Computer-Assisted and Data Driven Approaches for Surveillance, Drug Discovery, and Vaccine Design for the Zika Virus

 , ,
, , 
Abstract
“A sickly season,” the merchant said,“The town I left was filled with dead,and everywhere these queer red fliescrawled upon the corpses’ eyes,eating them away.”A Medieval Song about the Plague (http://www.historyofpainters.com/plague_art.htm)
“How many valiant men, how many fair ladies, breakfast with their kinfolkand the same night supped with their ancestors in the next world!”Giovanni Boccaccio, Of the Black Death
1. Discovery and Brief History
2. Virology
2.1. Structure
2.2. Evolution and Spread
3. Infection
3.1. Clinical Symptoms and Complications
3.2. Modes of Transmission
- (a)
- Through mosquito bites: ZIKV is transmitted to people primarily through the bite of infected mosquitoes (A. aegypti or A. albopictus). Mosquitoes become infected when they feed on a person already infected with the virus. Infected mosquitoes can then spread the virus to other people through bites.
- (b)
- From mother to child: A pregnant woman can pass ZIKV to her fetus during pregnancy.
- (c)
- Through sex: ZIKV can be transmitted through sex from a person who has Zika to his or her partners. The virus can be passed through sex, even if the infected person does not have symptoms at the time. It can be passed from a person with Zika before their symptoms start, while they have symptoms, and after their symptoms end.
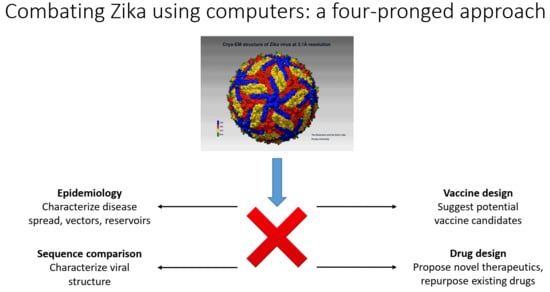
4. Mathematical/Computational Analysis and Results in ZIKV Virology, Peptide Vaccine Design, and Anti-Zika Drug Design
- (a)
- Epidemiological approaches for the characterization of reservoirs of next possible emerging pathogens;
- (b)
- Fast computational sequence comparison methods for the characterization of the emerging pathogens to understand how novel or severe they could be;
- (c)
- Once the sequences of the dominant strains have been determined, computer-aided vaccine design (CAVD) methods can be used to produce a set of probable vaccine candidates for quick synthesis/production and testing in the laboratory;
- (d)
- Computer-assisted design of novel therapeutics and testing of new drugs or repurposing already existing FDA-approved drugs.
4.1. Quantitative Epidemiological Modelling Strategies to Prevent Zika
4.2. Computer-Assisted Peptide Vaccine Design for Zika Virus
Peptide Vaccines
4.3. Use of Sequence (Structure)-Property Similarity Principle and Alignment Free Sequence Descriptors in the Characterization of ZIKV Sequences
“All cases are unique and very similar to others.”T. S. Eliot, The Cocktail Party
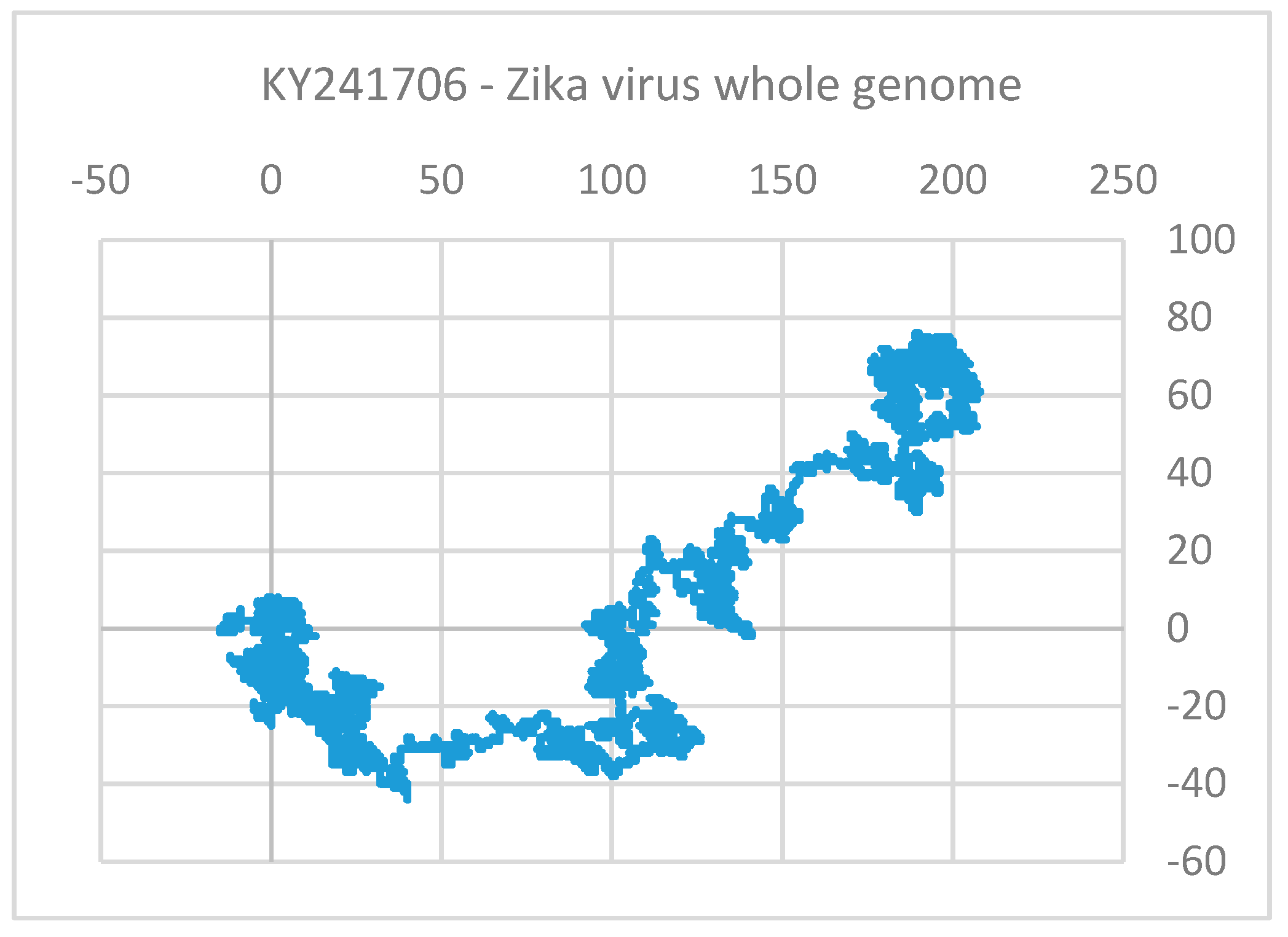
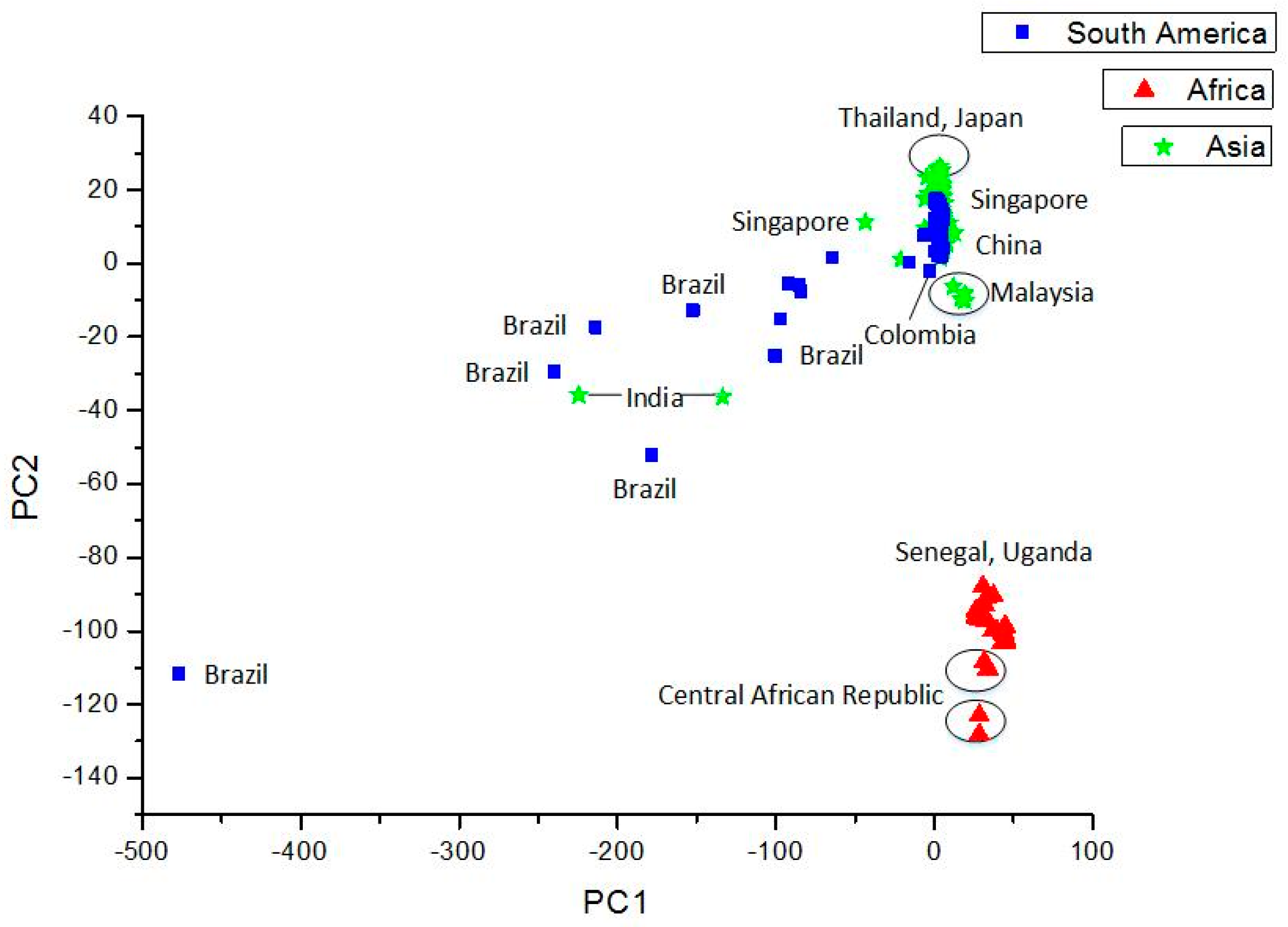
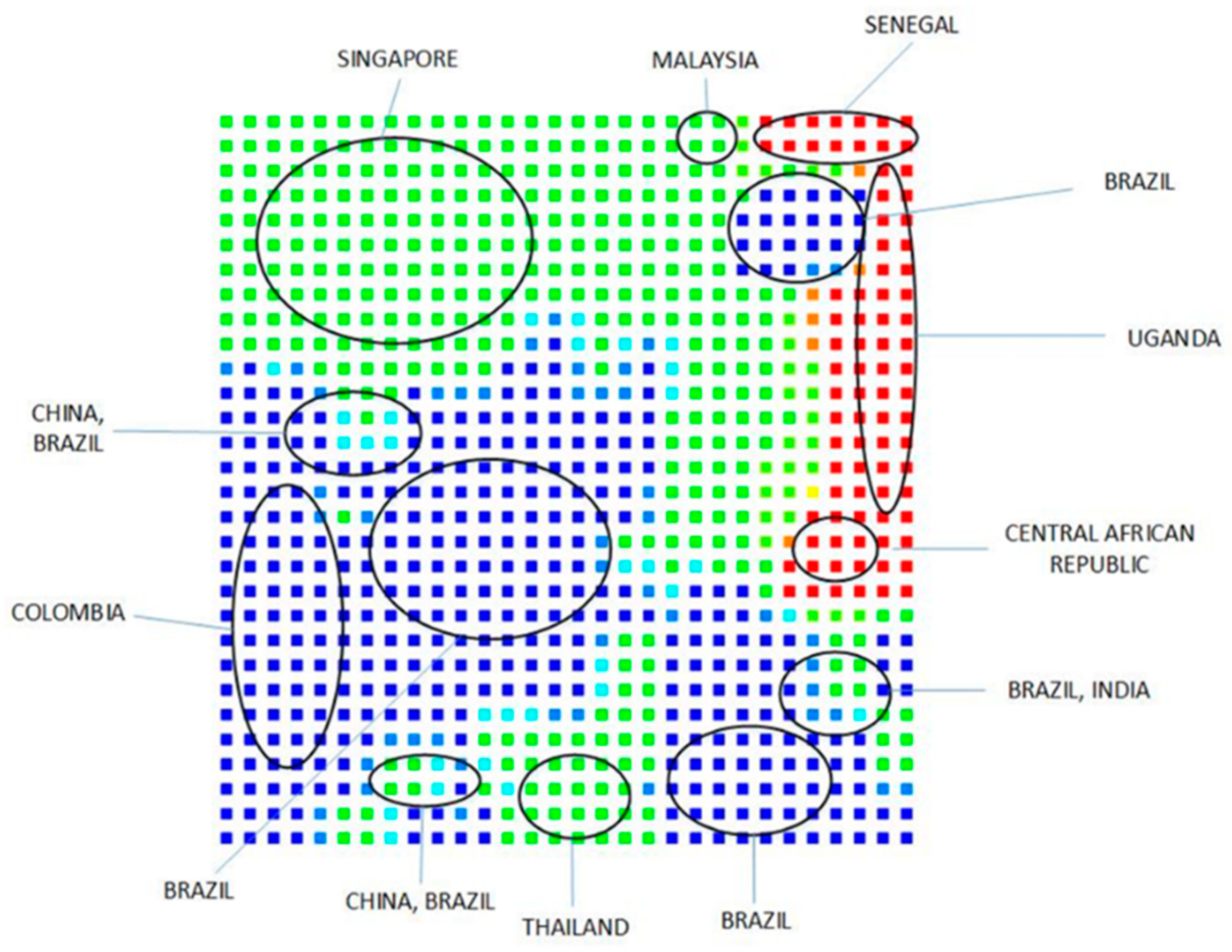
Clustering and Analysis of ZIKV Sequences
4.4. Discovery of Potential Anti-ZIKV Drugs from Literature including Computer-Assisted Approaches
“Computers are incredibly fast, accurate, and stupid.Human beings are incredibly slow, inaccurate, and brilliant.Together they are powerful beyond imagination.”Albert Einstein
4.4.1. Potential Anti-Zika Targets
4.4.2. Other Approaches
5. Discussion
“Those alone are wise who act after investigation.”Charaka, Sutrasthana 10:5
“We haven’t got the money, so we’ve got to think.”Ernest Rutherford
- (a)
- External characterization: modelling of disease spread mechanisms, vectors, and reservoirs of ZIKV (and emerging pathogens in general);
- (b)
- Internal characterization: bioinformatics-based sequence comparison methods to compare the genetic material of emerging strains/pathogens with the existing ones;
- (c)
- Vaccine design: computer-aided vaccine design methods that are applied on important sequences detected in the previous step to produce potential vaccine candidates for quick synthesis/production/testing;
- (d)
- Drug design: computer-assisted methods to propose and validate novel therapeutic molecules that may be precursors of new anti-Zika drugs or repurposing of already approved drugs.
5.1. Quantitative Characterization of ZIKV Infection
5.2. Sequence Comparison Methods
5.3. Computer-Assisted Vaccine Design
5.4. Anti-Zika Drug Discovery
Author Contributions
Funding
Conflicts of Interest
Appendix A. Technical Details of PCA Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | % Variance Explained |
|---|---|
| PC1 | 61.72276 |
| PC2 | 23.67231 |
| PC3 | 3.80329 |
| PC4 | 3.54875 |
| PC5 | 1.46926 |
| PC6 | 1.03775 |

| Descriptor | PC1 | PC2 | PC3 | PC4 | PC5 | PC6 |
|---|---|---|---|---|---|---|
| A | 0.37525 | −0.19154 | −0.30383 | −0.14333 | −0.03459 | −0.0214 |
| C | 0.30264 | 0.30112 | 0.02034 | −0.24198 | 0.05169 | 0.08496 |
| G | 0.45499 | 0.08624 | 0.27048 | 0.08604 | 0.10882 | −0.04221 |
| T | 0.30835 | −0.05888 | −0.20629 | 0.32184 | -0.15941 | 0.00631 |
| 1. neigh.AA | 0.08084 | 0.14915 | −0.32239 | 0.06124 | −0.0785 | 0.00945 |
| AC | 0.06785 | 0.01709 | −0.01214 | −0.18303 | 0.0477 | −0.05584 |
| AG | 0.14214 | −0.20504 | 0.05524 | −0.05171 | −0.00698 | −0.07525 |
| AT | 0.09397 | −0.15383 | −0.02502 | 0.02867 | 0.00811 | 0.10061 |
| CA | 0.11279 | −0.15156 | 0.12023 | −0.04033 | −0.08225 | 0.00573 |
| CC | 0.08502 | 0.05509 | 0.11682 | −0.11607 | −0.1074 | −0.18759 |
| CG | 0.03336 | 0.23813 | −0.04533 | 0.06401 | 0.31908 | −0.02018 |
| CT | 0.07977 | 0.16021 | −0.16937 | −0.14672 | −0.06374 | 0.28541 |
| GA | 0.15174 | −0.22587 | 0.0435 | −0.17946 | 0.07887 | −0.05781 |
| GC | 0.0845 | 0.14153 | 0.01696 | 0.10111 | 0.03199 | 0.03393 |
| GG | 0.15024 | 0.18539 | 0.15847 | 0.11032 | 0.1174 | 0.05727 |
| GT | 0.08098 | −0.01346 | 0.04668 | 0.05517 | −0.11229 | −0.07188 |
| TA | 0.04247 | 0.03678 | −0.14869 | 0.01361 | 0.05463 | 0.02565 |
| TC | 0.07274 | 0.0886 | −0.09937 | −0.04316 | 0.08842 | 0.29342 |
| TG | 0.13751 | −0.13156 | 0.10156 | −0.03741 | −0.31189 | −0.00237 |
| TT | 0.06248 | −0.05234 | −0.06042 | 0.38732 | 0.01392 | −0.30868 |
| 2. neigh.AA | 0.09635 | −0.09113 | −0.18414 | −0.01641 | −0.01696 | 0.08857 |
| AC | 0.08684 | −0.01636 | −0.01001 | −0.02046 | 0.24963 | 0.0758 |
| AG | 0.12814 | −0.07697 | 0.12034 | −0.0776 | −0.09957 | −0.17652 |
| AT | 0.07514 | −0.00809 | −0.22651 | −0.02836 | −0.15486 | −0.00966 |
| CA | 0.06435 | 0.17475 | −0.05956 | 0.06259 | −0.12713 | 0.20447 |
| CC | 0.06424 | 0.17368 | −0.03028 | −0.03357 | −0.01968 | −0.21684 |
| CG | 0.08972 | 0.11317 | 0.0235 | −0.31158 | −0.06387 | −0.04091 |
| CT | 0.09397 | −0.15892 | 0.09194 | 0.04293 | 0.27383 | 0.13541 |
| GA | 0.14944 | −0.19937 | 0.00492 | −0.24278 | 0.00391 | −0.15607 |
| GC | 0.0848 | 0.12145 | 0.00406 | −0.07691 | 0.15546 | −0.08149 |
| GG | 0.14281 | 0.1253 | 0.10387 | 0.22558 | 0.21362 | 0.09455 |
| GT | 0.09975 | 0.04099 | 0.15838 | 0.18232 | −0.24926 | 0.10006 |
| TA | 0.07649 | −0.07442 | −0.06657 | 0.05378 | 0.11689 | −0.15562 |
| TC | 0.08115 | 0.02246 | 0.06024 | −0.10957 | −0.32184 | 0.30228 |
| TG | 0.11086 | −0.07303 | 0.02508 | 0.25033 | 0.07385 | 0.08249 |
| TT | 0.05183 | 0.0677 | −0.23082 | 0.12703 | −0.02029 | −0.22091 |
| 3. neig.AA | 0.11116 | −0.08791 | −0.23494 | 0.00908 | 0.02267 | −0.02569 |
| AC | 0.08419 | −0.12537 | −0.00149 | −0.14264 | 0.23392 | 0.04934 |
| AG | 0.12295 | −0.03915 | 0.09917 | −0.05518 | −0.10449 | 0.08412 |
| AT | 0.07376 | 0.06077 | −0.16567 | 0.04808 | −0.16838 | −0.12946 |
| CA | 0.07496 | 0.11183 | 0.11847 | -0.0885 | −0.16906 | −0.14642 |
| CC | 0.08748 | 0.05298 | 0.09463 | 1.18E-4 | −0.06922 | 0.22857 |
| CG | 0.09406 | −0.01214 | −0.03707 | −0.06271 | 0.16529 | 0.07823 |
| CT | 0.05919 | 0.14918 | −0.15259 | −0.08828 | 0.13657 | −0.08248 |
| GA | 0.12164 | −0.16697 | −0.05858 | −0.08846 | 0.15677 | 0.08178 |
| GC | 0.08017 | 0.40191 | 0.01602 | −0.02209 | −0.10253 | −0.04902 |
| GG | 0.17269 | 0.09535 | 0.17819 | 0.01763 | 0.05321 | −0.28992 |
| GT | 0.10798 | −0.24128 | 0.14189 | 0.18252 | 0.01912 | 0.20649 |
| TA | 0.07981 | −0.04709 | −0.12778 | 0.02289 | −0.03166 | 0.06871 |
| TC | 0.06893 | −0.02786 | −0.0826 | −0.07336 | 0.00371 | −0.14839 |
| TG | 0.09038 | 0.04417 | 0.03068 | 0.19045 | 0.0128 | 0.08684 |
| TT | 0.08232 | −0.02577 | −0.02793 | 0.18266 | −0.13476 | 0.00604 |
| rG | 0.05465 | 0.02002 | 0.34324 | −0.0189 | −0.07488 | −0.03129 |
References
- World Health Organization (WHO) Report on Zika Virus. Available online: http://www.who.int/emergencies/zika-virus/mediacentre/press-releases/en/ (accessed on 7 July 2019).
- MacNamara, F.N. Zika virus: A report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1954, 48, 139–145. [Google Scholar] [CrossRef]
- Fagbami, A.H. Zika virus infection in Nigeria: Virology and seroepidemological investigation in Oyo state. J. Hyg. 1979, 83, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika Virus: History, Emergence, Biology, and Prospects for Control. Antiviral Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Marchette, N.J.; Garcia, R.; Rudnick, A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am. J. Trop. Med. Hyg. 1969, 18, 411–415. [Google Scholar] [CrossRef]
- Olson, J.G.; Ksiazek, T.G.; Triwibowo, S. Zika virus, a cause of fever in Central Java, Indonesia. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 389–393. [Google Scholar] [CrossRef]
- Liang, D.; Leung, R.K.K.; Lee, S.S.; Kam, K.M. Insights into intercontinental spread of Zika virus. PLoS ONE 2017, 12, e0176710. [Google Scholar] [CrossRef]
- WHO Report on Zika VIRUS, Microcephaly and Guillain–Barré Syndrome. Available online: http://apps.who.int/iris/bitstream/handle/10665/204609/zikasitrep_10Mar2016_eng.pdf;jsessionid=F9E4895E6176562081D34C0DED1A6550?sequence=1 (accessed on 7 July 2019).
- Nandy, A.; Basak, S.C. The Epidemic that Shook the World—The Zika Virus Rampage. Explor. Res. Hypothesis Med. 2017, 2, 43–56. [Google Scholar] [CrossRef]
- Armstrong, N.; Hou, W.; Tang, Q. Biological and historical overview of Zika virus. World J. Virol. 2017, 6, 1–8. [Google Scholar] [CrossRef]
- Faye, O.; Freire, C.C.M.; Iamarino, A.; Faye, O.; de Oliviera, J.V.C.; Diallo, M.; Zanotto, P.M.A.; Sall, A.A. Molecular Evolution of Zika Virus during Its Emergence in the 20th Century. PLoS Negl. Trop. Dis. 2017, 8, e2636. [Google Scholar] [CrossRef]
- Rather, I.A.; Lone, J.B.; Bajpai, V.K.; Paek, W.K.; Lim, J. Zika Virus: An Emerging Worldwide Threat. Front. Microbiol. 2017, 8, 1417. [Google Scholar] [CrossRef]
- Okafor, I.I.; Ezugwu, F.O.; Ekwochi, U. Zika Virus: The Emerging Global Health Challenge. Divers. Equal. Health Care 2016, 13, 394–401. [Google Scholar] [CrossRef][Green Version]
- Clarke, B.D.; Roby, J.A.; Slonchak, A.; Khromykh, A.A. Functional non-coding RNAs derived from the flavivirus 3′ untranslated region. Virus Res. 2015, 206, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Trevisan, M.; Sinigaglia, A.; Lavezzo, E.; Palu, G. Zika virus: From pathogenesis to disease control. FEMS Microbiol. Lett. 2016, 363, fnw202. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Nandy, A.; Dey, S.; Basak, S.C.; Bielinska-Waz, D.; Waz, P. Characterizing the Zika Virus Genome—A Bioinformatics Study. Curr. Comp. Aided Drug Des. 2016, 12, 87–97. [Google Scholar] [CrossRef]
- Kindhauser, M.K.; Allen, T.; Frank, V.; Santhana, R.S.; Dye, C. Zika: The origin and spread of a mosquito-borne virus. Bull. World Health Organ. 2016, 94, 675–686. [Google Scholar] [CrossRef]
- WHO Situation Report on Zika Virus, 9th March 2017. Available online: http://apps.who.int/iris/bitstream/10665/254714/1/zikasitrep10Mar17-eng.pdf (accessed on 7 July 2019).
- Centers for Disease Control and Prevention (CDC). Available online: https://www.cdc.gov/zika/hc-providers/preparing-for-zika/clinicalevaluationdisease.html (accessed on 7 July 2019).
- Mayo Clinic. Zika Virus: Symptoms and Causes. Available online: https://www.mayoclinic.org/diseases-conditions/zika-virus/symptoms-causes/syc-20353639 (accessed on 3 September 2019).
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jorge, A.; Jimenez-Arango, J.A.; Zea-Vara, A.F.; Gonzalez-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuniga, G.; Lopez-Gonzalez, R.; et al. Guillain–Barré Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Zika Virus transmission Methods, CDC. Available online: https://www.cdc.gov/zika/prevention/transmission-methods.html (accessed on 7 July 2019).
- Verity Murphy, V. Past Pandemics that Ravaged Europe. Available online: http://news.bbc.co.uk/2/hi/health/4381924.stm (accessed on 7 July 2019).
- Pringle, H. How Europeans Brought Sickness to the New World. Available online: https://www.sciencemag.org/news/2015/06/how-europeans-brought-sickness-new-world (accessed on 7 July 2019).
- Morens, D.M.; Fauci, A.S. Pandemic Zika: A Formidable Challenge to Medicine and Public Health. J. Infect. Dis. 2017, 216, S857–S859. [Google Scholar] [CrossRef]
- Zika Virus Surveillance, Vaccinology, and Anti-Zika Drug Discovery: Computer-Assisted Strategies to Combat the Menace; Basak, S.C., Bhattacharjee, A.K., Nandy, A., Eds.; Nova: New York, NY, USA, 2019; ISBN 978-1-53614-970-8. [Google Scholar]
- Hethcote, H. The Mathematics of Infectious Diseases. SIAM Rev. 2000, 42, 599–653. [Google Scholar] [CrossRef]
- Suparit, P.; Wiratsudakul, A.; Modchang, C. A mathematical model for Zika virus transmission dynamics with a time-dependent mosquito biting rate. Theor. Biol. Med. Model. 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Nishiura, H.; Mizumoto, K.; Villamil-Gomez, W.E.; Rodriguez-Morales, A.J. Preliminary estimation of the basic reproduction number of Zika virus infection during Colombia epidemic, 2015–2016. Trav. Med. Infect. Dis. 2016, 14, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Pritelj, M.; Mraz, J.; Kolenc, J.; Rus, K.R.; Vipotnik, T.V.; Vodusek, V.F.; et al. Zika virus associated with microcephaly. N. Eng. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Agusto, F.B.; Bewick, S.; Fagan, W.F. Mathematical model of Zika virus with vertical transmission. Infect. Dis. Model. 2017, 23, 244–267. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, P.; Seshaiyer, P.; Castillo-Chavez, C. Mathematical modeling, analysis and simulation of the spread of Zika with influence of sexual transmission and preventive measures. Lett. Bioinform. 2017, 4, 148–166. [Google Scholar] [CrossRef]
- Majumdar, S. Data-driven Strategies to Model and Mitigate the Threat of Zika. In Zika Virus Surveillance, Vaccinology, and Anti-Zika Drug Discovery: Computer-Assisted Strategies to Combat the Menace; Basak, S.C., Bhattacharjee, A.K., Nandy, A., Eds.; Nova: New York, NY, USA, 2019; pp. 129–152. [Google Scholar]
- Wiratsudakul, A.; Suparit, P.; Modchang, C. Dynamics of Zika virus outbreaks: An overview of mathematical modeling approaches. PeerJ 2018, 6, e4526. [Google Scholar] [CrossRef]
- Han, B.; Majumdar, S.; Calmon, F.P.; Glicksberg, B.S.; Horesh, R.; Kumar, A.; Perer, A.; von Marschall, E.B.; Wei, D.; Mojsilovic, A.; et al. Confronting data sparsity to identify potential sources of Zika virus spillover infection among primates. Epidemics 2019, 27, 59–65. [Google Scholar] [CrossRef]
- Sahoo, M.; Jena, L.; Daf, S.; Kumar, S. Virtual Screening for Potential Inhibitors of NS3 Protein of Zika Virus. Genomics Inform. 2016, 14, 104–111. [Google Scholar] [CrossRef]
- Singh, A.; Jana, N.K. Discovery of potential Zika virus RNA polymerase inhibitors by docking-based virtual screening. Comp. Biol. Chem. 2017, 71, 144–151. [Google Scholar] [CrossRef]
- Ramharack, P.; Soliman, M.E.S. Zika virus NS5 protein potential inhibitors: An enhanced in silico approach in drug discovery. J. Biomol. Struct. Dyn. 2018, 36, 1118–1133. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Teramoto, T.; Kulkarni, A.A.; Bhattacharjee, A.K.; Padmanabhan, R. Antiviral activities of selected antimalarials against dengue virus type 2 and Zika virus. Antivir. Res. 2017, 137, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Sinigaglia, A.; Riccetti, S.; Trevisan, M.; Barzon, L. In silico approaches to Zika virus drug discovery. Expert Opin. Drug Discov. 2018, 13, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Luca, S.; Mihaescu, T. History of BCG Vaccine. Mædica 2013, 8, 53–58. [Google Scholar] [PubMed]
- Maassab, H.F.; DeBorde, D.C. Development and characterization of cold-adapted viruses for use as live virus vaccines. Vaccine 1985, 3, 355–369. [Google Scholar] [CrossRef]
- Cate, T.R.; Couch, R.B.; Kasel, J.A.; Six, H.R. Clinical trials of monovalent influenza A/New Jersey/76 virus vaccines in adults: Reactogenicity, antibody response, and antibody persistence. J. Infect. Dis. 1977, 136, S450–S455. [Google Scholar] [CrossRef] [PubMed]
- Wiwanitkit, V. Development of a vaccine to prevent Japanese encephalitis: A brief review. Int. J. Gen. Med. 2009, 2, 195–200. [Google Scholar] [CrossRef]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of YFV 17D-based chimeric vaccines against dengue, West Nile and Japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef]
- WHO Vaccine Pipeline Tracker. Available online: https://docs.google.com/spreadsheets/d/19otvINcayJURCMg76xWO4KvuyedYbMZDcXqbyJGdcZM/pubhtml# (accessed on 15 June 2019).
- Kularatne, S. Dengue fever. BMJ 2015, 351, h4661. [Google Scholar] [CrossRef]
- Logan, I.S. ZIKA—How fast does this virus mutate? Zool. Res. 2016, 37, 110–115. [Google Scholar] [CrossRef]
- Badawi, M.M.; Osman, M.M.; Alla, A.A.E.F.; Ahmedani, A.M.; Abdalla, M.H.; Gasmelseed, M.M.; Elsayed, A.A.; Salih, M.A. Highly Conserved Epitopes of ZIKA Envelope Glycoprotein May Act as a Novel Peptide Vaccine with High Coverage: Immunoinformatics Approach. Am. J. Biomed. Res. 2016, 4, 46–60. [Google Scholar] [CrossRef]
- Dikhit, M.R.; Ansari, M.Y.; Vijaymahantesh; Kalyani; Mansuri, R.; Sahoo, B.R.; Dehury, B.; Amit, A.; Topno, R.K.; Sahoo, G.C.; et al. Computational prediction and analysis of potential antigenic CTL epitopes in Zika virus: A first step towards vaccine development. Infect. Genet. Evol. 2016, 45, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Shawan, M.M.A.K.; Mahmud, H.A.; Hasan, M.M.; Parvin, A.; Rahman, M.N.; Rahman, S.M.B. In Silico Modeling and Immunoinformatics Probing Disclose the Epitope Based Peptide Vaccine Against Zika Virus Envelope Glycoprotein. Ind. J. Pharma. Biol. Res. 2014, 2, 44–57. [Google Scholar]
- Mirza, M.U.; Rafique, S.; Ali, A.; Munir, M.; Ikram, N.; Manan, A.; Salo-Ahen, O.M.H.; Idrees, M. Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci. Rep. 2016, 6, 37313. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Das, S.; Nandy, A. Characterization of Zika and Other Human Infecting Flavivirus Envelope Proteins and Determination of Common Conserved Epitope Regions. EC Microbiol. 2017, 8, 29–46. [Google Scholar]
- Dar, H.; Zaheer, T.; Rehman, M.T.; Ali, A.; Javed, A.; Khan, G.A.; Babar, M.M.; Waheed, Y. Prediction of promiscuous T-cell epitopes in the Zika virus polyprotein: An in silico approach. Asian Pac. J. Trop. Med. 2016, 9, 844–850. [Google Scholar] [CrossRef]
- Dos Santos Franco, L.; Oliveira Vidal, P.; Amorim, J.H. In silico design of a Zika virus non-structural protein 5 aiming vaccine protection against zika and dengue in different human populations. J. Biomed. Sci. 2017, 24, 88. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA vaccines protect against Zika virus infection. Cell 2017, 168, 1114–1125. [Google Scholar] [CrossRef]
- Nandy, A.; Ghosh, A.; Nandy, P. Numerical characterization of protein sequences and application to voltage-gated sodium channel alpha subunit phylogeny. Silico Biol. 2009, 9, 77–87. [Google Scholar] [CrossRef]
- Ghosh, A.; Chattopadhyay, S.; Chawla-Sarkar, M.; Nandy, P.; Nandy, A. In Silico Study of Rotavirus VP7 Surface Accessible Conserved Regions for Antiviral Drug/Vaccine Design. PLoS ONE 2012, 7, e40749. [Google Scholar] [CrossRef]
- Dey, S.; De, A.; Nandy, A. Rational Design of Peptide Vaccines Against Multiple Types of Human Papillomavirus. Cancer Inform. 2016, 15, 1–16. [Google Scholar] [CrossRef]
- Dey, S.; Nandy, A.; Basak, S.C.; Nandy, P.; Das, S. A Bioinformatics approach to designing a Zika virus vaccine. Comput. Biol. Chem. 2017, 68, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Cellis, E. Getting peptide vaccines to work: Just a matter of quality control? J. Clin. Investig. 2002, 110, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Feuer, R.; Hassett, D.E.; Whitton, J.L. Peptide vaccination of mice immune to LCMV or vaccinia virus causes serious CD8+ T cell mediated, TNF-dependent immunopathology. J. Clin. Investig. 2006, 116, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Forecasting the Convergence of Artificial Intelligence and Precision Medicine. Available online: https://vector.childrenshospital.org/2018/06/bio-2018-artificial-intelligence-precision-medicine (accessed on 26 May 2019).
- Pioneering a New Era in Human Health. Available online: https://www.humanvaccinesproject.org/vision (accessed on 26 June 2019).
- Zika spreads rapidly in India, with 94 cases confirmed. Available online: https://www.cnn.com/2018/10/17/health/india-jaipur-zika-outbreak-rapid-increase-intl (accessed on 15 October 2019).
- Rooney, B.V.; Crucian, B.E.; Pierson, D.L.; Laudenslager, M.; Mehta, S.K. Herpes Virus Reactivation in Astronauts During Spaceflight and Its Application on Earth. Front. Microbiol. 2019, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D., III; Patterson, D.E.; Bunce, J.D. Comparative Molecular Field Analysis (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Ai, C.; Wang, Y. In Silico Prediction of Estrogen Receptor Subtype Binding Affinity and Selectivity Using Statistical Methods and Molecular Docking with 2-Arylnaphthalenes and 2-Arylquinolines. Int. J. Mol. Sci. 2010, 11, 3434–3458. [Google Scholar] [CrossRef]
- Basak, S.C.; Zhu, Q.; Mills, D. Prediction of anticancer activity of 2-phenylindoles: Comparative molecular field analysis versus ridge regression using mathematical molecular descriptors. Acta Chim. Slov. 2010, 57, 541–550. [Google Scholar]
- Basak, S.C. Mathematical descriptors in the prediction of property, bioactivity, and toxicity of chemicals from their structure: A chemical-cum-biochemical Approach. Curr. Comput. Aided Drug Des. 2013, 9, 449–462. [Google Scholar] [CrossRef]
- Basak, S.C.; Mills, D. Quantitative Structure-Activity Relationship Studies of Boron-Containing Dipeptide Proteasome Inhibitors Using Calculated Mathematical Descriptors. J. Math. Chem. 2011, 49, 185–200. [Google Scholar] [CrossRef]
- Nandy, A.; Harle, M.; Basak, S.C. Mathematical descriptors of DNA sequences: Development and applications. ARKIVOC 2006, 9, 211–238. [Google Scholar] [CrossRef]
- Sen, D.; Dasgupta, S.; Pal, I. Intercorrelation of major DNA/RNA sequence descriptors—A preliminary study. Curr. Comput. Aided Drug Des. 2016, 12, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhury., C.; Nandy, A. Indexing Scheme and Similarity Measures for Macromolecular Sequences. J. Chem. Inf. Comput. Sci. 1999, 39, 243–247. [Google Scholar] [CrossRef]
- Randić, M.; Vracko, M.; Novic, M.; Plavsic, D. Spectrum-Like Graphical Representation of RNA Secondary Structure. Int. J. Quantum Chem. 2009, 109, 2982–2995. [Google Scholar] [CrossRef]
- Johnson, M.; Basak, S.C.; Maggiora, G. A characterization of molecular similarity methods for property prediction. Math. Comput. Model. 1988, 11, 630–634. [Google Scholar] [CrossRef]
- Roy Choudhury, A.; Zhukov, N.; Vracko, M. Mathematical Characterization of Protein Transmembrane Regions. Sci. World. J. 2013, 2013, 607830. [Google Scholar] [CrossRef] [PubMed]
- Vracko, M.; Basak, S.C.; Nandy, A.; Sen, D. Clustering of Zika viruses originating from different geographical regions using computational sequence descriptors. Curr. Comput. Aided Drug Des. 2019. submitted. [Google Scholar]
- Drgan, V.; Zuperl, S.; Vracko, M.; Cappelli, C.I.; Novic, M. CPANNatNIC software for counter-propagation neural network to assist in read-across. J. Cheminform. 2017, 9, 30. [Google Scholar] [CrossRef]
- Mahalanobis, P.C. On the generalized distance in statistics. Proc. Nat. Inst. Sci. India 1936, 2, 49–55. [Google Scholar]
- Lednicky, J.; Beau De Rochars, V.M.; El Badry, M.; Loeb, J.; Telisma, T.; Chavannes, S.; Anilis, G.; Cella, E.; Ciccozzi, M.; Rashid, M.; et al. Zika Virus Outbreak in Haiti in 2014: Molecular and Clinical Data. PLoS Negl. Trop. Dis. 2016, 10, e0004687. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K. Discovery of anti-zika drugs using in silico pharmacophore modeling. In Zika Virus Surveillance, Vaccinology, and Anti-zika Drug Discovery; Basak, S.C., Bhattacharjee, A.K., Nandy, A., Eds.; Nova: New York, NY, USA, 2019; pp. 39–73. ISBN 978-1-53614-970-8. [Google Scholar]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.K. Pharmacophore modeling applied to mosquito-borne diseases. In Computational Design of Chemicals for the Control of Mosquitoes and Their Diseases; Devillers, J., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 139–169. ISBN 978-1-49874-180-4. [Google Scholar]
- Kier, L.B. Molecular orbital calculation of preferred conformations of acetylcholine, muscarine, and muscarone. Mol. Pharmacol. 1967, 3, 487–494. [Google Scholar] [PubMed]
- Podlogar, B.L.; Muegge, I.; Brice, L.J. Computational methods to estimate drug development parameters. Curr. Opin. Drug Discov. 2001, 12, 102–109. [Google Scholar] [CrossRef]
- Mottin, M.; Borba, J.V.V.B.; Braga, R.C.; Torres, P.H.M.; Martini, M.C.; Proenca-Modena, J.L.; Judice, C.C.; Costa, F.T.M.; Ekins, S.; Perryman, A.L.; et al. The A–Z of Zika drug discovery. Drug Discov. Today 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Watterson, D.; Modhiran, N.; Young, P.R. The many faces of the flavivirus NS1 protein offer a multitude of options for inhibitor design. Antivir. Res. 2016, 130, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Barrows, N.J.; Campos, R.K.; Powell, S.T.; Prasanth, K.R.; Schott-Lerner, G.; Soto-Acosta, R.; Galarza-Muñoz, G.; McGrath, E.L.; Urrabaz-Garza, R.; Gao, J.; et al. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe 2016, 20, 259–270. [Google Scholar] [CrossRef]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Pascoalino, B.S.; Courtemanche, G.; Cordeiro, M.T.; Gil, L.H.; Freitas-Junior, L. Zika antiviral chemotherapy: Identification of drugs and promising starting points for drug discovery from an FDA-approved library. F1000Research 2016, 5, 2523. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, C.; Huang, L.S.; Zhang, X.Q.; Xu, Y.; Fang, X.; Zhou, J.; Wu, M.; Schooley, R.T.; Huang, Z.; et al. Discovery and Computational Analyses of Novel Small Molecule Zika Virus Inhibitors. Molecules 2019, 24, 1465. [Google Scholar] [CrossRef]
- Wilder-Smith, A.; Vannice, K.; Durbin, A.; Hombach, J.; Thomas, S.J.; Thevarjan, I.; Simmons, C.P. Zika vaccines and therapeutics: Landscape analysis and challenges ahead. BMC Med. 2018, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, R.; Higa, L.M.; Pezzuto, P.; Valadao, A.L.; Garcez, P.P.; Monteiro, F.L.; Loiola, E.C.; Dias, A.A.; Silva, F.J.M.; Aliota, M.T.; et al. Chloroquine, an Endocytosis Blocking Agent, Inhibits Zika Virus Infection in Different Cell Models. Viruses 2016, 8, 322. [Google Scholar] [CrossRef] [PubMed]
- Devillers, J. Repurposing drugs for use against Zika virus infection. SAR QSAR Environ. Res. 2018, 29, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Oguntade, S.; Ramharack, P.; Soliman, M.E.S. Characterizing the ligand-binding landscape of Zika NS3 helicase-promising lead compounds as potential inhibitors. Future Virol. 2017, 12, 261–273. [Google Scholar] [CrossRef]
- Hercík, K.; Kozak, J.; Sala, M.; Dejmek, M.; Xu, L.; Bei, Z.; Zhang, D.; Dou, Y.; Wang, H. Viral polymerase inhibitors T-705 and T-1105 are potential inhibitors of Zika virus replication. Antivir. Res. 2017, 137, 131–133. [Google Scholar] [CrossRef]
- Munjal, A.; Khandia, R.; Dhama, K.; Sachan, S.; Karthik, K.; Tiwari, R.; Malik, Y.S.; Kumar, D.; Singh, R.K.; Iqbal, H.M.N.; et al. Advances in Developing Therapies to Combat Zika Virus: Current Knowledge and Future Perspectives. Front. Microbiol. 2017, 8, 1469. [Google Scholar] [CrossRef]
- Ekins, S.; Mietchen, D.; Coffee, M.; Stratton, T.P.; Freundlich, J.S.; Freitas-Junior, L.; Muratov, E.; Siqueira-Neto, J.; Williams, A.J.; Andrade, C. Open drug discovery fore the Zika virus. F1000Research 2016, 5, 150. [Google Scholar] [CrossRef]
- Micewicz, E.D.; Khachatoorian, R.; French, S.W.; Ruchala, P. Identification of novel small-molecule inhibitors of Zika virus infection. Bioorg. Med. Chem. Lett. 2018, 28, 452–458. [Google Scholar] [CrossRef]
- Ncube, N.B.; Ramharack, P.; Soliman, M.E.S. An “All-In-One” Pharmacophoric Architecture for the Discovery of Potential Broad-Spectrum Anti-Flavivirus Drugs. Appl. Biochem. Biotechnol. 2018, 185, 799–814. [Google Scholar] [CrossRef]
- Sarukhanyan, E.; Shityakov, S.; Dandekar, T. In Silico Designed Axl Receptor Blocking Drug Candidates Against Zika Virus. ACS Omega 2018, 3, 5281–5290. [Google Scholar] [CrossRef]
- Pan, T.; Peng, Z.; Tan, L.; Zou, F.; Zhou, N.; Liu, B.; Liang, L.; Chen, C.; Liu, J.; Wu, L. Nonsteroidal Anti-inflammatory Drugs Potently Inhibit the Replication of Zika Viruses by Inducing the Degradation of AXL. J. Virol. 2018, 26, 92. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Nguyen, T.T.; Kim, N.M.; Park, J.S.; Jang, T.J.; Kim, D. Inhibitory effect of flavonoids against NS2B-NS3 protease of ZIKA virus and their structure activity relationship. Biotechnol. Lett. 2017, 39, 415–442. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Coloma, J.; Garcia-Sastre, A.; Aggarwal, A.K. Structure of the NS3 helicase from Zika virus. Nat. Struct. Mol. Biol. 2016, 23, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Eyer, L.; Nencka, R.; Huvarova, I.; Palus, M.; Joao Alves, M.; Gould, E.A.; De Clercq, E.; Růžek, D. Nucleoside Inhibitors of Zika Virus. J. Infect. Dis. 2016, 214, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Hercik, K.; Brynda, J.; Nencka, R.; Boura, E. Structural basis of Zika virus methyltransferase inhibition by sinefungin. Arch. Virol. 2017, 162, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Adcock, R.S.; Chu, Y.K.; Golden, J.E.; Chung, D.H. Evaluation of anti-Zika virus activities of broad-spectrum antivirals and NIH clinical collection compounds using a cell-based, high-throughput screen assay. Antivir. Res. 2017, 138, 47–56. [Google Scholar] [CrossRef]
- Han, Y.; Mesplède, T. Investigational drugs for the treatment of Zika virus infection: A preclinical and clinical update. Expert Opin. Investig. Drugs 2018, 27, 951–962. [Google Scholar] [CrossRef]
- Carneiro, B.M.; Batista, M.N.; Braga, A.C.S.; Nogueira, M.L.; Rahal, P. The green tea molecule EGCG inhibits Zika virus entry. Virology 2016, 496, 215–218. [Google Scholar] [CrossRef]
- Roy, A.; Lim, L.; Srivastava, S.; Lu, Y.; Song, J. Solution conformations of Zika NS2B-NS3pro and its inhibition by natural products from edible plants. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Lim, S.P.; Wang, Q.Y.; Noble, C.G.; Chen, Y.L.; Dong, H.; Zou, B.; Yokokawa, F.; Nilar, S.; Smith, P.; Beer, D.; et al. Ten years of dengue drug discovery: Progress and prospects. Antivir. Res. 2013, 100, 500–519. [Google Scholar] [CrossRef]
- Xie, X.; Zou, J.; Wang, Q.Y.; Shi, P.Y. Targeting dengue virus NS4B protein for drug discovery. Antivir. Res. 2015, 118, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, Q.Y.; Xu, H.Y.; Qing, M.; Kramer, L.; Yuan, Z.; Shi, P.Y. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 2011, 85, 11183–11195. [Google Scholar] [CrossRef] [PubMed]
- van Cleef, K.W.; Overheul, G.J.; Thomassen, M.C.; Kaptein, S.J.; Davidsonm, A.D.; Jacobs, M.; Neyts, J.; van Kuppeveld, F.J.; van Rij, R.P. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antivir. Res. 2013, 99, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Stephen, P.; Baz, M.; Bolvin, G.; Lin, S.X. Structural insight into NS5 of Zika virus leading to the discovery of MTase inhibitors. J. Am. Chem. Soc. 2016, 138, 16212–16215. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.J.; Gahan, M.E.; Mahalingam, S.; Keller, P.A. The medicinal chemistry of dengue fever. J. Med. Chem. 2009, 52, 7911–7926. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.P.; Noble, C.G.; Shi, P.Y. The dengue virus NS5 protein as a target for drug discovery. Antivir. Res. 2015, 119, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Bekerman, E.; Einav, S. Combating emerging viral threats. Science 2015, 348, 282–283. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Lucas-Hourani, M.; Dauzonne, D.; Jorda, P.; Cousin, G.; Lupan, A.; Helynck, O.; Caignard, G.; Janvier, G.; André-Leroux, G.; Khiar, S.; et al. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLoS Pathog. 2013, 9, e1003678. [Google Scholar] [CrossRef]
- Crance, J.M.; Scaramozzino, N.; Jouan, A.; Garin, D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: Antiviral compounds active against pathogenic flaviviruses. Antivir. Res. 2003, 58, 73–79. [Google Scholar] [CrossRef]
- Xie, Y.; Ogah, C.A.; Jiang, X.; Li, J.; Shen, J. Nucleoside inhibitors of hepatitis C virus NS5B polymerase: A systematic review. Curr. Drug Targets 2016, 17, 1560–1576. [Google Scholar] [CrossRef] [PubMed]
- Julander, J.G.; Siddharthan, V.; Evans, J.; Taylor, R.; Tolbert, K.; Apuli, C.; Stewart, J.; Collins, P.; Gebre, M.; Neilson, S.; et al. Efficacy of the broad-spectrum antiviral compound BCX4430 against Zika virus in cell culture and in a mouse model. Antivir. Res. 2017, 137, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Q.; Zhang, N.N.; Li, C.F.; Tian, M.; Hao, J.-N.; Xie, X.-P.; Shi, P.-Y.; Qin, C.-F. Adenosine analog NITD008 is a potent inhibitor of Zika virus. Open Forum Infect. Dis. 2016, 3, ofw175. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, A.; Palermo, N.; Sahoo, B.R.; Yuan, Z.; Hu, D.; Annamalai, A.S.; Vu, H.L.X.; Correas, I.; Prathipati, P.K.; Destache, C.J.; et al. Discovery of a non-nucleoside RNA polymerase inhibitor for blocking Zika virus replication through in silico screening. Antivir. Res. 2018, 151, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xu, M.; Lee, E.M.; Shiryaev, S.A.; He, S.; Sun, W.; Cheng, Y.-S.; Hu, X.; Tharappel, A.M.; Lu, B.; et al. Emetine inhibits Zika and Ebola virus infections through two molecular mechanisms: Inhibiting viral replication and decreasing viral entry. Cell Discov. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.; Riley, C.; Isaac, G.; Hopf-Jannasch, A.S.; Moore, R.J.; Weitz, K.W.; Pasa-Tolic, L.; Metz, T.O.; Adamec, J.; Kuhn, R.J. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, B.; Choy, H.A.; Pinne, M.; Rotondi, M.L.; Miller, M.C.; Demoll, E.; Kraiczy, P.; Cooley, A.E.; Creamer, T.P.; Suchard, M.A.; et al. Leptospira interrogans endostatin-like outer membrane proteins bind host fibronectin, laminin and regulators of complement. PLoS ONE 2007, 2, e1188. [Google Scholar] [CrossRef]
- Kanno, T.; Kobuchi, H.; Kajitani, N.; Utsumi, T.; Yano, H.; Horton, A.A.; Yasuda, T.; Utsumi, K. Mevastatin, an inhibitor of HMG-CoA reductase, induces apoptosis, differentiation and Rap1 expression in HL-60 cells. Physiol. Chem. Phys. Med. NMR 2002, 34, 1–15. [Google Scholar]
- Mohr, E.L.; McMullan, L.K.; Lo, M.K.; Spengler, J.R.; Bergeron, É.; Albariño, C.G.; Shrivastava-Ranjan, P.; Chiang, C.F.; Nichol, S.T.; Spiropoulou, C.F.; et al. Inhibitors of cellular kinases with broad-spectrum antiviral activity for hemorrhagic fever viruses. Antivir. Res. 2015, 120, 40–47. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K.; Geyer, J.A.; Woodard, C.L.; Kathcart, A.K.; Nichols, D.A.; Prigge, S.T.; Li, Z.; Mott, B.T.; Waters, N.C. A three dimensional in silico pharmacophore model for inhibition of Plasmodium falciparum cyclin dependent kinases and discovery of different classes of novel Pfmrk specific inhibitors. J. Med. Chem. 2004, 47, 5418–5426. [Google Scholar] [CrossRef]
- Meertens, L.; Labeau, A.; Dejamac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Wang, Z.; Zhen, Z.D.; Feng, K.H.; Guo, J.; Gao, N.; Fan, D.Y.; Han, D.S.; Wang, P.G.; An, J. Axl is not an indispensable factor for zika virus infection in mice. J. Gen. Virol. 2017, 98, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.-C.; Martin-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, S.; Bespalov, M.M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antivir. Res. 2017, 139, 117–128. [Google Scholar] [CrossRef]
- Jurgeit, A.; McDowell, R.; Moese, S.; Meldrum, E.; Schwendener, R.; Greber, U.F. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog. 2012, 8, e1002976. [Google Scholar] [CrossRef]
- Cheng, F.; Murray, J.L.; Rubin, D.H. Drug repurposing: New treatments for Zika virus infection? Trends Mol. Med. 2016, 22, 919–921. [Google Scholar] [CrossRef]
- Yang, H.T.; Ju, J.H.; Wong, Y.T.; Shmulevich, I.; Chiang, J.H. Literature-based discovery of new candidates for drug repurposing. Brief Bioinform. 2017, 18, 488–497. [Google Scholar] [CrossRef]
- Giulietti, M.; Righetti, A.; Cianfruglia, L.; Šabanović, B.; Armeni, T.; Principato, G.; Piva, F. To accelerate the Zika beat: Candidate design for RNA interference-based therapy. Virus Res. 2018, 255, 133–140. [Google Scholar] [CrossRef]
- Meng, X.Y.; Luo, Y.; Anwar, M.N.; Sun, Y.; Gao, Y.; Zhang, H.; Munir, M.; Qiu, H.-J. Long non-coding RNAs: Emerging and versatile regulators in host-virus interactions. Front. Immunol. 2017, 8, 1663. [Google Scholar] [CrossRef]
- Bruzzoni-Giovanelli, H.; Alezra, V.; Wolff, N.; Dong, C.Z.; Tuffery, P.; Rebollo, A. Interfering peptides targeting protein-protein interactions: The next generation of drugs? Drug Discov. Today 2018, 3, 272–285. [Google Scholar] [CrossRef]
- Kazmirchuk, T.; Dick, K.; Burnside, D.J.; Barnes, B.; Moteshareie, H.; Hajikarimlou, M.; Omidi, K.; Ahmed, D.; Low, A.; Lettl, C. Designing anti-Zika virus peptides derived from predicted human-Zika virus protein-protein interactions. Comput. Biol. Chem. 2017, 71, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Goertz, G.P.; Abbo, S.R.; Fros, J.J.; Pijlman, G.P. Functional RNA during Zika virus 980 infection. Virus Res. 2017, 254, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Hermann, T. Small molecules targeting viral RNA. Wiley Interdiscip. Rev. RNA 2016, 7, 726–743. [Google Scholar] [CrossRef] [PubMed]
- Connelly, C.M.; Moon, M.H.; Schneekloth, J.S., Jr. The emerging role of RNA as a therapeutic target for small molecules. Cell Chem. Biol. 2016, 23, 1077–1090. [Google Scholar] [CrossRef]
- Sperandio, S.; Barat, C.; Cabrita, M.A.; Gargaun, A.; Berezovski, M.V.; Tremblay, M.J.; de Belle, I. TOE1 is an inhibitor of HIV-1 replication with cell-penetrating capability. Proc. Natl. Acad. Sci. USA 2015, 112, E3392–E3401. [Google Scholar] [CrossRef]
- Martinez, J.P.; Sasse, F.; Bronstrup, M.; Diez, J.; Meyerhans, A. Antiviral drug discovery: Broad-spectrum drugs from nature. Nat. Prod. Rep. 2015, 32, 29–48. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Teixeira, R.R.; Oliveira, A.S.; Souza, A.P.; Silva, M.L.; Paula, S.O. Potential antivirals: Natural products targeting replication enzymes of dengue and chikungunya viruses. Molecules 2017, 22, 505. [Google Scholar] [CrossRef]
- Valadao, A.L.; Aguiar, R.S.; de Arruda, L.B. Interplay between inflammation and cellular stress triggered by Flaviviridae viruses. Front. Microbiol. 2016, 7, 1233. [Google Scholar] [CrossRef]
- Li, C.; Zhu, X.; Ji, X.; Quanquin, N.; Deng, Y.-Q.; Tian, M.; Aliyari, R.; Zuo, X.; Yuan, L.; Afridi, S.K.; et al. Chloroquine, a FDA-approved drug, prevents Zika Virus infection and its associated congenital microcephaly in mice. EBioMedicine 2017, 24, 189–194. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Mesci, P.; Pinto, A.; Fernandes, I.; Sheets, N.; Shresta, S.; Farhy, C.; Huang, C.-T.; Strongin, A.Y.; Muotri, A.R.; et al. Repurposing of the anti-malaria drug chloroquine for Zika virus treatment and prophylaxis. Sci. Rep. 2017, 7, 15771. [Google Scholar] [CrossRef]
- Han, Y.; Mesplede, T.; Xu, H.; Quan, Y.; Wainberg, M.A. The antimalarial drug amodiaquine possesses anti-ZIKA virus activities. J. Med. Virol. 2018, 90, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Tan, L.; Cederquist, G.Y.; Fan, Y.; Hartley, B.J.; Mukherjee, S.; Tomishima, M.; Brennand, K.J.; Zhang, Q.; Schwartz, R.E.; et al. High-content screening in hPSC-neural progenitors identifies drug candidates that inhibit Zika virus infection in fetal-like organoids and adult brain. Cell Stem Cell 2017, 21, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Warfield, K.L.; Warren, T.K.; Qiu, X.; Wells, J.; Mire, C.E.; Geisbert, J.B.; Stuthman, K.S.; Garza, N.L.; Van Tongeren, S.A.; Shurtleff, A.C.; et al. Assessment of the potential for host-targeted iminosugars UV-4 and UV-5 activity against filovirus infections in vitro and in vivo. Antivir. Res. 2017, 138, 22–31. [Google Scholar] [CrossRef]
- Adolf-Bryfogle, J.; Kalyuzhniy, O.; Kubitz, M.; Weitzner, B.D.; Hu, X.; Adachi, Y.; Schief, W.R.; Dunbrack, R.L., Jr. RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PLoS Comput. Biol. 2018, 14, e1006112. [Google Scholar] [CrossRef]
- North, B.; Lehmann, A.; Dunbrack, R.L. A new clustering of antibody CDR loop conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef]
- Chowdhury, R.; Allan, M.F.; Maranas, C.D. OptMAVEn-2.0: De novo design of variable antibody regions against targeted antigen epitopes. Antibodies 2018, 7, 23. [Google Scholar] [CrossRef]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore super positioning and pattern matching in computational drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef]
- Hufnagel, L.; Brockmann, D.; Geisel, T. Forecast and control of epidemics in a globalized world. Proc. Natl. Acad. Sci. USA 2004, 101, 15124–15129. [Google Scholar] [CrossRef]
- May, R.M. Simple mathematical models with very complicated dynamics. Nature 1976, 261, 459–467. [Google Scholar] [CrossRef]
- Heesterbeek, J.A.P.; Roberts, M.G. How mathematical epidemiology became a field of biology: A commentary on Anderson and May (1981) ‘The population dynamics of microparasites and their invertebrate hosts’. Phil. Trans. R. Soc. B 2014, 370. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Lou, Y.; He, D.; Porco, T.C.; Kuang, Y.; Chowell, G.; Ruan, S. Prevention and Control of Zika as a Mosquito-Borne and Sexually Transmitted Disease: A Mathematical Modeling Analysis. Sci. Rep. 2016, 6, 28070. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, R.M.; Ravantti, J.J.; Bamford, D.H. Nucleic and Amino Acid Sequences Support Structure-Based Viral Classification. J. Virol. 2017, 91, e02275-16. [Google Scholar] [CrossRef] [PubMed]
- van Hemert, F.; Jebbink, M.; van der Ark, A.; Scholer, F.; Berkhout, B. Euclidean Distance Analysis Enables Nucleotide Skew Analysis in Viral Genomes. Comp. Math. Meth. Med. 2018, 2018, 6490647. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bai, X.; Lu, Y.Y.; Tang, K.; Wang, Y.; Reinert, G.; Sun, F. Alignment-Free Sequence Analysis and Applications. Annu. Rev. Biomed. Data Sci. 2018, 1, 93–114. [Google Scholar] [CrossRef]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215. [Google Scholar] [CrossRef]
- Basak, S.C.; Magnuson, V.R.; Niemi, G.J.; Regal, R.R. Determining structural similarity of chemicals using graph-theoretic indices. Discr. Appl. Math. 1988, 19, 17–44. [Google Scholar] [CrossRef]
- Ebola in the Democratic Republic of the Congo: Health Emergency Update. Available online: https://www.who.int/emergencies/diseases/ebola/drc-2019 (accessed on 1 August 2019).





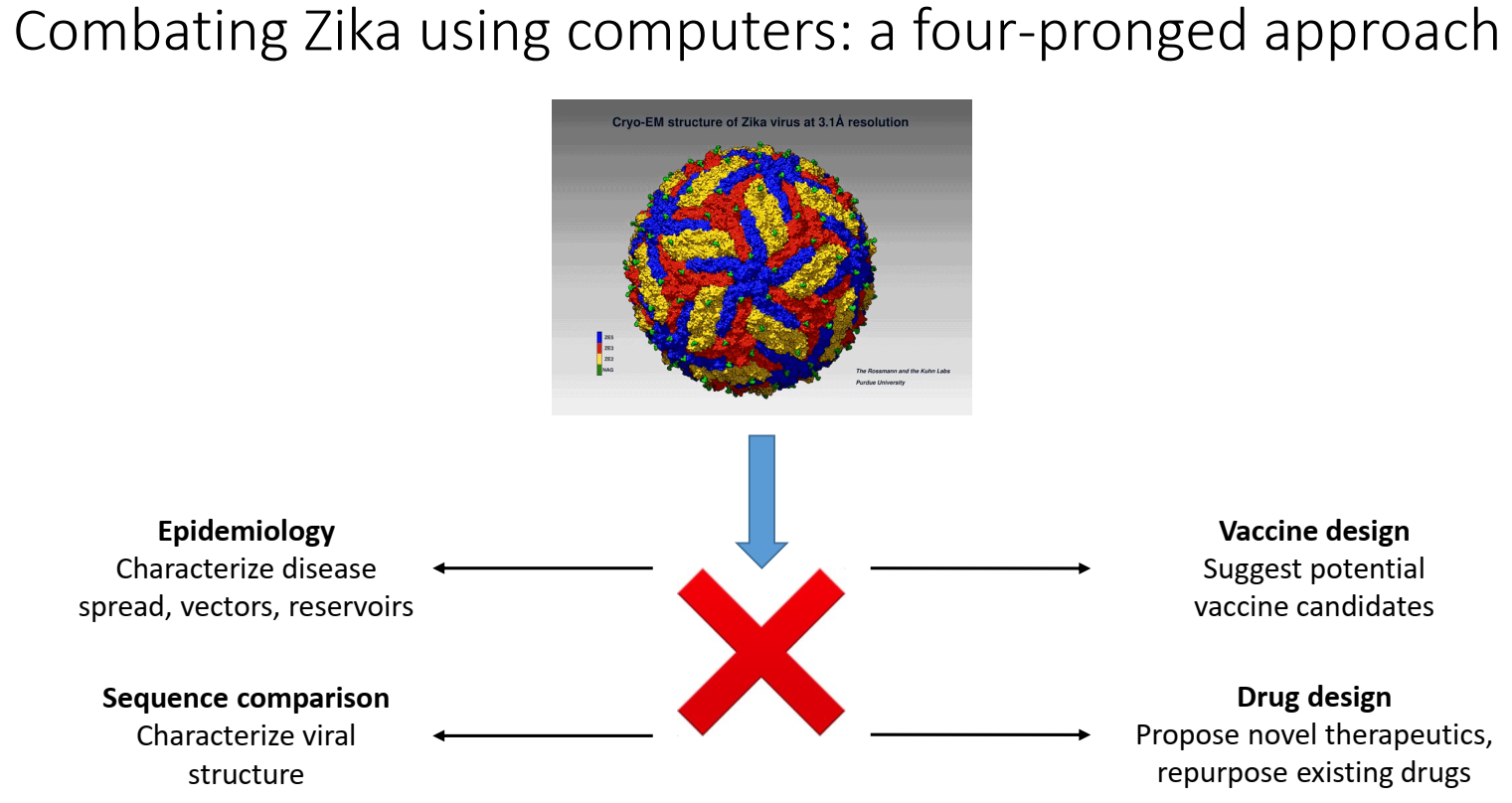
| Gene | Sequence Span, nt | Sequence Length | Protein/Biological Function |
|---|---|---|---|
| 5′-UTR | 1–107 | 107 | Encodes regions essential for genome cyclization/replication. |
| Capsid | 108–473 | 366 | Virion structure. |
| prM/M | 474–977 | 504 | prM forms heterodimers with E to form immature virion. prM then cleaved and mature virions formed with M. |
| E | 978–2489 | 1512 | Viral entry into host cell. |
| NS1 | 2490–3545 | 1056 | Viral replication, immune evasion, genome synthesis. |
| NS2A | 3546–4223 | 678 | Transmembrane protein, part of replication complex; assembly/secretion of virus particles. |
| NS2B | 4224–4613 | 390 | Cofactor for proteinase domain of NS3; proteolytic processing. |
| NS3 | 4614–6464 | 1851 | Protease/helicase. |
| NS4A | 6465–6914 | 381 | Viral RNA replication and amplification. |
| 2K | 6846–6914 | 69 | Peptide generated by cleavage at the N terminus of the NS4B signal sequence. |
| NS4B | 6915–7667 | 753 | Facilitates viral replication complexes; counteracts innate immune responses. |
| NS5 | 7668–10376 | 2709 | Methyltransferase; RNA-dependent RNA polymerase. |
| 3′-UTR | 10380–10807 | 427 | Facilitates viral replication and translation. |
| Year | Country | Remarks |
|---|---|---|
| 1947 | Uganda | First isolation and identification of ZIKV. Found in rhesus monkey, R766, caged in Zika forest. |
| 1948 | Uganda | Detected in A. africanus mosquitoes. Found in Zika forest. |
| 1951 | Nigeria | First instance of ZIKV antibodies in human blood, found in children. |
| 1952 | Uganda, Tanganyika | First human cases of ZIKV infection detected. |
| India | ZIKV antibodies found in human blood. | |
| 1953 | Malaya, North Borneo, Philippines | ZIKV antibodies found in residents. |
| Nigeria | ZIKV infection detected in three persons. | |
| 1954 | Egypt, Vietnam | ZIKV antibodies found in few residents. |
| 1955 | Nigeria | ZIKV antibodies found in human blood. |
| 1957 | Mozambique | ZIKV antibodies found in human blood. |
| 1958 | Uganda | Two strains of ZIKV found in A. aegypti mosquitoes in Zika forest. |
| 1960 | Angola | ZIKV antibodies found in indigenous residents. |
| 1961–1962 | Central African Republic | ZIKV antibodies found in human blood. |
| 1961–1964 | Ethiopia | ZIKV antibodies found in human blood. |
| 1962 | Senegal | ZIKV antibodies found in human blood. |
| 1963–1964 | Central African Republic, Burkina-Faso | ZIKV antibodies found in human blood. |
| 1963–1965 | Ivory Coast | ZIKV antibodies found in human blood. |
| 1964 | Uganda | First confirmation that ZIKV causes human disease. Clinical features reported. |
| 1964–1965 | Guinea-Bissau | ZIKV antibodies found in human blood. |
| 1964–1966 | Togo, Cameroon | ZIKV antibodies found in human blood. |
| 1965 | Niger | ZIKV antibodies found in human blood. |
| 1965–1967 | Nigeria | ZIKV antibodies found in human blood. |
| 1967 | Benin, Gabon, Liberia | ZIKV antibodies found in human blood. |
| 1966–1967 | Uganda, Kenya, Somalia, Morocco | ZIKV antibodies found in human blood. |
| 1967–1969 | Uganda | ZIKV antibodies found in human blood. |
| 1968 | Kenya | ZIKV antibodies found in human blood. |
| 1969–1972 | Nigeria | ZIKV antibodies found in human blood. |
| 1969 | Malaysia | ZIKV found in A. aegypti mosquitoes. |
| 1969–1983 | Indonesia, Malaysia, Pakistan | ZIKV found in mosquitoes. Sporadic human infections. |
| 1970 | Nigeria | ZIKV antibodies found in human blood. |
| 1971–1972 | Angola | ZIKV antibodies found in human blood. |
| 1972,1975, 1988,1990 | Senegal | ZIKV antibodies found in human blood. |
| 1979 | Central African Republic | ZIKV antibodies found in human blood. |
| 1980 | Nigeria | ZIKV antibodies found in human blood. |
| 1984 | Uganda | ZIKV antibodies found in human blood. |
| 1996–1997 | Malaysia | ZIKV antibodies found in human blood. |
| 1999 | Ivory Coast | ZIKV antibodies found in human blood. |
| Year | Country | Remarks |
|---|---|---|
| 2007 | Yap Island, Micronesia | First outbreak reported in humans. |
| 2008 | Senegal | First reported case of traveler infected in Senegal returning to home country and passing infection through sexual contact. |
| 2010 | Cameroon | ZIKV antibodies found in human blood. |
| 2010–2015 | Cambodia, Indonesia, Malaysia, Philippines, Thailand, Maldives | Mosquito transmission of ZIKV in these countries to travelers who then carried the infection to their home countries. |
| 2011–2014 | French Polynesia | Second reported outbreak of ZIKV infections. Connection with microcephaly and neurological disorders established later. |
| 2013–2014 | Chile, Cook Islands, New Caledonia | ZIKV outbreak. |
| 2013 | Tahiti | ZIKV isolated from patient’s semen showing sexual transmissibility. |
| 2014 | Zambia | ZIKV antibodies found in human blood. |
| 2015 April/May | Brazil, Bahia state | National Reference Laboratory, Brazil confirmed, by PCR, ZIKV infections, for the first time in the Americas. |
| 2015 July | Brazil | Zika cases confirmed by laboratory tests in 12 states. Neurological disorders associated with prior viral infections detected primarily in the Bahia region. |
| 2015 October | South America | Colombia, Republic of Cabo Verde report confirmed cases of ZIKV infections. Brazil reported unusual increase in the number of cases of neonatal microcephaly. |
| 2015 November | Central and South America | Brazil reported 141 suspected cases of microcephaly and declared a national public health emergency. Brazil reported detection of ZIKV in amniotic fluid of fetuses with confirmed microcephaly. Suriname, Panama, El Salvador, Guatemala, and Paraguay confirmed cases of ZIKV infection. The Pan American Health Organization and WHO issued an epidemiological alert. |
| 2015 November | Mexico | Three cases of ZIKV infection confirmed by PCR. |
| 2015 November | French Polynesia | Retrospective analysis reveals unusually large number of central nervous system malformations in fetuses and infants in 2014–2015. |
| 2015 December | Central and South America | Honduras, French Guiana, and Martinique reported confirmed cases of ZIKV infections. |
| 2015 December | Puerto Rico | First confirmed case of Zika infection reported. |
| 2016 | Maldives | Finnish national working in Maldivestested positive for Zika after return to Finland. |
| 2016 January | Americas | Guyana reported the first PCR-confirmed case of locally acquired Zika infection. Ecuador, Bolivia, Barbados, Haiti, Dominican Republic, Nicaragua, Curacao and Jamaica reported the first confirmed cases of Zika infections. First case of Zika in St. Martin reported. |
| 2016 January | US Virgin Islands | First confirmed case of Zika in St. Croix reported. |
| 2016 February | Americas | First confirmed case of ZIKV infection in Chile reported. First case of sexually transmitted Zika infection in Texas, USA reported. |
| 2016 | Various countries | Angola, Antigua, British Virgin Islands, Trinidad and Tobago, Guadulope, Fiji, Marshall Islands, Papua New Guinea, and other countries report first cases of ZIKV infections. |
| 2016 | Singapore | ZIKV infection reported. |
| 2016/2017 | India | Three cases of ZIKV infection reported in Ahmedabad. |
| 2017 | Singapore | Several cases of locally transmitted ZIKV confirmed. |
| Location | No of Seqs | Average gR | Std Dev | Change | Hosts |
|---|---|---|---|---|---|
| Africa | 7 | 100.80 | 0.58 | - | Aedes africanus, A. taylori |
| Asia | 106 | 89.08 | 3.95 | −11.63% | Homo sapiens |
| South America | 103 | 85.92 | 3.31 | −4.55% | Homo sapiens |
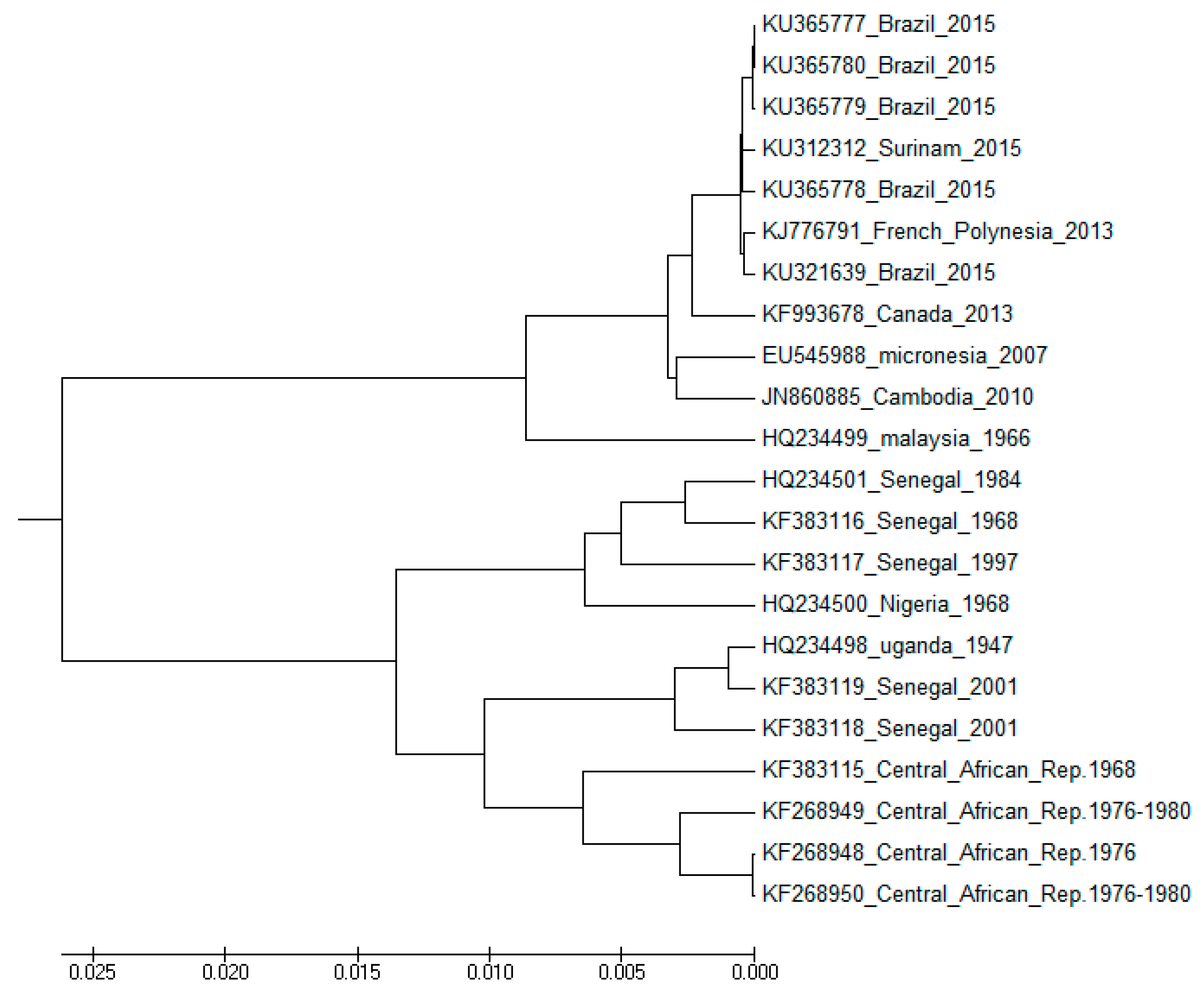
| Continent | d | Locus ID | Year | Country | Host |
|---|---|---|---|---|---|
| Africa | 83.1238 | KF268949 | 1980 | Central African Republic | Aedes opok |
| 226.8671 | KF383115 | 1968 | Central African Republic | Aedes africanus | |
| 187.0571 | KF383116 | 1968 | Senegal | Aedes luteocephalus | |
| 204.2905 | KF383118 | 2001 | Senegal | Aedes dalzieli | |
| Asia | 219.0294 | KY241697 | 2016 | Singapore | Homo sapiens |
| 258.6828 | KY241700 | 2016 | Singapore | Homo sapiens | |
| 224.1343 | KY241704 | 2016 | Singapore | Homo sapiens | |
| 286.1132 | KY241766 | 2016 | Singapore | Homo sapiens | |
| 261.6707 | MK238035 | 2018 | India | Homo sapiens | |
| 261.6707 | MK238038 | 2018 | India | Homo sapiens | |
| South America | 220.3336 | KY559005 | 2018 | Brazil | Homo sapiens |
| 261.9161 | KY559027 | 2018 | Brazil | Homo sapiens | |
| 242.1343 | KY785427 | 2018 | Brazil | Homo sapiens | |
| 264.0801 | KY785429 | 2018 | Brazil | Homo sapiens | |
| 185.0375 | KY785433 | 2018 | Brazil | Homo sapiens | |
| 283.8677 | KY785456 | 2018 | Brazil | Homo sapiens | |
| 294.1674 | MH882537 | 2018 | Brazil | Homo sapiens |
| Compounds | Derivatives | Reference |
|---|---|---|
| Chloroquine | Derivatives particularly at the C-4 position of N-(2-arylmethylimino)ethyl-7-chloroquinolin-4-amine derivatives | [98] |
| Quinacrine (QC), Mefloquine (MQ), and GSK369796 | Antimalarial aminoquinoline derivatives | [41,87] |
| PHA-690509 | Cyclin dependent kinase (CDK) inhibitor | [99] |
| Lapachol, HMC-HO1α and Ivermectin | Hybrid drugs against co-infections of ZIKV, dengue and chikungunya | [100] |
| 20-Cmethylated nucleosides | Inhibitors of RNA-dependent RNA polymerase (RdRp) | [101] |
| NS3 inhibitors | Covalent inhibitors of a viral protein and anti-Toll-like receptor molecules | [102] |
| FDA-approved drugs | In vitro screening of 774 compounds led to twenty compounds that were found to reduce ZIKV infection | [93] |
| NIH clinical library of compounds | By screening 725 chemically diverse compounds from the library, 22 compounds were reported to have potent anti-ZIKV activity of which five were found promising. These are Lovastatin (Pubchem CID: 53232), 5-Fluorouracil (Pubchem CID: 3385); 6-Azauridine (Pubchem CID: 5901); Palonosetron (Pubchem CID: 6337614) and Kitasamycin (Pubchem CID: 44634697). | [103] |
| A limited proprietary library of small organic compounds | Anti-ZIKV activity through screening and confirming potent anti-ZIKV activity in in vitro plaque assay | [104] |
| NITD008 | A type of nucleoside adenosine analog | [105] |
| Warfarin and a few similar structural analogues | Inhibitors of dimerization of Axl receptor (a tyrosine kinase) | [106] |
| Nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, ibuprofen, naproxen, acetaminophen, and lornoxicam, potently inhibited the entry of Zika virus Env/HIV-1-pseudotyped viruses | Inhibited replication of wild-type ZIKV both in cell lines and in primary human fetal endothelial cells. Interestingly, the NSAIDs exerted this inhibitory effect by potently reducing the expression of AXL, the entry cofactor of ZIKV. Further studies showed that the NSAIDs downregulated the prostaglandin E2/prostaglandin E receptor 2 (EP2)/cAMP/protein kinase A (PKA) signaling pathway and reduced PKA-dependent CDC37 phosphorylation and the interaction between CDC37 and HSP90, which subsequently facilitated CHIP/ubiquitination/proteasome-mediated AXL degradation. | [107] |
| Nanchangmycin | Envelope glycoprotein inhibitor | [90] |
| Temoporfin, NSC157058 | NS2B-NS3protease inhibitors | [108] |
| Suramin | NS3 polymerase inhibitors | [109] |
| Sofosbuvir, 2′-C-ethynyl-UTP and DMB213 | NS5 polymerase inhibitors | [110] |
| Sinefungin | NS5 methyltransferase inhibitor | [111] |
| 6-azauridine and 5-fluorouracil | Pyrimidine biosynthesis inhibitors | [93,95] |
| Lovastatin and Mevastatin | HMG-CoA reductase inhibitor | [112] |
| BCX4430 | An adenosine nucleoside analog, functions as a selective inhibitor of viral RNA-dependent RNA polymerase (RdRp). It was found that BCX4430 had EC50 values in the range 3.8–18.2 μg/mL in vitro, with favorable selective index (SI) values. In a mouse model of ZIKV infection (300 mg/kg/d), treatment with BCX4430 showed promising results. The protective effect of BCX4430 was observed to continue for 24 h even after virus challenge. | [113] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basak, S.C.; Majumdar, S.; Nandy, A.; Roy, P.; Dutta, T.; Vracko, M.; Bhattacharjee, A.K. Computer-Assisted and Data Driven Approaches for Surveillance, Drug Discovery, and Vaccine Design for the Zika Virus. Pharmaceuticals 2019, 12, 157. https://doi.org/10.3390/ph12040157
Basak SC, Majumdar S, Nandy A, Roy P, Dutta T, Vracko M, Bhattacharjee AK. Computer-Assisted and Data Driven Approaches for Surveillance, Drug Discovery, and Vaccine Design for the Zika Virus. Pharmaceuticals. 2019; 12(4):157. https://doi.org/10.3390/ph12040157
Chicago/Turabian StyleBasak, Subhash C., Subhabrata Majumdar, Ashesh Nandy, Proyasha Roy, Tathagata Dutta, Marjan Vracko, and Apurba K. Bhattacharjee. 2019. "Computer-Assisted and Data Driven Approaches for Surveillance, Drug Discovery, and Vaccine Design for the Zika Virus" Pharmaceuticals 12, no. 4: 157. https://doi.org/10.3390/ph12040157
APA StyleBasak, S. C., Majumdar, S., Nandy, A., Roy, P., Dutta, T., Vracko, M., & Bhattacharjee, A. K. (2019). Computer-Assisted and Data Driven Approaches for Surveillance, Drug Discovery, and Vaccine Design for the Zika Virus. Pharmaceuticals, 12(4), 157. https://doi.org/10.3390/ph12040157