Inhibition of DOT1L by Half-Selenopsammaplin A Analogs Suppresses Tumor Growth and EMT-Mediated Metastasis in Triple-Negative Breast Cancer


Abstract
1. Introduction
2. Results
2.1. Preparation of Half-Selenopsammaplin A (HSPA) Analogs
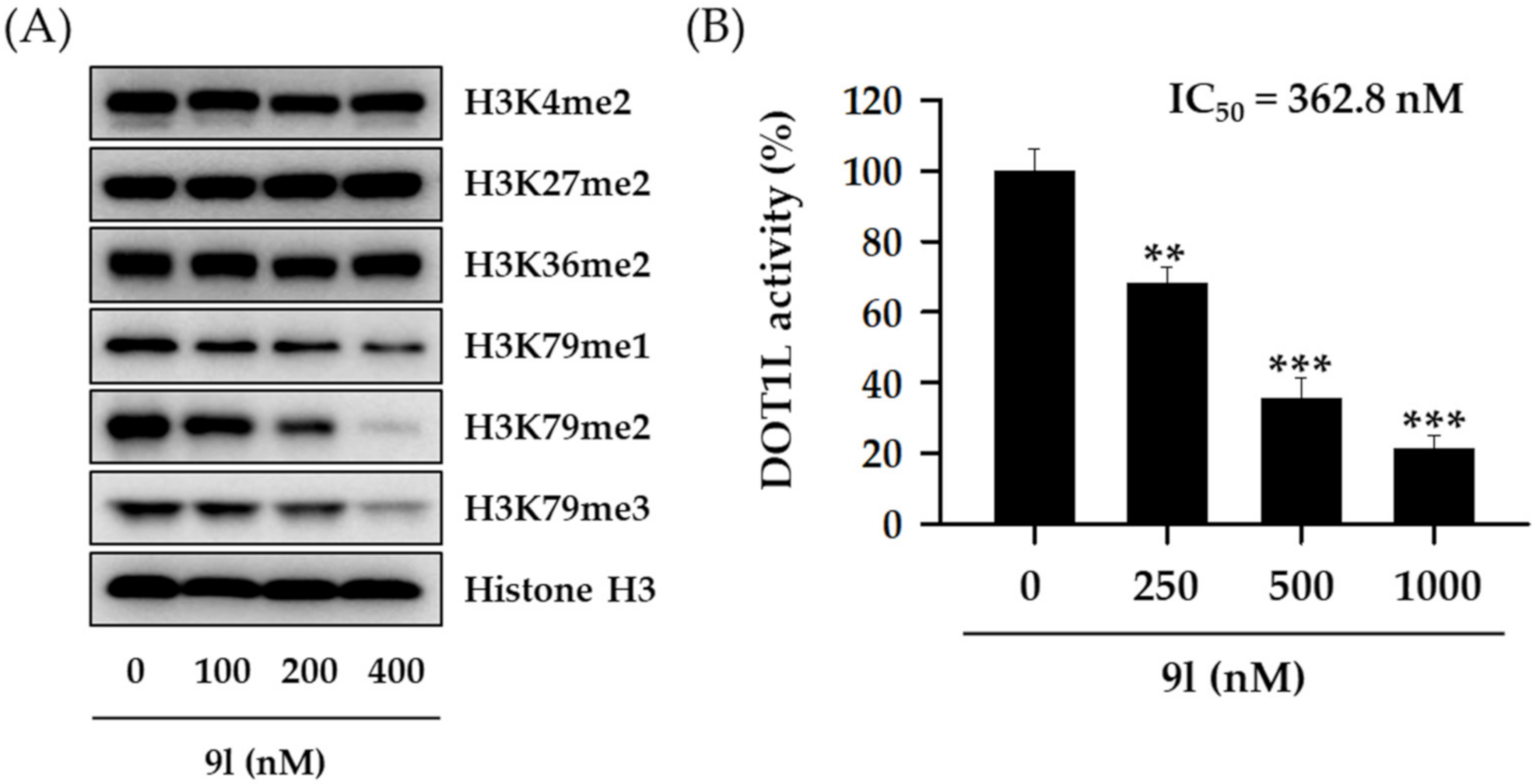
2.2. DOT1L Inhibitory Activity of HSPA Analogs and Expression Level of DOT1L in Human TNBC Cells
2.3. Antiproliferative Activity of HSPA Analogs and Selective Inhibition of H3K79 Methylation in Human TNBC Cells
2.4. 9l Inhibits Cell Invasion and Migration by Regulating Epithelial-Mesenchymal Transition (EMT) Biomarker Expression in Human TNBC Cells
2.5. Antitumor Activity of 9l in MDA-MB-231/Luc Cells-Implanted Orthotopic Mouse Model
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. DOT1L Enzyme Activity Assay
4.3. Cell Culture and Chemicals
4.4. Western Blotting Analysis
4.5. SRB Assay (Cell Proliferation Assay)
4.6. Transwell Cell Invasion Assay
4.7. Wound Healing Assay (Cell Migration Assay)
4.8. In Vivo Orthotopic Mouse Tumor Model (Mammary Fat Pad Model)
4.9. Ex Vivo Biochemical Analyses of Tumors
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Portolani, N.; Coniglio, A.; Ghidoni, S.; Giovanelli, M.; Benetti, A.; Tiberio, G.A.M.; Giulini, S.M. Early and late recurrence after liver resection for hepatocellular carcinoma: Prognostic and therapeutic implications. Ann. Surg. 2006, 243, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.-J.; Yeh, Y.-C.; Jeng, W.-J.; Chien, H.-C.; Wu, Y.-C.; Chou, T.-Y.; Hsu, W.-H. Prognostic Factors of Survival after Recurrence in Patients with Resected Lung Adenocarcinoma. J. Thorac Oncol. 2015, 10, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Pronzato, P.; Rondini, M. First line chemotherapy of metastatic breast cancer. Ann. Oncol. 2006, 17, 165–168. [Google Scholar] [CrossRef]
- Chan, A. A review of the use of trastuzumab (Herceptin) plus vinorelbine in metastatic breast cancer. Ann. Oncol. 2007, 18, 1152–1158. [Google Scholar] [CrossRef]
- Park, S.K.; Byun, W.S.; Lee, S.; Han, Y.T.; Jeong, Y.-S.; Jang, K.; Chung, S.-J.; Lee, J.; Suh, Y.-G.; Lee, S.K. A novel small molecule STAT3 inhibitor SLSI-1216 suppresses proliferation and tumor growth of triple-negative breast cancer cells through apoptotic induction. Biochem. Pharmacol. 2020, 178, 114053–114067. [Google Scholar] [CrossRef]
- Mayer, E.L.; Burstein, H.J. Chemotherapy for Triple-Negative Breast Cancer: Is More Better? J. Clin. Oncol. 2016, 34, 3369–3371. [Google Scholar] [CrossRef]
- Poggio, F.; Bruzzone, M.; Ceppi, M.; Pondé, N.F.; la Valle, G.; del Mastro, L.; de Azambuja, E.; Lambertini, M. Platinum-based neoadjuvant chemotherapy in triple-negative breast cancer: A systematic review and meta-analysis. Ann. Oncol. 2018, 29, 1497–1508. [Google Scholar] [CrossRef]
- Chung, D. Histone modification: The next wave in cancer therapeutics. Trends Mol. Med. 2002, 8, 10–11. [Google Scholar] [CrossRef]
- Kurdistani, S.K. Histone modifications as markers of cancer prognosis: A cellular view. Br. J. Cancer 2007, 97, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Samec, M.; Liskova, A.; Koklesova, L.; Mestanova, V.; Franekova, M.; Kassayova, M.; Bojkova, B.; Uramova, S.; Zubor, P.; Janikova, K.; et al. Fluctuations of Histone Chemical Modifications in Breast, Prostate, and Colorectal Cancer: An Implication of Phytochemicals as Defenders of Chromatin Equilibrium. Biomolecules 2019, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Park, J.H.; Choi, H.J.; Park, M.K.; Won, H.Y.; Park, Y.J.; Lee, C.H.; Oh, S.H.; Song, Y.S.; Kim, H.S.; et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 2015, 6, 7821–7834. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.R.; Zhou, L.H.; Hu, J.X.; Liu, L.M.; Wan, H.P.; Zhang, X.Q. UNC0638, a G9a inhibitor, suppresses epithelial-mesenchymal transition-mediated cellular migration and invasion in triple negative breast cancer. Mol. Med. Rep. 2018, 17, 2239–2244. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kong, G. DOT1L: A new therapeutic target for aggressive breast cancer. Oncotarget 2015, 6, 30451–30452. [Google Scholar] [CrossRef]
- Hong, S.; Shin, Y.; Jung, M.; Ha, M.W.; Park, Y.; Lee, Y.J.; Shin, J.; Oh, K.B.; Lee, S.K.; Park, H.-G. Efficient synthesis and biological activity of Psammaplin A and its analogues as antitumor agents. Eur. J. Med. Chem. 2015, 96, 218–230. [Google Scholar] [CrossRef]
- Byun, W.S.; Kim, W.K.; Han, H.J.; Chung, H.-J.; Jang, K.; Kim, H.S.; Kim, S.; Kim, D.; Bae, E.S.; Park, S.; et al. Targeting histone methyltransferase DOT1L by a novel psammaplin A analog inhibits growth and metastasis of triple-negative breast cancer. Mol. Ther. Oncolytics 2019, 15, 140–152. [Google Scholar] [CrossRef]
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERα gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, 5590–5613. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Mohammed, S. Breast cancer metastasis and the lymphatic system. Oncol. Lett. 2015, 10, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Oktyabri, D.; Ishimura, A.; Tange, S.; Terashima, M.; Suzuki, T. DOT1L histone methyltransferase regulates the expression of BCAT1 and is involved in sphere formation and cell migration of breast cancer cell lines. Biochimie 2016, 123, 20–31. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, R.; El Jamal, L.; El Iskandarani, S.; Kort, J.; Abdel Salam, M.; Assi, H. Endocrine and Targeted Therapy for Hormone-Receptor-Positive, HER2-Negative Advanced Breast Cancer: Insights to Sequencing Treatment and Overcoming Resistance Based on Clinical Trials. Front. Oncol. 2019, 9, 510–532. [Google Scholar] [CrossRef]
- Lima, Z.S.; Ghadamzadeh, M.; Arashloo, F.T.; Amjad, G.; Ebadi, M.R.; Younesi, L. Recent advances of therapeutic targets based on the molecular signature in breast cancer: Genetic mutations and implications for current treatment paradigms. J. Hematol. Oncol. 2019, 12, 38–62. [Google Scholar] [CrossRef]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer—Current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61–73. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. Ther. 2019, 4, 62–100. [Google Scholar] [CrossRef]
- Cohen, I.; Poręba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef]
- Choi, D.K.; Kim, Y.K.; Park, S.W.; Lee, H.; Lee, S.; Kim, S.A.; Kim, S.J.; Lee, J.; Kim, W.; Min, S.-H.; et al. The histone lysine methyltransferase SETD8 regulates angiogenesis through HES-1 in human umbilical vein endothelial cells. Sci. Rep. 2020, 10, 12089–12099. [Google Scholar] [CrossRef]
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal. Transduct Target. Ther. 2020, 5, 143–155. [Google Scholar] [CrossRef]
- Shukla, N.; Wetmore, C.; O’Brien, M.M.; Silverman, L.B.; Brown, P.; Cooper, T.M.; Thomson, B.; Blakemore, S.J.; Daigle, S.; Suttle, B.; et al. Final Report of Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Children with Relapsed or Refractory MLL-r Acute Leukemia. Blood 2016, 128, 2780. [Google Scholar] [CrossRef]
- Byun, W.S.; Kim, W.K.; Yoon, J.S.; Jarhad, D.B.; Jeong, L.S.; Lee, S.K. Antiproliferative and Antimigration Activities of Fluoro-Neplanocin A via Inhibition of Histone H3 Methylation in Triple-Negative Breast Cancer. Biomolecules 2020, 10, 530. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Byun, W.S.; Jin, M.; Yu, J.; Kim, W.K.; Song, J.; Chung, H.J.; Jeong, L.S.; Lee, S.K. A novel selenonucleoside suppresses tumor growth by targeting Skp2 degradation in paclitaxel-resistant prostate cancer. Biochem. Pharmacol. 2018, 158, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Byun, W.S.; Kim, S.; Shin, Y.H.; Kim, W.K.; Oh, D.-C.; Lee, S.K. Antitumor Activity of Ohmyungsamycin A through the Regulation of the Skp2-p27 Axis and MCM4 in Human Colorectal Cancer Cells. J. Nat. Prod. 2020, 83, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Ban, Y.H.; Byun, W.S.; Kim, D.; Jang, Y.-J.; An, J.S.; Shin, B.; Lee, S.K.; Shin, J.; Yoon, Y.J.; et al. Camporidines A and B: Antimetastatic and Anti-inflammatory Polyketide Alkaloids from a Gut Bacterium of Camponotus kiusiuensis. J. Nat. Prod. 2019, 82, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSPA Analog | Structure | HSPA Analog | Structure |
|---|---|---|---|
| 9a |  | 9h |  |
| 9b |  | 9i |  |
| 9c |  | 9j |  |
| 9d |  | 9k |  |
| 9e |  | 9l |  |
| 9f |  | 9m |  |
| 9g |  | 9n |  |
| Analogs | MDA-MB-231 IC50 (μM) a | MCF10A IC50 (μM) a | Therapeutic Index b |
|---|---|---|---|
| Psammaplin A (PsA) | 2.82 | 32.70 | 11.60 |
| PsA-3091 | 1.98 | 136.60 | 68.99 |
| 9a | 0.21 | 17.24 | 82.10 |
| 9b | 0.25 | 21.52 | 86.08 |
| 9c | 0.46 | 28.20 | 61.30 |
| 9d | 0.39 | 26.04 | 66.77 |
| 9e | 0.43 | 23.32 | 54.23 |
| 9f | 0.47 | 27.07 | 57.60 |
| 9g | 0.33 | 22.66 | 68.67 |
| 9h | 0.40 | 29.14 | 72.85 |
| 9i | 0.17 | 21.02 | 123.65 |
| 9j | 0.18 | 19.48 | 108.22 |
| 9k | 0.19 | 18.15 | 95.53 |
| 9l | 0.16 | 25.86 | 161.63 |
| 9m | 0.25 | 17.02 | 68.08 |
| 9n | 0.23 | 18.51 | 80.48 |
| EPZ-5676 | >50 | >50 | - |
| Paclitaxel c | 0.01 | 0.198 | 19.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byun, W.S.; Lee, G.H.; Park, H.-g.; Lee, S.K. Inhibition of DOT1L by Half-Selenopsammaplin A Analogs Suppresses Tumor Growth and EMT-Mediated Metastasis in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 18. https://doi.org/10.3390/ph14010018
Byun WS, Lee GH, Park H-g, Lee SK. Inhibition of DOT1L by Half-Selenopsammaplin A Analogs Suppresses Tumor Growth and EMT-Mediated Metastasis in Triple-Negative Breast Cancer. Pharmaceuticals. 2021; 14(1):18. https://doi.org/10.3390/ph14010018
Chicago/Turabian StyleByun, Woong Sub, Gyu Ho Lee, Hyeung-geun Park, and Sang Kook Lee. 2021. "Inhibition of DOT1L by Half-Selenopsammaplin A Analogs Suppresses Tumor Growth and EMT-Mediated Metastasis in Triple-Negative Breast Cancer" Pharmaceuticals 14, no. 1: 18. https://doi.org/10.3390/ph14010018
APA StyleByun, W. S., Lee, G. H., Park, H.-g., & Lee, S. K. (2021). Inhibition of DOT1L by Half-Selenopsammaplin A Analogs Suppresses Tumor Growth and EMT-Mediated Metastasis in Triple-Negative Breast Cancer. Pharmaceuticals, 14(1), 18. https://doi.org/10.3390/ph14010018