Metformin Reduces NGF-Induced Tumour Promoter Effects in Epithelial Ovarian Cancer Cells

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results
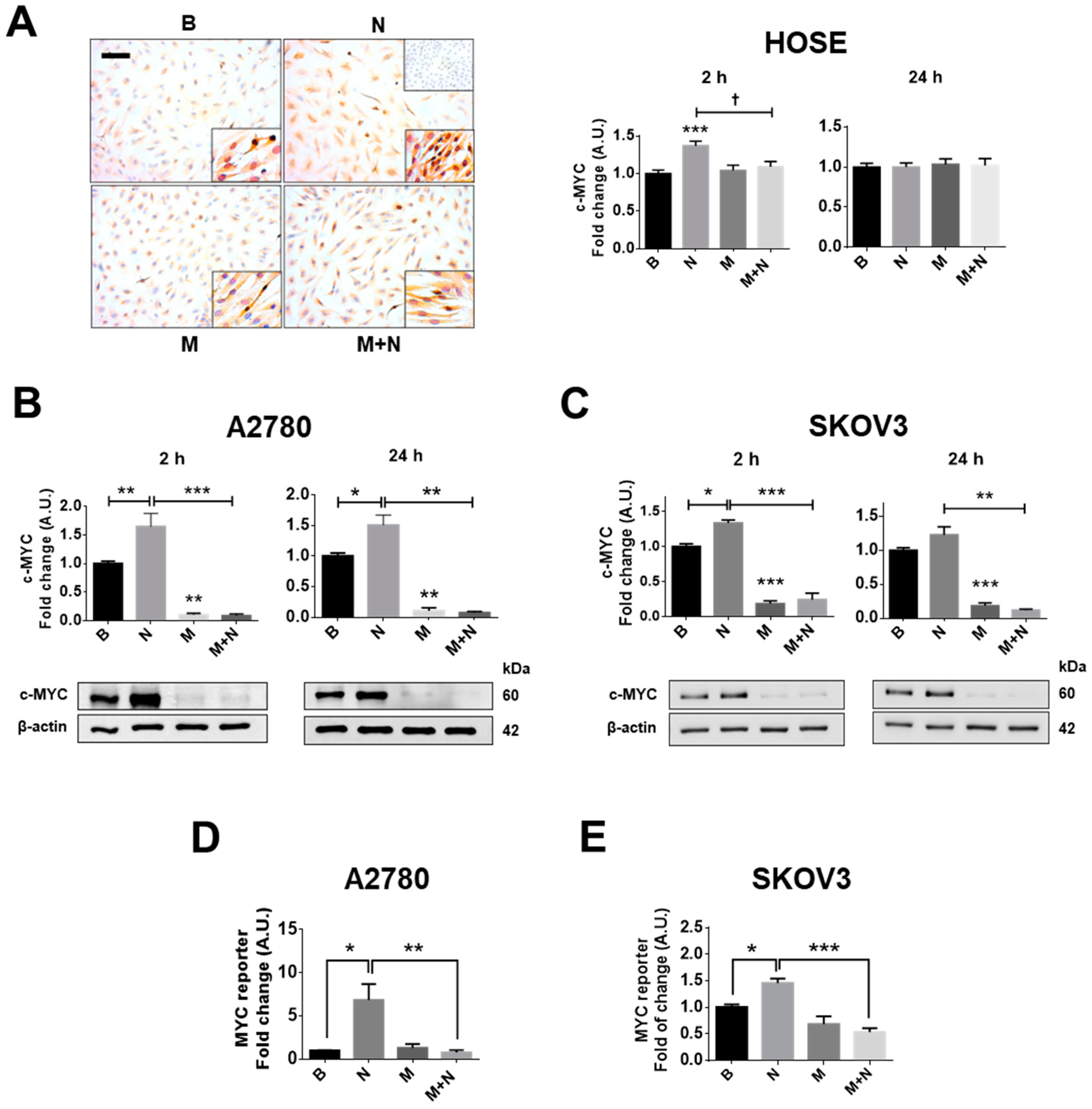
2.1. Metformin Blocks the NGF-Induced Increase in c-MYC Protein Levels and Transcriptional Activity in Ovarian Cancer Cells
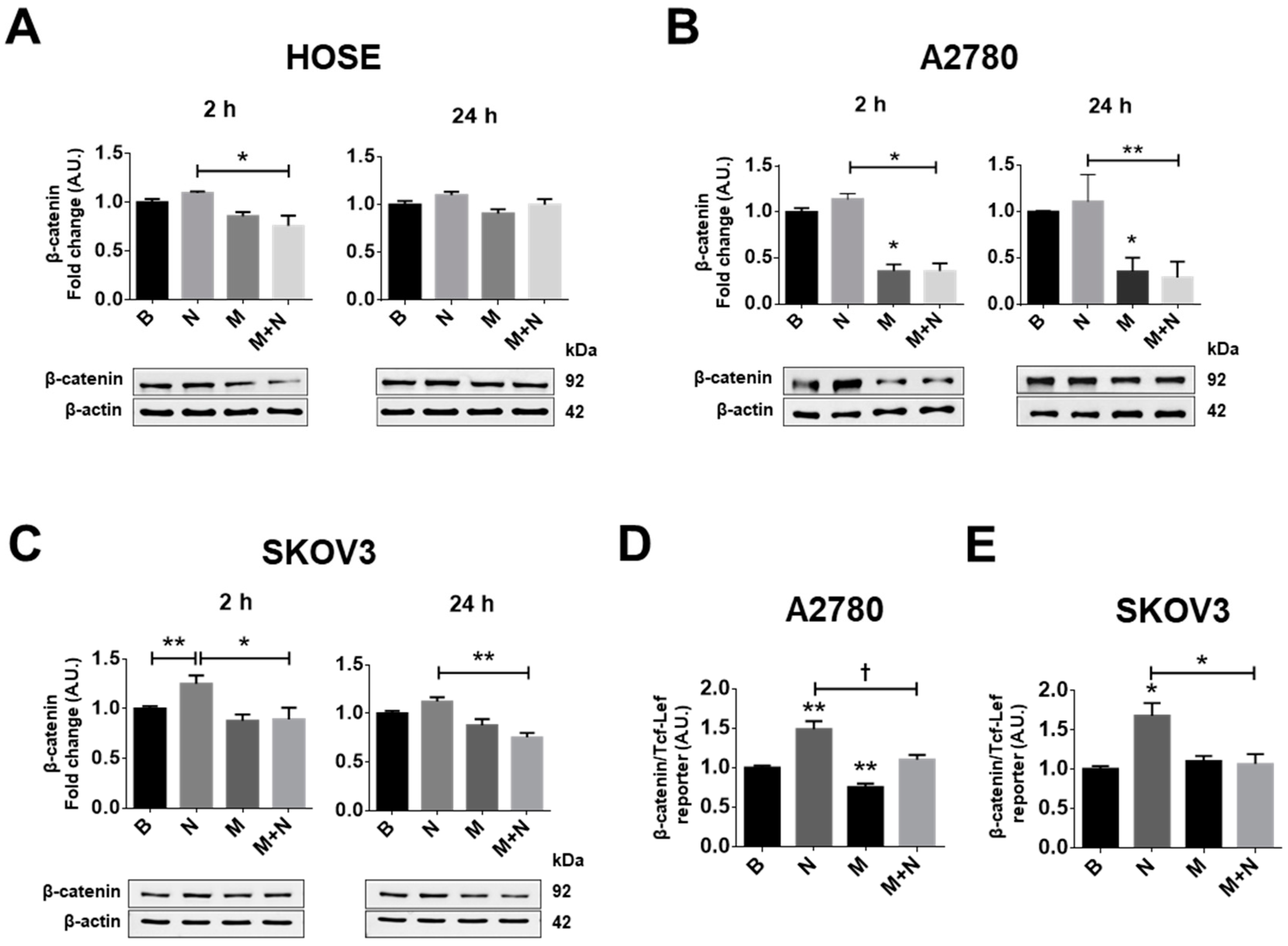
2.2. Metformin Treatment Prevents the Increase in β-Catenin/TCF-Lef Transcriptional Activity Induced by NGF in Ovarian Cancer Cells
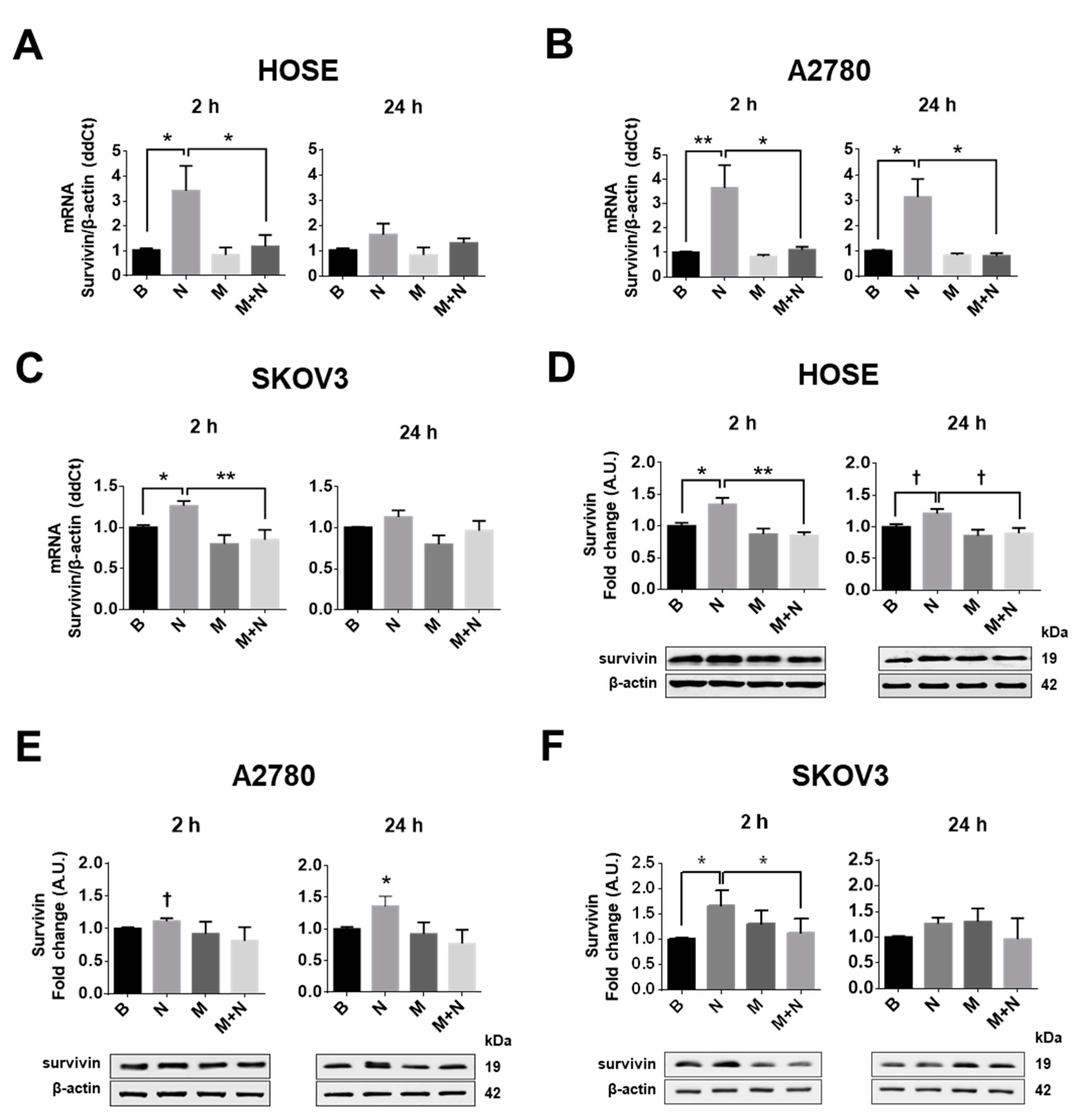
2.3. Metformin Decreases NGF-Induced Survivin Levels in Ovarian Cells
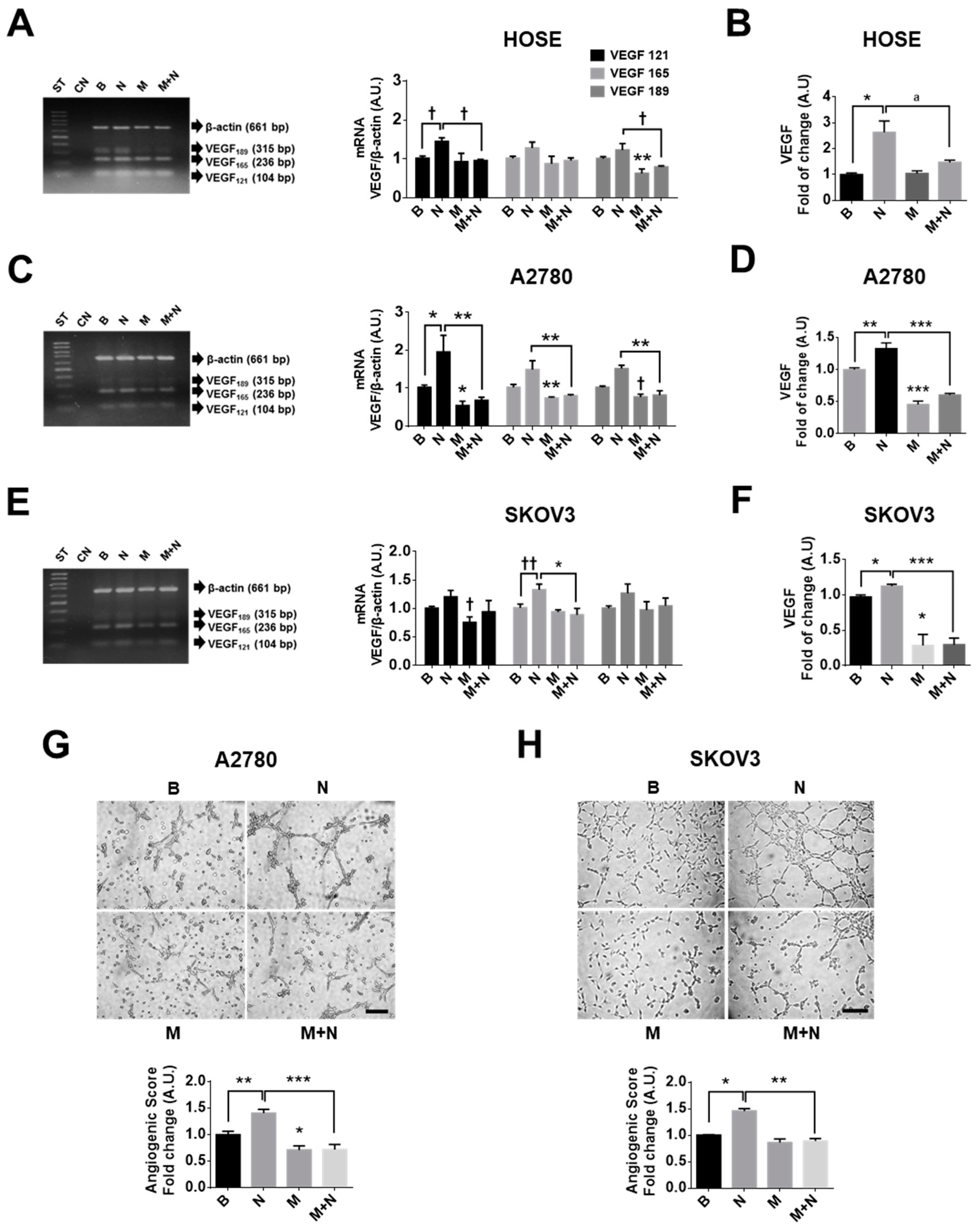
2.4. Metformin Decreases NGF-Induced VEGF Expression in Ovarian Cancer Cells
2.5. Metformin Decreases the Vasculogenic Potential of Ovarian Cancer Cells Induced by NGF
2.6. Metformin Increases Levels of miR-23b and miR-145 and Prevents the NGF-Induced Decreases of These miRs in EOC Cells
3. Discussion
4. Material and Methods
4.1. Cell Culture and Treatments
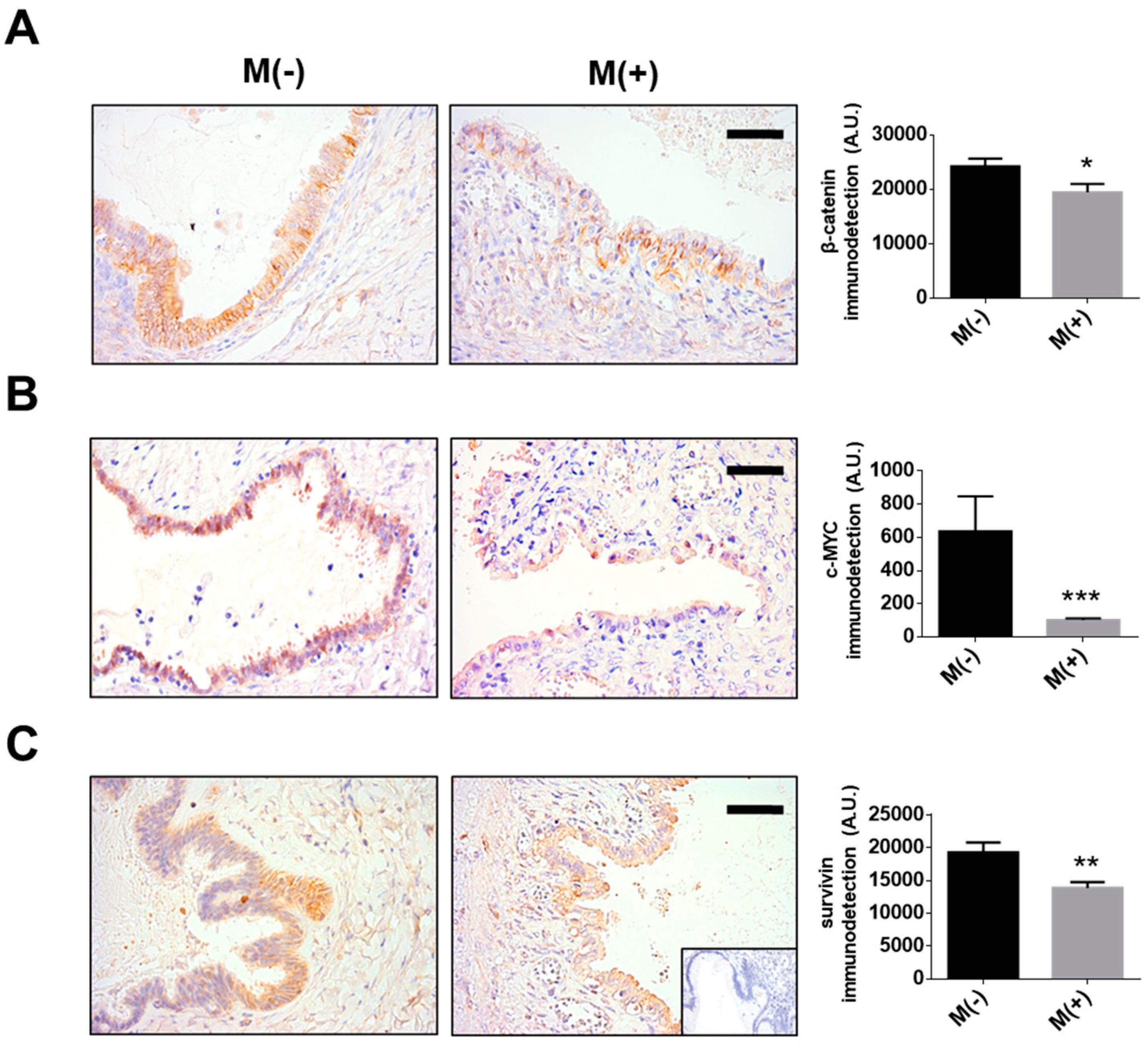
4.2. Tissue Samples
4.3. Protein Detection by Western Blotting
4.4. Immunohistochemistry
4.5. Immunocytochemistry
4.6. VEGF Determination
4.7. Total RNA Extraction and RT-PCR
4.8. β-Catenin/Tcf-Lef Reporter Assay
4.9. MYC Reporter Assay
4.10. miR Extraction and qPCR
4.11. Invasion Assays
4.12. Vasculogenesis Assays
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [PubMed]
- American Cancer Society. Survival Rates for Ovarian Cancer, by Stage. Available online: https://www.cancer.org/cancer/ovarian-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 21 September 2020).
- Cancer Research UK. Ovarian Cancer. Available online: https://www.cancerresearchuk.org/about-cancer/ovarian-cancer/survival (accessed on 21 September 2020).
- Tapia, V.; Gabler, F.; Munoz, M.; Yazigi, R.; Paredes, A.; Selman, A.; Vega, M.; Romero, C. Tyrosine kinase a receptor (trka): A potential marker in epithelial ovarian cancer. Gynecol. Oncol. 2011, 121, 13–23. [Google Scholar] [CrossRef]
- Duncan, W.C.; Nio-Kobayashi, J. Targeting angiogenesis in the pathological ovary. Reprod. Fertil. Dev. 2013, 25, 362–371. [Google Scholar] [CrossRef]
- Doubeni, C.A.; Doubeni, A.R.; Myers, A.E. Diagnosis and management of ovarian cancer. Am. Fam. Physician 2016, 93, 937–944. [Google Scholar] [PubMed]
- Urzua, U.; Tapia, V.; Geraldo, M.P.; Selman, A.; Vega, M.; Romero, C. Nerve growth factor stimulates cellular proliferation of human epithelial ovarian cancer. Horm. Metab. Res. 2012, 44, 656–661. [Google Scholar] [CrossRef]
- Campos, X.; Munoz, Y.; Selman, A.; Yazigi, R.; Moyano, L.; Weinstein-Oppenheimer, C.; Lara, H.E.; Romero, C. Nerve growth factor and its high-affinity receptor trka participate in the control of vascular endothelial growth factor expression in epithelial ovarian cancer. Gynecol. Oncol. 2007, 104, 168–175. [Google Scholar] [CrossRef]
- Garrido, M.P.; Torres, I.; Vega, M.; Romero, C. Angiogenesis in gynecological cancers: Role of neurotrophins. Front. Oncol. 2019, 9, 913. [Google Scholar] [CrossRef]
- Garrido, M.P.; Hurtado, I.; Valenzuela-Valderrama, M.; Salvatierra, R.; Hernandez, A.; Vega, M.; Selman, A.; Quest, A.F.G.; Romero, C. Ngf-enhanced vasculogenic properties of epithelial ovarian cancer cells is reduced by inhibition of the cox-2/pge2 signaling axis. Cancers 2019, 11, 1970. [Google Scholar] [CrossRef]
- Zhang, J.; Shemezis, J.R.; McQuinn, E.R.; Wang, J.; Sverdlov, M.; Chenn, A. Akt activation by n-cadherin regulates beta-catenin signaling and neuronal differentiation during cortical development. Neural. Dev. 2013, 8, 7. [Google Scholar] [CrossRef]
- Brown, J.B.; Cheresh, P.; Goretsky, T.; Managlia, E.; Grimm, G.R.; Ryu, H.; Zadeh, M.; Dirisina, R.; Barrett, T.A. Epithelial phosphatidylinositol-3-kinase signaling is required for beta-catenin activation and host defense against citrobacter rodentium infection. Infect. Immun. 2011, 79, 1863–1872. [Google Scholar] [CrossRef]
- Krejci, P.; Aklian, A.; Kaucka, M.; Sevcikova, E.; Prochazkova, J.; Masek, J.K.; Mikolka, P.; Pospisilova, T.; Spoustova, T.; Weis, M.; et al. Receptor tyrosine kinases activate canonical wnt/beta-catenin signaling via map kinase/lrp6 pathway and direct beta-catenin phosphorylation. PLoS ONE 2012, 7, e35826. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Meng, Q.; Liu, L.Z.; You, Y.P.; Liu, N.; Jiang, B.H. Regulation of survivin by pi3k/akt/p70s6k1 pathway. Biochem. Biophys. Res. Commun. 2010, 395, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Cai, W.; Zheng, Y.; Evers, B.M.; She, Q.B. Erk and akt signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene 2014, 33, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Li, G.; Wen, X.; Lv, Y.; Zhang, M.; Wei, Y.; Xie, W.; Zhong, P.; Pang, L. Prognostic significance of beta-catenin expression in patients with ovarian cancer: A meta-analysis. Gene 2018, 678, 270–279. [Google Scholar] [CrossRef]
- Liguang, Z.; Peishu, L.; Hongluan, M.; Hong, J.; Rong, W.; Wachtel, M.S.; Frezza, E.E. Survivin expression in ovarian cancer. Exp. Oncol. 2007, 29, 121–125. [Google Scholar]
- Chiou, S.K.; Jones, M.K.; Tarnawski, A.S. Survivin-an anti-apoptosis protein: Its biological roles and implications for cancer and beyond. Med. Sci. Monit. 2003, 9, PI25–PI29. [Google Scholar]
- Fukuda, S.; Pelus, L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther. 2006, 5, 1087–1098. [Google Scholar] [CrossRef]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and tumorigenesis: Molecular mechanisms and therapeutic strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef]
- Asanuma, H.; Torigoe, T.; Kamiguchi, K.; Hirohashi, Y.; Ohmura, T.; Hirata, K.; Sato, M.; Sato, N. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/akt signaling pathway in breast cancer cells. Cancer Res. 2005, 65, 11018–11025. [Google Scholar] [CrossRef]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Sanhueza, C.; Wehinger, S.; Castillo Bennett, J.; Valenzuela, M.; Owen, G.I.; Quest, A.F. The twisted survivin connection to angiogenesis. Mol. Cancer 2015, 14, 198. [Google Scholar] [CrossRef]
- Cohen, C.; Lohmann, C.M.; Cotsonis, G.; Lawson, D.; Santoianni, R. Survivin expression in ovarian carcinoma: Correlation with apoptotic markers and prognosis. Mod. Pathol. 2003, 16, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.G.; Rodriguez, D.A.; Valenzuela, M.; Calderon, C.; Urzua, U.; Munroe, D.; Rosas, C.; Lemus, D.; Diaz, N.; Wright, M.C.; et al. Survivin expression promotes vegf-induced tumor angiogenesis via pi3k/akt enhanced beta-catenin/tcf-lef dependent transcription. Mol. Cancer 2014, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- McCrea, P.D.; Turck, C.W.; Gumbiner, B. A homolog of the armadillo protein in drosophila (plakoglobin) associated with e-cadherin. Science 1991, 254, 1359–1361. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor lef-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Brunner, E.; Peter, O.; Schweizer, L.; Basler, K. Pangolin encodes a lef-1 homologue that acts downstream of armadillo to transduce the wingless signal in drosophila. Nature 1997, 385, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical wnt signals in development and disease: Conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Howe, L.R.; Watanabe, O.; Leonard, J.; Brown, A.M. Twist is up-regulated in response to wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003, 63, 1906–1913. [Google Scholar]
- Bu, H.; Liu, D.; Cui, J.; Cai, K.; Shen, F. Wnt/b-catenin signaling pathway is involved in induction of apoptosis by oridonin in colon cancer colo205 cells. Transl. Cancer Res. 2019, 8, 1782–1794. [Google Scholar] [CrossRef]
- Pak, S.; Park, S.; Kim, Y.; Park, J.H.; Park, C.H.; Lee, K.J.; Kim, C.S.; Ahn, H. The small molecule wnt/beta-catenin inhibitor cwp232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J. Exp. Clin. Cancer Res. 2019, 38, 342. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Retamales-Ortega, R.; Orostica, L.; Vera, C.; Cuevas, P.; Hernandez, A.; Hurtado, I.; Vega, M.; Romero, C. Role of nerve growth factor (ngf) and mirnas in epithelial ovarian cancer. Int. J. Mol. Sci. 2017, 18, 507. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alexander, C.J.; Yano, J.; Trivax, B.; Azziz, R. Use of metformin in polycystic ovary syndrome. Am. J. Obstet. Gynecol. 2008, 199, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Scarpello, J.H.; Howlett, H.C. Metformin therapy and clinical uses. Diabetes Vasc. Dis. Res. 2008, 5, 157–167. [Google Scholar] [CrossRef]
- Kelley, K.W.; Carroll, D.G.; Meyer, A. A review of current treatment strategies for gestational diabetes mellitus. Drugs Context 2015, 4, 212282. [Google Scholar] [CrossRef]
- Bodmer, M.; Becker, C.; Meier, C.; Jick, S.S.; Meier, C.R. Use of metformin and the risk of ovarian cancer: A case-control analysis. Gynecol. Oncol. 2011, 123, 200–204. [Google Scholar] [CrossRef]
- Shi, J.; Liu, B.; Wang, H.; Zhang, T.; Yang, L. Association of metformin use with ovarian cancer incidence and prognosis: A systematic review and meta-analysis. Int. J. Gynecol. Cancer 2019, 29, 140–146. [Google Scholar] [CrossRef]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the h19/sahh axis. Oncogene 2016, 36, 2345–2354. [Google Scholar] [CrossRef]
- Tang, G.; Guo, J.; Zhu, Y.; Huang, Z.; Liu, T.; Cai, J.; Yu, L.; Wang, Z. Metformin inhibits ovarian cancer via decreasing h3k27 trimethylation. Int. J. Oncol. 2018, 52, 1899–1911. [Google Scholar] [CrossRef]
- Do, M.T.; Kim, H.G.; Choi, J.H.; Jeong, H.G. Metformin induces microrna-34a to downregulate the sirt1/pgc-1alpha/nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic. Biol. Med. 2014, 74, 21–34. [Google Scholar]
- Xie, W.; Wang, L.; Sheng, H.; Qiu, J.; Zhang, D.; Zhang, L.; Yang, F.; Tang, D.; Zhang, K. Metformin induces growth inhibition and cell cycle arrest by upregulating microrna34a in renal cancer cells. Med. Sci. Monit. 2017, 23, 29–37. [Google Scholar] [CrossRef]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating csc function mediated by deregulating mirnas in pancreatic cancer cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Garrido, M.P.; Vera, C.; Vega, M.; Quest, A.F.G.; Romero, C. Metformin prevents nerve growth factor-dependent proliferative and proangiogenic effects in epithelial ovarian cancer cells and endothelial cells. Ther. Adv. Med. Oncol. 2018, 10, 1758835918770984. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by akt promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Yan, X.; Lyu, T.; Jia, N.; Yu, Y.; Hua, K.; Feng, W. Huaier aqueous extract inhibits ovarian cancer cell motility via the akt/gsk3beta/beta-catenin pathway. PLoS ONE 2013, 8, e63731. [Google Scholar]
- The European Collection of Authenticated Cell Cultures (ECACC). Cell Line Profile: Ovarian Cancer Cell Line a2780. Available online: https://www.phe-culturecollections.org.uk/media/113526/a2780-cell-line-profile.pdf (accessed on 21 September 2020).
- American Type Culture Collection (ATCC). Sk-ov-3 [skov-3; skov3] (atcc® htb-77™). Available online: https://www.atcc.org/Products/All/HTB-77.aspx#documentation (accessed on 21 September 2020).
- Hallas-Potts, A.; Dawson, J.C.; Herrington, C.S. Ovarian cancer cell lines derived from non-serous carcinomas migrate and invade more aggressively than those derived from high-grade serous carcinomas. Sci. Rep. 2019, 9, 5515. [Google Scholar] [CrossRef] [PubMed]
- Geva, E.; Jaffe, R.B. Role of vascular endothelial growth factor in ovarian physiology and pathology. Fertil. Steril. 2000, 74, 429–438. [Google Scholar] [CrossRef]
- Stimpfl, M.; Tong, D.; Fasching, B.; Schuster, E.; Obermair, A.; Leodolter, S.; Zeillinger, R. Vascular endothelial growth factor splice variants and their prognostic value in breast and ovarian cancer. Clin. Cancer Res. 2002, 8, 2253–2259. [Google Scholar] [PubMed]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by micrornas. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of micrornas on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- World Ovarian Cancer Coalition. The World Ovarian Cancer Coalition Atlas. Global Trends in Incidence, Mortality and Survival. 2018. Available online: https://worldovariancancercoalition.org/wp-content/uploads/2018/10/THE-WORLD-OVARIAN-CANCER-COALITION-ATLAS-2018.pdf (accessed on 28 September 2020).
- Cai, X.; Hu, X.; Cai, B.; Wang, Q.; Li, Y.; Tan, X.; Hu, H.; Chen, X.; Huang, J.; Cheng, J.; et al. Metformin suppresses hepatocellular carcinoma cell growth through induction of cell cycle g1/g0 phase arrest and p21cip and p27kip expression and downregulation of cyclin d1 in vitro and in vivo. Oncol. Rep. 2013, 30, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Pius, C.; Nabeel, M.; Nair, M.; Vishwanatha, J.K.; Ahmad, S.; Basha, R. Ovarian cancer: Current status and strategies for improving therapeutic outcomes. Cancer Med. 2019, 8, 7018–7031. [Google Scholar] [CrossRef] [PubMed]
- Duong, J.K.; Kumar, S.S.; Furlong, T.J.; Kirkpatrick, C.M.; Graham, G.G.; Greenfield, J.R.; Williams, K.M.; Day, R.O. The pharmacokinetics of metformin and concentrations of haemoglobin a1c and lactate in indigenous and non-indigenous australians with type 2 diabetes mellitus. Br. J. Clin. Pharmacol. 2014, 79, 617–623. [Google Scholar] [CrossRef] [PubMed]
- FDA. Glumetza, 500 mg (Metformin Hydrochloride Extended Release Tablets). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/021748s005lbl.pdf (accessed on 14 July 2020).
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef]
- Tria, M.A.; Fusco, M.; Vantini, G.; Mariot, R. Pharmacokinetics of nerve growth factor (ngf) following different routes of administration to adult rats. Exp. Neurol. 1994, 127, 178–183. [Google Scholar] [CrossRef]
- Tseng, C.H. Metformin reduces ovarian cancer risk in taiwanese women with type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 2015, 31, 619–626. [Google Scholar] [CrossRef]
- Shin, H.Y.; Yang, W.; Lee, E.J.; Han, G.H.; Cho, H.; Chay, D.B.; Kim, J.H. Establishment of five immortalized human ovarian surface epithelial cell lines via sv40 t antigen or hpv e6/e7 expression. PLoS ONE 2018, 13, e0205297. [Google Scholar] [CrossRef]
- Maines-Bandiera, S.L.; Kruk, P.A.; Auersperg, N. Simian virus 40-transformed human ovarian surface epithelial cells escape normal growth controls but retain morphogenetic responses to extracellular matrix. Am. J. Obstet. Gynecol. 1992, 167, 729–735. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van, I.W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (occp): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhu, J.; Zhang, H.; Liu, Y.; Sun, H. Metformin inhibits ovarian cancer growth and migration in vitro and in vivo by enhancing cisplatin cytotoxicity. Am. J. Transl. Res. 2018, 10, 3086–3098. [Google Scholar] [PubMed]
- Zhou, C.; Dickens, H.; Robinson, J.; Gehrig, P.A.; Bae-Jump, V.L. Abstract 4080: Metformin and rapamycin inhibit proliferation and alter glucose metabolism in endometrial and ovarian cancer cell lines. Cancer Res. 2011, 71, 4080. [Google Scholar]
- Xu, W.X.; Liu, Z.; Deng, F.; Wang, D.D.; Li, X.W.; Tian, T.; Zhang, J.; Tang, J.H. Mir-145: A potential biomarker of cancer migration and invasion. Am. J. Transl. Res. 2019, 11, 6739–6753. [Google Scholar] [PubMed]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. 2016, 175, 92–102.e102. [Google Scholar] [CrossRef]
- Demont, Y.; Corbet, C.; Page, A.; Ataman-Onal, Y.; Choquet-Kastylevsky, G.; Fliniaux, I.; Le Bourhis, X.; Toillon, R.A.; Bradshaw, R.A.; Hondermarck, H. Pro-nerve growth factor induces autocrine stimulation of breast cancer cell invasion through tropomyosin-related kinase a (trka) and sortilin protein. J. Biol. Chem. 2012, 287, 1923–1931. [Google Scholar] [CrossRef]
- Truzzi, F.; Marconi, A.; Lotti, R.; Dallaglio, K.; French, L.E.; Hempstead, B.L.; Pincelli, C. Neurotrophins and their receptors stimulate melanoma cell proliferation and migration. J. Investig. Dermatol. 2008, 128, 2031–2040. [Google Scholar] [CrossRef]
- Li, B.; Cai, S.; Zhao, Y.; He, Q.; Yu, X.; Cheng, L.; Zhang, Y.; Hu, X.; Ke, M.; Chen, S.; et al. Nerve growth factor modulates the tumor cells migration in ovarian cancer through the wnt/beta-catenin pathway. Oncotarget 2016, 7, 81026–81048. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, N.; Li, D.; Zou, G.; Chen, Y. Metformin improves the sensitivity of ovarian cancer cells to chemotherapeutic agents. Oncol. Lett. 2019, 18, 2404–2411. [Google Scholar] [CrossRef]
- Haenisch, S.; Werk, A.N.; Cascorbi, I. Micrornas and their relevance to abc transporters. Br. J. Clin. Pharmacol. 2014, 77, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, K.; Yamamoto, M.; Miyazaki, S.; Mizutani, H.; Iwamoto, T.; Okuda, M. Microrna-145 post-transcriptionally regulates the expression and function of p-glycoprotein in intestinal epithelial cells. Mol. Pharmacol. 2013, 83, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc deletion rescues apc deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Garinet, S.; Pasmant, E. Ras-mapk pathway epigenetic activation in cancer: Mirnas in action. Oncotarget 2016, 7, 38892–38907. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Liu, Q.; Cao, J.; Lu, Z.; Mo, Y.Y. Negative regulation of mir-145 by c/ebp-beta through the akt pathway in cancer cells. Nucleic Acids Res. 2012, 40, 6683–6692. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-mediated increase in dicer1 regulates microrna expression and cellular senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef]
- Leclerc, G.M.; Leclerc, G.J.; Kuznetsov, J.N.; DeSalvo, J.; Barredo, J.C. Metformin induces apoptosis through ampk-dependent inhibition of upr signaling in all lymphoblasts. PLoS ONE 2013, 8, e74420. [Google Scholar] [CrossRef]
- Testa, J.R.; Tsichlis, P.N. Akt signaling in normal and malignant cells. Oncogene 2005, 24, 7391–7393. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Mechanisms of mtor inhibitor resistance in cancer therapy. Target. Oncol. 2011, 6, 17–27. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Gao, Z.Y.; Liu, Z.; Bi, M.H.; Zhang, J.J.; Han, Z.Q.; Han, X.; Wang, H.Y.; Sun, G.P.; Liu, H. Metformin induces apoptosis via a mitochondria-mediated pathway in human breast cancer cells in vitro. Exp. Ther. Med. 2016, 11, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- Park, D. Metformin induces oxidative stress-mediated apoptosis without the blockade of glycolysis in h4iie hepatocellular carcinoma cells. Biol. Pharm. Bull. 2019, 42, 2002–2008. [Google Scholar] [CrossRef] [PubMed]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either ampk activation or increased ros production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Gaglio, D.; Bianco, M.R.; Aprea, F.; Virtuoso, A.; Bonanomi, M.; Alberghina, L.; Papa, M.; Colangelo, A.M. Differentiation by nerve growth factor (ngf) involves mechanisms of crosstalk between energy homeostasis and mitochondrial remodeling. Cell Death Dis. 2018, 9, 391. [Google Scholar] [CrossRef]
- He, X.J.; Zhang, Q.; Ma, L.P.; Li, N.; Chang, X.H.; Zhang, Y.J. Aberrant alternative polyadenylation is responsible for survivin up-regulation in ovarian cancer. Chin. Med. J. 2016, 129, 1140–1146. [Google Scholar] [CrossRef]
- Kohan-Ivani, K.; Gabler, F.; Selman, A.; Vega, M.; Romero, C. Role of dihydrotestosterone (dht) on tgf-beta1 signaling pathway in epithelial ovarian cancer cells. J. Cancer Res. Clin. Oncol. 2016, 142, 47–58. [Google Scholar] [CrossRef]
- Rodriguez, D.A.; Tapia, J.C.; Fernandez, J.G.; Torres, V.A.; Munoz, N.; Galleguillos, D.; Leyton, L.; Quest, A.F. Caveolin-1-mediated suppression of cyclooxygenase-2 via a beta-catenin-tcf/lef-dependent transcriptional mechanism reduced prostaglandin e2 production and survivin expression. Mol. Biol. Cell 2009, 20, 2297–2310. [Google Scholar] [CrossRef]
- Aranda, E.; Owen, G.I. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the ea.Hy926 endothelial cell line. Biol. Res. 2009, 42, 377–389. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Base | miR-145 | miR-23b |
|---|---|---|
| Microrna.org | BIRC5, VEGFA, c-MYC | CTNNB1, VEGFA |
| Targetscan | BIRC5 | - |
| TarBase v.8 | CTNNB1 | CTNNB1, VEGFA, BIRC5 |
| miRtarBase | c-MYC, VEGFA | - |
| MirWalk | c-MYC, VEGFA | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido, M.P.; Salvatierra, R.; Valenzuela-Valderrama, M.; Vallejos, C.; Bruneau, N.; Hernández, A.; Vega, M.; Selman, A.; Quest, A.F.G.; Romero, C. Metformin Reduces NGF-Induced Tumour Promoter Effects in Epithelial Ovarian Cancer Cells. Pharmaceuticals 2020, 13, 315. https://doi.org/10.3390/ph13100315
Garrido MP, Salvatierra R, Valenzuela-Valderrama M, Vallejos C, Bruneau N, Hernández A, Vega M, Selman A, Quest AFG, Romero C. Metformin Reduces NGF-Induced Tumour Promoter Effects in Epithelial Ovarian Cancer Cells. Pharmaceuticals. 2020; 13(10):315. https://doi.org/10.3390/ph13100315
Chicago/Turabian StyleGarrido, Maritza P., Renato Salvatierra, Manuel Valenzuela-Valderrama, Christopher Vallejos, Nicole Bruneau, Andrea Hernández, Margarita Vega, Alberto Selman, Andrew F. G. Quest, and Carmen Romero. 2020. "Metformin Reduces NGF-Induced Tumour Promoter Effects in Epithelial Ovarian Cancer Cells" Pharmaceuticals 13, no. 10: 315. https://doi.org/10.3390/ph13100315
APA StyleGarrido, M. P., Salvatierra, R., Valenzuela-Valderrama, M., Vallejos, C., Bruneau, N., Hernández, A., Vega, M., Selman, A., Quest, A. F. G., & Romero, C. (2020). Metformin Reduces NGF-Induced Tumour Promoter Effects in Epithelial Ovarian Cancer Cells. Pharmaceuticals, 13(10), 315. https://doi.org/10.3390/ph13100315