Expanding the Structural Diversity of DNA Methyltransferase Inhibitors

 ,
,  , ,
, ,  and
and 
Abstract
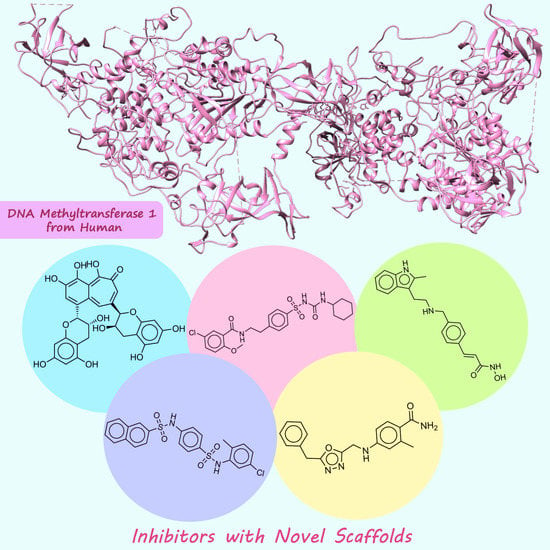
1. Introduction
2. Results
2.1. Biochemical DNMT Assays
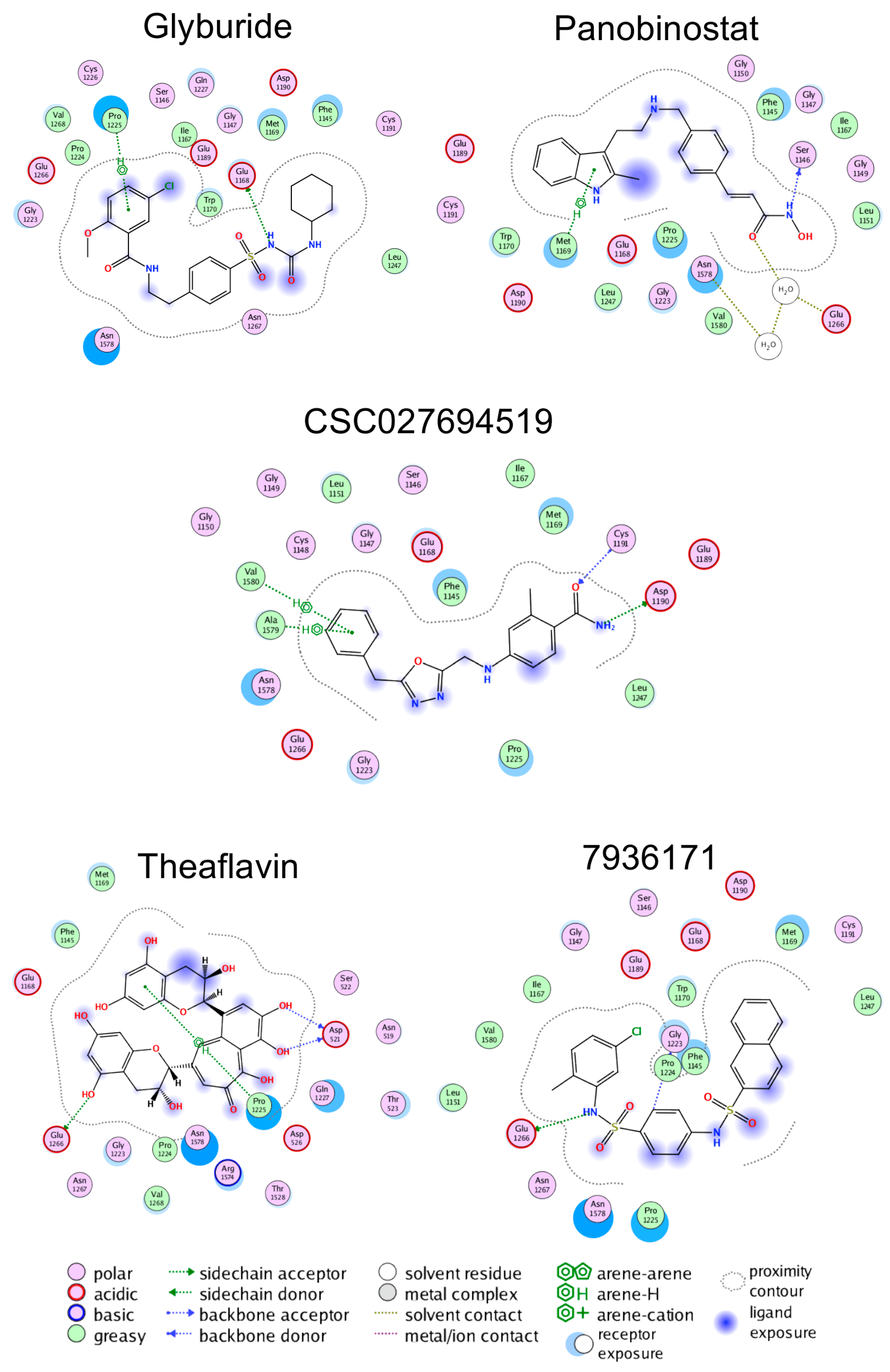
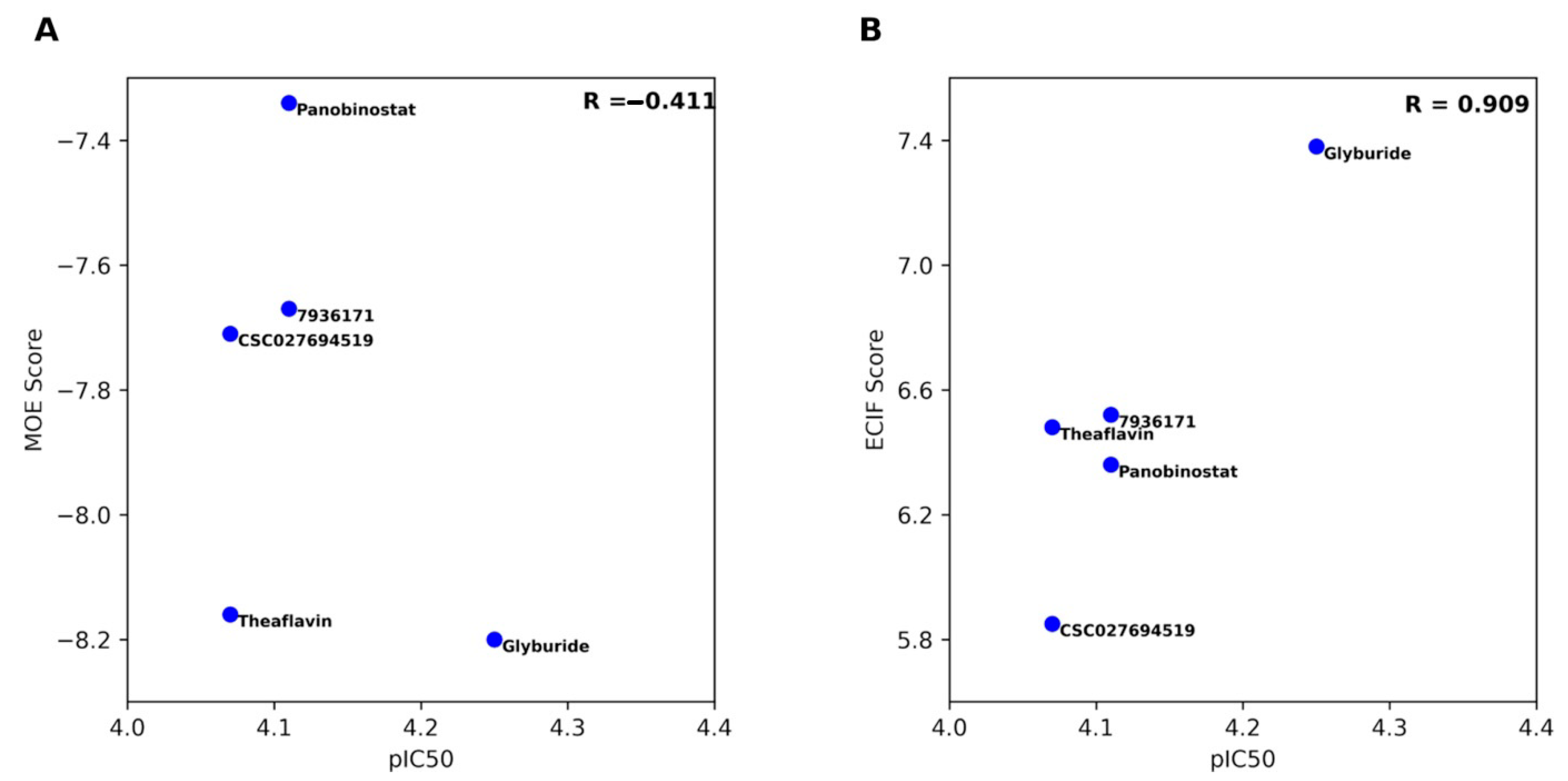
2.2. Molecular Docking and Re-Scoring
2.3. Molecular Dynamics and Adaptive Sampling
3. Discussion
3.1. Biochemical DNMT Assays
3.2. Computational Studies
4. Materials and Methods
4.1. Compounds
4.2. Biochemical DNMT Assays
4.3. Molecular Docking and Re-Scoring
4.4. Probing the Putative Binding Mode of Glyburide
Adaptive Sampling and End-Point Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waddington, C.H. The epigenotype, endeavor, 1942, vol. 1 (pg. 18-20) reprinted in. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Greally, J.M. A user’s guide to the ambiguous word ‘epigenetics’. Nat. Rev. Mol. Cell Biol. 2018, 19, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef]
- Sessions, Z.; Sánchez-Cruz, N.; Prieto-Martínez, F.D.; Alves, V.M.; Santos, H.P., Jr.; Muratov, E.; Tropsha, A.; Medina-Franco, J.L. Recent progress on cheminformatics approaches to epigenetic drug discovery. Drug Discov. Today 2020, 25, 2268–2276. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2017, 19, 81. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA methyltransferases in cancer: Biology, paradox, aberrations, and targeted therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef]
- Estey, E.H. Epigenetics in clinical practice: The examples of azacitidine and decitabine in myelodysplasia and acute myeloid leukemia. Leukemia 2013, 27, 1803–1812. [Google Scholar] [CrossRef]
- Yu, J.; Xie, T.; Wang, Z.; Wang, X.; Zeng, S.; Kang, Y.; Hou, T. DNA methyltransferases: Emerging targets for the discovery of inhibitors as potent anticancer drugs. Drug Discov. Today 2019, 24, 2323–2331. [Google Scholar] [CrossRef]
- Arguelles, A.O.; Meruvu, S.; Bowman, J.D.; Choudhury, M. Are epigenetic drugs for diabetes and obesity at our door step? Drug Discovery Today 2016, 21, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Teitell, M.; Richardson, B. Dna methylation in the immune system. Clin. Immunol. 2003, 109, 2–5. [Google Scholar] [CrossRef]
- Weng, Y.-L.; An, R.; Shin, J.; Song, H.; Ming, G.-L. DNA modifications and neurological disorders. Neurotherapeutics 2013, 10, 556–567. [Google Scholar] [CrossRef] [PubMed]
- de Lera, A.R.; Ganesan, A. Two-hit wonders: The expanding universe of multitargeting epigenetic agents. Curr. Opin. Chem. Biol. 2020, 57, 135–154. [Google Scholar] [CrossRef]
- Rabal, O.; San José-Enériz, E.; Agirre, X.; Sánchez-Arias, J.A.; Vilas-Zornoza, A.; Ugarte, A.; de Miguel, I.; Miranda, E.; Garate, L.; Fraga, M.; et al. Discovery of reversible DNA methyltransferase and lysine methyltransferase g9a inhibitors with antitumoral in vivo efficacy. J. Med. Chem. 2018, 61, 6518–6545. [Google Scholar] [CrossRef]
- López-López, E.; Rabal, O.; Oyarzabal, J.; Medina-Franco, J.L. Towards the understanding of the activity of g9a inhibitors: An activity landscape and molecular modeling approach. J. Comp. Aided Mol. Des. 2020, 34, 659–669. [Google Scholar] [CrossRef]
- Yuan, Z.; Chen, S.; Gao, C.; Dai, Q.; Zhang, C.; Sun, Q.; Lin, J.-S.; Guo, C.; Chen, Y.; Jiang, Y. Development of a versatile DNMT and HDAC inhibitor C02S modulating multiple cancer hallmarks for breast cancer therapy. Bioorg. Chem. 2019, 87, 200–208. [Google Scholar] [CrossRef]
- Arce, C.; Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Candelaria, M.; Dueñnas-Gonzalez, A. Hydralazine target: From blood vessels to the epigenome. J. Transl. Med. 2006, 4, 10. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.; Chie, E.; DaYoung, P.; Kim, I.; Kim, I. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat. Oncol. 2012, 7, 39. [Google Scholar] [CrossRef]
- Schcolnik-Cabrera, A.; Domínguez-Gómez, G.; Dueñas-González, A. Comparison of DNA demethylating and histone deacetylase inhibitors hydralazine-valproate versus vorinostat-decitabine incutaneous t-cell lymphoma in HUT78 cells. Am. J. Blood Res. 2018, 8, 5–16. [Google Scholar]
- Singh, N.; Dueñas-González, A.; Lyko, F.; Medina-Franco, J.L. Molecular modeling and dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem 2009, 4, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Kuck, D.; Singh, N.; Lyko, F.; Medina-Franco, J.L. Novel and selective DNA methyltransferase inhibitors: Docking-based virtual screening and experimental evaluation. Bioorg. Med. Chem. 2010, 18, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Kuck, D.; Caulfield, T.; Lyko, F.; Medina-Franco, J.L. Nanaomycin a selectively inhibits DNMT3b and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010, 9, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Medina-Franco, J.L. Trimethylaurintricarboxylic acid inhibits human DNA methyltransferase 1: Insights from enzymatic and molecular modeling studies. J. Mol. Model. 2012, 18, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Kim, J.H.; Robertson, K.D.; Medina-Franco, J.L. Molecular modeling of inhibitors of human DNA methyltransferase with a crystal structure: Discovery of a novel DNMT1 inhibitor. Adv. Protein Chem. Struct. Biol. 2012, 87, 219–247. [Google Scholar] [PubMed]
- Méndez-Lucio, O.; Tran, J.; Medina-Franco, J.L.; Meurice, N.; Muller, M. Towards drug repurposing in epigenetics: Olsalazine as a novel hypomethylating compound active in a cellular context. ChemMedChem 2014, 9, 560–565. [Google Scholar] [CrossRef]
- Aldawsari, F.S.; Aguayo-Ortiz, R.; Kapilashrami, K.; Yoo, J.; Luo, M.; Medina-Franco, J.L.; Velázquez-Martínez, C.A. Resveratrol-salicylate derivatives as selective DNMT3 inhibitors and anticancer agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 695–703. [Google Scholar] [CrossRef]
- Davide, G.; Sandra, A.; Emily, B.; Mattia, C.; Marta, G.; Annalisa, C.; Livio, S.; Gianluca, M.; Chiara, C.; Eli, F.-d.G.; et al. Design and synthesis of N-benzoyl amino acid derivatives as DNA methylation inhibitors. Chem. Biol. Drug Des. 2016, 88, 664–676. [Google Scholar]
- Palomino-Hernandez, O.; Jardinez-Vera, A.; Medina-Franco, J. Progress on the computational development of epigenetic modulators of dna methyltransferases 3A and 3B. J. Mex. Chem. Soc. 2017, 61, 266–272. [Google Scholar] [CrossRef]
- Pechalrieu, D.; Dauzonne, D.; Arimondo, P.B.; Lopez, M. Synthesis of novel 3-halo-3-nitroflavanones and their activities as DNA methyltransferase inhibitors in cancer cells. Eur. J. Med. Chem. 2020, 186, 111829. [Google Scholar] [CrossRef]
- Shao, Z.; Xu, P.; Xu, W.; Li, L.; Liu, S.; Zhang, R.; Liu, Y.-C.; Zhang, C.; Chen, S.; Luo, C. Discovery of novel DNA methyltransferase 3a inhibitors via structure-based virtual screening and biological assays. Bioorg. Med. Chem. Lett. 2017, 27, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Shukla, S.; Lakra, A.D.; Meeran, S.M.; Siddiqi, M.I. Identification of potent inhibitors of DNA methyltransferase 1 (DNMT1) through a pharmacophore-based virtual screening approach. J. Mol. Graph. Model. 2017, 75, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, A.; Arimondo, P.B.; Guianvarc’h, D. Structure-guided optimization of DNA methyltransferase inhibitors. In Epi-Informatics; Medina-Franco, J.L., Ed.; Academic Press: London, UK, 2016; pp. 53–74. [Google Scholar]
- Joshi, M.; Rajpathak, S.N.; Narwade, S.C.; Deobagkar, D. Ensemble-based virtual screening and experimental validation of inhibitors targeting a novel site of human DNMT1. Chem. Biol. Drug Des. 2016, 88, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Kabro, A.; Lachance, H.; Marcoux-Archambault, I.; Perrier, V.; Dore, V.; Gros, C.; Masson, V.; Gregoire, J.M.; Ausseil, F.; Cheishvili, D.; et al. Preparation of phenylethylbenzamide derivatives as modulators of dnmt3 activity. MedChemComm 2013, 4, 1562–1570. [Google Scholar] [CrossRef][Green Version]
- Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; López-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J.L.; Sbardella, G. Synthesis and biochemical evaluation of δ2-isoxazoline derivatives as DNA methyltransferase 1 inhibitors. J. Med. Chem. 2011, 54, 7663–7677. [Google Scholar] [CrossRef]
- Newton, A.S.; Faver, J.C.; Micevic, G.; Muthusamy, V.; Kudalkar, S.N.; Bertoletti, N.; Anderson, K.S.; Bosenberg, M.W.; Jorgensen, W.L. Structure-guided identification of DNMT3b inhibitors. ACS Med. Chem. Lett. 2020, 11, 971–976. [Google Scholar] [CrossRef]
- Naveja, J.J.; Medina-Franco, J.L. Insights from pharmacological similarity of epigenetic targets in epipolypharmacology. Drug Discov. Today 2018, 23, 141–150. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; López-Vallejo, F.; Kuck, D.; Lyko, F. Natural products as DNA methyltransferase inhibitors: A computer-aided discovery approach. Mol. Divers. 2011, 15, 293–304. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Gómez-García, A.; Chávez-Ponce de León, D.E.; Sánchez-Cruz, N.; Ruiz-Rios, J.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Inhibitors of DNA methyltransferases from natural sources: A computational perspective. Front. Pharmacol. 2018, 9, 1144. [Google Scholar] [CrossRef]
- Akone, S.H.; Ntie-Kang, F.; Stuhldreier, F.; Ewonkem, M.B.; Noah, A.M.; Mouelle, S.E.M.; Müller, R. Natural products impacting DNA methyltransferases and histone deacetylases. Front. Pharmacol. 2020, 11, 992. [Google Scholar] [CrossRef]
- Lee, W.J.; Shim, J.Y.; Zhu, B.T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Zhu, B.T. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis 2006, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Mayorga, K.; Montes, C.P. The role of nutrition in epigenetics and recent advances of in silico studies. In Epi-Informatics; Medina-Franco, J.L., Ed.; Academic Press: London, UK, 2016; pp. 385–398. [Google Scholar]
- Prieto-Martínez, F.; Peña-Castillo, A.; Méndez-Lucio, O.; Fernández-de Gortari, E.; Medina-Franco, J.L. Molecular modeling and chemoinformatics to advance the development of modulators of epigenetic targets: A focus on DNA methyltransferases. Adv. Protein Chem. Struct. Biol. 2016, 105, 1–26. [Google Scholar] [PubMed]
- Medina-Franco, J.L.; Méndez-Lucio, O.; Yoo, J.; Dueñas, A. Discovery and development of DNA methyltransferase inhibitors using in silico approaches. Drug Discov. Today 2015, 20, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Sun, Q.; Li, D.; Miao, S.; Chen, S.; Song, L.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Design, synthesis and anticancer potential of NSC-319745 hydroxamic acid derivatives as DNMT and HDAC inhibitors. Eur. J. Med. Chem. 2017, 134, 281–292. [Google Scholar] [CrossRef]
- Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P.B. Targeting DNA methylation with small molecules: What’s next? J. Med. Chem. 2014, 58, 2569–2583. [Google Scholar] [CrossRef]
- Gros, C.; Fleury, L.; Nahoum, V.; Faux, C.; Valente, S.; Labella, D.; Cantagrel, F.; Rilova, E.; Bouhlel, M.A.; David-Cordonnier, M.-H.; et al. New insights on the mechanism of quinoline-based DNA methyltransferase inhibitors. J. Biol. Chem. 2015, 290, 6293–6302. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Drozdetski, A.; Walker, R.C.; Onufriev, A.V. Speed of conformational change: Comparing explicit and implicit solvent molecular dynamics simulations. Biophys. J. 2015, 108, 1153–1164. [Google Scholar] [CrossRef]
- Röblitz, S.; Weber, M. Fuzzy spectral clustering by PCCA+: Application to Markov state models and data classification. Adv. Data Anal. Classif. 2013, 7, 147–179. [Google Scholar] [CrossRef]
- Podvinec, M.; Lim, S.P.; Schmidt, T.; Scarsi, M.; Wen, D.; Sonntag, L.S.; Sanschagrin, P.; Shenkin, P.S.; Schwede, T. Novel inhibitors of dengue virus methyltransferase: Discovery by in vitro-driven virtual screening on a desktop computer grid. J. Med. Chem. 2010, 53, 1483–1495. [Google Scholar] [CrossRef]
- Guianvarc’h, D.; Arimondo, P.B. Challenges in developing novel DNA methyltransferases inhibitors for cancer therapy. Fut. Med. Chem. 2014, 6, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hu, G.; Luo, C.; Liang, Z. DNA methyltransferase inhibitors: An updated patent review (2012–2015). Expert Opin. Ther. Pat. 2016, 26, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Seefeldt, T.; Young, A.; Zhang, X.; Guan, X. Design, synthesis, and biological evaluation of n-acetyl-s-(p-chlorophenylcarbamoyl)cysteine and its analogs as a novel class of anticancer agents. Bioorg. Med. Chem. 2011, 19, 287–294. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, H.-B.; Shi, X.-F.; Song, Y. Conventional hypoglycaemic agents and the risk of lung cancer in patients with diabetes: A meta-analysis. PLoS ONE 2014, 9, e99577. [Google Scholar] [CrossRef]
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (lbh589): A potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.A.; Lee, C.; Kwak, P.A.; Park, C.-K.; Wang, K.-C.; Phi, J.H.; Lee, J.Y.; Chong, S.; Kim, S.-K. Histone deacetylase inhibitor panobinostat potentiates the anti-cancer effects of mesenchymal stem cell-based strail gene therapy against malignant glioma. Cancer Lett. 2019, 442, 161–169. [Google Scholar] [CrossRef]
- Jung, M.; Kim, S.; Lee, J.-K.; Yoon, S.O.; Park, H.S.; Hong, S.W.; Park, W.-S.; Kim, J.E.; Kim, J.; Keam, B.; et al. Clinicopathological and preclinical findings of nut carcinoma: A multicenter study. Oncologist 2019, 24, e740–e748. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Yamauchi, T.; Hosono, N.; Uzui, K.; Negoro, E.; Morinaga, K.; Nishi, R.; Yoshida, A.; Kimura, S.; Maekawa, T.; et al. Combination of panobinostat with ponatinib synergistically overcomes imatinib-resistant CML cells. Cancer Sci. 2016, 107, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Zopf, S.; Ocker, M.; Neureiter, D.; Alinger, B.; Gahr, S.; Neurath, M.F.; Di Fazio, P. Inhibition of DNA methyltransferase activity and expression by treatment with the pan-deacetylase inhibitor panobinostat in hepatocellular carcinoma cell lines. BMC Cancer 2012, 12, 386. [Google Scholar] [CrossRef]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef]
- Min, H.-Y.; Lee, S.-C.; Woo, J.K.; Jung, H.J.; Park, K.H.; Jeong, H.M.; Hyun, S.Y.; Cho, J.; Lee, W.; Park, J.E.; et al. Essential role of DNA methyltransferase 1–mediated transcription of insulin-like growth factor 2 in resistance to histone deacetylase inhibitors. Clin. Cancer Res. 2017, 23, 1299. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev.Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Rajavelu, A.; Tulyasheva, Z.; Jaiswal, R.; Jeltsch, A.; Kuhnert, N. The inhibition of the mammalian DNA methyltransferase 3a (DNMT3a) by dietary black tea and coffee polyphenols. BMC Biochem. 2011, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.; Goossens, L. DNA methylation targeting: The DNMT/HMT crosstalk challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on g9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef]
- Prinz, J.H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schütte, C.; Noé, F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134, 174105. [Google Scholar] [CrossRef]
- López-López, E.; Barrientos-Salcedo, C.; Prieto-Martínez, F.D.; Medina-Franco, J.L. In silico tools to study molecular targets of neglected diseases: Inhibition of TcSir2rp3, an epigenetic enzyme of Trypanosoma cruzi. Adv. Protein Chem. Struct. Biol. 2020, 122, 203–229. [Google Scholar]
- Sánchez-Cruz, N.; Medina-Franco, J.L.; Mestres, J.; Barril, X. Extended connectivity interaction features: Improving binding affinity prediction through chemical description. Bioinformatic 2020, in press. [Google Scholar] [CrossRef]
- Reaction Biology Corporation. Available online: http://www.reactionbiology.com (accessed on 12 December 2020).
- Molecular Operating Environment (MOE), Version 2018.08, Chemical Computing Group Inc.: Montreal, QC, Canada. Available online: http://www.chemcomp.com (accessed on 12 December 2020).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Zhang, Z.-M.; Liu, S.; Lin, K.; Luo, Y.; Perry, J.J.; Wang, Y.; Song, J. Crystal structure of human DNA methyltransferase 1. J. Mol. Biol. 2015, 427, 2520–2531. [Google Scholar] [CrossRef]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comp. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Ren, X.; Shi, Y.S.; Zhang, Y.; Liu, B.; Zhang, L.H.; Peng, Y.B.; Zeng, R. Novel consensus docking strategy to improve ligand pose prediction. J. Chem. Inf. Model. 2018, 58, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14sb: Improving the accuracy of protein side chain and backbone parameters from ff99sb. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Aaqvist, J.; Warshel, A. Free energy relationships in metalloenzyme-catalyzed reactions. Calculations of the effects of metal ion substitutions in staphylococcal nuclease. J. Am. Chem. Soc. 1990, 112, 2860–2868. [Google Scholar] [CrossRef]
- Liao, Q.; Pabis, A.; Strodel, B.; Kamerlin, S.C.L. Extending the nonbonded cationic dummy model to account for ion-induced dipole interactions. J. Phys. Chem. Lett. 2017, 8, 5408–5414. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. Openmm 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. Mdtraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Hagberg, A.; Schult, D.; Swart, P. Exploring network structure, dynamics, and function using networkx. In Proceedings of the 7th Python in Science Conference (SciPy 2008); Varoquaux, G., Vaught, T., Millman, J., Eds.; SciPy Conference: Pasadena, CA, USA, 2008; pp. 11–15. [Google Scholar]
- Swinburne, T.D.; Wales, D.J. Defining, calculating, and converging observables of a kinetic transition network. J. Chem. Theory Comput. 2020, 16, 2661–2679. [Google Scholar] [CrossRef]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.-H.; Noé, F. Pyemma 2: A software package for estimation, validation, and analysis of markov models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef] [PubMed]
- Plattner, N.; Noé, F. Protein conformational plasticity and complex ligand-binding kinetics explored by atomistic simulations and Markov models. Nat. Commun. 2015, 6, 7653. [Google Scholar] [CrossRef] [PubMed]
- Husic, B.E.; Pande, V.S. Markov state models: From an art to a science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Set | Compound | DNMT1 | DNMT3B |
|---|---|---|---|
| Approved drug | Glyburide | 40.04 (±0.78) | 95.97 (±4.76) |
| Approved drug | Panobinostat | 37.31 (±1.80) | 103.05 (±0.59) |
| Dietary component | Theaflavin | 34.62 (±0.06) | 66.75 (±1.11) |
| Inhibitor of the viral NS5 RNA methyltransferase | 7936171 | 37.74 (±2.15) | 78.20 (±0.30) |
| DNMT-focused library | CSC027480404 | 70.63 (±0.19) | 96.39 (±0.28) |
| DNMT-focused library | CSC026286840 | 73.45 (±3.06) | 90.59 (±2.49) |
| DNMT-focused library | CSC027694519 | 35.42 (±1.78) | 97.42 (±2.49) |
| DNMT-focused library | 6631802 | 93.26 (±7.97) | 93.54 (±0.54) |
| DNMT-focused library | CSC027796832 | 107.25 (±0.59) | 95.28 (±0.47) |
| DNMT-focused library | CSC027083851 | 105.71 (±1.27) | 97.85 (±5.66) |
| Set | Compound | DNMT1 (IC50 μM) |
|---|---|---|
| Approved drug | Glyburide | 55.85 (±1.11) |
| Approved drug | Panobinostat | 76.78 (±0.23) |
| Dietary component | Theaflavin b | 85.33 (±0.14) |
| Inhibitor of the viral NS5 RNA methyltransferase | 7936171 | 78.53 (±8.60) |
| DNMT-focused library | CSC027694519 | 85.11 (±4.10) |
| ID | ECIF Score a | MOE Score | pIC50 | % Inhibition |
|---|---|---|---|---|
| Glyburide | 7.38 | −8.20 | 4.25 | 59.96 |
| Panobinostat | 6.36 | −7.34 | 4.11 | 62.69 |
| Theaflavin | 6.48 | −8.16 | 4.07 | 65.38 |
| 7936171 | 6.52 | −7.67 | 4.11 | 62.26 |
| CSC027694519 | 5.85 | −7.71 | 4.07 | 64.58 |
| 6631802 | 7.26 | −8.35 | ND b | 6.74 |
| CSC027796832 | 5.50 | −7.09 | ND | −7.25 |
| CSC027480404 | 6.62 | −8.15 | ND | 29.97 |
| CSC026286840 | 6.69 | −8.28 | ND | 26.55 |
| CSC027083851 | 6.81 | −7.42 | ND | −5.71 |
| SAH | 5.25 | −9.48 | 6.59 | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juárez-Mercado, K.E.; Prieto-Martínez, F.D.; Sánchez-Cruz, N.; Peña-Castillo, A.; Prada-Gracia, D.; Medina-Franco, J.L. Expanding the Structural Diversity of DNA Methyltransferase Inhibitors. Pharmaceuticals 2021, 14, 17. https://doi.org/10.3390/ph14010017
Juárez-Mercado KE, Prieto-Martínez FD, Sánchez-Cruz N, Peña-Castillo A, Prada-Gracia D, Medina-Franco JL. Expanding the Structural Diversity of DNA Methyltransferase Inhibitors. Pharmaceuticals. 2021; 14(1):17. https://doi.org/10.3390/ph14010017
Chicago/Turabian StyleJuárez-Mercado, K. Eurídice, Fernando D. Prieto-Martínez, Norberto Sánchez-Cruz, Andrea Peña-Castillo, Diego Prada-Gracia, and José L. Medina-Franco. 2021. "Expanding the Structural Diversity of DNA Methyltransferase Inhibitors" Pharmaceuticals 14, no. 1: 17. https://doi.org/10.3390/ph14010017
APA StyleJuárez-Mercado, K. E., Prieto-Martínez, F. D., Sánchez-Cruz, N., Peña-Castillo, A., Prada-Gracia, D., & Medina-Franco, J. L. (2021). Expanding the Structural Diversity of DNA Methyltransferase Inhibitors. Pharmaceuticals, 14(1), 17. https://doi.org/10.3390/ph14010017