitó, M.; Muñoz-Aguayo, D.; Covas Planells, M.I.; Farré, M.; et al. Unsaturated Fatty Alcohol Derivatives of Olive Oil Phenolic Compounds with Potential Low-Density Lipoprotein (LDL) Antioxidant and Antiobesity Properties. J. Agric. Food Chem. 2012, 60, 1067–1074. [Google Scholar] [CrossRef]
- Decara, J.M.; Javier, F.; Pavón, P.; Suárezsuárez, J.; Romero-Cuevas, M.; Baixeras, E.; Vázquezvázquez, M.; Rivera, P.; Gavito, A.L.; Almeida, B.; et al. Treatment with a novel oleic-acid-dihydroxyamphetamine conjugation ameliorates non-alcoholic fatty liver disease in obese Zucker rats. Dis. Models Mech. 2015, 8, 1213–1225. [Google Scholar] [CrossRef]
- Loverme, J.; Duranti, A.; Tontini, A.; Spadoni, G.; Mor, M.; Rivara, S.; Stella, N.; Xu, C.; Tarzia, G.; Piomelli, D. Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg. Med. Chem. Lett. 2009, 19, 639–643. [Google Scholar] [CrossRef]
- Silvestri, C.; Paris, I.; Martella, A.; Melck, D.; Guadagnino, I.; Cawthorne, M.; Motta, A.; di Marzo, V. Two non-psychoactive cannabinoids reduce intracellular lipid levels and inhibit hepatosteatosis. J. Hepatol. 2015, 62, 1382–1390. [Google Scholar] [CrossRef]
- Leone, S.; Ferrante, C.; Recinella, L.; Chiavaroli, A.; Mollica, A.; Tömböly, C.; Stefanucci, A.; Dimmito, M.; Dvorácskó, S.; Verratti, V.; et al. Effects of RVD-hemopressin (α) on feeding and body weight after standard or cafeteria diet in rats. Neuropeptides 2018, 72, 38–46. [Google Scholar] [CrossRef]
- González-Mariscal, I.; Carmona-Hidalgo, B.; Winkler, M.; Unciti-Broceta, J.D.; Escamilla, A.; Gómez-Cañas, M.; Fernández-Ruiz, J.; Fiebich, B.L.; Romero-Zerbo, S.-Y.; Bermúdez-Silva, F.J.; et al. (+)-trans-Cannabidiol-2-hydroxy pentyl is a dual CB 1 R antagonist/CB 2 R agonist that prevents diabetic nephropathy in mice. Pharmacol. Res. 2021, 169, 1043–6618. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Bellini, G.; Luongo, L.; Manzo, I.; Tolone, S.; Tortora, C.; Bernardo, M.E.; Grandone, A.; Conforti, A.; Docimo, L.; et al. Cannabinoid Receptor 2 as Antiobesity Target: Inflammation, Fat Storage, and Browning Modulation. J. Clin. Endocrinol. Metab. 2016, 101, 3469. [Google Scholar] [CrossRef] [PubMed]
- Palomares, B.; Ruiz-Pino, F.; Navarrete, C.; Velasco, I.; Sánchez-Garrido, M.; Jimenez-Jimenez, C.; Pavicic, C.; Vazquez, M.; Appendino, G.; Bellido, M.; et al. VCE-004.8, A Multitarget Cannabinoquinone, Attenuates Adipogenesis and Prevents Diet-Induced Obesity. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Mahadevan, A.; Coccurello, R.; Chang, J.W.; Allarà, M.; Chen, Y.; Giacovazzo, G.; Lichtman, A.; Cravatt, B.; Moles, A.; et al. A novel fluorophosphonate inhibitor of the biosynthesis pf the endocannabinoid 2-arachidonoylglycerol with potential anti-obesity effects. J. Pharmacol. 2013, 169, 784–793. [Google Scholar] [CrossRef]
- Tovar, R.; Gavito, A.L.; Vargas, A.; Soverchia, L.; Hernandez-Folgado, L.; Jagerovic, N.; Baixeras, E.; Ciccocioppo, R.; de Fonseca, F.; Decara, J. Palmitoleoylethanolamide Is an Efficient Anti-Obesity Endogenous Compound: Comparison with Oleylethanolamide in Diet-Induced Obesity. Nutrients 2021, 13, 2589. [Google Scholar] [CrossRef]
- Matias, I.; Gonthier, M.-P.; Petrosino, S.; Docimo, L.; Capasso, R.; Hoareau, L.; Monteleone, P.; Roche, R.; Izzo, A.A.; di Marzo, V. Role and regulation of acylethanolamides in energy balance: Focus on adipocytes and β-cells. Br. J. Pharmacol. 2009, 152, 676–690. [Google Scholar] [CrossRef]
- Grevengoed, T.J.; Trammell, S.A.J.; McKinney, M.K.; Petersen, N.; Cardone, R.L.; Svenningsen, J.S.; Ogasawara, D.; Nexøe-Larsen, C.C.; Knop, F.K.; Schwartz, T.W.; et al. N-acyl taurines are endogenous lipid messengers that improve glucose homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 24770–24778. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Héaulme, M.; Alonso, R.; Shire, D.; Congy, C.; Soubrié, P.; Brelière, J.C.; le Fur, G. Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci. 1995, 56, 1941–1947. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacodynamics | Drug | Effects | References |
|---|---|---|---|
| Central CB1 inverse antagonists | Rimonabant, AM251 | Reduced food intake, body weight gain, metabolic effects | [151,205] |
| Taranabant | Increased energy expenditure, decreased caloric intake | [149] | |
| Peripheral CB1 inverse antagonists | LH-21, TM38837, Compound 6a, BMS-811064, BMS-812204 | Reduced food intake, body weight gain | [155,157,165,166] |
| AJ5012, AJ5018 | Reduced body weight gain, adipose tissue inflammation | [158,159] | |
| JD5037 | Reduced food intake, body weight gain, metabolic effects | [160] | |
| BPR0912 | Reduced body weight gain, thermogenesis modulation | [161] | |
| BPR697, TXX-522 | Reduced body weight gain, metabolic effects | [162,163] | |
| ENP11 | Reduced food intake, thermogenesis | [164] | |
| Compound 2p | Reduction in plasma glucose | [170] | |
| CB1 agonists | WIN 55212, CP-55940 | Reduced body weight gain | [122,172] |
| CBDD | Metabolic effects | [173] | |
| THC | Reduce food intake, body weight gain, fat mass | [175] | |
| CB1 neutral antagonists | AM6545 | Metabolic effects | [176] |
| AM4113, THCV | Reduced food intake, body weight gain | [150,178] | |
| NESS06SM | Reduced body weight gain, fat mass | [181] | |
| SM-11 | Reduced food intake, self-administration of palatable food | [182] | |
| PIMSR | Reduced hepatic steatosis | [183] | |
| BAR-1 | Reduced body weight gain, metabolic effects | [184] | |
| Compound10q, Compound2c, Compound5, Compound13, Compound6a, CompoundD4 | Reduced food intake | [165,172,185,186,187,188] | |
| AVE-1625, SLV-319 | Reduced body weight gain | [174,189] | |
| CB1 antibodies | / | / | [190] |
| CB1 inverse agonist/iNOS inhibitor | MRI-1569 | Reduced food intake, body weight gain, hepatic steatosis, metabolic effects | [191] |
| MRI-1867 | Attenuates obesity-induced chronic kidney disease | [191] | |
| CB1/PPAR-α antagonists | Compound7 | Metabolic effects | [193] |
| OLHHA | Reduced food intake, metabolic effect, ameliorate non-alcoholic fatty liver disease | [194] | |
| CB1 antagonist/CB2 agonist | URB-447, CBD | Reduced body weight gain | [195,196] |
| RVD-hemopressin(α) | Reduced food intake, metabolic effects | [197] | |
| CB2 agonists | JWH-105 | Reduced body weight gain | [174] |
| JWH-133 | Reduced adipose tissue inflammation | [199] | |
| PPAR-γ/ CB2 agonist | VCE 004.8 | Reduced body weight gain, fat mass, metabolic effects | [200] |
| DAGL-α inhibitor | O-7640 | Reduced food intake, body weight gain | [201] |
| Endocannabinoid-like molecules acting on other receptors | PEA, C18:1 NAT | Reduced food intake | [203,204] |
| OEA | Reduced food intake, body weight gain, metabolic effects | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Ceglia, M.; Decara, J.; Gaetani, S.; Rodríguez de Fonseca, F. Obesity as a Condition Determined by Food Addiction: Should Brain Endocannabinoid System Alterations Be the Cause and Its Modulation the Solution? Pharmaceuticals 2021, 14, 1002. https://doi.org/10.3390/ph14101002
de Ceglia M, Decara J, Gaetani S, Rodríguez de Fonseca F. Obesity as a Condition Determined by Food Addiction: Should Brain Endocannabinoid System Alterations Be the Cause and Its Modulation the Solution? Pharmaceuticals. 2021; 14(10):1002. https://doi.org/10.3390/ph14101002
Chicago/Turabian Stylede Ceglia, Marialuisa, Juan Decara, Silvana Gaetani, and Fernando Rodríguez de Fonseca. 2021. "Obesity as a Condition Determined by Food Addiction: Should Brain Endocannabinoid System Alterations Be the Cause and Its Modulation the Solution?" Pharmaceuticals 14, no. 10: 1002. https://doi.org/10.3390/ph14101002
APA Stylede Ceglia, M., Decara, J., Gaetani, S., & Rodríguez de Fonseca, F. (2021). Obesity as a Condition Determined by Food Addiction: Should Brain Endocannabinoid System Alterations Be the Cause and Its Modulation the Solution? Pharmaceuticals, 14(10), 1002. https://doi.org/10.3390/ph14101002