Bioisosteric Replacement as a Tool in Anti-HIV Drug Design


Abstract
1. Introduction
2. Principle of Bioisosterism and Historical Background
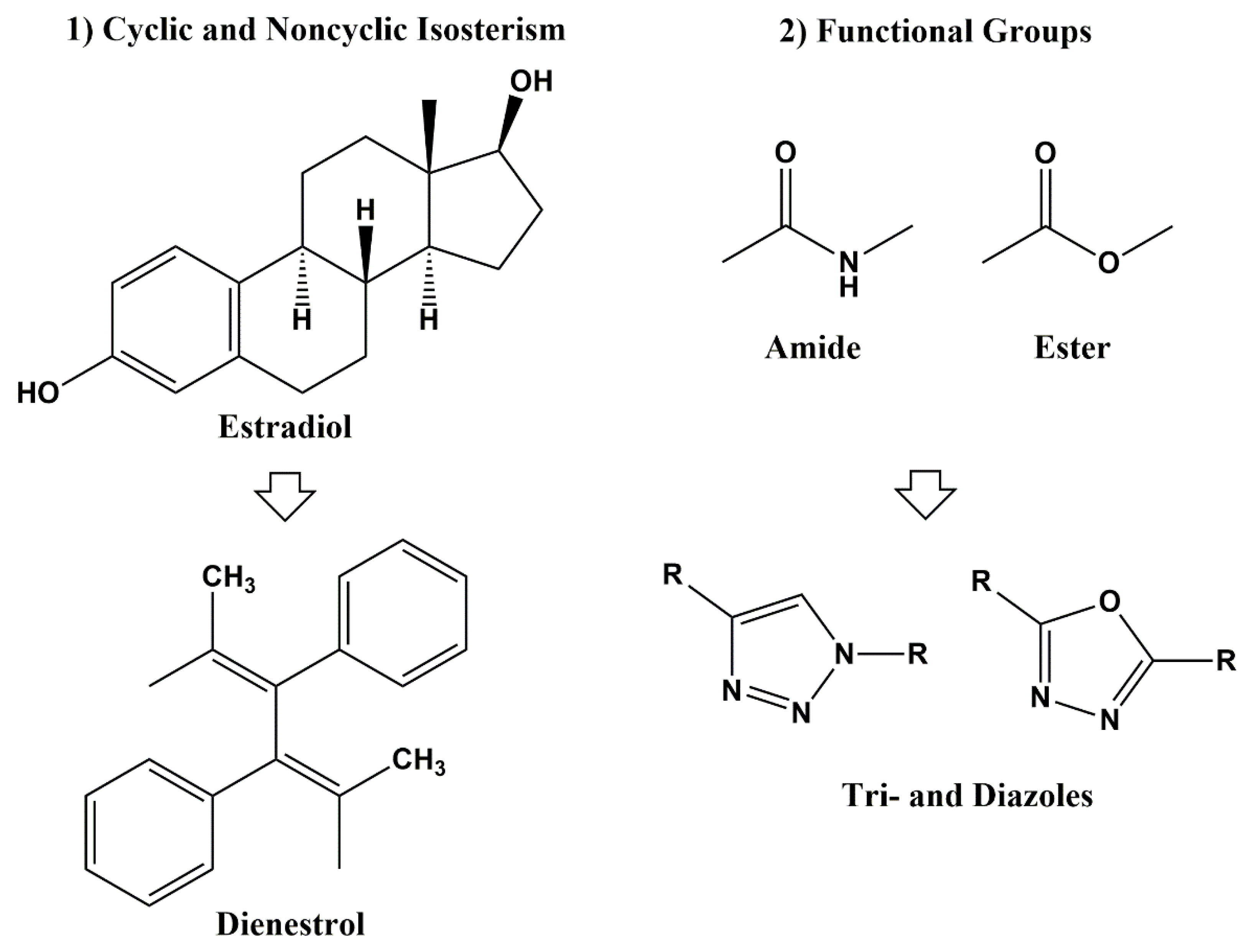
3. Classical and Non-Classical Bioisosteres
3.1. Recent Applications for Classical Bioisosteres in Anti-HIV Drug Design and Development
3.1.1. Monovalent Bioisosteres
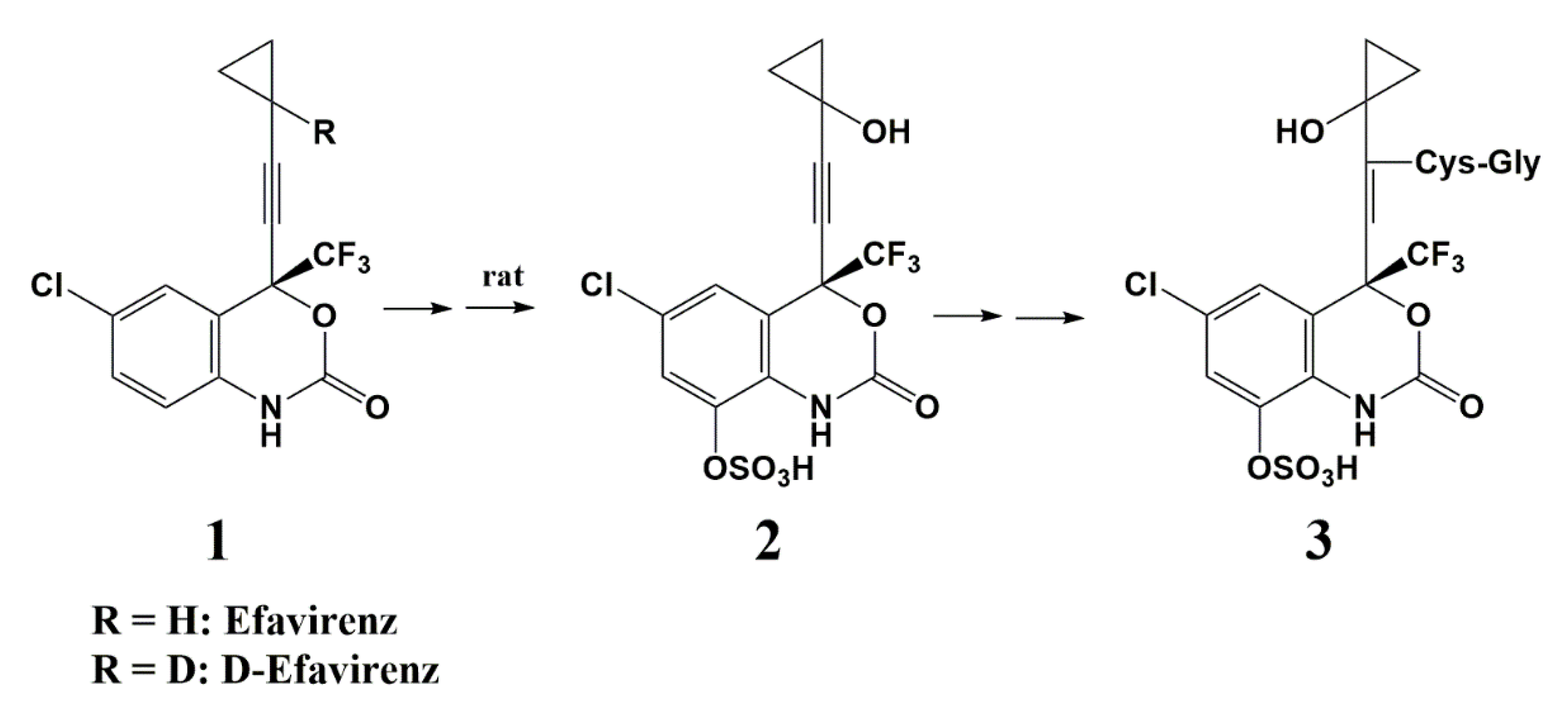
Deuterium as a Hydrogen Isostere in HIV-1 Reverse Transcriptase Inhibitors
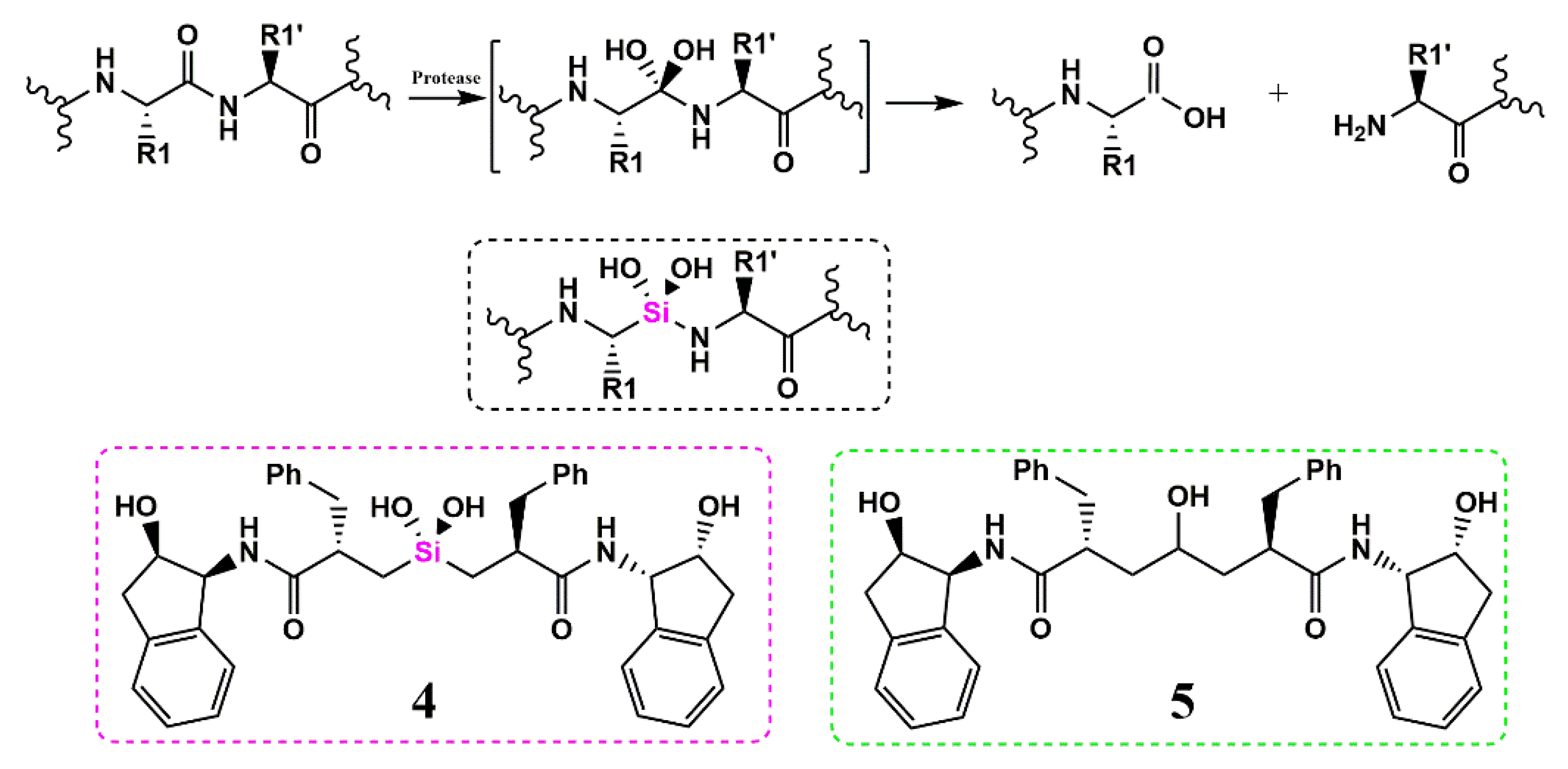
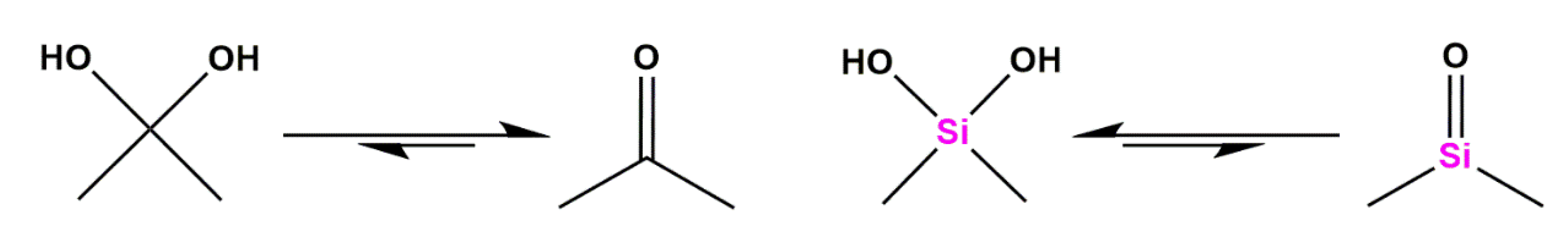
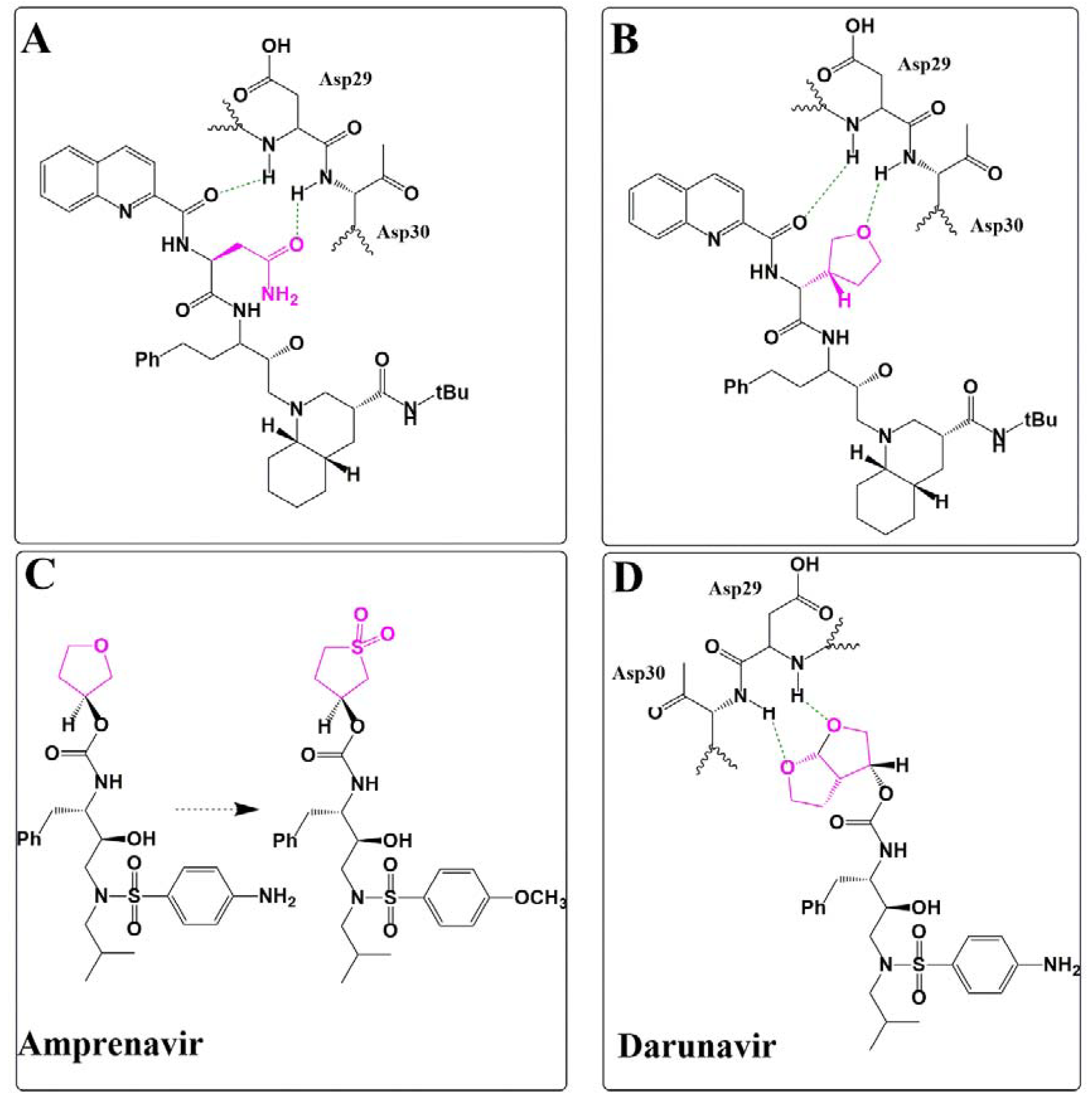
Silicon as a Carbon Isostere in HIV-1 Protease Inhibitors
3.1.2. Divalent Bioisosteres
Ether/Sulfone Substitution in HIV-1 Protease Inhibitor Design
3.2. Non-Classical Bioisosteres in Anti-HIV-1 Drug Design and Development
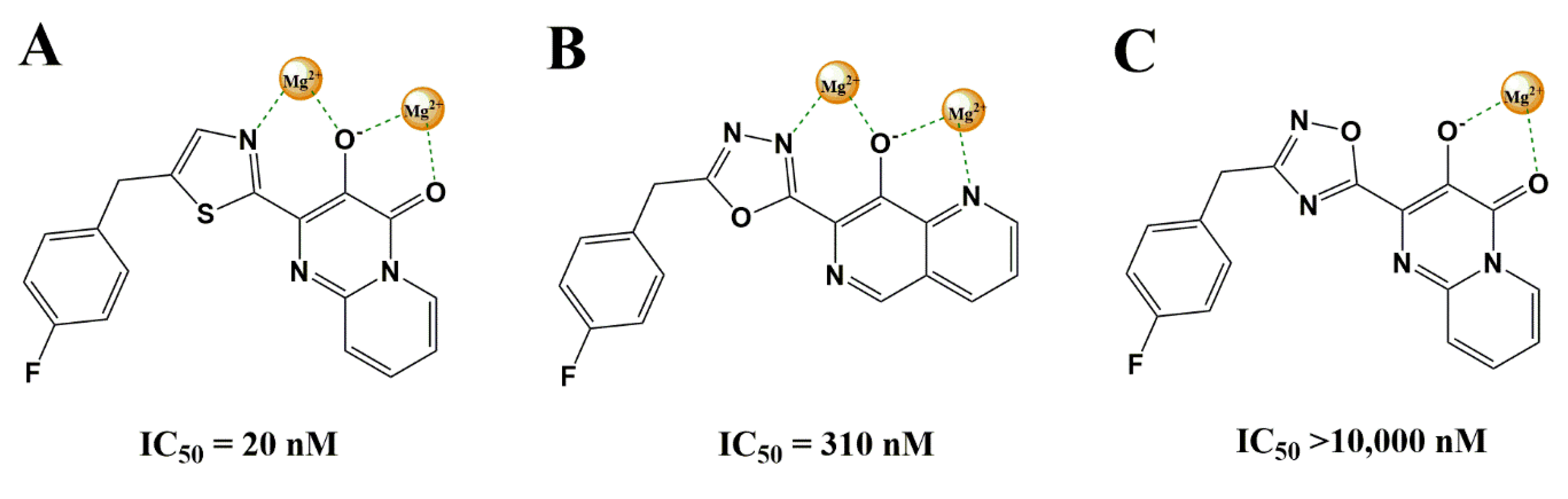
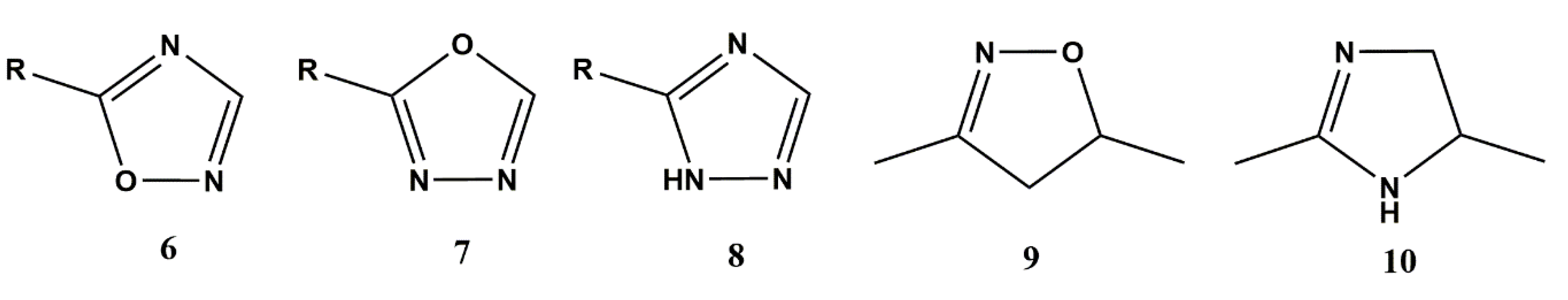
3.2.1. Heterocyclic Bioisosterism
3.2.2. Bioisosterism of Functional Groups
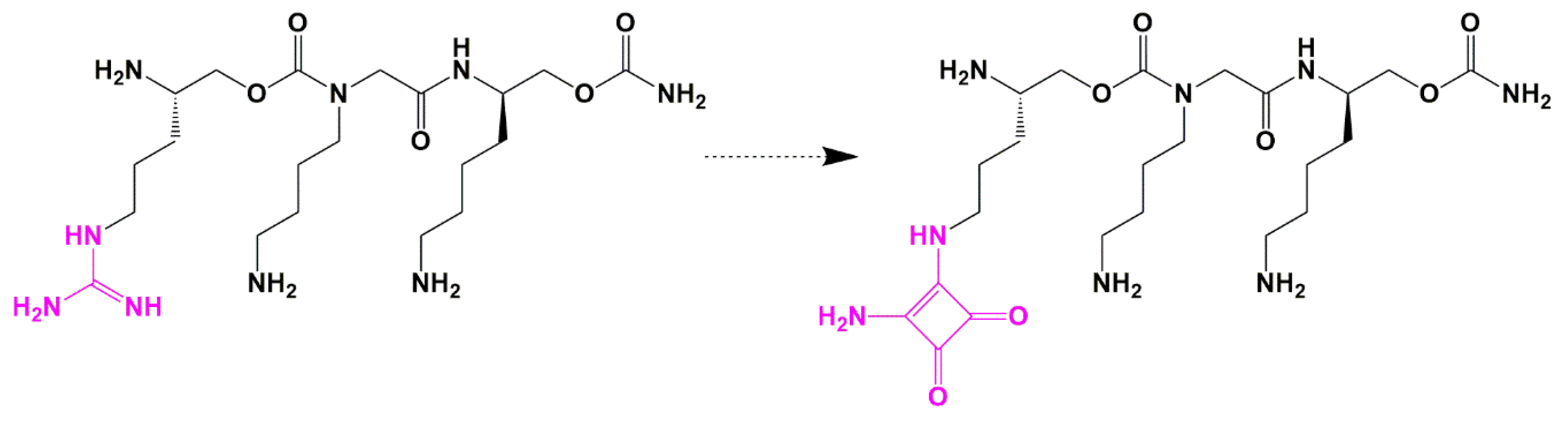
Guanidine Bioisosteres
Amide and Ester Bioisosterism
4. In Silico Molecular Field-Based Scaffold Hopping for Non-Classical Bioisostere Identification
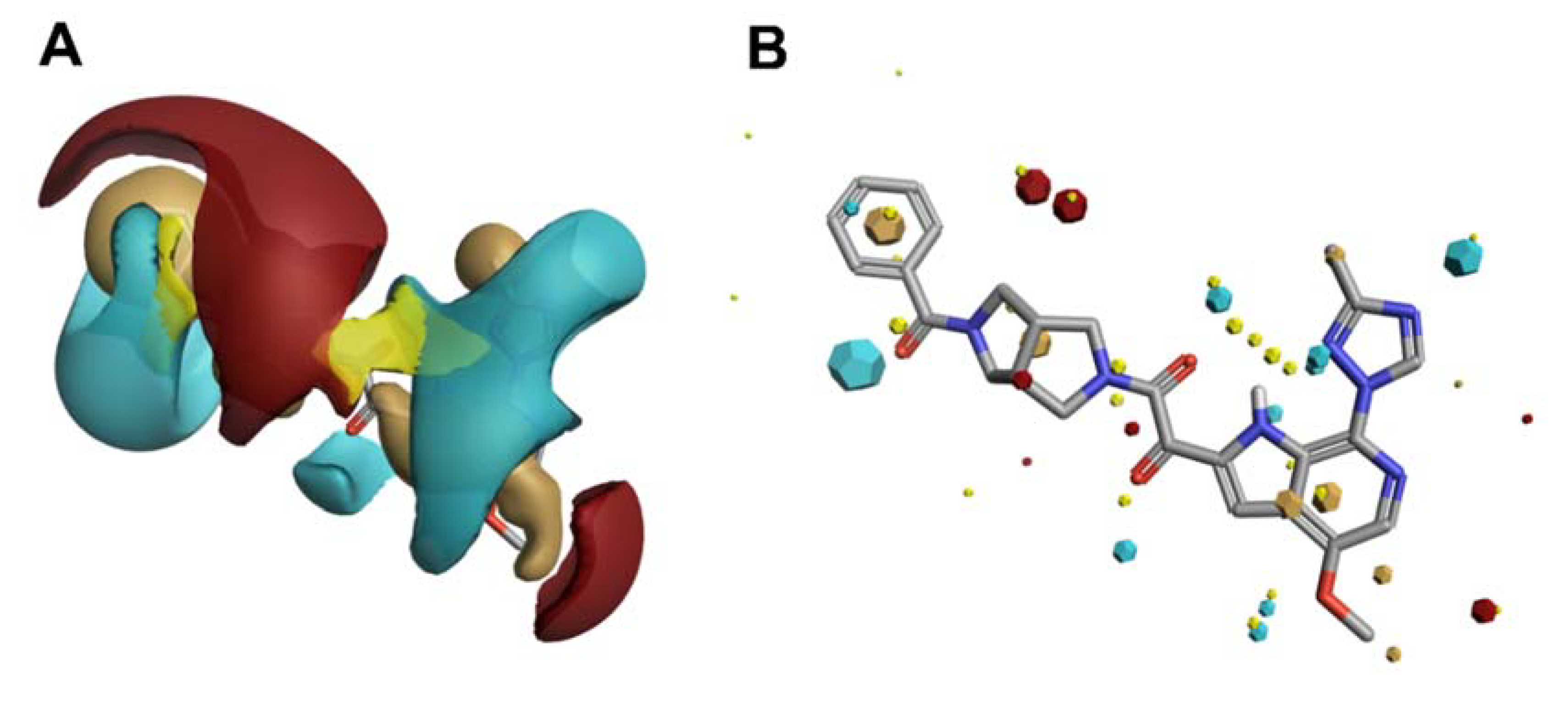
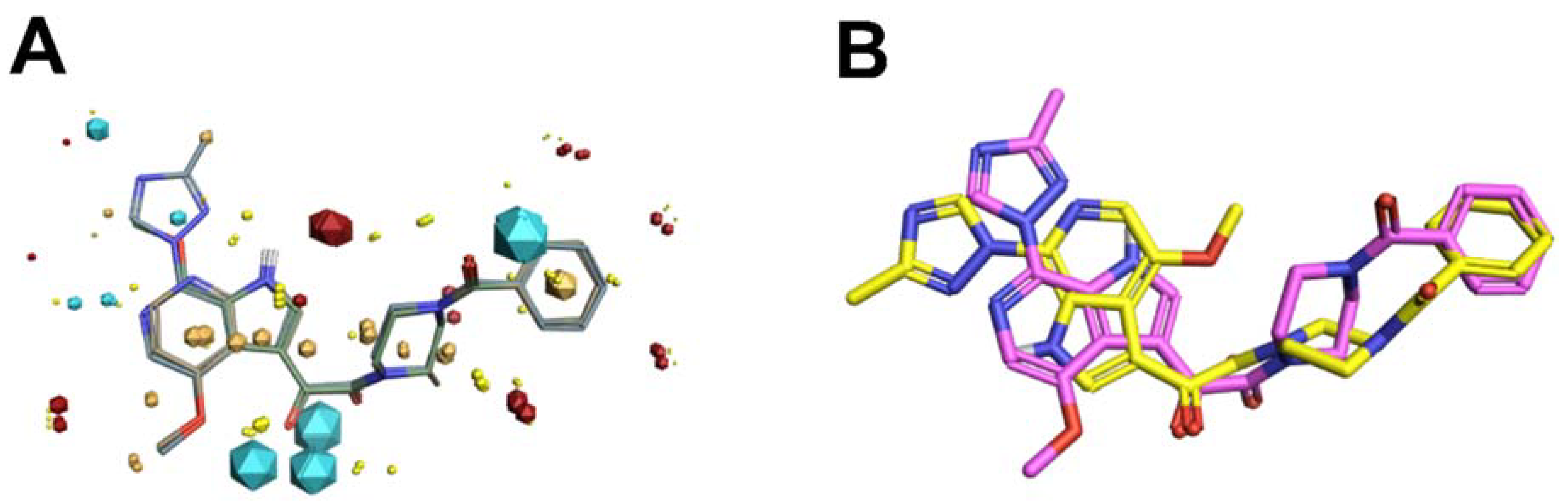
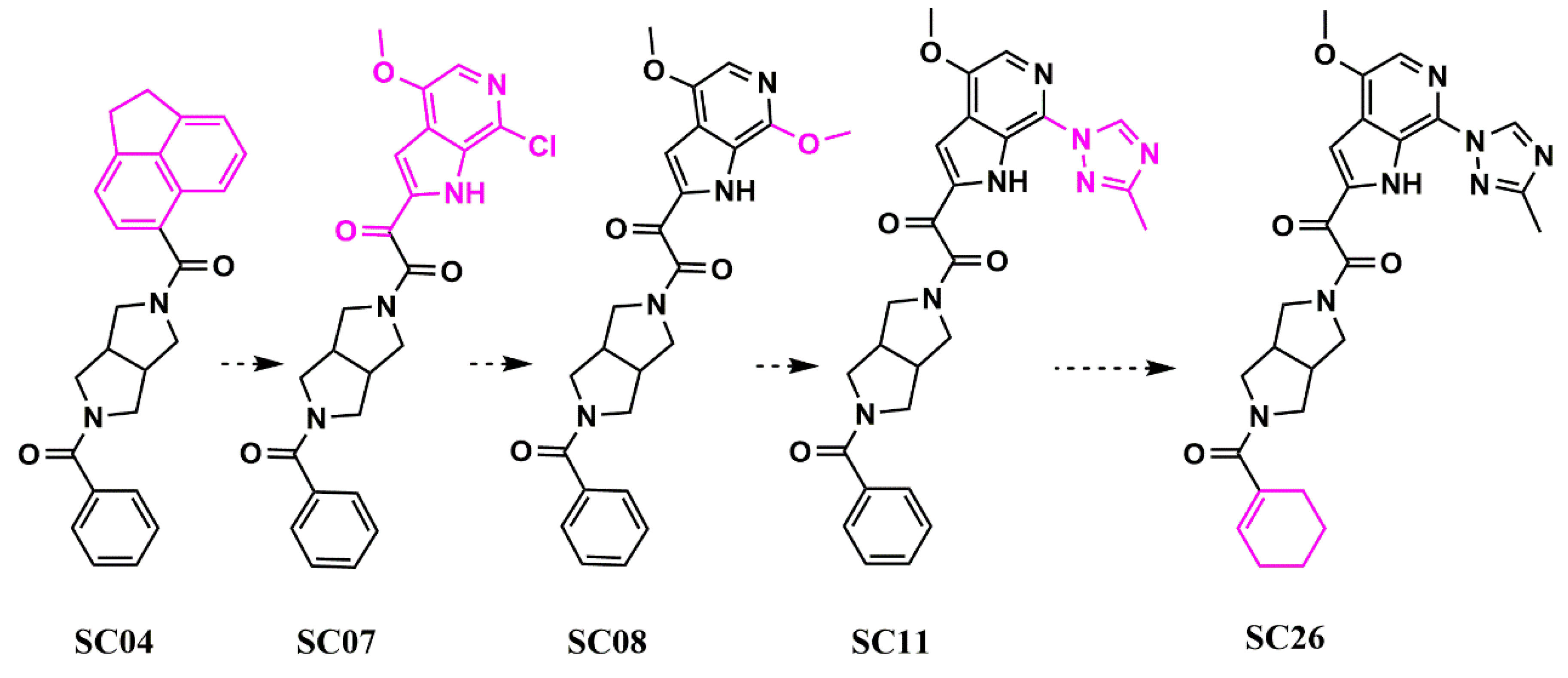
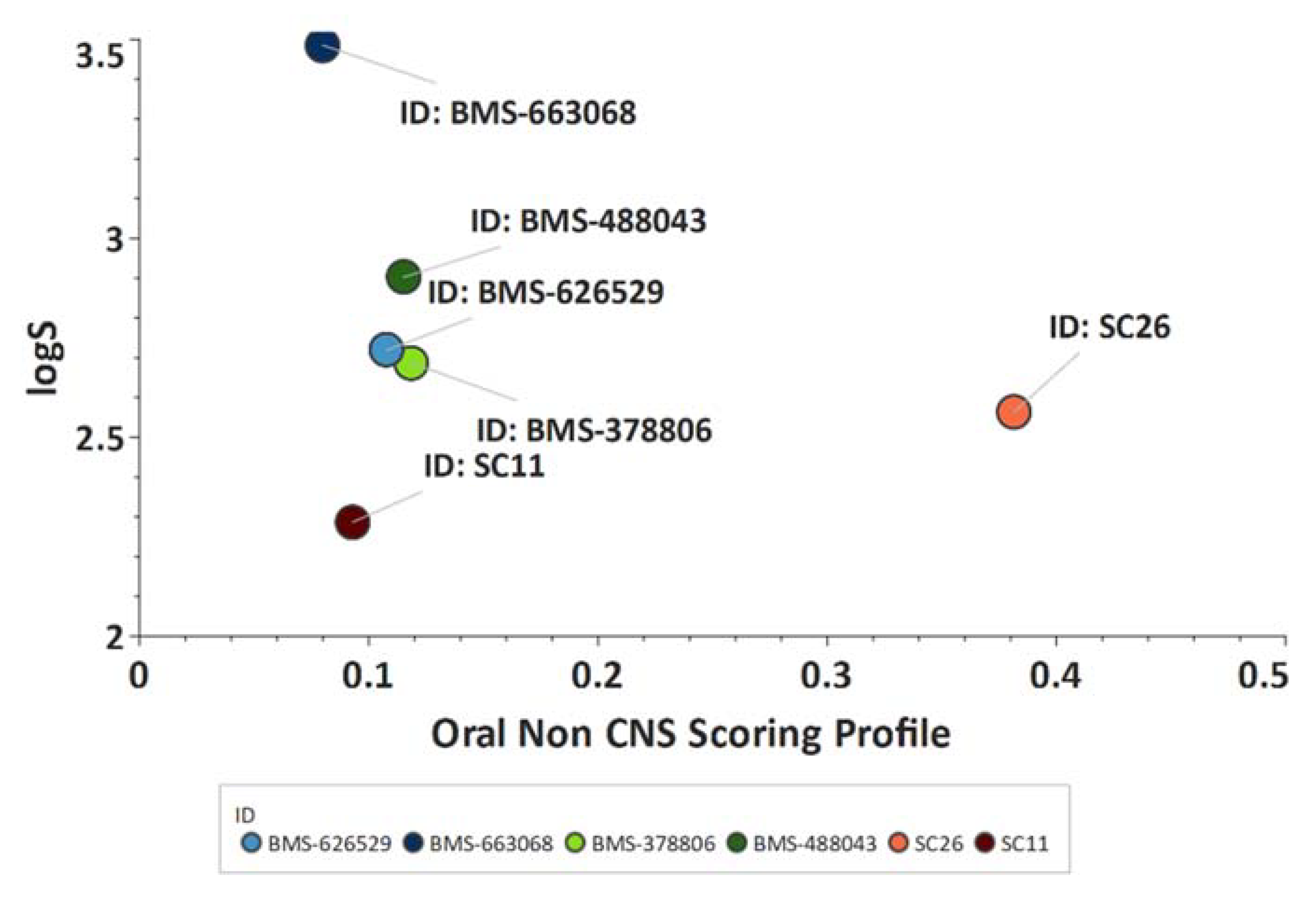
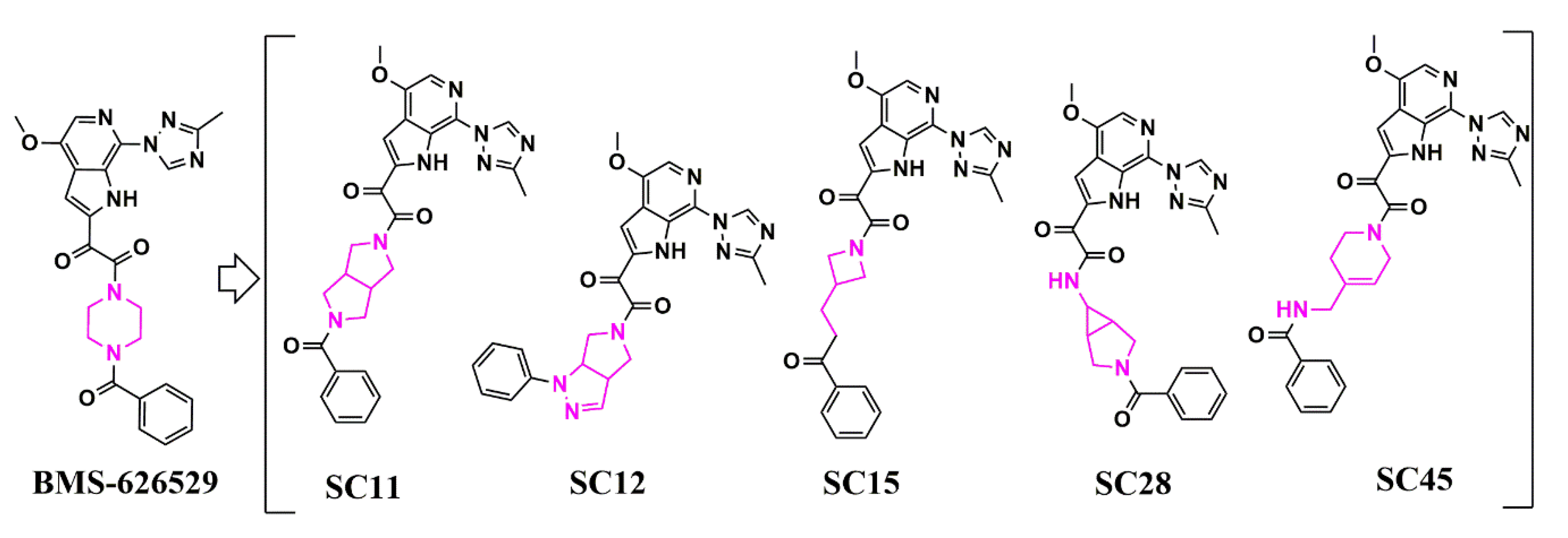
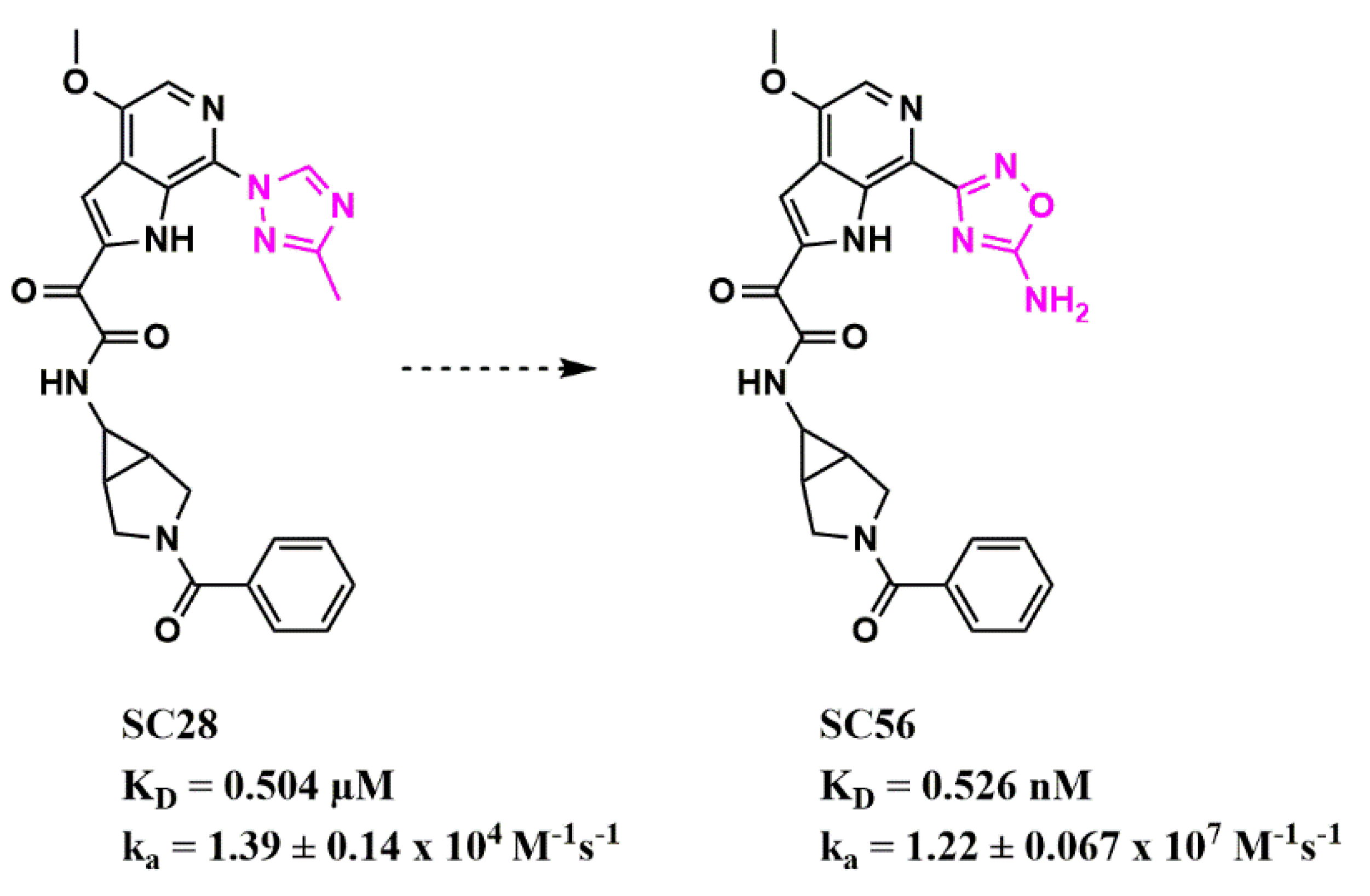
Scaffold Hopping for Potency and ADME Improvement of HIV-1 Entry Leads
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Langmuir, I. Isomorphism, Isosterism and Covalence. J. Am. Chem. Soc. 1919, 41, 1543–1559. [Google Scholar] [CrossRef]
- Grimm, H.G. Structure and Size of the Non-metallic Hydrides. Z. Electrochem. 1925, 31, 474–480. [Google Scholar]
- Grimm, H.G. On the Systematic Arrangement of Chemical Compounds from the Perspective of Research on Atomic Composition; and on Some Challenges in Experimental Chemistry. Naturwissenschaften 1929, 17, 557–564. [Google Scholar] [CrossRef]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef]
- Friedman, H.L. Influence of Isosteric Replacements upon Biological Activity. Nasnrs 1951, 206, 295–358. [Google Scholar]
- Burger, A. Isosterism and Bioisosterism in Drug Design. In Progress in Drug Research/Fortschritte der Arzneimittelforschung/Progrès des Recherches Pharmaceutiques; Birkhäuser: Basel, Switzerland, 1991. [Google Scholar]
- Doak, G.O.; Freedman, L.E. Medicinal Chemistry, 3rd ed.; Wiley-Interscience: New York, NY, USA, 1970. [Google Scholar]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef]
- Wade, D. Deuterium isotope effects on noncovalent interactions between molecules. Chem. Biol. Interact. 1999, 117, 191–217. [Google Scholar] [CrossRef]
- Hu, W.S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef]
- Mutlib, A.E.; Gerson, R.J.; Meunier, P.C.; Haley, P.J.; Chen, H.; Gan, L.S.; Davies, M.H.; Gemzik, B.; Christ, D.D.; Krahn, D.F.; et al. The species-dependent metabolism of efavirenz produces a nephrotoxic glutathione conjugate in rats. Toxicol. Appl. Pharmacol. 2000, 169, 102–113. [Google Scholar] [CrossRef]
- Bertrand, G. The modest undressing of a silicon center. Science 2004, 305, 783–785. [Google Scholar] [CrossRef]
- Franz, A.K.; Wilson, S.O. Organosilicon molecules with medicinal applications. J. Med. Chem. 2013, 56, 388–405. [Google Scholar] [CrossRef] [PubMed]
- West, R.; Baney, R.H. Hydrogen Bonding Studies. II. The Acidity and Basicity of Silanols Compared to Alcohols. J. Am. Chem. Soc. 1959, 81, 6145–6148. [Google Scholar] [CrossRef]
- Sieburth, S.M.; Chen, C.A. Silanediol protease inhibitors: From conception to validation. Eur. J. Org. Chem. 2006, 2006, 311–322. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104. [Google Scholar]
- Chen, C.A.; Sieburth, S.M.; Glekas, A.; Hewitt, G.W.; Trainor, G.L.; Erickson-Viitanen, S.; Garber, S.S.; Cordova, B.; Jeffry, S.; Klabe, R.M. Drug design with a new transition state analog of the hydrated carbonyl: Silicon-based inhibitors of the HIV protease. Chem. Biol. 2001, 8, 1161–1166. [Google Scholar] [CrossRef]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2004/021785lbl.pdf (accessed on 29 February 2019).
- Ghosh, A.K.; Chapsal, B.D.; Weber, I.T.; Mitsuya, H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 2008, 41, 78–86. [Google Scholar] [CrossRef]
- Ghosh, A.K. Harnessing nature’s insight: Design of aspartyl protease inhibitors from treatment of drug-resistant HIV to Alzheimer’s disease. J. Med. Chem. 2009, 52, 2163–2176. [Google Scholar] [CrossRef]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021976s003s004lbl.pdf (accessed on 29 February 2019).
- Craigie, R. The molecular biology of HIV integrase. Future Virol. 2012, 7, 679–686. [Google Scholar] [CrossRef]
- Le, G.; Vandegraaff, N.; Rhodes, D.I.; Jones, E.D.; Coates, J.A.; Lu, L.; Li, X.; Yu, C.; Feng, X.; Deadman, J.J. Discovery of potent HIV integrase inhibitors active against raltegravir resistant viruses. Bioorganic Med. Chem. Lett. 2010, 20, 5013–5018. [Google Scholar] [CrossRef]
- Le, G.; Vandegraaff, N.; Rhodes, D.I.; Jones, E.D.; Coates, J.A.; Thienthong, N.; Winfield, L.J.; Lu, L.; Li, X.; Yu, C.; et al. Design of a series of bicyclic HIV-1 integrase inhibitors. Part 2: Azoles: Effective metal chelators. Bioorganic Med. Chem. Lett. 2010, 20, 5909–5912. [Google Scholar] [CrossRef]
- Johns, B.A.; Weatherhead, J.G.; Allen, S.H.; Thompson, J.B.; Garvey, E.P.; Foster, S.A.; Jeffrey, J.L.; Miller, W.H. The use of oxadiazole and triazole substituted naphthyridines as HIV-1 integrase inhibitors. Part 1: Establishing the pharmacophore. Bioorganic Med. Chem. Lett. 2009, 19, 1802–1806. [Google Scholar] [CrossRef] [PubMed]
- Johns, B.A.; Weatherhead, J.G.; Allen, S.H.; Thompson, J.B.; Garvey, E.P.; Foster, S.A.; Jeffrey, J.L.; Miller, W.H. 1, 3, 4-Oxadiazole substituted naphthyridines as HIV-1 integrase inhibitors. Part 2: SAR of the C5 position. Bioorganic Med. Chem. Lett. 2009, 19, 1807–1810. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.D.; Vandegraaff, N.; Le, G.; Choi, N.; Issa, W.; Macfarlane, K.; Thienthong, N.; Winfield, L.J.; Coates, J.A.; Lu, L.; et al. Design of a series of bicyclic HIV-1 integrase inhibitors. Part 1: Selection of the scaffold. Bioorganic Med. Chem. Lett. 2010, 20, 5913–5917. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Sodroski, J.; Patarca, R.; Rosen, C.; Wong-Staal, F.; Haseltine, W. Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science 1985, 229, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Holland, E.C. HIV-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167. [Google Scholar] [CrossRef]
- Lee, C.W.; Cao, H.; Ichiyama, K.; Rana, T.M. Design and synthesis of a novel peptidomimetic inhibitor of HIV-1 Tat-TAR interactions: Squaryldiamide as a new potential bioisostere of unsubstituted guanidine. Bioorganic Med. Chem. Lett. 2005, 15, 4243–4246. [Google Scholar] [CrossRef]
- Strebel, K.; Daugherty, D.; Clouse, K.; Cohen, D.; Folks, T.; Martin, M.A. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature 1987, 328, 728–730. [Google Scholar] [CrossRef]
- Gabuzda, D.H.; Lawrence, K.; Langhoff, E.; Terwilliger, E.; Dorfman, T.; Haseltine, W.A.; Sodroski, J. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 1992, 66, 6489–6495. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Nathans, R.; Cao, H.; Sharova, N.; Ali, A.; Sharkey, M.; Stranska, R.; Stevenson, M.; Rana, T.M. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 2008, 26, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Wang, J.; Nathans, R.S.; Cao, H.; Sharova, N.; Stevenson, M.; Rana, T.M. Synthesis and structure-activity relationship studies of HIV-1 virion infectivity factor (Vif) inhibitors that block viral replication. ChemMedChem 2012, 7, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
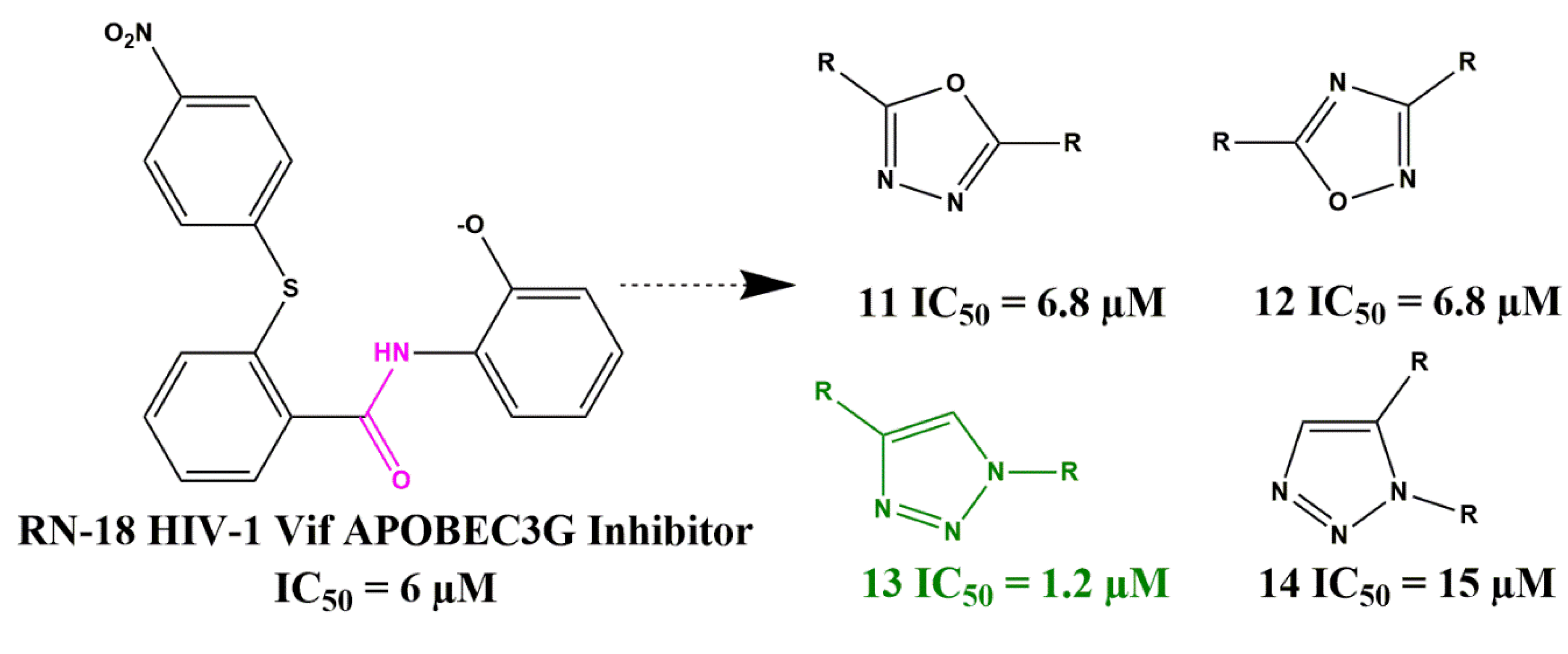
- Mohammed, I.; Parai, M.K.; Jiang, X.; Sharova, N.; Singh, G.; Stevenson, M.; Rana, T.M. SAR and Lead Optimization of an HIV-1 Vif-APOBEC3G Axis Inhibitor. ACS Med. Chem. Lett. 2012, 3, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Kummetha, I.R.; Singh, G.; Sharova, N.; Lichinchi, G.; Dang, J.; Stevenson, M.; Rana, T.M. 1, 2, 3-Triazoles as Amide Bioisosteres: Discovery of a New Class of Potent HIV-1 Vif Antagonists. J. Med. Chem. 2016, 59, 7677–7682. [Google Scholar] [CrossRef]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Modeling 2006, 46, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field technology applied to virtual screening and finding the bioactive conformation. Expert Opin. Drug Discov. 2007, 2, 131–144. [Google Scholar] [CrossRef]
- Cheeseright, T.J.; Holm, M.; Lehmann, F.; Luik, S.; Gottert, M.; Melville, J.L.; Laufer, S. Novel lead structures for p38 MAP kinase via FieldScreen virtual screening. J. Med. Chem. 2009, 52, 4200–4209. [Google Scholar] [CrossRef]
- Cheeseright, T.J.; Mackey, M.D.; Melville, J.L.; Vinter, J.G. FieldScreen: Virtual screening using molecular fields. Application to the DUD data set. J. Chem. Inf. Modeling 2008, 48, 2108–2117. [Google Scholar] [CrossRef]
- Cheeseright, T.J.; Mackey, M.D.; Scoffin, R.A. High content pharmacophores from molecular fields: A biologically relevant method for comparing and understanding ligands. Curr. Comput. Aided Drug Des. 2011, 7, 190–205. [Google Scholar] [CrossRef]
- Apaya, R.P.; Lucchese, B.; Price, S.L.; Vinter, J.G. The matching of electrostatic extrema: A useful method in drug design? A study of phosphodiesterase III inhibitors. J. Comput. Aided Mol. Des. 1995, 9, 33–43. [Google Scholar] [CrossRef]
- Vinter, J.G. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J. Comput. Aided Mol. Des. 1994, 8, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Ketas, T.J.; Schader, S.M.; Zurita, J.; Teo, E.; Polonis, V.; Lu, M.; Klasse, P.J.; Moore, J.P. Entry inhibitor-based microbicides are active in vitro against HIV-1 isolates from multiple genetic subtypes. Virology 2007, 364, 431–440. [Google Scholar] [CrossRef][Green Version]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H- pyrrolo[2, 3-b]pyridin-3-yl)oxoacetyl]-2- (R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef] [PubMed]
- Landry, I.; Zhu, L.; Tarif, M.A.; Hruska, M.; Sadler, B.M.; Pitsiu, M.; Joshi, S.; Hanna, G.J.; Lataillade, M.; Boulton, D.W.; et al. Model-Based Phase 3 Dose Selection for HIV-1 Attachment Inhibitor Prodrug BMS-663068 in HIV-1-Infected Patients: Population Pharmacokinetics/Pharmacodynamics of the Active Moiety, BMS-626529. Antimicrob. Agents Chemother. 2016, 60, 2782–2789. [Google Scholar] [CrossRef] [PubMed]
- Nowicka-Sans, B.; Gong, Y.F.; McAuliffe, B.; Dicker, I.; Ho, H.T.; Zhou, N.; Eggers, B.; Lin, P.F.; Ray, N.; Wind-Rotolo, M.; et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob. Agents Chemother. 2012, 56, 3498–3507. [Google Scholar] [CrossRef] [PubMed]
- Tuyishime, M.; Danish, M.; Princiotto, A.; Mankowski, M.K.; Lawrence, R.; Lombart, H.G.; Esikov, K.; Berniac, J.; Liang, K.; Ji, J.; et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorganic Med. Chem. Lett. 2014, 24, 5439–5445. [Google Scholar] [CrossRef]
- Lin, P.F.; Blair, W.; Wang, T.; Spicer, T.; Guo, Q.; Zhou, N.; Gong, Y.F.; Wang, H.G.; Rose, R.; Yamanaka, G.; et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. USA 2003, 100, 11013–11018. [Google Scholar] [CrossRef]
- Yang, Z.; Zadjura, L.M.; Marino, A.M.; D’Arienzo, C.J.; Malinowski, J.; Gesenberg, C.; Lin, P.F.; Colonno, R.J.; Wang, T.; Kadow, J.F.; et al. Utilization of in vitro Caco-2 permeability and liver microsomal half-life screens in discovering BMS-488043, a novel HIV-1 attachment inhibitor with improved pharmacokinetic properties. J. Pharm. Sci. 2010, 99, 2135–2152. [Google Scholar] [CrossRef]
- Tuyishime, M.; Lawrence, R.; Cocklin, S. Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement. Bioorganic Med. Chem. Lett. 2016, 26, 228–234. [Google Scholar] [CrossRef]
- Meuser, M.E.; Rashad, A.A.; Ozorowski, G.; Dick, A.; Ward, A.B.; Cocklin, S. Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor. Molecules 2019, 24, 1581. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | N | O | F | Ne | Na+ |
|---|---|---|---|---|---|
| CH | NH | OH | FH | - | |
| CH2 | NH2 | OH2 | FH2+ | ||
| CH3 | NH3 | OH3+ | |||
| CH4 | NH4+ |
| Number of Valence Electrons | ||||
|---|---|---|---|---|
| 4 | 5 | 6 | 7 | 8 |
| N+ | P | S | Cl | ClH |
| P+ | As | Se | Br | BrH |
| S+ | Sb | Te | I | IH |
| As+ | PH | SH | SH2 | |
| Sb+ | PH2 | PH3 | ||
| Compound | IC50 JR-CSF | IC50 B41 | IC50 HxBc2 | IC50 AMLV |
|---|---|---|---|---|
| SC11 | 0.0008 ± 0.0001 | 0.002 ± 0.0002 | 0.001 ± 0.0001 | N.A. |
| SC12 | 0.008 ± 0.002 | 0.006 ± 0.003 | 0.080 ± 0.020 | N.A. |
| SC15 | 0.003 ± 0.001 | 0.007 ± 0.001 | 0.009 ± 0.001 | N.A. |
| SC28 | 0.096 ± 0.019 | 0.085 ± 0.03 | 0.069 ± 0.014 | N.A. |
| SC45 | 0.224 ± 0.017 | 0.350 ± 0.030 | 0.380 ± 0.030 | N.A. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dick, A.; Cocklin, S. Bioisosteric Replacement as a Tool in Anti-HIV Drug Design. Pharmaceuticals 2020, 13, 36. https://doi.org/10.3390/ph13030036
Dick A, Cocklin S. Bioisosteric Replacement as a Tool in Anti-HIV Drug Design. Pharmaceuticals. 2020; 13(3):36. https://doi.org/10.3390/ph13030036
Chicago/Turabian StyleDick, Alexej, and Simon Cocklin. 2020. "Bioisosteric Replacement as a Tool in Anti-HIV Drug Design" Pharmaceuticals 13, no. 3: 36. https://doi.org/10.3390/ph13030036
APA StyleDick, A., & Cocklin, S. (2020). Bioisosteric Replacement as a Tool in Anti-HIV Drug Design. Pharmaceuticals, 13(3), 36. https://doi.org/10.3390/ph13030036