Antiproliferative and Carbonic Anhydrase II Inhibitory Potential of Chemical Constituents from Lycium shawii and Aloe vera: Evidence from In Silico Target Fishing and In Vitro Testing

 ,
, 

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Phytochemical Investigation
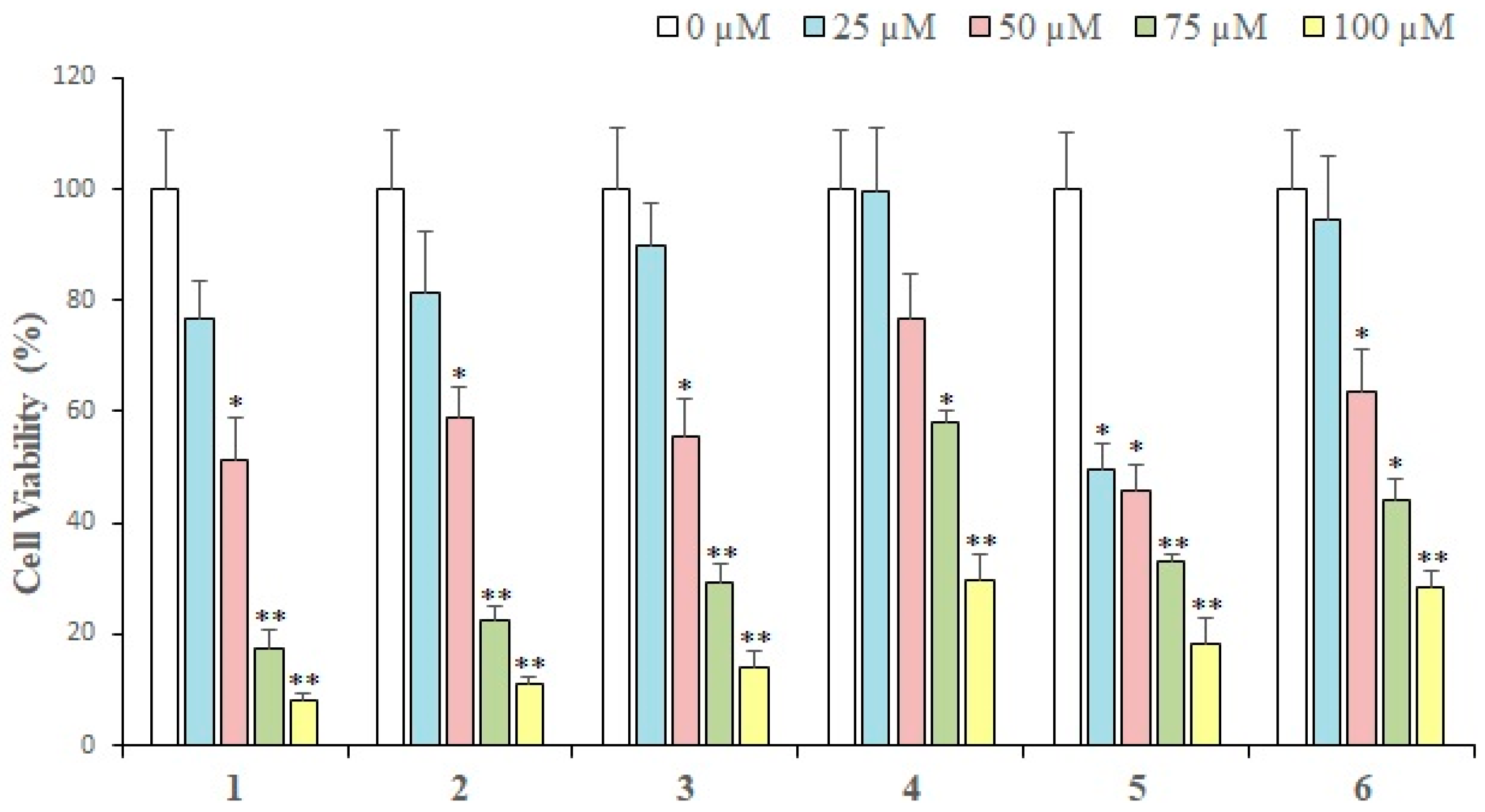
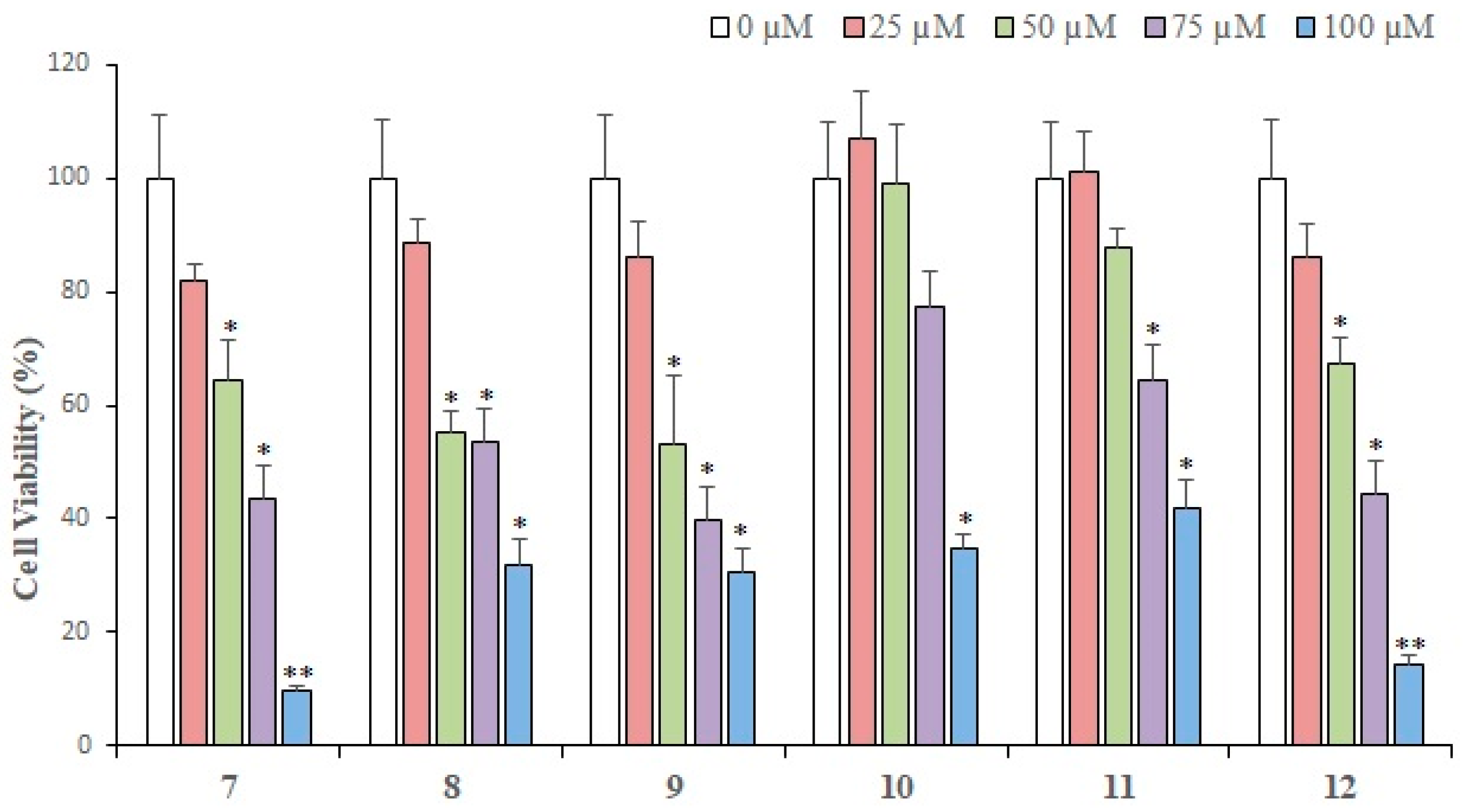
2.2. Cytotoxic Activity
2.3. Antioxidant Activity
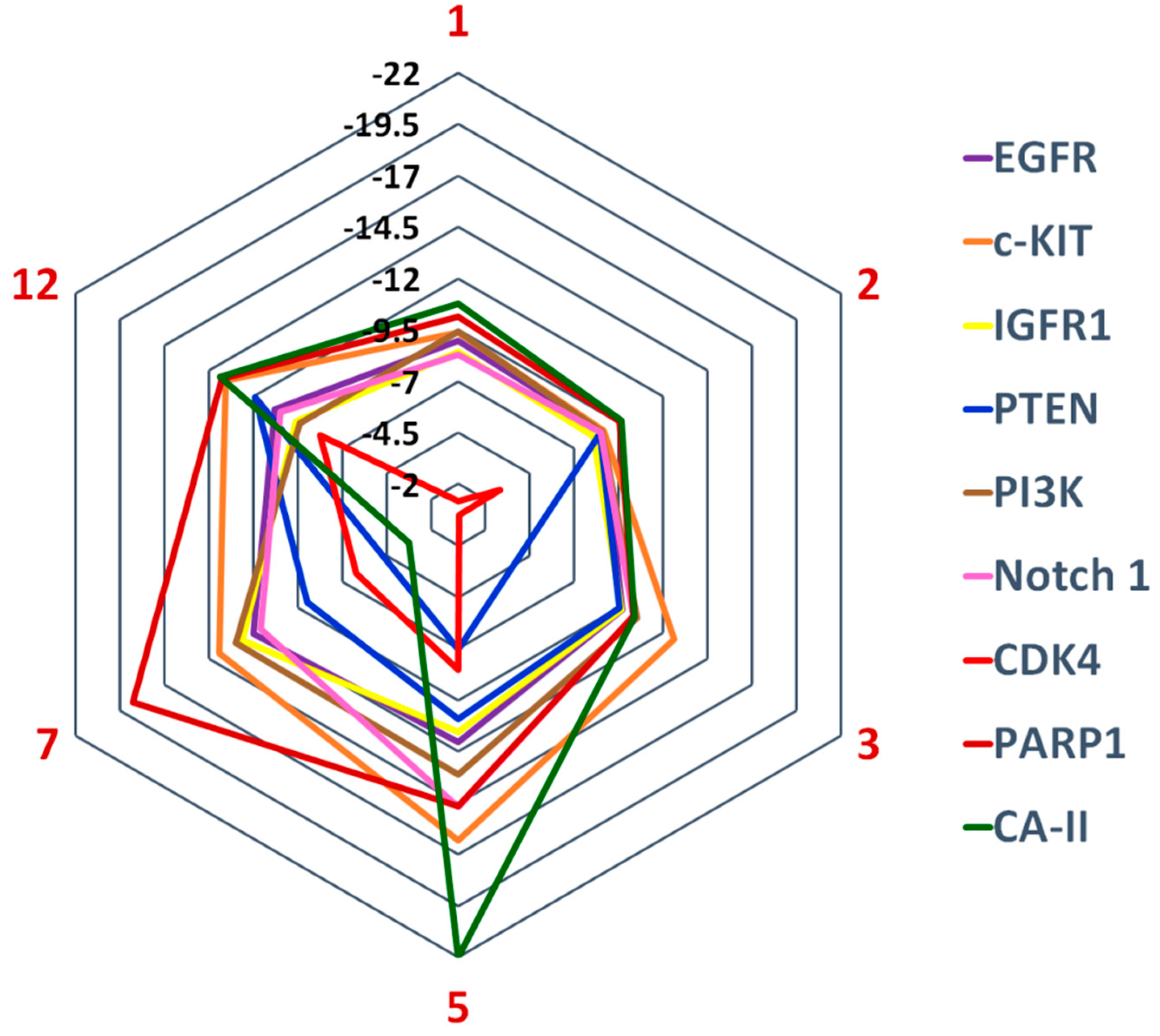
2.4. Human Intracellular Drug Targets
2.5. Pharmacophore Modeling
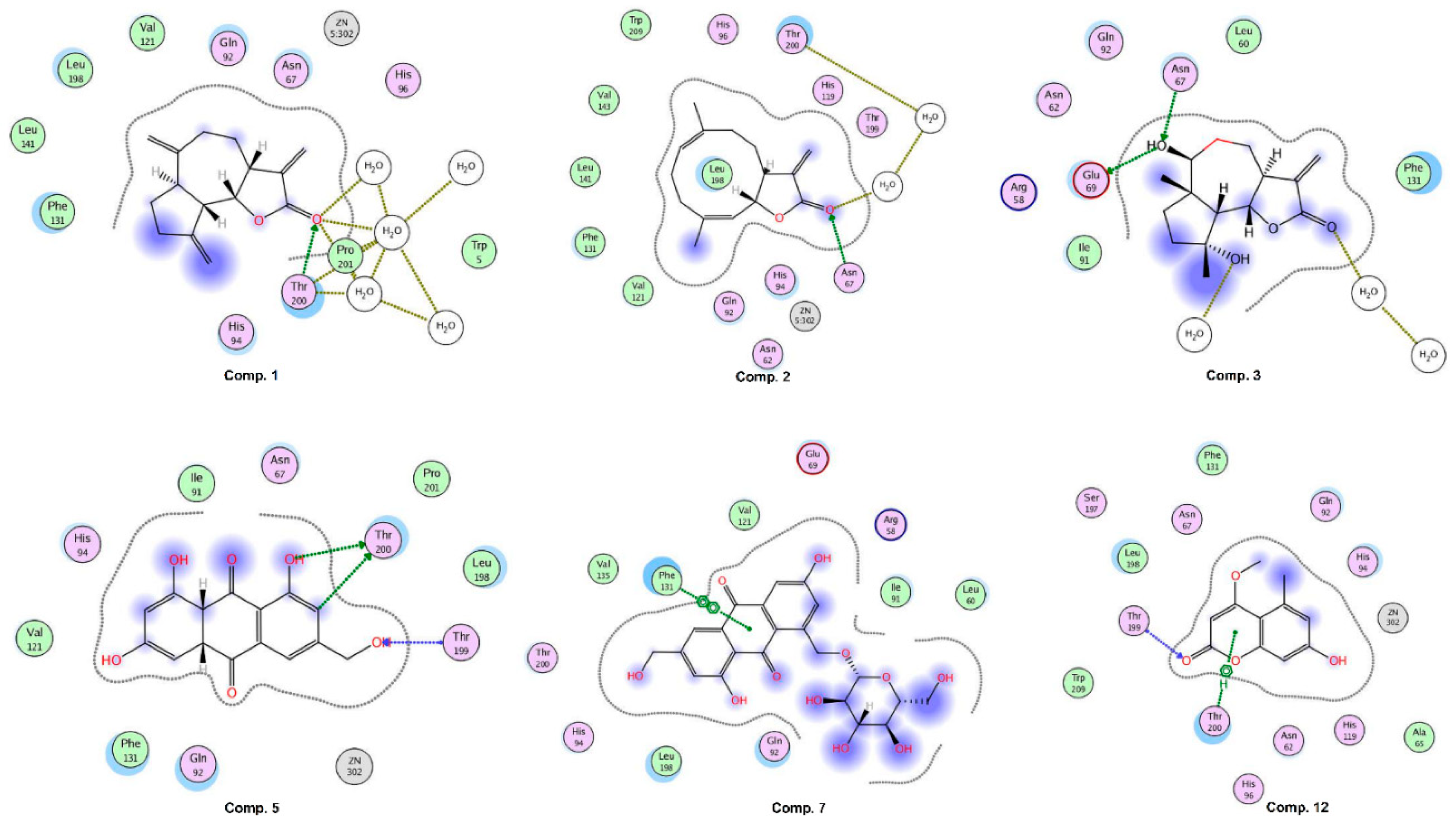
2.6. Molecular Docking Studies
2.7. Inhibition of Carbonic Anhydrase II (CA-II) by Compounds 1, 2, 5, 7, and 12
3. Materials and Methods
3.1. General Instrumentation
3.2. Plant Material and Identification
3.3. Extraction, Fractionation, and Isolation of Bioactive Compounds
3.4. Assay Protocol for Cytotoxic Activity
3.5. Assay Protocol for DPPH Radical Scavenging Activity
3.6. In Silico Target Fishing
3.7. Selection of Known Drugs
3.8. Pharmacophore Modeling
3.9. Molecular Docking
3.10. Carbonic Anhydrase II Inhibition
3.11. Statistical Analysis for Cytotoxic Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roy, N.K.; Deka, A.; Bordoloi, D.; Mishra, S.; Kumar, A.P.; Sethi, G.; Kunnumakkara, A.B. The potential role of boswellic acids in cancer prevention and treatment. Cancer Lett. 2016, 377, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.; Gordon, M.C. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2012, 79, 629–661. [Google Scholar] [CrossRef]
- Grover, J.K.; Yadav, S.; Vats, V. Medicinal plants of India with anti-diabetic potential. J. Ethnopharmacol. 2002, 81, 81–100. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Gu, H.; Xiong, X.; Ao, H.; Cao, J.; Lin, W.; Yu, M.; Lin, J.; Cui, Q. MicroRNAs involved in carcinogenesis, prognosis, therapeutic resistance and applications in human triple-negative breast cancer. Cells 2019, 8, 1492. [Google Scholar] [CrossRef]
- Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; Anderson, B.O.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2016: A systematic analysis for the global burden of disease study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar]
- Xiao, Y.; Humphries, B.; Yang, C.; Wang, Z. MiR-205 Dysregulations in Breast Cancer: The Complexity and Opportunities. Non-Coding RNA. 2019, 5, 53. [Google Scholar] [CrossRef]
- Carpenter, K.J.; Valfort, A.C.; Steinauer, N.; Chatterjee, A.; Abuirqeba, S.; Majidi, S.; Sengupta, M.; Di Paolo, R.J.; Shornick, L.P.; Zhang, J.; et al. LXR-inverse agonism stimulates immune-mediated tumor destruction by enhancing CD8 T-cell activity in triple negative breast cancer. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Alizart, M.; Saunus, J.; Cummings, M.; Lakhani, S.R. Molecular classification of breast carcinoma. Diagn. Histopathol. 2012, 18, 97–103. [Google Scholar] [CrossRef]
- Huynh, M.M.; Jayanthan, A.; Pambid, M.R.; Los, G.; Dunn, S.E. RSK2: A promising therapeutic target for the treatment of triple-negative breast cancer. Expert Opin. Ther. Targets 2020, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A. Molecular portraits of human breast tumours. Nature 2000, 406, 747. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, W.; Hu, P.; Lakowski, T.M. Integrative analysis reveals subtype-specific regulatory determinants in triple negative breast cancer. Cancers 2019, 11, 507. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, Y.X.; Qi, Z.; Tsang, S.Y. TRPC3 Regulates the proliferation and apoptosis resistance of triple negative breast cancer cells through the TRPC3/RASA4/MAPK pathway. Cancers 2019, 11, 558. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106. [Google Scholar]
- Mehanna, J.; Haddad, F.G.; Eid, R.; Lambertini, M.; Kouriem, H.R. Triple-negative breast cancer: Current perspective on the evolving therapeutic landscape. Int. J. Womens Health 2019, 11, 431. [Google Scholar] [CrossRef]
- Tripp, B.C.; Smith, K.; Ferry, J.G. Carbonic anhydrase: New insights for an ancient enzyme. J. Biol. Chem. 2001, 276, 48615–48618. [Google Scholar] [CrossRef]
- Chegwidden, W.R.; Dodgson, S.J.; Spencer, I.M. The roles of carbonic anhydrase in metabolism, cell growth and cancer in animals. In The Carbonic Anhydrases: New Horizons; Chegwidden, W.R., Carter, N.D., Edwards, Y.H., Eds.; Springer: Basel, Switzerland, 2000; pp. 343–361. [Google Scholar]
- Venta, P.J. Carbonic anhydrases in mammalian cell culture and tumors. In The Carbonic Anhydrases: Cellular Physiology and Molecular Genetics; Dodgson, S.J., Tashian, R.E., Gros, G., Carter, N.D., Eds.; Plenum Press: New York, NY, USA, 1991; pp. 71–78. [Google Scholar]
- Webb, S.D.; Sherratt, J.A.; Fish, R.G. Mathematical modelling of tumour acidity: Regulation of intracellular pH. J. Theor. Biol. 1999, 196, 237–250. [Google Scholar] [CrossRef]
- Gerweck, L.E. Tumor pH: Implications for treatment and novel drug design. Semin. Radiat. Oncol. 1998, 8, 176–182. [Google Scholar] [CrossRef]
- Montcourrier, P.; Silver, I.; Farnoud, R.; Bird, I.; Rochefort, H. Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin. Exp. Metastasis 1997, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Tannock, I.F. Heterogeneity of intracellular pH and of mechanisms that regulate intracellular pH in populations of cultured cells. Cancer Res. 1998, 58, 1901–1908. [Google Scholar] [PubMed]
- Parkkila, A.K.; Herva, R.; Parkkila, S.; Rajaniemi, H. Immunohistochemical demonstration of human carbonic anhydrase isoenzymes I and II in brain tumours. Histochem. J. 1995, 27, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Pastoreková, S.; Parkkila, S.; Parkkila, A.K.; Opavský, R.; Zelník, V.; Saarnio, J.; Pastorek, J. Carbonic anhydrase IX, MN/CA IX: Analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology 1997, 112, 398–408. [Google Scholar] [CrossRef]
- Frazier, M.L.; Lilly, B.J.; Wu, E.F.; Ota, T.; Hewett-Emmett, D. Carbonic anhydrase II gene expression in cell lines from human pancreatic adenocarcinoma. Pancreas 1990, 5, 507–514. [Google Scholar] [CrossRef]
- Parkkila, S.; Parkkila, A.K.; Juvonen, T.; Lehto, V.P.; Rajaniemi, H. Immunohistochemical demonstration of human carbonic anhydrase isoenzymes I and II in pancreatic tumours. Histochem. J. 1995, 27, 133–138. [Google Scholar]
- Parkkila, S.; Rajaniemi, H.; Parkkila, A.K.; Kivelä, J.; Waheed, A.; Pastoreková, S.; Pastorek, J.; Sly, W. Carbonic anhydrase inhibitor suppresses invasion of renal cancer cells in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 2220–2224. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Prochazkova, D.; Bousova, I.; Wilhelmova, N. Antioxidant and prooxidant properties of flavonoids. Fitoterapia 2011, 82, 513–523. [Google Scholar] [CrossRef]
- Li, Y.X.; Kim, S.K. Utilization of seaweed derived ingredients as potential antioxidants and functional ingredients in the food industry: An Overview. Food Sci. Biotechnol. 2011, 20, 1461–1466. [Google Scholar] [CrossRef]
- Mahdi-Pour, B.; Jothy, S.L.; Latha, L.Y.; Chen, Y.; Sasidharan, S. Antioxidant activity of methanol extracts of different parts of Lantana camara. Asian Pac. J. Trop. Biomed. 2012, 2, 960–965. [Google Scholar] [CrossRef]
- Ali, S.S.; El-Zawawy, N.A.; Al-Tohamy, R.; El-Sapagh, S.; Mustafa, A.M.; Sun, J. Lycium shawii Roem. & Schult.: A new bioactive antimicrobial and antioxidant agent to combat multi-drug/pan-drug resistant pathogens of wound burn infections. J. Tradit. Complement. Med. 2020, 10, 13–25. [Google Scholar] [PubMed]
- Rehman, N.U.; Hussain, H.; Al-Riyami, S.A.; Csuk, R.; Khiat, M.; Abbas, G.; Al-Rawahi, A.; Green, I.R.; Ahmed, I.; Al-Harrasi, A. Lyciumaside and lyciumate: A new diacylglycoside and sesquiterpene lactone from Lycium shawii. Helv. Chim. Acta 2016, 99, 632–635. [Google Scholar] [CrossRef]
- Rehman, N.U.; Al-Riyami, S.A.; Hussain, H.; Ali, A.; Khan, A.L.; Al-Harrasi, A. Secondary metabolites from the resins of Aloe vera and Commiphora mukul mitigate lipid peroxidation. Acta Pharmaceutica. 2019, 69, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Taukoorah, U.; Mahomoodally, M.F. Crude Aloe vera gel shows antioxidant propensities and inhibits pancreatic lipase and glucose movement in vitro. Adv. Pharmacol. Sci. 2016, 2016, 3720850. [Google Scholar] [PubMed]
- Rauwald, H.W.; Lohse, K. Structure revision of 4-hydroxyaloin: 10-hydroxyaloins A and B as main in vitro-oxidation products of the diastereomeric aloins. Planta Med. 1992, 58, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, G.; Dai, R.; Bi, K. Isolation of aloinoside B and metabolism by rat intestinal Bacteria. Pharm. Biol. 2004, 42, 581–587. [Google Scholar] [CrossRef]
- Girol, C.G.; Fisch, K.M.; Heinekamp, T.; Günther, S.; Hüttel, W.; Piel, J.; Brakhage, A.A.; Müller, M. Regio- and stereoselective oxidative phenol coupling in Aspergillus nige. Angew. Chem. Int. Ed. Engl. 2012, 51, 9788–9791. [Google Scholar] [CrossRef]
- Heerden, F.R.; Viljoen, A.M.; Wyk, B.E. 6′-O-Coumaroylaloesin from Aloe castanea—A taxonomic marker for Aloe section Anguialoe. Phytochemistry 2000, 55, 117–120. [Google Scholar] [CrossRef]
- Speranza, G.; Manitto, P.; Monti, D.; Lianza, F. Feroxidin, a novel 1-methyltetralin derivative isolated from Cape aloe. Tetrahedron Lett. 1990, 31, 3077–3080. [Google Scholar] [CrossRef]
- Zakir, H.A.; Subbarao, G.V.; Pearse, S.J.; Gopalakrishnan, S.; Ito, O.; Ishikawa, T.; Kawano, N.; Nakahara, K.; Yoshihashi, T.; Ono, H.; et al. Detection, isolation and characterization of a root-exuded compound, methyl 3-(4-hydroxyphenyl) propionate, responsible for biological nitrification inhibition by sorghum (Sorghum bicolor). New Phytol. 2008, 180, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, H.; Tajimi, A.T.; Ichihara, A. Phytotoxic metabolites isolated from Scolecotrichum graminis Fuckel. Biosci. Biotech. Biochem. 1994, 58, 1956–1959. [Google Scholar] [CrossRef]
- Harish, R.; Divakar, S.; Srivastava, A.; Shivanandappa, T. Isolation of antioxidant compounds from the methanolic extract of the roots of Decalepis hamiltonii (Wight and Arn.). J. Agric. Food Chem. 2005, 53, 7709–7714. [Google Scholar] [CrossRef] [PubMed]
- Poh, B.L.; Lim, C.H.; Tan, C.M.; Wong, W. 1H NMR study on the complexation of phenols with cyclotetrachromotropylene in aqueous solution. Tetrahedron 1993, 49, 7259–7266. [Google Scholar] [CrossRef]
- He, Z.H.; Huang, Y.Q.; Weng, S.F.; Tan, Y.R.; He, T.P.; Qin, Y.M.; Liang, N.C. Effect of Aloe emodin on invasion and metastasis of high metastatic breast cancer MDA-MB-231 cells. Zhong Yao Cai 2013, 36, 1481–1485. [Google Scholar] [PubMed]
- Huang, P.H.; Huang, C.Y.; Chen, M.C.; Lee, Y.T.; Yue, C.H.; Wang, H.Y.; Lin, H. Emodin and aloe-emodin suppress breast cancer cell proliferation through ERα inhibition. Evid. Based Complement. Alternat. Med. 2013, 2013, 376123. [Google Scholar] [CrossRef] [PubMed]
- Gaweesh, A.; Sengab, A.E.N.B.; Osman, H.M.H.S.M.; Abdou, A.M. Phytoconstituents, cytotoxic, antioxidant and hepatoprotective activities of the aerial parts of Lycium shawii R. growing in Egypt. Med. Aromat Plants 2015, 4, 1–7. [Google Scholar]
- Muhammad, A.; Tel-Çayan, G.; Öztürk, M.; Duru, M.E.; Nadeem, S.; Anis, I.; Ng, S.W.; Shah, M.R. Phytochemicals from and their antioxidant and anticholinesterase activities with structure-activity relationships. Pharm. Biol. 2016, 11, 1–7. [Google Scholar]
- Moumbock, A.F.; Li, J.; Mishra, P.; Gao, M.; Günther, S. Current computational methods for predicting protein interactions of natural products. Comput. Struct. Biotechnol. J. 2019, 17, 1367–1376. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. Tools for in silico target fishing. Methods 2015, 71, 98–103. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Bender, A.; Davies, J.W. In silico target fishing: Predicting biological targets from chemical structure. Drug Discov. Today Technol. 2006, 3, 413–421. [Google Scholar] [CrossRef]
- Akhoon, B.A.; Tiwari, H.; Nargotra, A. In Silico Drug Design Methods for Drug Repurposing. In In Silico Drug Design; Academic Press: Cambridge, MA, USA, 2019; pp. 47–84. [Google Scholar]
- Rehman, N.U.; Hussain, H.; Al-Riyami, S.A.; Green, I.R.; Al-Harrasi, A. Chemical constituents isolated from Lycium shawii and their chemotaxonomic significance. Rec. Nat. Prod. 2018, 12, 380–384. [Google Scholar] [CrossRef]
- Raees, M.A.; Hussain, H.; Al-Rawahi, A.; Csuk, R.; Muhammad, S.A.; Khan, H.Y.; Rehman, N.U.; Abbas, G.; Al-Broumi, M.A.; Green, I.R.; et al. Anti-proliferative and computational studies of two new pregnane glycosides from Desmidorchis flava. Bioorg. Chem. 2016, 67, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Rehman, N.U.; Hussain, H.; Khan, H.Y.; Csuk, R.; Abbas, G.; Green, I.R.; Al-Harrasi, A. A norterpenoid and tripenoids from Commiphora mukul: Isolation and biological activity. Zeitschrift für Naturforschung B 2017, 72, 11–15. [Google Scholar] [CrossRef]
- Mills, N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www.cambridgesoft.com. Commercial Price: $1910 for download, $2150 for CD-ROM; Academic Price: $710 for download, $800 for CD-ROM. J. Am. Chem. Soc. 2006, 128, 13649–13650. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) (2013.08); Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Cisek, K.; Cooper, G.L.; Huseby, C.J.; Kuret, J. Structure and mechanism of action of tau aggregation inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. [Google Scholar] [CrossRef]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Wu, J.C.; Christensen, J.; Deshmukh, G.D.; Diehl, W.; DiNitto, J.P.; English, J.M.; Greig, M.J.; He, Y.A.; Jacques, S.L.; et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. USA 2009, 106, 1542–1547. [Google Scholar] [CrossRef]
- Miller, L.M.; Mayer, S.C.; Berger, D.M.; Boschelli, D.H.; Boschelli, F.; Di, L.; Du, X.; Dutia, M.; Floyd, M.B.; Johnson, M.; et al. Lead identification to generate 3-cyanoquinoline inhibitors of insulin-like growth factor receptor (IGF-1R) for potential use in cancer treatment. Bioorg. Med. Chem. Lett. 2009, 19, 62–66. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Lee, C.U.; Hahne, G.; Hanske, J.; Bange, T.; Bier, D.; Rademacher, C.; Hennig, S.; Grossmann, T.N. Redox modulation of PTEN phosphatase activity by hydrogen peroxide and bisperoxidovanadium complexes. Angew. Chem. Int. Ed. 2015, 54, 13796–13800. [Google Scholar] [CrossRef]
- Hoegenauer, K.; Soldermann, N.; Stauffer, F.; Furet, P.; Graveleau, N.; Smith, A.B.; Hebach, C.; Hollingworth, G.J.; Lewis, I.; Gutmann, S.; et al. Discovery and pharmacological characterization of novel quinazoline-based PI3K delta-selective inhibitors. ACS Med. Chem. Lett. 2016, 7, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Day, P.J.; Cleasby, A.; Tickle, I.J.; O’Reilly, M.; Coyle, J.E.; Holding, F.P.; McMenamin, R.L.; Yon, J.; Chopra, R.; Lengauer, C.; et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. USA 2009, 106, 4166–4170. [Google Scholar] [CrossRef] [PubMed]
- Thorsell, A.G.; Ekblad, T.; Karlberg, T.; Löw, M.; Pinto, A.F.; Trésaugues, L.; Moche, M.; Cohen, M.S.; Schüler, H. Structural basis for potency and promiscuity in poly (ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J. Med. Chem. 2016, 60, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; McKenna, R.; Supuran, C.T. Effect of incorporating a thiophene tail in the scaffold of acetazolamide on the inhibition of human carbonic anhydrase isoforms I, II, IX and XII. Bioorg. Med. Chem. Lett. 2013, 23, 5646–5649. [Google Scholar] [CrossRef] [PubMed]
- Shank, R.P.; Doose, D.R.; Streeter, A.J.; Bialer, M. Plasma and whole blood pharmacokinetics of topiramate: The role of carbonic anhydrase. Epilepsy Res. 2005, 63, 103–112. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (Mean ± SEM) (μM) |
|---|---|
| 1 | 35.36 ± 2.56 |
| 2 | 42.08 ± 2.98 |
| 3 | 49.39 ± 3.73 |
| 4 | 101.4 ± 7.09 |
| 5 | 31.36 ± 2.44 |
| 6 | 73.47 ± 5.89 |
| 7 | 57.32 ± 4.14 |
| 8 | 72.21 ± 6.29 |
| 9 | 76.9 ± 7.04 |
| 10 | 142.8 ± 12.66 |
| 11 | 140.4 ± 13.1 |
| 12 | 60.09 ± 4.82 |
| Doxorubicin (+ve control) | 3.31 ± 0.19 |
| Antioxidant % Inhibition (IC50 ± SEM) | |||
|---|---|---|---|
| Code | L. shawii | Code | A. vera |
| BF | 72 (650 ± 1.50) | MF | NA |
| MF | 50 | EF | 51 |
| WF | NA | DF | 42 |
| HF | NA | BF | 32 |
| EF | 76 (378 ± 1.50) | WF | 35 |
| DF | 60 (735 ± 2.00) | HF | NA |
| Ascorbic acid | 90 (53 ± 1.32) | ||
| Numbering | % Inhibition (1 mM) | IC50 ± SEM (μM) |
|---|---|---|
| 4 | 78 | 55 ± 2.0 |
| 6 | 71 | 645 ± 1.5 |
| 13 | 73 | 762 ± 2.0 |
| 14 | 80 | 241 ± 1.5 |
| Compounds | Docking Score | % Inhibition | IC50 (µM) ± (SEM) |
|---|---|---|---|
| 1 | −10.75 | 33 | NA |
| 2 | −9.64 | 84.7 | 24.4 |
| 3 | −10.39 | NT | NT |
| 5 | −21.98 | 86.3 | 14.4 ± 1.14 |
| 7 | −3.26 | 37.5 | NA |
| 12 | −13.88 | 91.2 | 23.3 ± 1.63 |
| Target | Drugs |
|---|---|
| EGFR | Afatinib, Canertinib dihydrochloride, Dacomitinib, Erlotinib, Gefitinib, Icotinib, Lapatinib, Lifirafenib, Masoprocol, Mavelertinib, Naquotinib, Nazartinib, Neratinib, Olmutinib, Osimertinib, Pelitinib, Rociletinib, Vandetanib, Varlitinib |
| c-KIT | Amuvatinib, Ancestim, Avapritinib, Cabozantinib, Dasatinib, Dovitinib lactate, Imatinib, Masitinib, Midostaurin, Motesanib, Nilotinib, Pazopanib, Regorafenib, Ripretinib, Semaxanib, Sorafenib, Sunitinib, Tandutinib, Toceranib, Vatalanib |
| IGFR1 | Ibutamoren mesylate, Linsitinib, Mecasermin, Mecasermin rinfabate, Toremifene |
| Notch 1 | Crenigacestat |
| PI3K | Apitolisib, Bimiralisib, Buparlisib, Dactolisib, Gedatolisib, Leniolisib, Omipalisib, Pictilisib, Samotolisib |
| CDK2 | Omacetaxine mepesuccinate |
| PARP | Olaparib, Niraparib, Rucaparib, Talazoparib, Veliparib |
| CA-II | Acetazolamide |
| S # | Target | PDB ID | Ligand ID | Resolution (Å) | References |
|---|---|---|---|---|---|
| 1 | EGFR | 2G5J | 0WN (Afatinib) | 2.8 | [61] |
| 2 | c- Kit | 3G0E | B49 (Sunitinib) | 1.6 | [62] |
| 3 | IGFR1 | 3F5P | 741 (3-Cyanoquinoline) | 2.9 | [63] |
| 4 | Notch 1 | 3L95 | Antibody FAB fragment | 2.19 | [64] |
| 5 | PTEN | 5BZX | VO4 (bisperoxovanadium complex) | 2.5 | [65] |
| 6 | PI3K | 5ITD | 6CY (5-{4-[3-(4-acetylpiperazine-1-carbonyl)phenyl]quinazolin-6-yl}-2-methoxypyridine-3-carbonitrile) | 3.02 | [66] |
| 7 | CDK4 | 2W9Z | Cyclin D | 2.45 | [67] |
| 8 | PARP1 | 4R6E | 3JD (Niraparib) | 2.2 | [68] |
| 9 | CA-II | 4IWZ | 1GO (acetazolamide derivative) | 1.598 | [69] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ur Rehman, N.; Halim, S.A.; Khan, M.; Hussain, H.; Yar Khan, H.; Khan, A.; Abbas, G.; Rafiq, K.; Al-Harrasi, A. Antiproliferative and Carbonic Anhydrase II Inhibitory Potential of Chemical Constituents from Lycium shawii and Aloe vera: Evidence from In Silico Target Fishing and In Vitro Testing. Pharmaceuticals 2020, 13, 94. https://doi.org/10.3390/ph13050094
Ur Rehman N, Halim SA, Khan M, Hussain H, Yar Khan H, Khan A, Abbas G, Rafiq K, Al-Harrasi A. Antiproliferative and Carbonic Anhydrase II Inhibitory Potential of Chemical Constituents from Lycium shawii and Aloe vera: Evidence from In Silico Target Fishing and In Vitro Testing. Pharmaceuticals. 2020; 13(5):94. https://doi.org/10.3390/ph13050094
Chicago/Turabian StyleUr Rehman, Najeeb, Sobia Ahsan Halim, Majid Khan, Hidayat Hussain, Husain Yar Khan, Ajmal Khan, Ghulam Abbas, Kashif Rafiq, and Ahmed Al-Harrasi. 2020. "Antiproliferative and Carbonic Anhydrase II Inhibitory Potential of Chemical Constituents from Lycium shawii and Aloe vera: Evidence from In Silico Target Fishing and In Vitro Testing" Pharmaceuticals 13, no. 5: 94. https://doi.org/10.3390/ph13050094
APA StyleUr Rehman, N., Halim, S. A., Khan, M., Hussain, H., Yar Khan, H., Khan, A., Abbas, G., Rafiq, K., & Al-Harrasi, A. (2020). Antiproliferative and Carbonic Anhydrase II Inhibitory Potential of Chemical Constituents from Lycium shawii and Aloe vera: Evidence from In Silico Target Fishing and In Vitro Testing. Pharmaceuticals, 13(5), 94. https://doi.org/10.3390/ph13050094