Myristic Acid Coated Protein Immobilised Mesoporous Silica Particles as pH Induced Oral Delivery System for the Delivery of Biomolecules


Abstract
1. Introduction
2. Results and Discussion
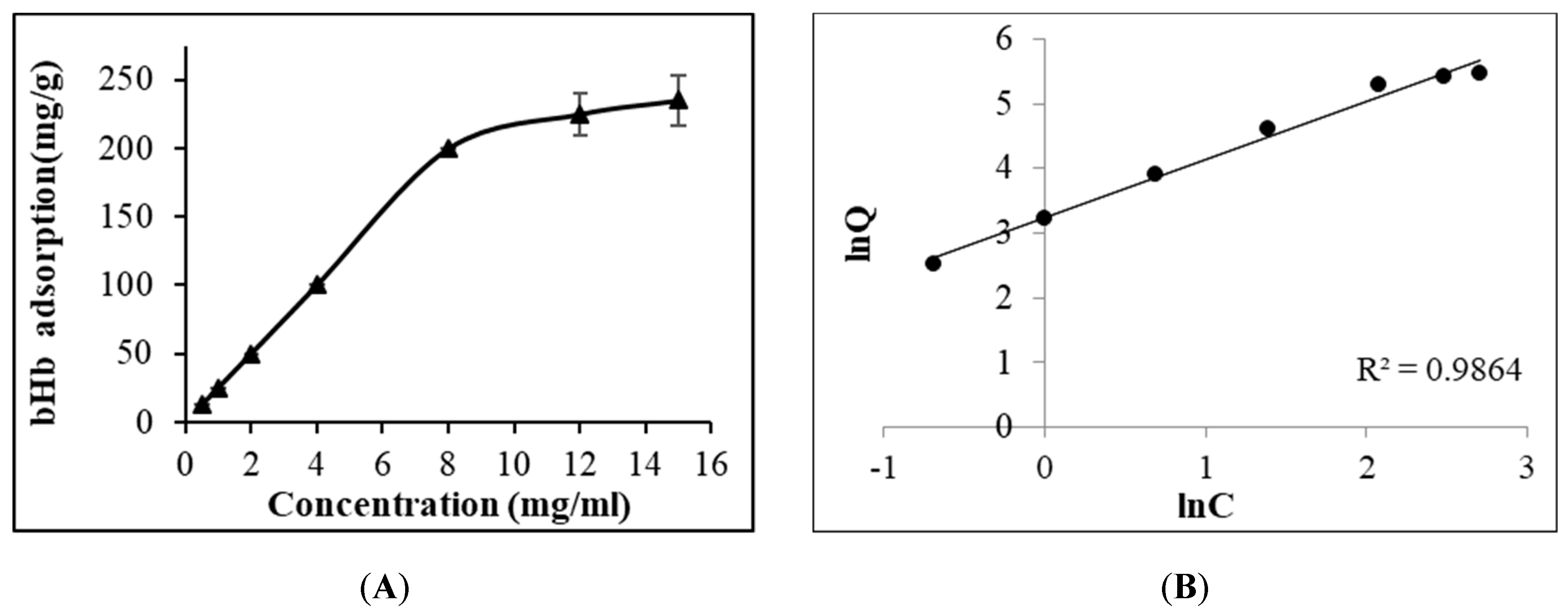
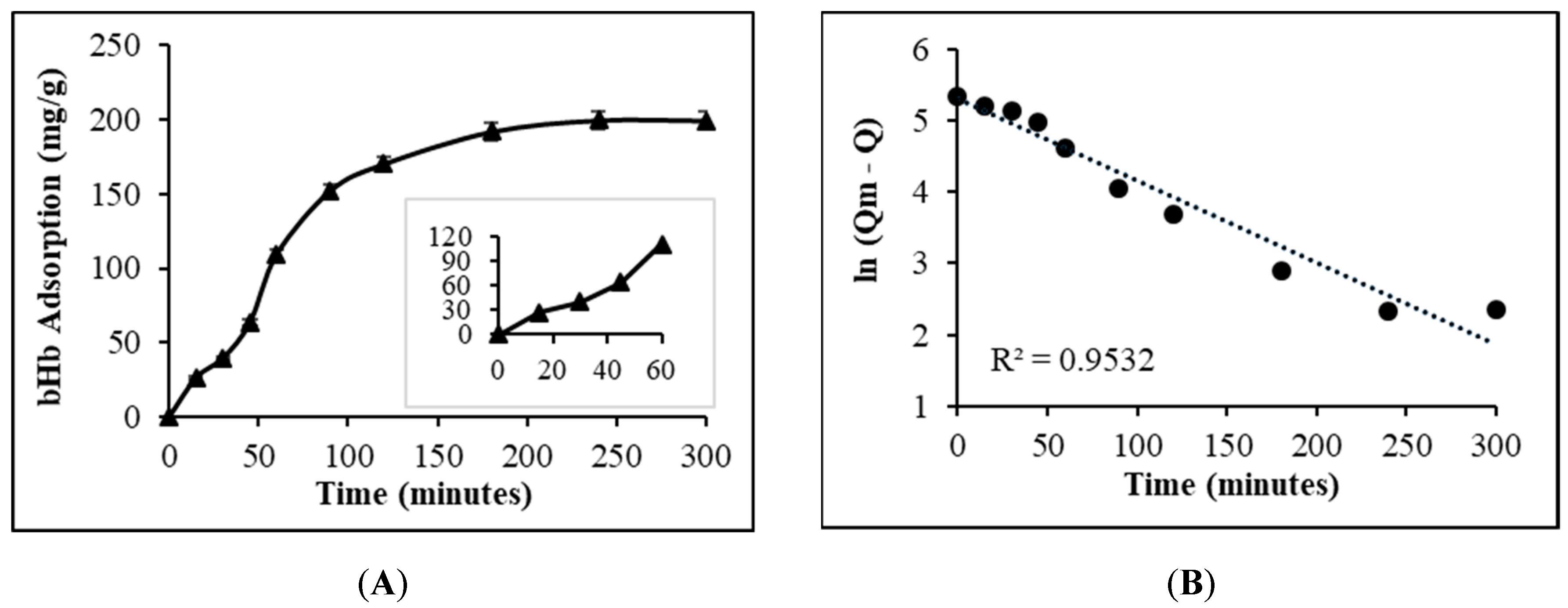
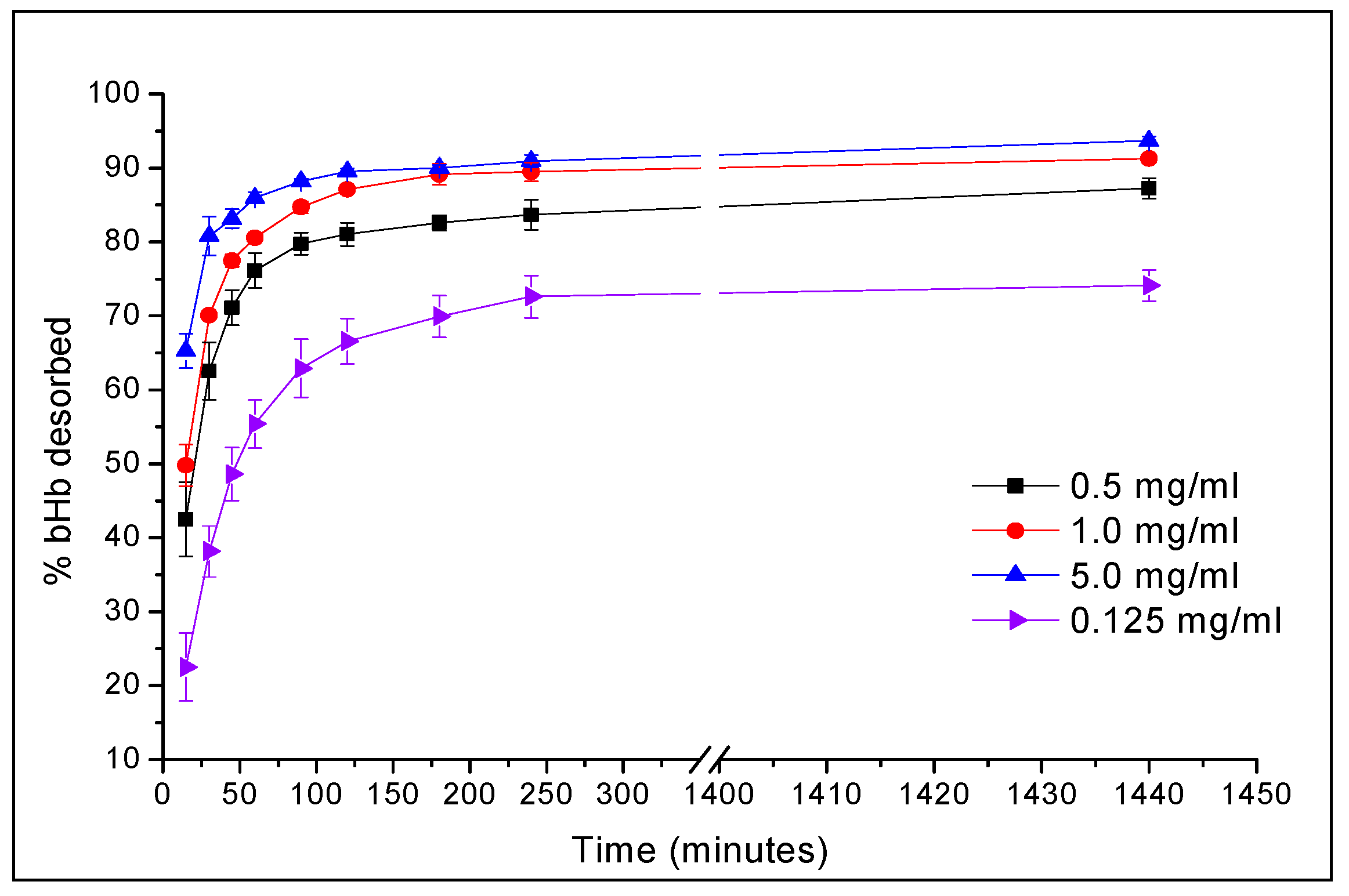
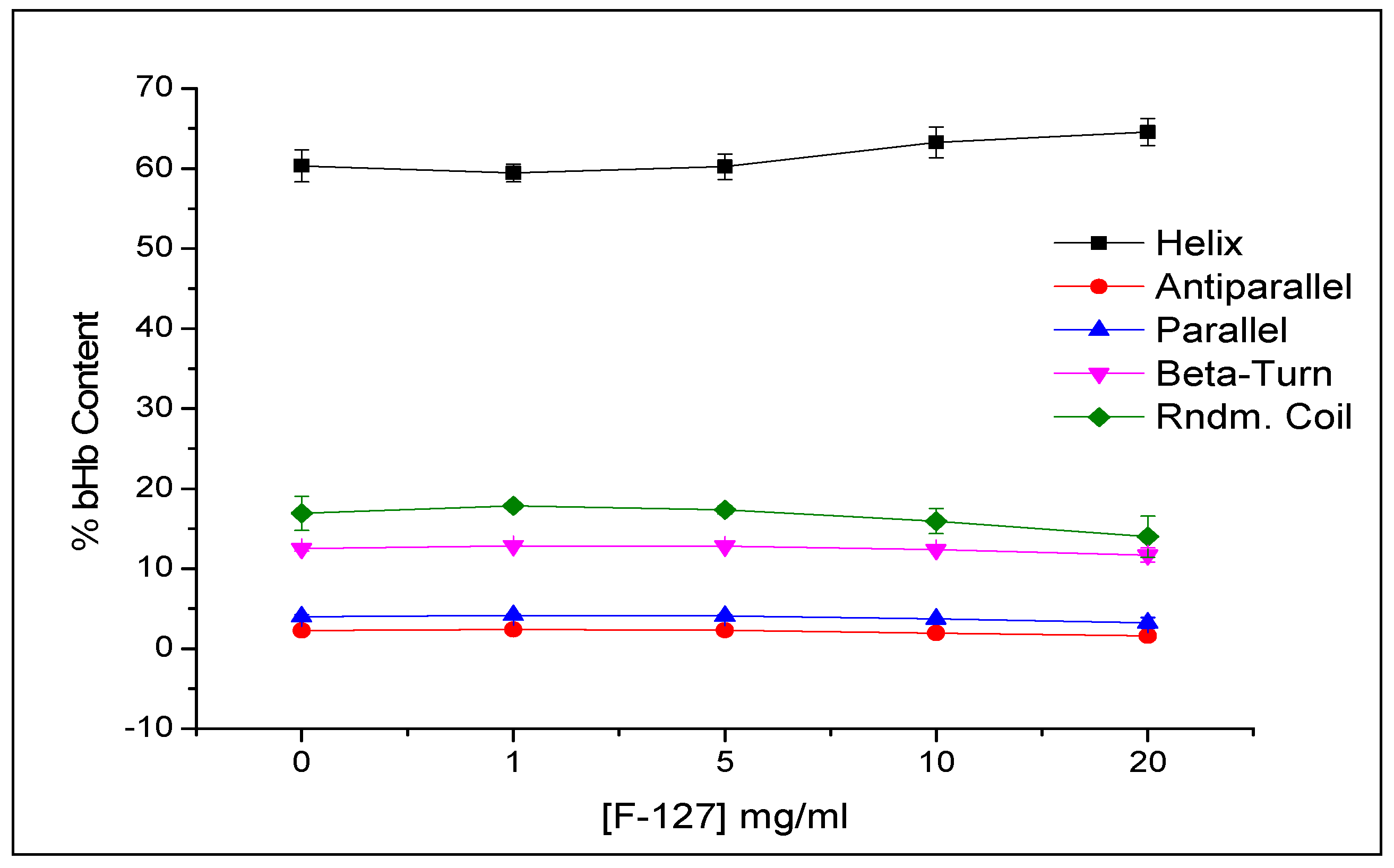
2.1. Maximum bHb Adsorption and Adsorption Kinetics
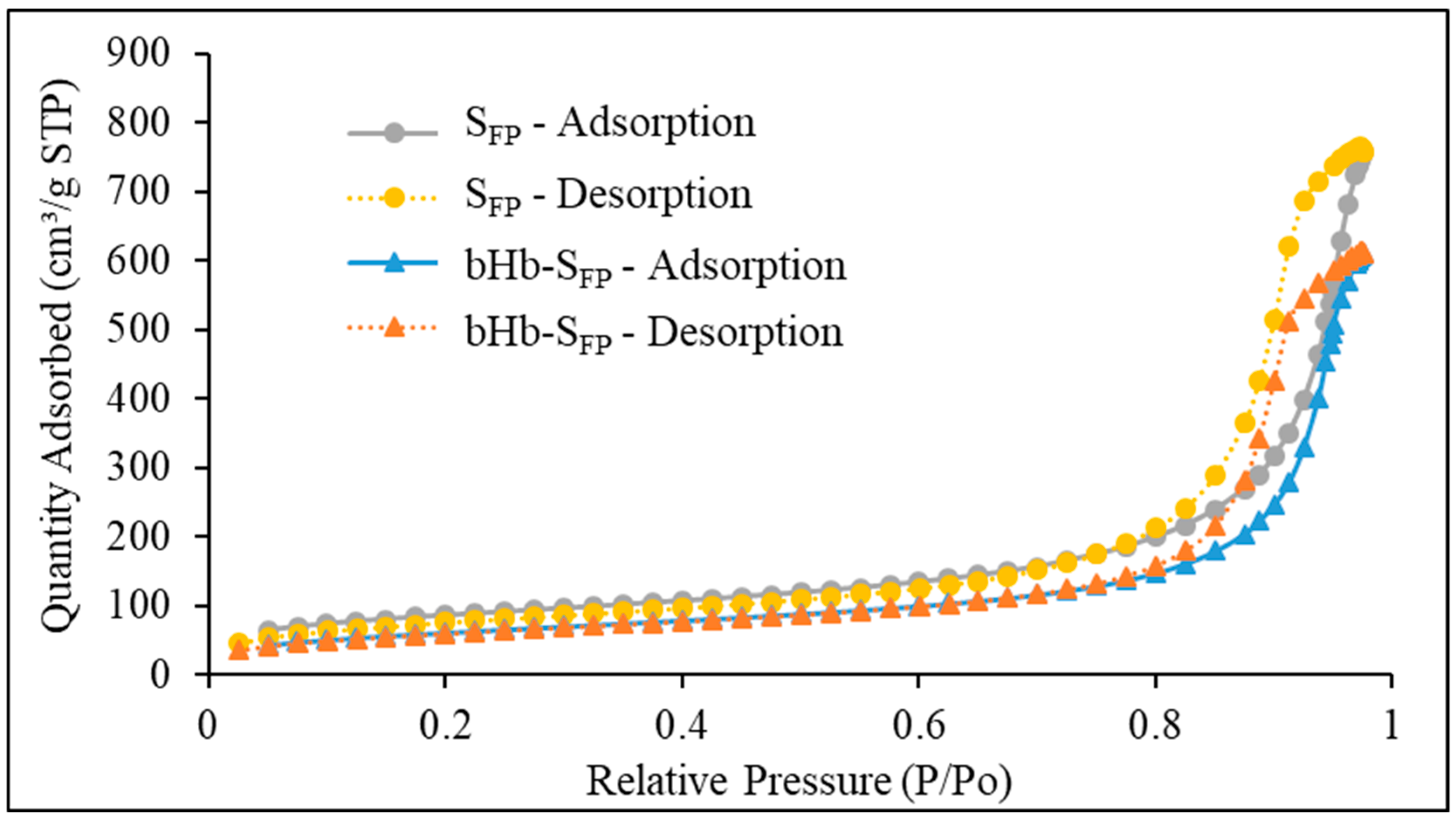
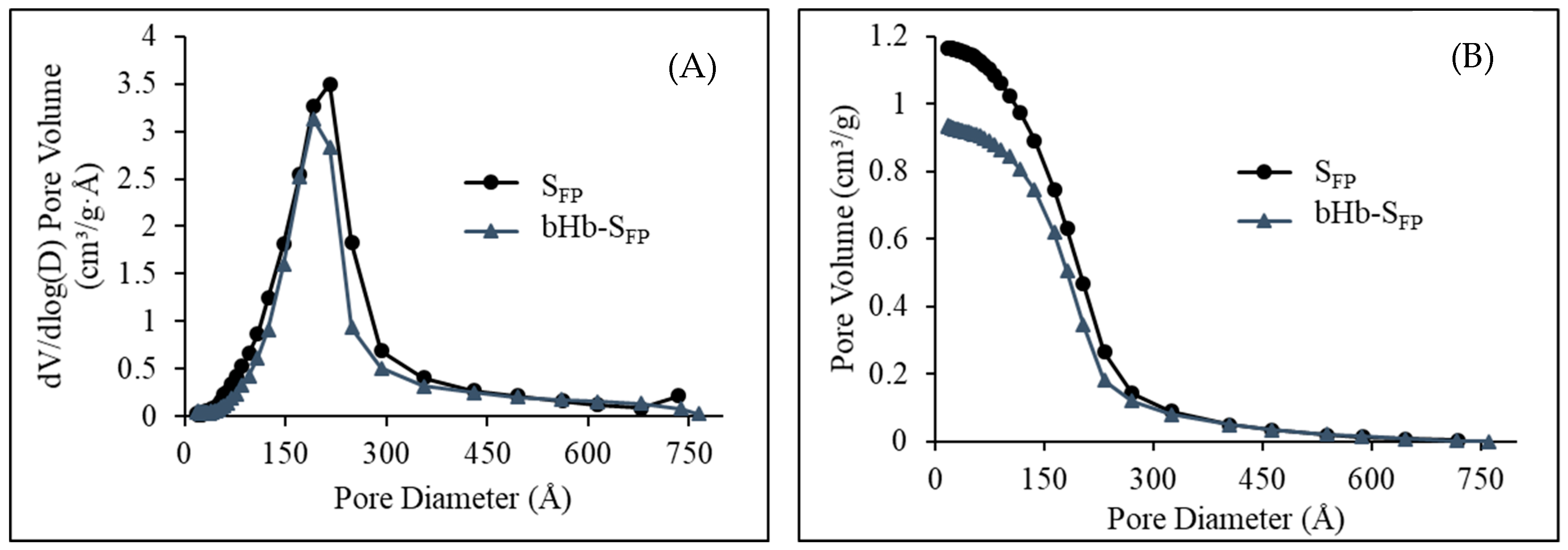
2.2. Effect of Protein Adsorption on Porosity and Pore Volume
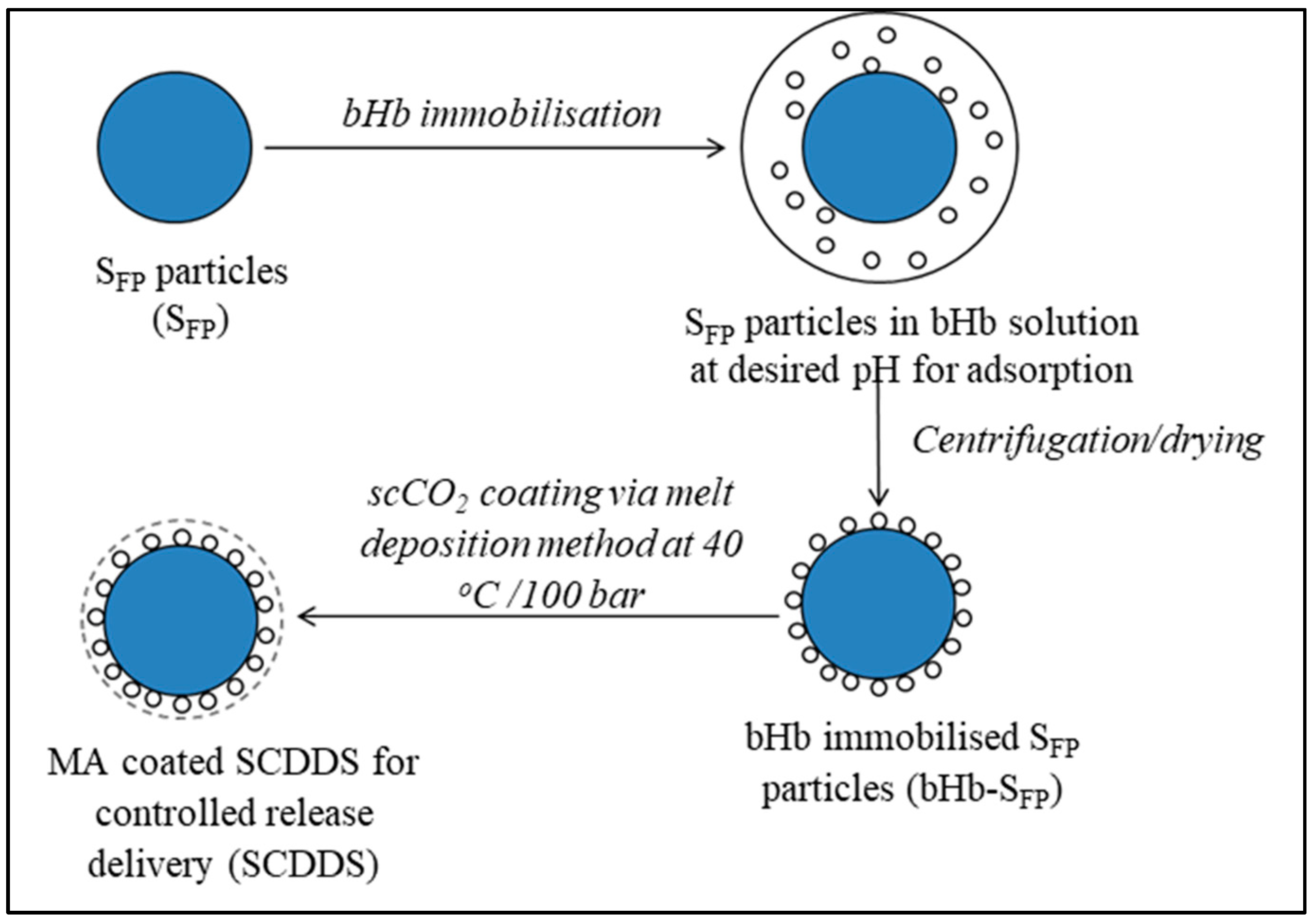
2.3. SCDDS Preparation
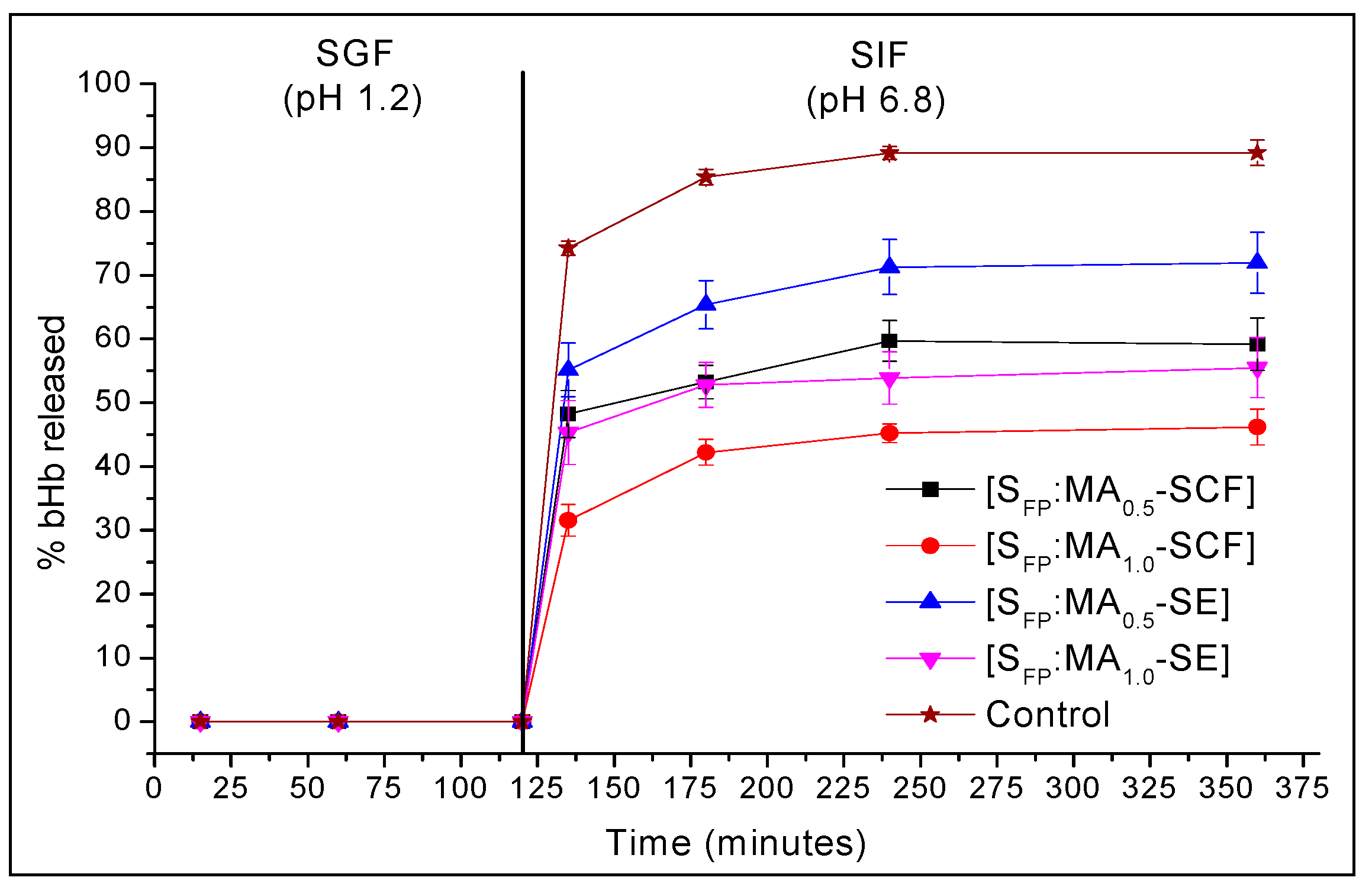
2.4. In-Vitro Release Studies at pH 1.2 and 6.8
3. Materials and Methods
3.1. Materials
3.2. bHb Adsorption and Kinetics
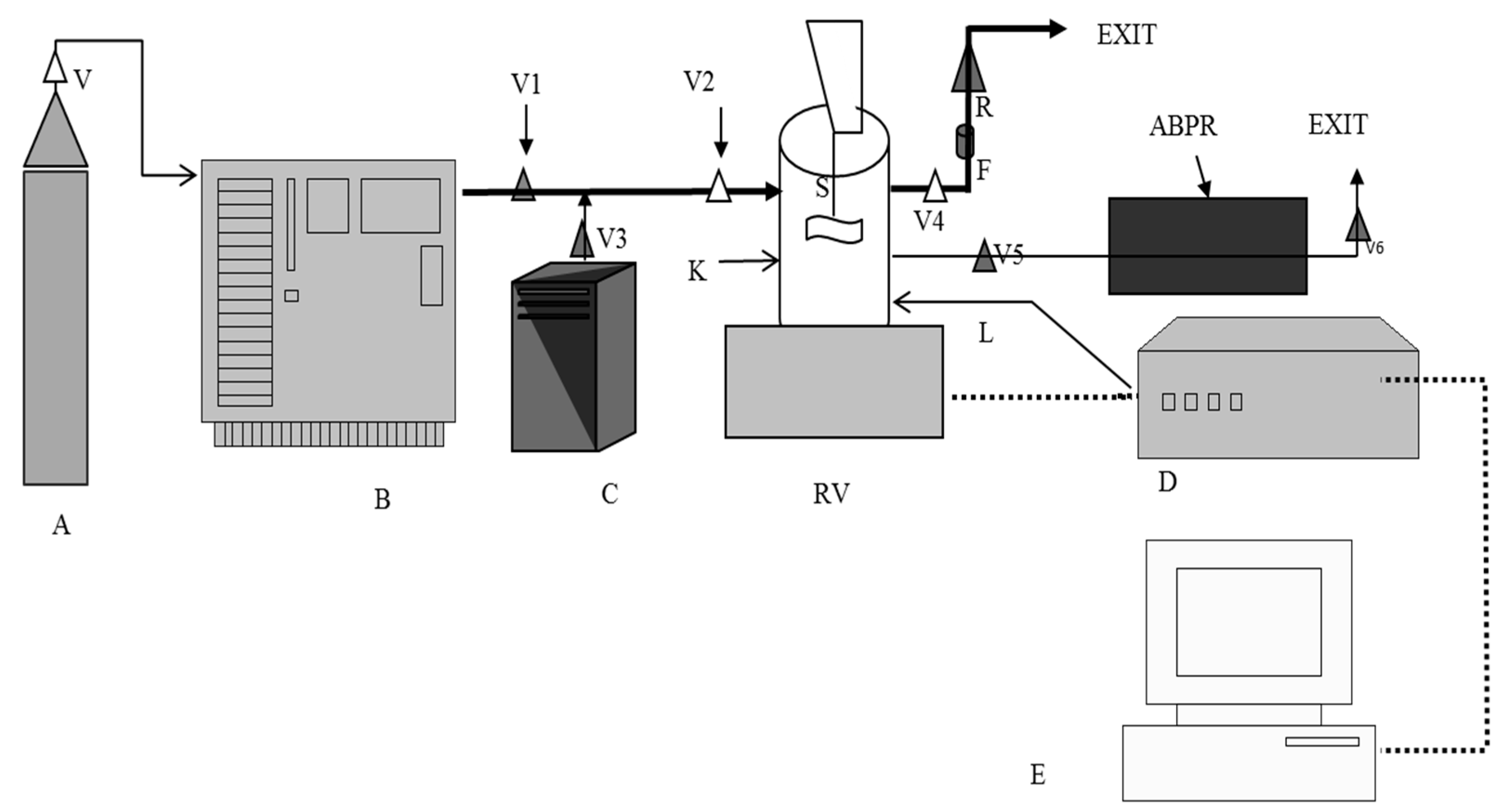
3.3. Coating of Silica Particles by SCF Processing
3.4. Coating of Silica Particles by Solvent Evaporation
3.5. Analysis
3.5.1. bHb Quantification
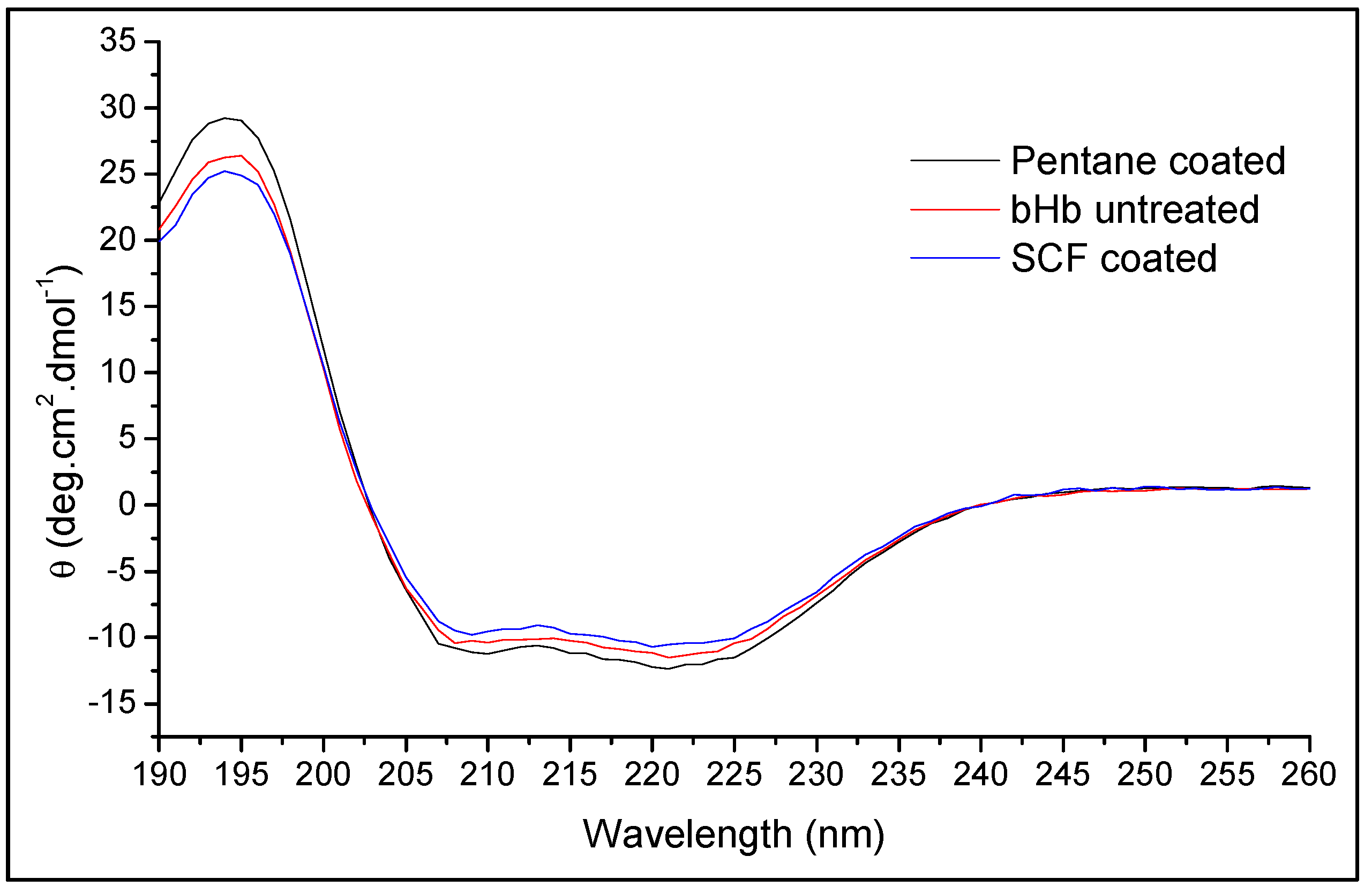
3.5.2. Conformational Integrity Determination
3.5.3. Nitrogen (N2) Sorption Analysis
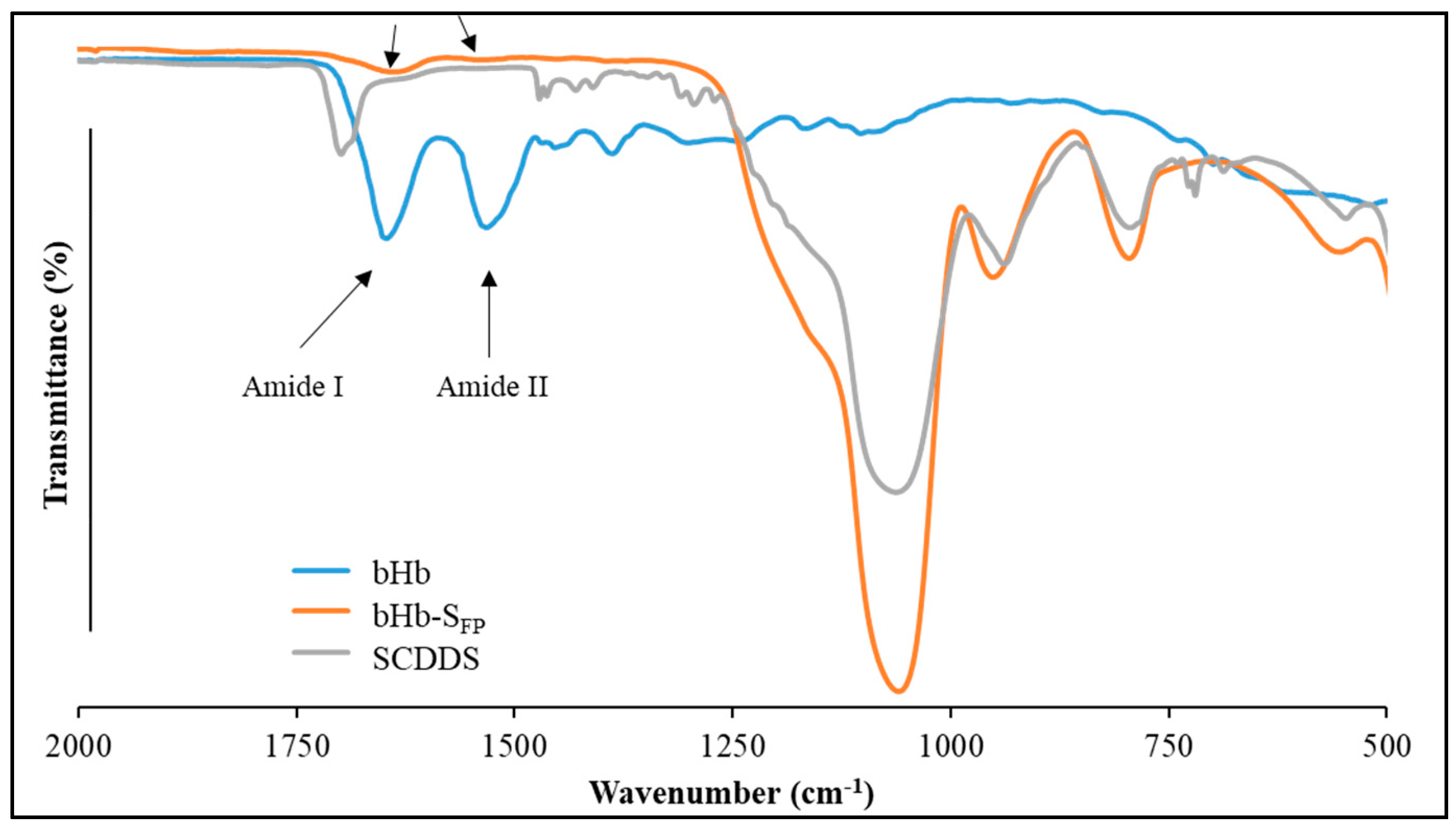
3.5.4. ATR-FTIR Analysis
3.6. In-vitro Release Studies in Simulated Gastric (SGF) and Intestine (SIF) Fluids
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andrade, F.; Antunes, F.; Nascimento, A.V.; da Silva, S.B.; das Neves, J.; Ferreira, D.; Sarmento, B. Chitosan formulations as carriers for therapeutic proteins. Curr. Drug Discov. Technol. 2011, 8, 157–172. [Google Scholar] [CrossRef]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery--liposomes versus lipid nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Yang, M.; Lai, S.K.; Wang, Y.; Zhong, W.; Happe, C.; Zhang, M.; Fu, J.; Hanes, J. Biodegradable nanoparticles composed entirely of safe materials that rapidly penetrate human mucus. Angew. Chem. Int. Ed. Engl. 2011, 50, 2597–2600. [Google Scholar] [CrossRef]
- Brown, L.R. Commercial challenges of protein drug delivery. Expert Opin. Drug Deliv. 2005, 2, 29–42. [Google Scholar] [CrossRef]
- Wang, W. Advanced protein formulations. Protein Sci. 2015, 24, 1031–1039. [Google Scholar] [CrossRef]
- Tabernero, A.; Martín del Valle, E.M.; Galán, M.A. Supercritical fluids for pharmaceutical particle engineering: Methods, basic fundamentals and modelling. Chem. Eng. Process. Process. Intensif. 2012, 60, 9–25. [Google Scholar] [CrossRef]
- Soon, H.S.; Lai, Y.L. Microencapsulation and nanoencapsulation using supercritical fluid (SCF) techniques. Pharmaceutics 2019, 11, 21–38. [Google Scholar]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.W.; Lin, V.S.Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Roggers, R.; Kanvinde, S.; Boonsith, S.; Oupický, D. The practicality of mesoporous silica nanoparticles as drug delivery devices and progress toward this goal. AAPS PharmSciTech 2014, 15, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Barbe, C.; Bartlett, J.; Kong, L.; Finnie, K.; Lin, H.Q.; Larkin, M.; Calleja, S.; Bush, A.; Calleja, G. Silica particles: A novel drug-delivery system. Adv. Mater. 2004, 16, 1959–1966. [Google Scholar] [CrossRef]
- Kwon, S.; Singh, R.K.; Perez, R.A.; Abou Neel, E.A.; Kim, H.W.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Areva, S.; Aäritalo, V.; Tuusa, S.; Jokinen, M.; Lindén, M.; Peltola, T. Sol-Gel-derived TiO2-SiO2 implant coatings for direct tissue attachment. Part II: Evaluation of cell response. J. Mater. Sci. Mater. Med. 2007, 18, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Huo, Q.; Liu, J.; Wang, L.; Jiang, Y.; Lambert, T.N.; Fang, E. A new class of silica cross-linked micellar core-shell nanoparticles. J. Am. Chem. Soc. 2006, 128, 6447–6453. [Google Scholar] [CrossRef] [PubMed]
- Allouche, J.; Boissire, M.; Helary, C.; Livage, J.; Coradin, T. Biomimetic core-shell gelatine/silica nanoparticles: A new example of biopolymer-based nanocomposites. J. Mater. Chem. 2006, 16, 3120. [Google Scholar] [CrossRef]
- Arruebo, M.; Galán, M.; Navascués, N.; Téllez, C.; Marquina, C.; Ibarra, M.R.; Santamaría, J. Development of magnetic nanostructured silica-based materials as potential vectors for drug-delivery applications. Chem. Mater. 2006, 18, 1911–1919. [Google Scholar] [CrossRef]
- Liu, Y.; Hamari, C.E.G.; Haifeng, Y.; Fussenegger, M. A synthetic free fatty acid-regulated transgene switch in mammalian cells and mice. Nucleic Acids Res. 2018, 46, 9864–9874. [Google Scholar] [CrossRef]
- Killen, B.U.; Corrigan, O.I. Factors influencing drug release from stearic acid based compacts. Int. J. Pharm. 2001, 228, 189–198. [Google Scholar] [CrossRef]
- Xie, S.; Zhu, L.; Dong, Z.; Wang, Y.; Wang, X.; Zhou, W. Preparation and evaluation of ofloxacin-loaded palmitic acid solid lipid nanoparticles. Int. J. Nanomed. 2011, 6, 547–555. [Google Scholar]
- Zhang, Q.; Yie, G.; Li, Y.; Yang, Q.; Nagai, T. Studies on the cyclosporin A loaded stearic acid nanoparticles. Int. J. Pharm. 2000, 200, 153–159. [Google Scholar] [CrossRef]
- Nam, J.P.; Park, S.C.; Kim, T.H.; Jang, J.Y.; Choi, C.; Jang, M.K.; Nah, J.W. Encapsulation of paclitaxel into lauric acid-O-carboxymethyl chitosan-transferrin micelles for hydrophobic drug delivery and site-specific targeted delivery. Int. J. Pharm. 2013, 457, 124–135. [Google Scholar] [CrossRef]
- Robson, H.J.; Craig, D.Q.; Deutsch, D. An investigation into the release of cefuroxime axetil from taste-masked stearic acid microspheres. Part 1: The influence of the dissolution medium on the drug release profile and the physical integrity of the microspheres. Int. J. Pharm. 1999, 190, 183–192. [Google Scholar] [CrossRef]
- Cerchiara, T.; Luppi, B.; Bigucci, F.; Petrachi, M.; Orienti, I.; Zecchi, V. Controlled release of vancomycin from freeze-dried chitosan salts coated with different fatty acids by spray-drying. J. Microencapsul. 2003, 20, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Urabe, Y.; Shiomi, T.; Itoh, T.; Kawai, A.; Tsunoda, T.; Mizukami, F.; Sakaguchi, K. Encapsulation of hemoglobin in mesoporous silica (FSM)-enhanced thermal stability and resistance to denaturants. Chembiochem 2007, 8, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.M.; Liu, J.; Nigra, M.; Coppens, M.O. Chaperonin-inspired pH protection by mesoporous silica SBA-15 on myoglobin and lysozyme. Langmuir 2016, 32, 9604–9610. [Google Scholar] [CrossRef]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [Google Scholar] [CrossRef]
- Zhan, G.; Li, C.; Luo, D. Electrochemical investigation of bovine hemoglobin at an acetylene black paste electrode in the presence of sodium dodecyl sulfate. Bull. Chem. Soc. 2007, 28, 1720–1724. [Google Scholar]
- Bhomia, R. Studies into Solid Core Drug Delivery System Using Haemoglobin as a Model Drug and Supercritical Fluid Processing for Encapsulation. Unpublished Ph.D. Thesis, University of Greenwich, Kent, UK, 2015. [Google Scholar]
- Bhomia, R.; Trivedi, V.; Coleman, N.J.; Mitchell, J.C. The thermal and storage stability of bovine haemoglobin by ultraviolet–visible and circular dichroism spectroscopies. J. Pharm. Anal. 2016, 6, 242–248. [Google Scholar] [CrossRef]
- Chung, H.K.; Kim, W.H.; Park, J.; Cho, J.; Jeong, T.Y.; Park, P.K. Application of langmuir and freundlich isotherms to predict adsorbate removal efficiency or required amount of adsorbent. J. Ind. Eng. Chem. 2015, 28, 241–246. [Google Scholar] [CrossRef]
- Kalavathy, M.H.; Swaroop, G.; Padmini, E.; Miranda, L.R. Moringa Oleifera, A biosorbent for resorcinol adsorption-isotherm and kinetic studies. Carbon Lett. 2009, 10, 23–32. [Google Scholar] [CrossRef]
- Waters, L.J.; Hussain, T.; Parkes, G.; Hanrahan, J.P.; Tobin, J.M. Inclusion of fenofibrate in a series of mesoporous silicas using microwave irradiation. Eu. J. Pharm. Biopharm. 2013, 85, 936–941. [Google Scholar] [CrossRef]
- Choudhari, Y.; Reddy, U.; Monsuur, F.; Pauly, T.; Hoefer, H.; McCarthy, W. Comparative evaluation of porous silica based carriers for lipids and liquid drug formulations. Mesoporous Biomater. 2014, 1, 61–74. [Google Scholar] [CrossRef]
- Multifunctional Excipients for the Pharmaceutical Industry. Available online: https://grace.com/pharma-and-biotech/en-us/Documents/Syloid/M309c_Syloid_FP_XDP_Tech_Note_0915.pdf (accessed on 27 September 2019).
- Sing, K.; Everett, D.; Haul, R.; Moscou, L.; Pierotti, R.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Ojeda, L.M.; Esparza, J.M.; Campero, A.; Cordero, S.; Kornhauser, I.; Rojas, F. On comparing BJH and NLDFT pore-size distributions determined from N2 sorption on SBA-15 substrata. Phys. Chem. Chem. Phys. 2003, 5, 1859–1866. [Google Scholar] [CrossRef]
- Pettit, M.W.; Dyer, P.D.R.; Mitchell, J.C.; Griffiths, P.C.; Alexander, B.; Cattoz, B.; Heenan, R.K.; King, S.M.; Schweins, R.; Pullen, F.; et al. Construction and physiochemical characterisation of a multi-composite, potential oral vaccine delivery system (VDS). Int. J. Pharm. 2014, 468, 264–271. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef]
- Norde, W.; MacRitchie, F.; Nowicka, G.; Lyklema, J. Protein adsorption at solid-liquid interfaces: Reversibility and conformation aspects. J. Colloid Interface Sci. 1986, 112, 447–456. [Google Scholar] [CrossRef]
- Rapoza, R.J.; Horbett, T.A. The effects of concentration and adsorption time on the elutability of adsorbed proteins in surfactant solutions of varying structures and concentrations. J. Colloid Interface Sci. 1990, 136, 480–493. [Google Scholar] [CrossRef]
- Bohnert, J.L.; Horbett, T.A. Changes in adsorbed fibrinogen and albumin interactions with polymers indicated by decreases in detergent elutability. J. Colloid Interface Sci. 1986, 111, 363–377. [Google Scholar] [CrossRef]
- Elwing, H.; Askendal, A.; Lundström, I. Desorption of fibrinogen and γ-globulin from solid surfaces induced by a nonionic detergent. J. Colloid Interface Sci. 1989, 128, 296–300. [Google Scholar] [CrossRef]
- Sarkar, D.; Chattoraj, D.K. Kinetics of desorption of proteins from the surface of protein-coated alumina by various desorbing reagents. J. Colloid Interface Sci. 1996, 178, 606–613. [Google Scholar] [CrossRef]
- Vasilescu, M.; Angelescu, D. Interactions of globular proteins with surfactants studied with fluorescence probe methods. Langmuir 1999, 15, 2635–2643. [Google Scholar] [CrossRef]
- Attiaa, M.A.; El-Gibalya, I.; Shaltoutb, S.E.; Fetiha, G.N. Transbuccal permeation, anti-inflammatory activity and clinical efficacy of piroxicam formulated in different gels. Int. J. Pharm. 2004, 276, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Vorum, H.; Brodersen, R.; Kragh-Hansen, U.; Pedersen, A.O. Solubility of long-chain fatty acids in phosphate buffer at pH 7.4. Biochim. Biophys. Acta-Lipids Lipid Metab. 1992, 1126, 135–142. [Google Scholar] [CrossRef]
- Kassaye, L.; Genete, G. Evaluation and comparison of in-vitro dissolution profiles for different brands of amoxicillin capsules. Afr. Health Sci. 2013, 13, 369–375. [Google Scholar] [CrossRef]
- Bryksa, B.C.; Yada, R.Y. Protein structure insights into the bilayer interactions of the saposin-like domain of Solanum tuberosum aspartic protease. Sci. Rep. 2017, 7, 1–19. [Google Scholar] [CrossRef]
- Trivedi, V.; Bhomia, R.; Mitchell, J.C.; Coleman, N.J.; Douroumis, D.; Snowden, M.J. Study of the effect of pressure on melting behavior of saturated fatty acids in liquid or supercritical carbon dioxide. J. Chem. Eng. Data 2013, 58, 1861–1866. [Google Scholar] [CrossRef]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | % Mean bHb Released ± S.D | ||||
|---|---|---|---|---|---|
| Control | SFP:MA0.5-SCF | SFP:MA1.0-SCF | SFP:MA0.5-PEN | SFP:MA1.0-SCF | |
| 135 | 74.3 ± 1.1 | 48.2 ± 3.7 | 31.6 ± 2.5 | 55.2 ± 4.2 | 45.3 ± 5.0 |
| 180 | 85.4 ± 1.2 | 53.3 ± 2.6 | 42.2 ± 2.0 | 65.4 ± 3.8 | 52.8 ± 3.5 |
| 240 | 89.2 ± 1.0 | 59.7 ± 3.2 | 45.2 ± 1.5 | 71.3 ± 4.3 | 53.9 ± 4.1 |
| 360 | 89.2 ± 2.0 | 59.2 ± 4.1 | 46.2 ± 2.8 | 71.9 ± 4.8 | 55.5 ± 4.7 |
| F1 | 35 | 51 | 22 | 39 | |
| F2 | 27 | 18 | 36 | 24 | |
| Secondary Structure | bHb (Untreated) | bHb Released from SFP Formulation | |
|---|---|---|---|
| SCF Coated | Pentane Coated | ||
| Helix | 60.5 ± 1.5% | 59.1 ± 0.4% | 60.7 ± 0.9% |
| Antiparallel | 0.7 ± 0.1% | 0.8 ± 0.0% | 0.7 ± 0.1% |
| Parallel | 4.9 ± 0.2% | 5.1 ± 0.1% | 4.9 ± 0.2% |
| Beta turns | 12.4 ± 0.2% | 12.5 ± 0.1% | 12.3 ± 0.1% |
| Random coil | 20.7 ± 1.0% | 21.2 ± 0.1% | 20.6 ± 0.7% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trivedi, V.; Bhomia, R.; Mitchell, J.C. Myristic Acid Coated Protein Immobilised Mesoporous Silica Particles as pH Induced Oral Delivery System for the Delivery of Biomolecules. Pharmaceuticals 2019, 12, 153. https://doi.org/10.3390/ph12040153
Trivedi V, Bhomia R, Mitchell JC. Myristic Acid Coated Protein Immobilised Mesoporous Silica Particles as pH Induced Oral Delivery System for the Delivery of Biomolecules. Pharmaceuticals. 2019; 12(4):153. https://doi.org/10.3390/ph12040153
Chicago/Turabian StyleTrivedi, Vivek, Ruchir Bhomia, and John C Mitchell. 2019. "Myristic Acid Coated Protein Immobilised Mesoporous Silica Particles as pH Induced Oral Delivery System for the Delivery of Biomolecules" Pharmaceuticals 12, no. 4: 153. https://doi.org/10.3390/ph12040153
APA StyleTrivedi, V., Bhomia, R., & Mitchell, J. C. (2019). Myristic Acid Coated Protein Immobilised Mesoporous Silica Particles as pH Induced Oral Delivery System for the Delivery of Biomolecules. Pharmaceuticals, 12(4), 153. https://doi.org/10.3390/ph12040153