Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity

 ,
,  ,
,  ,
, 


Abstract

1. Introduction
1.1. From Platinum(II) to Ruthenium(III)-based Complexes: The Importance of Nanoformulations in Metallo Drug Delivery
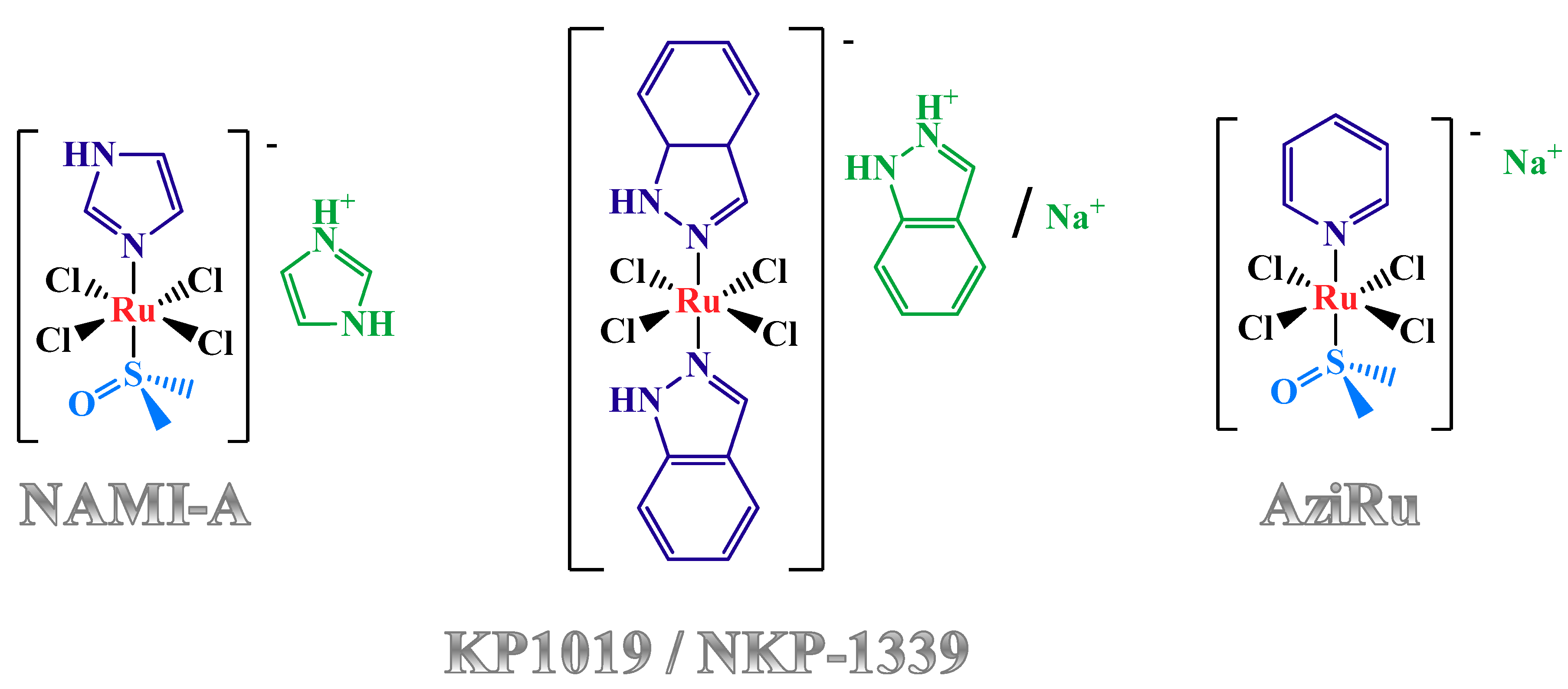
1.2. Anticancer Activity and Mechanism of Action of the Lead Low Molecular Weight Ru(III)-Based Compounds
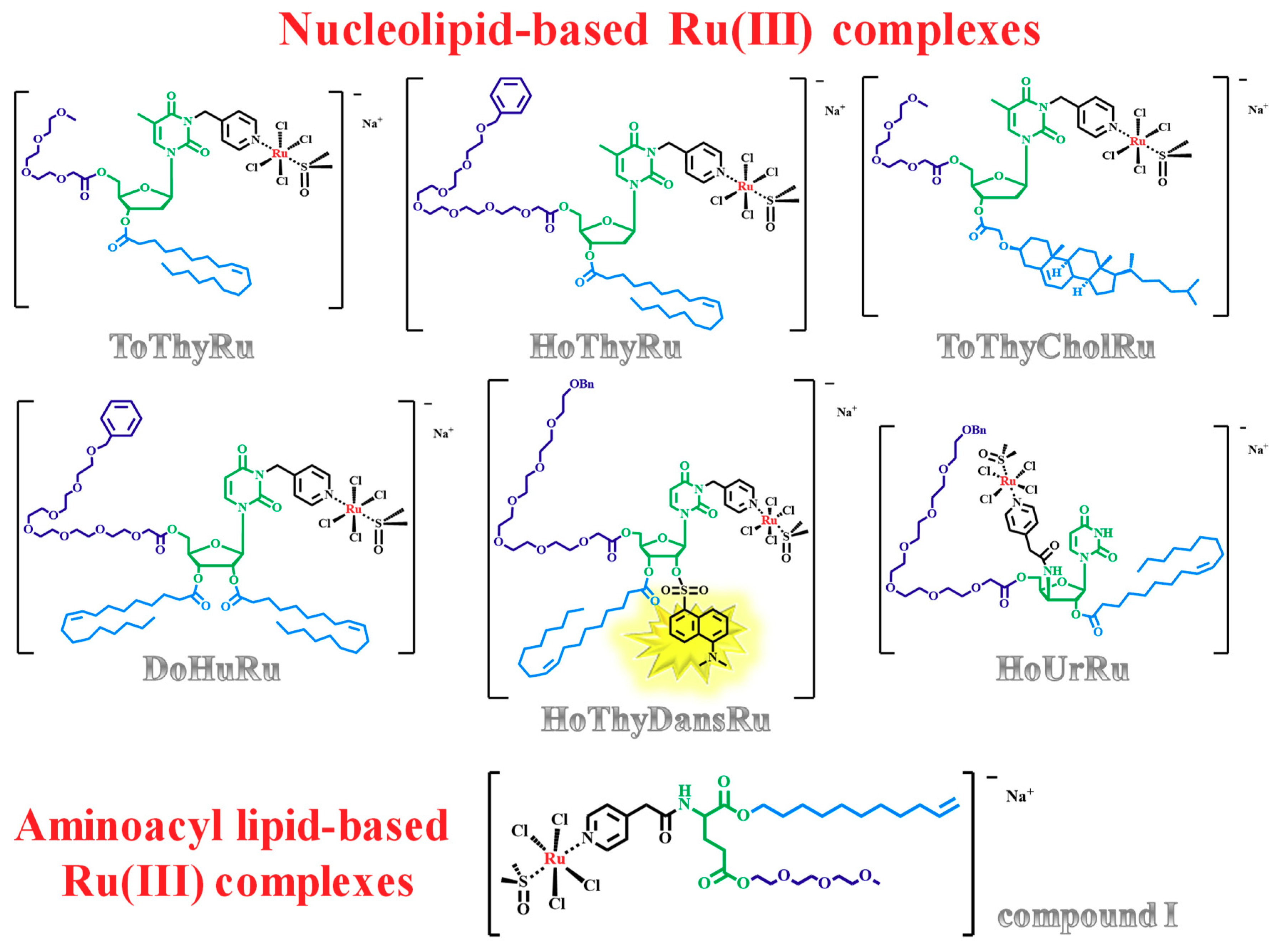
1.3. Nucleolipid and Aminoacyl Lipid-based Structures Incorporating AziRu, a NAMI-A-Like Ruthenium Compound
2. Ru(III)-Containing Formulations as Efficient Drug Delivery Systems
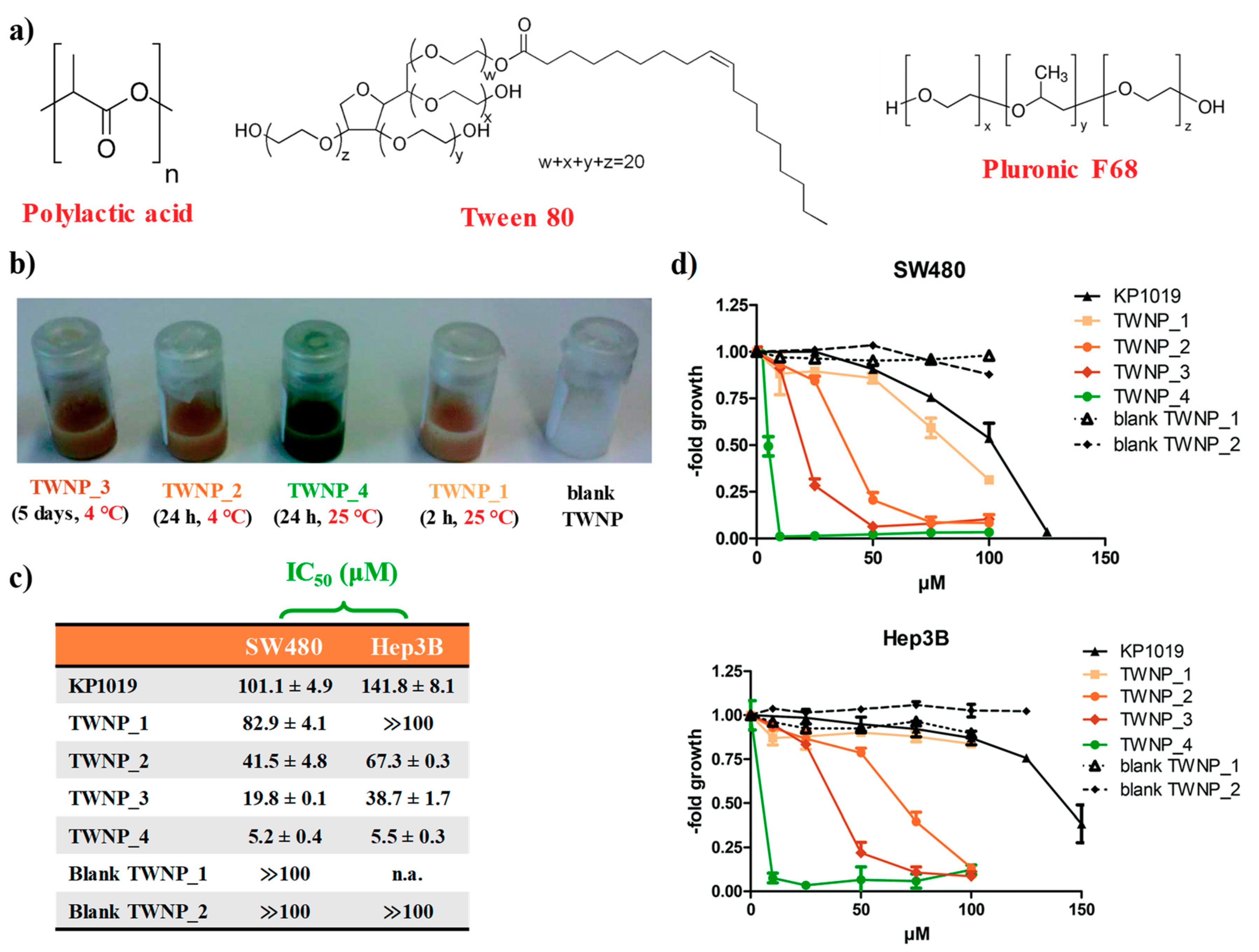
2.1. KP1019-Hosting Nanosystems
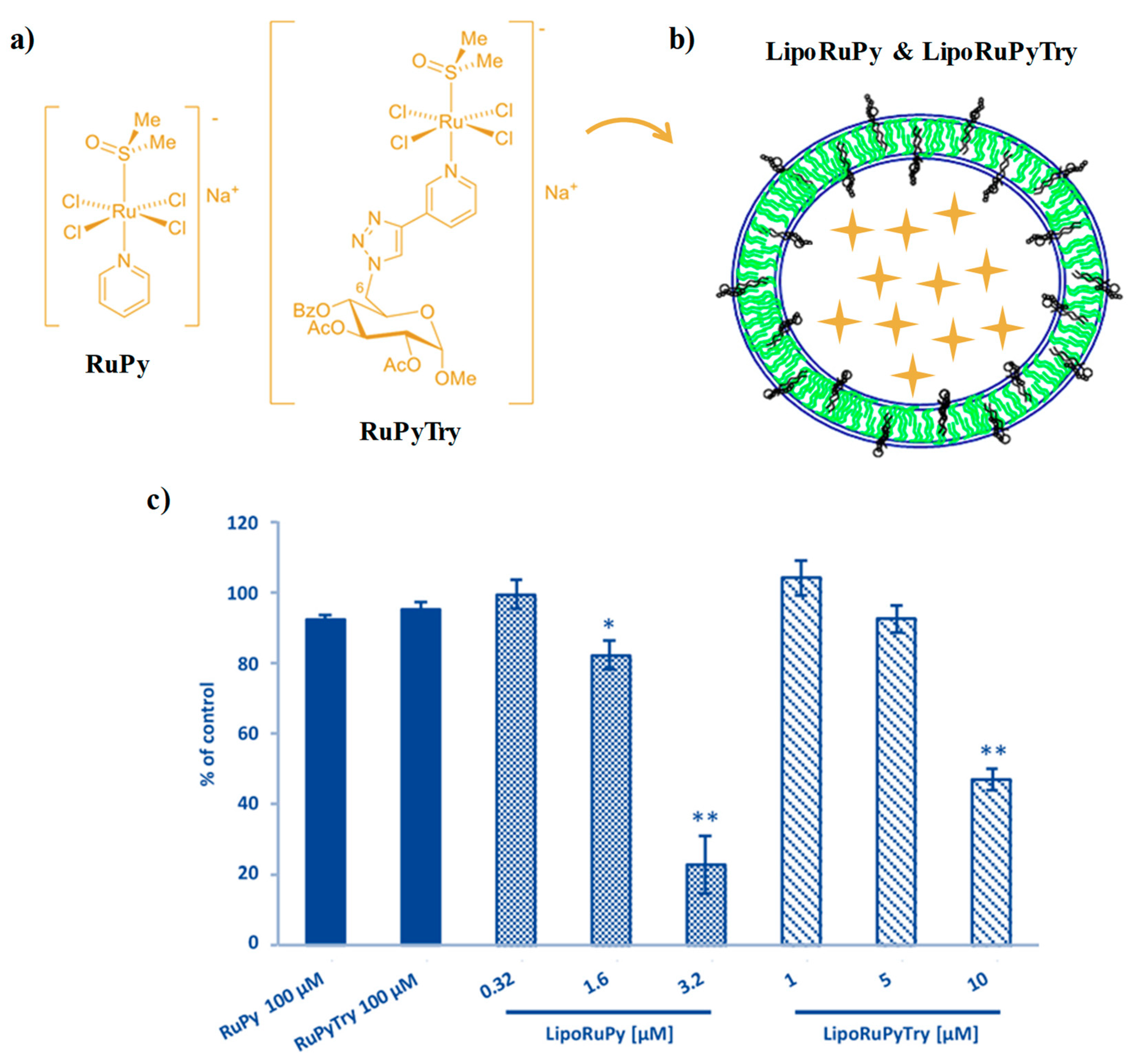
2.2. NAMI-A-Hosting Nanosystems
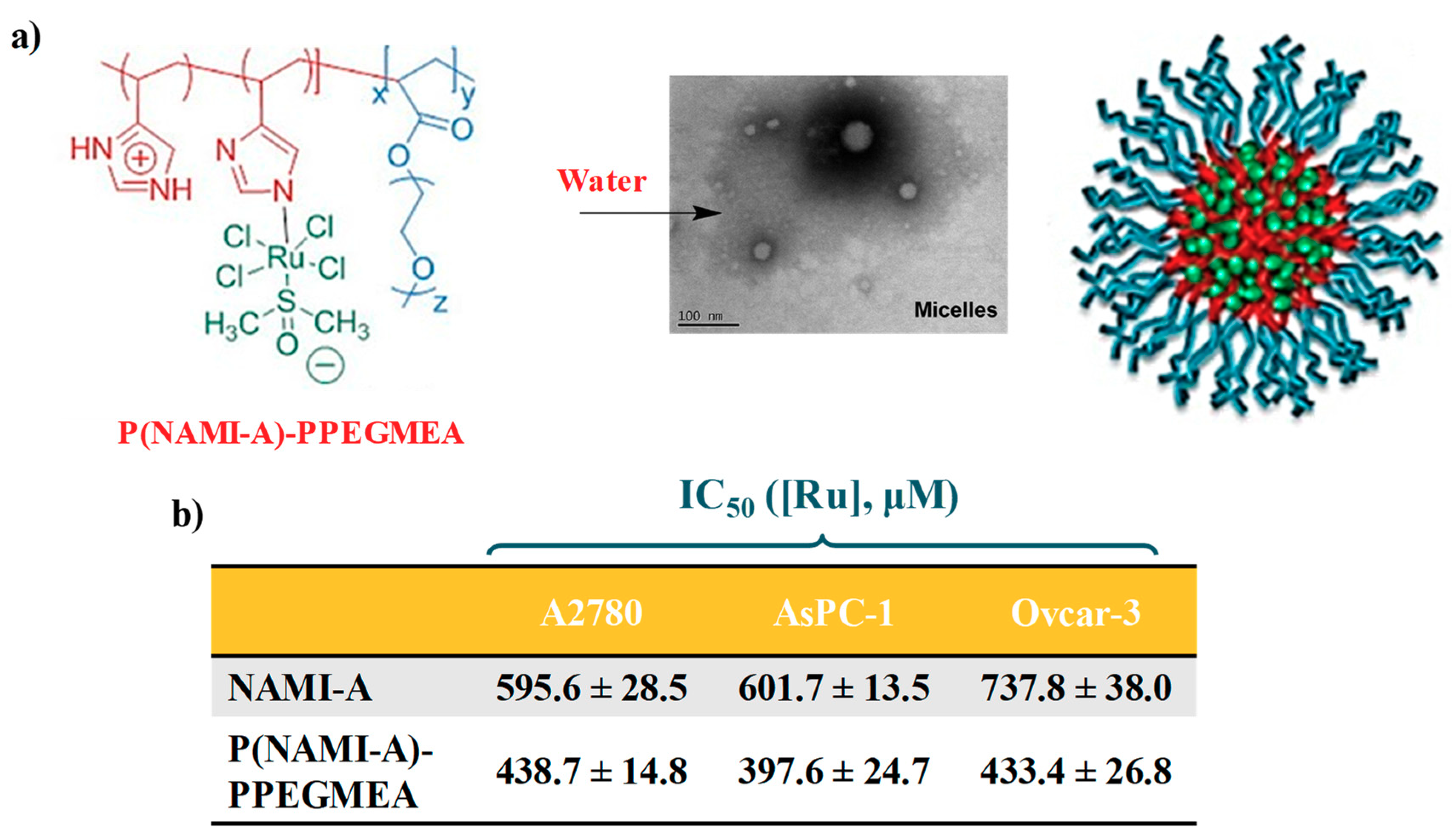
2.3. AziRu-Hosting Nanosystems
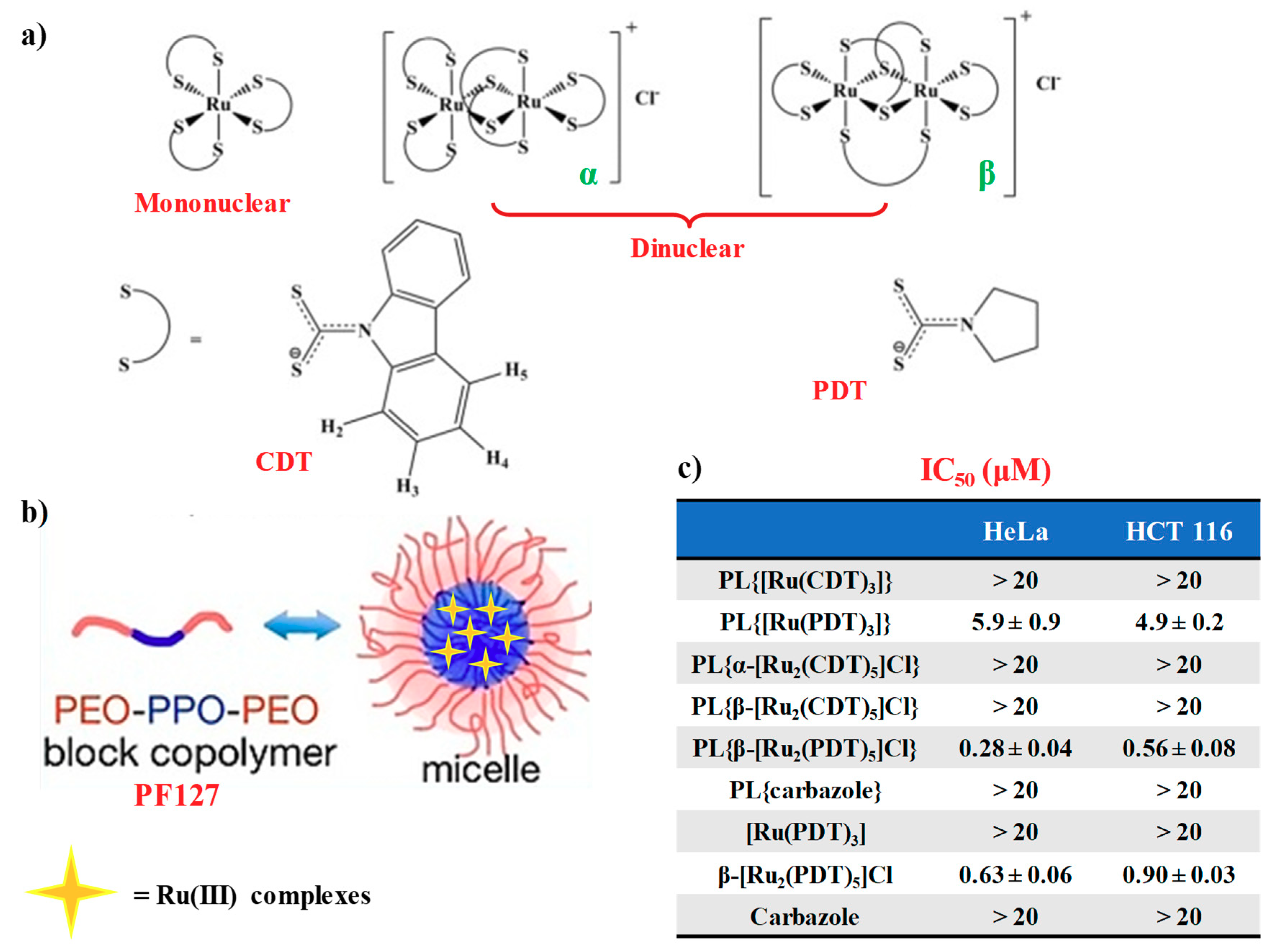
2.4. Mononuclear and Dinuclear Ru(III)-Dithiocarbamato Complexes Encapsulated in Nanosized Carriers
2.5. Liposome-Based Systems Containing Nucleolipid or Aminoacyl Lipid-Based Ru(III) Complexes
3. Antiproliferative Effects of Liposome-Based Systems Containing Nucleolipid or Aminoacyl Lipid-Based Ru(III) Complexes: Insight into Their Mode of Action
3.1. In Vitro Bioactivity
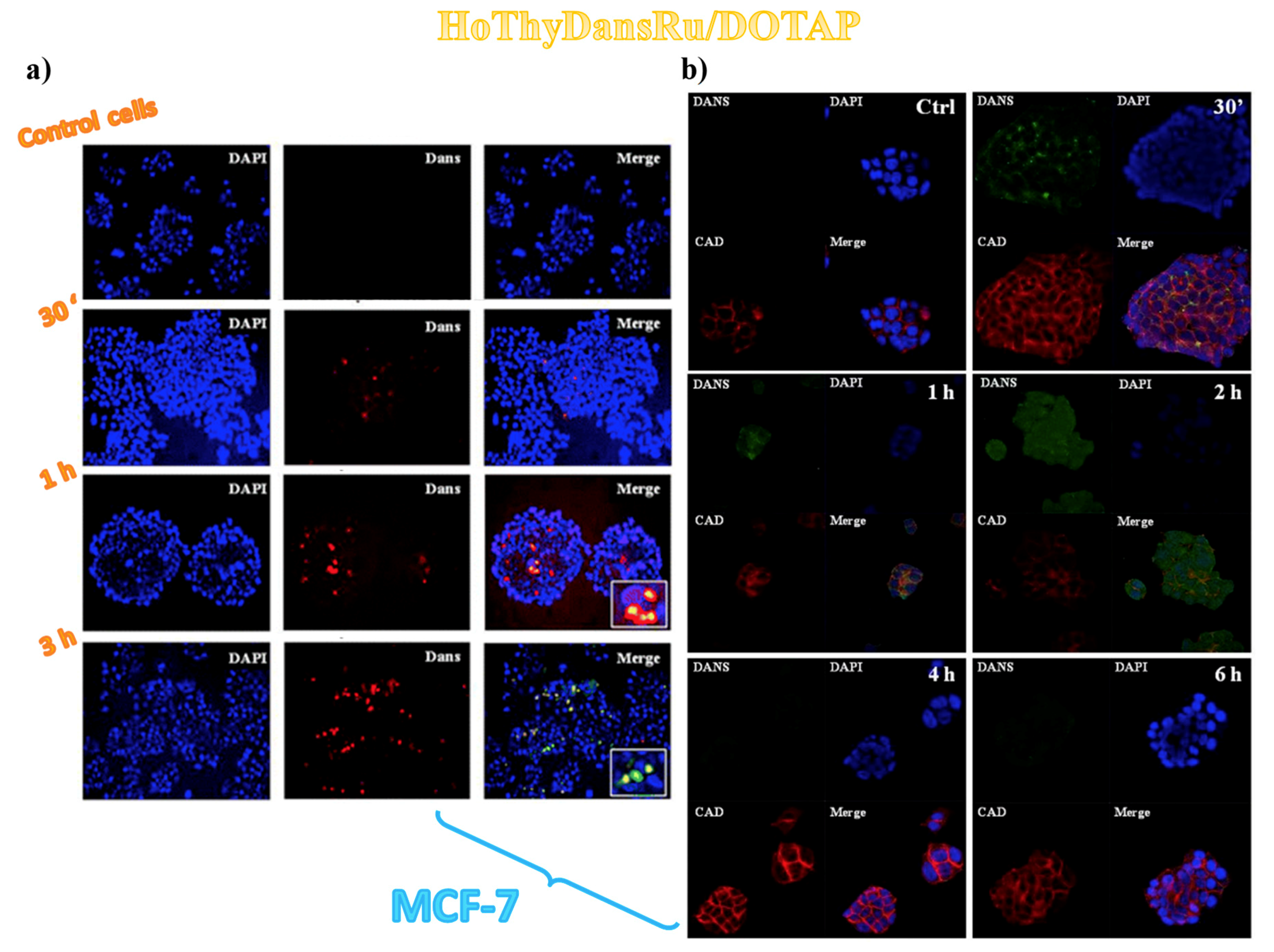
3.2. Cellular Uptake Studies on Ru(III)-Containing Liposomes by Fluorescence Microscopy
3.3. Sub-Cellular Accumulation of the Ru(III) Complexes
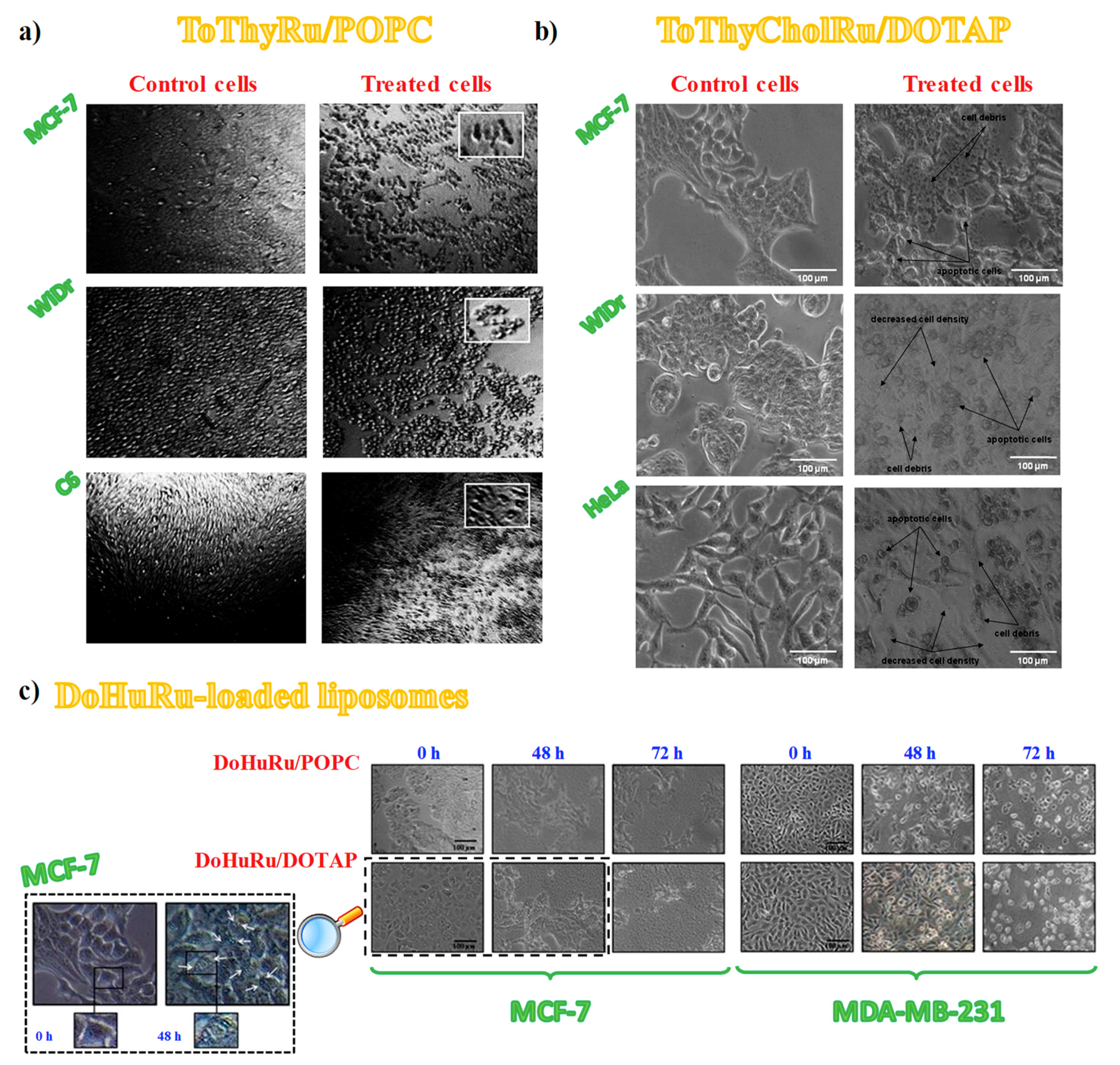
3.4. Cell Morphological Changes Induced by in Vitro Treatment with Ru(III)-Containing Liposomes
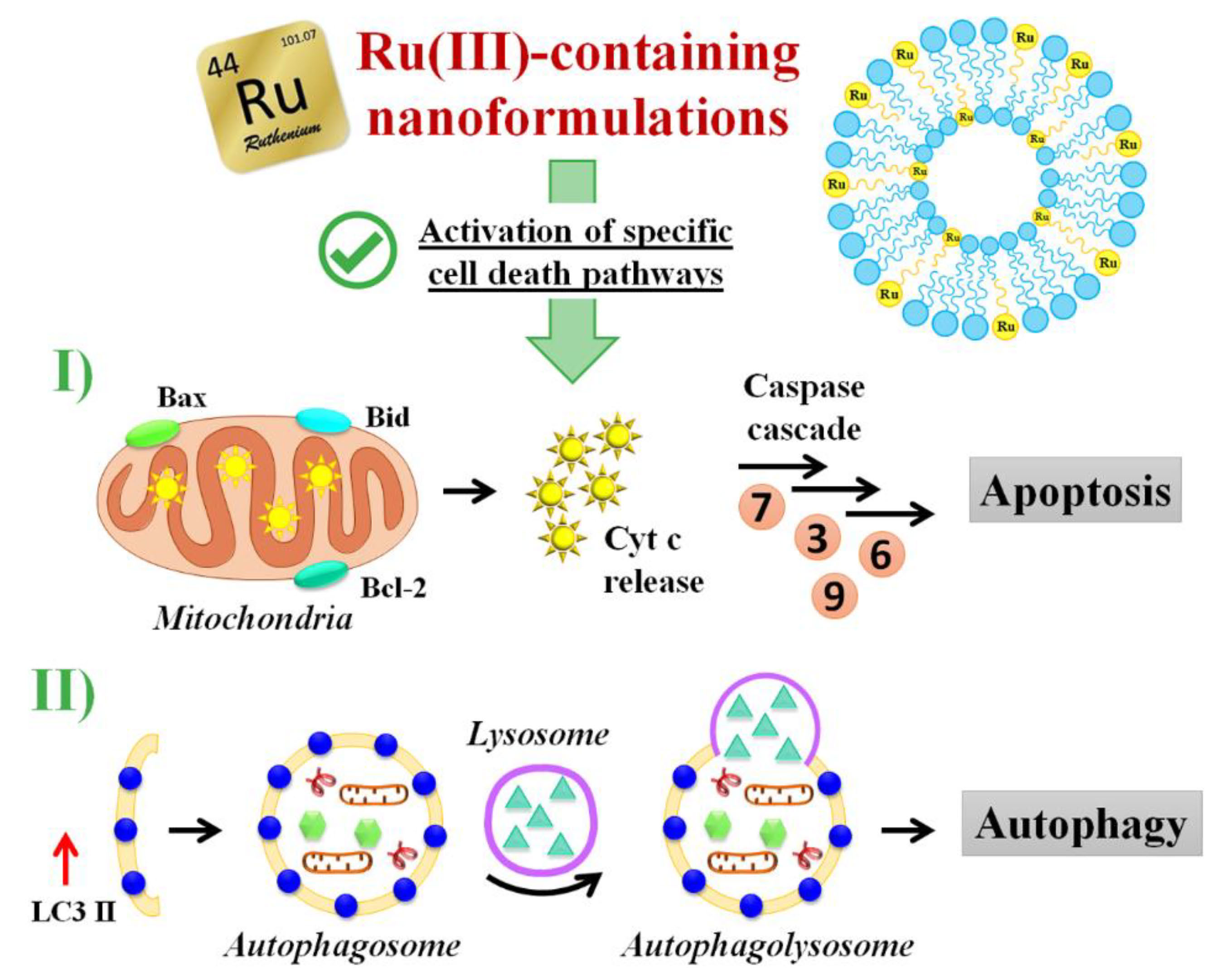
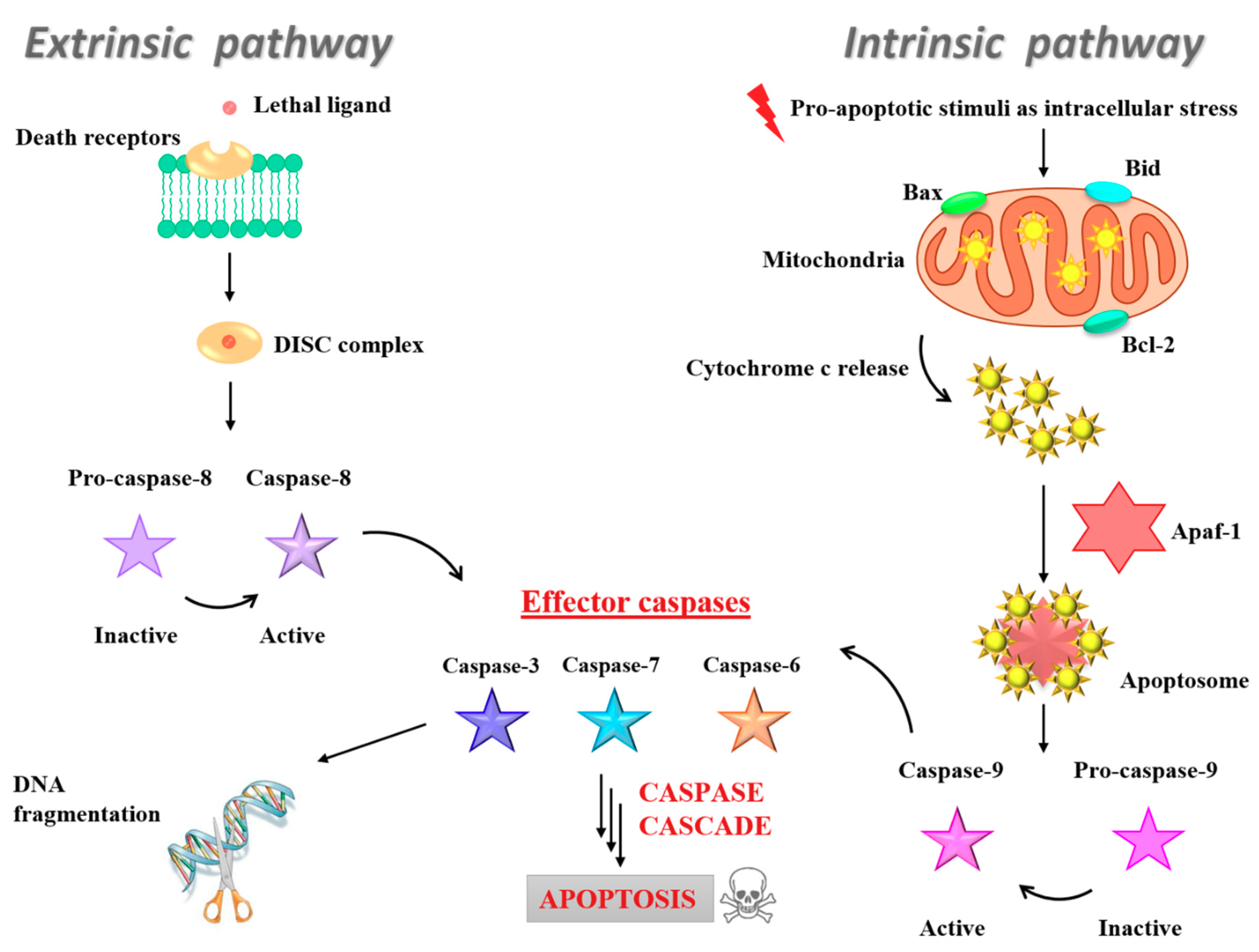
3.5. Insight into the Ru(III)-Containing Liposomes Mode of Action: Identification of Molecular Cell Death Pathways
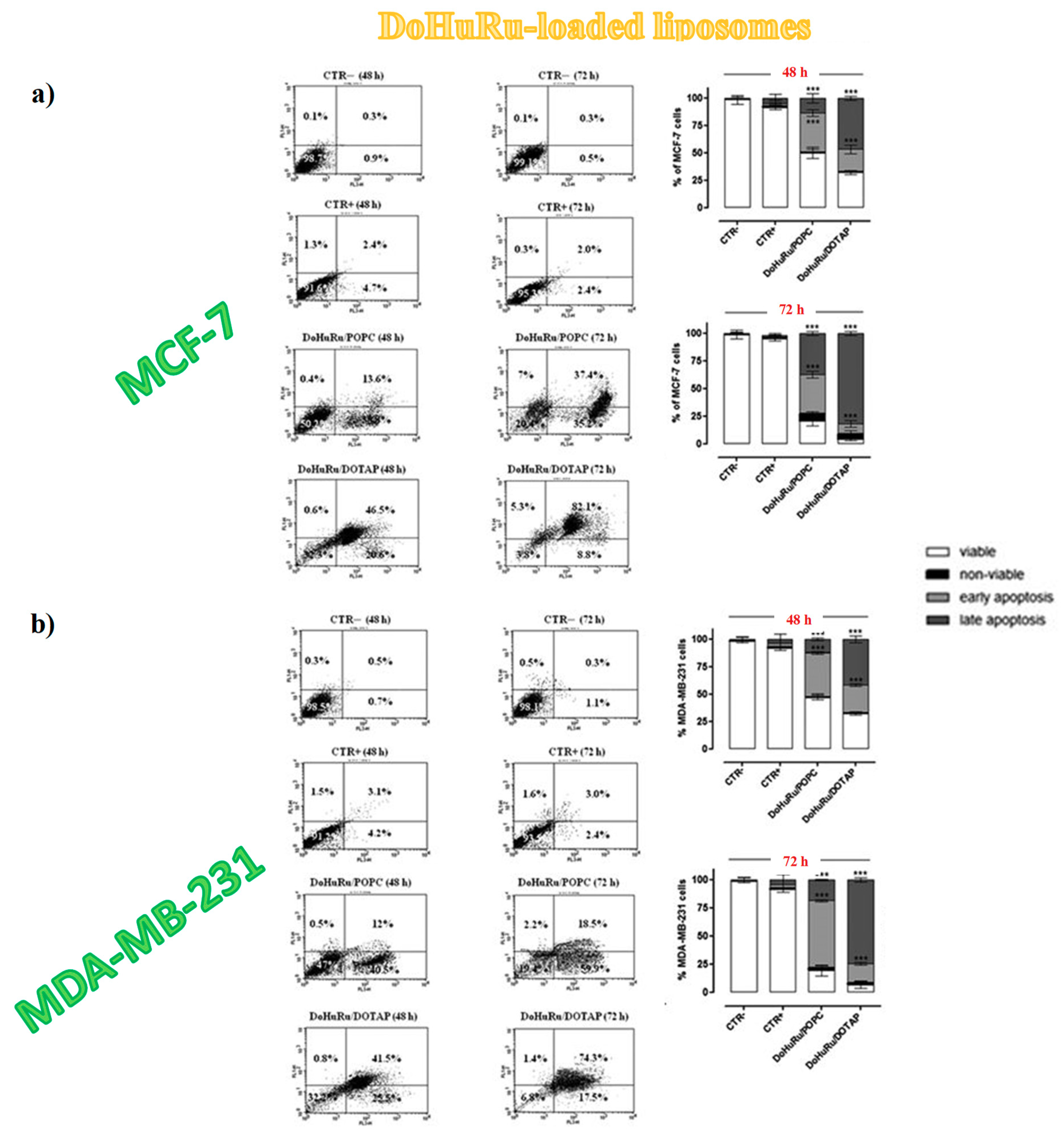
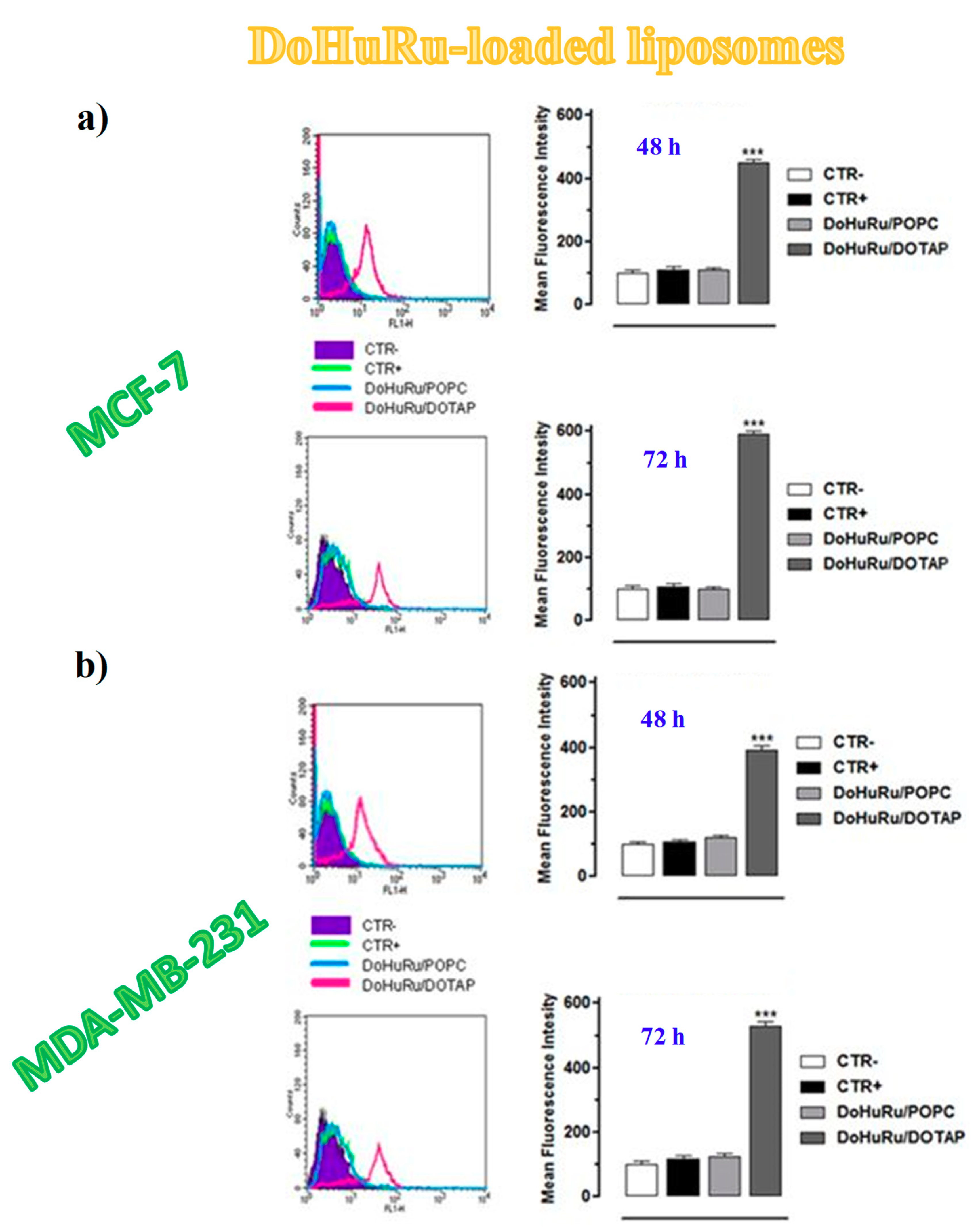
3.5.1. Pro-Apoptotic Effects in Breast Cancer Cells Evaluated by FACS Analysis
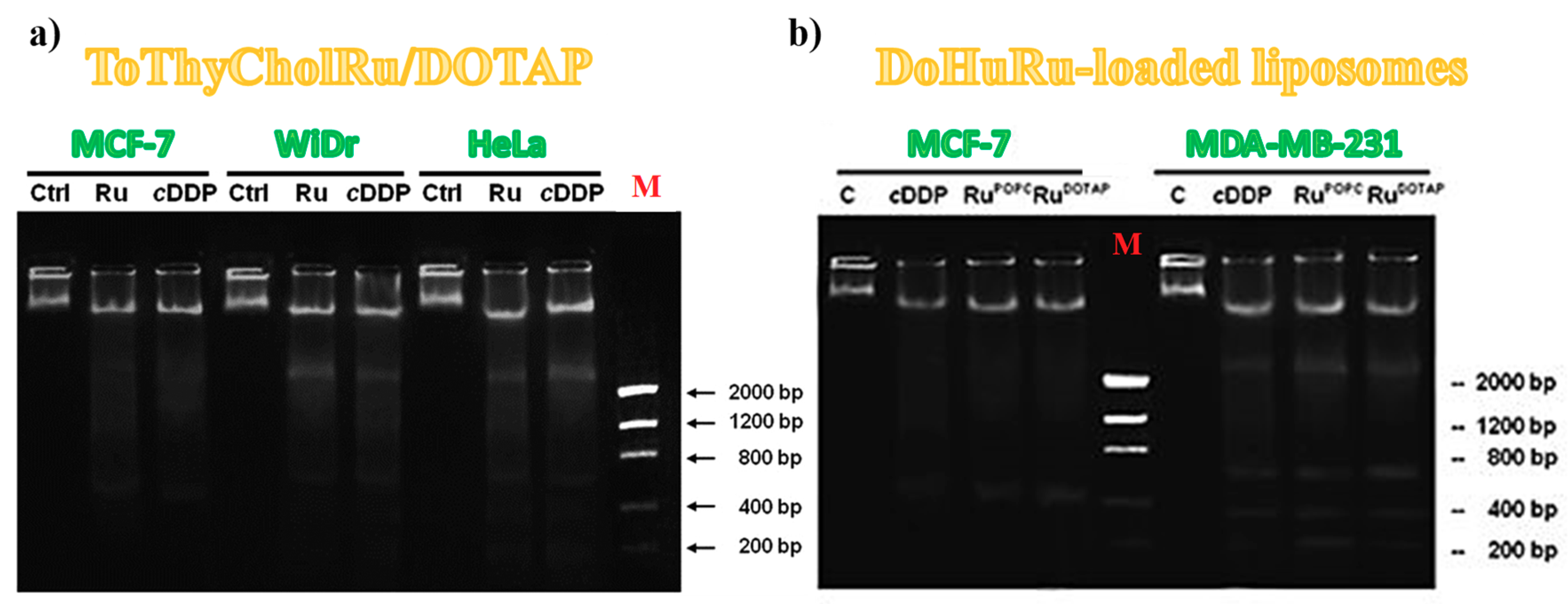
3.5.2. DNA Fragmentation Assay
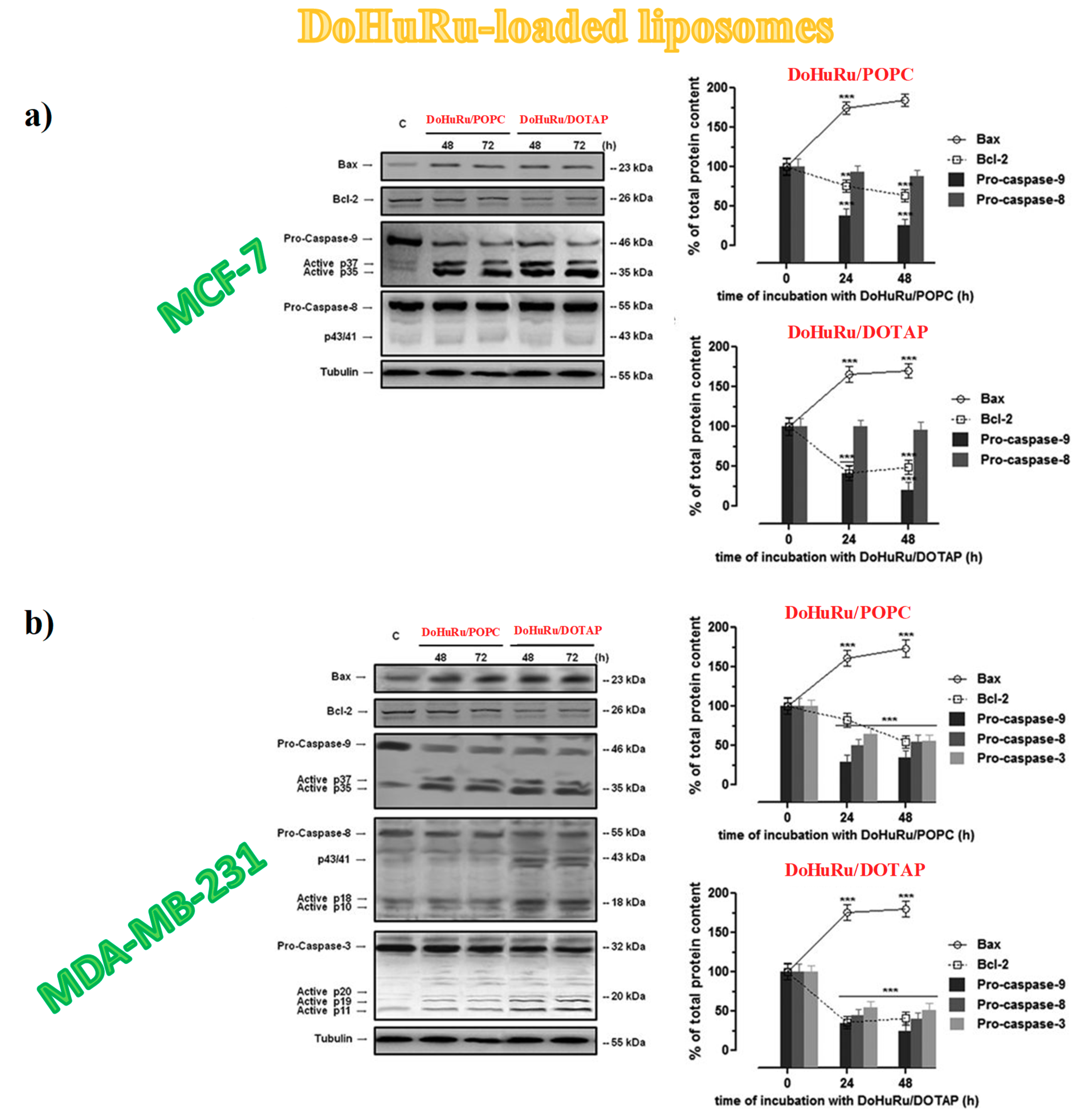
3.5.3. Apoptotic-Related Protein Expression in Breast Cancer Cells
3.5.4. Autophagy Activation in Breast Cancer Cells
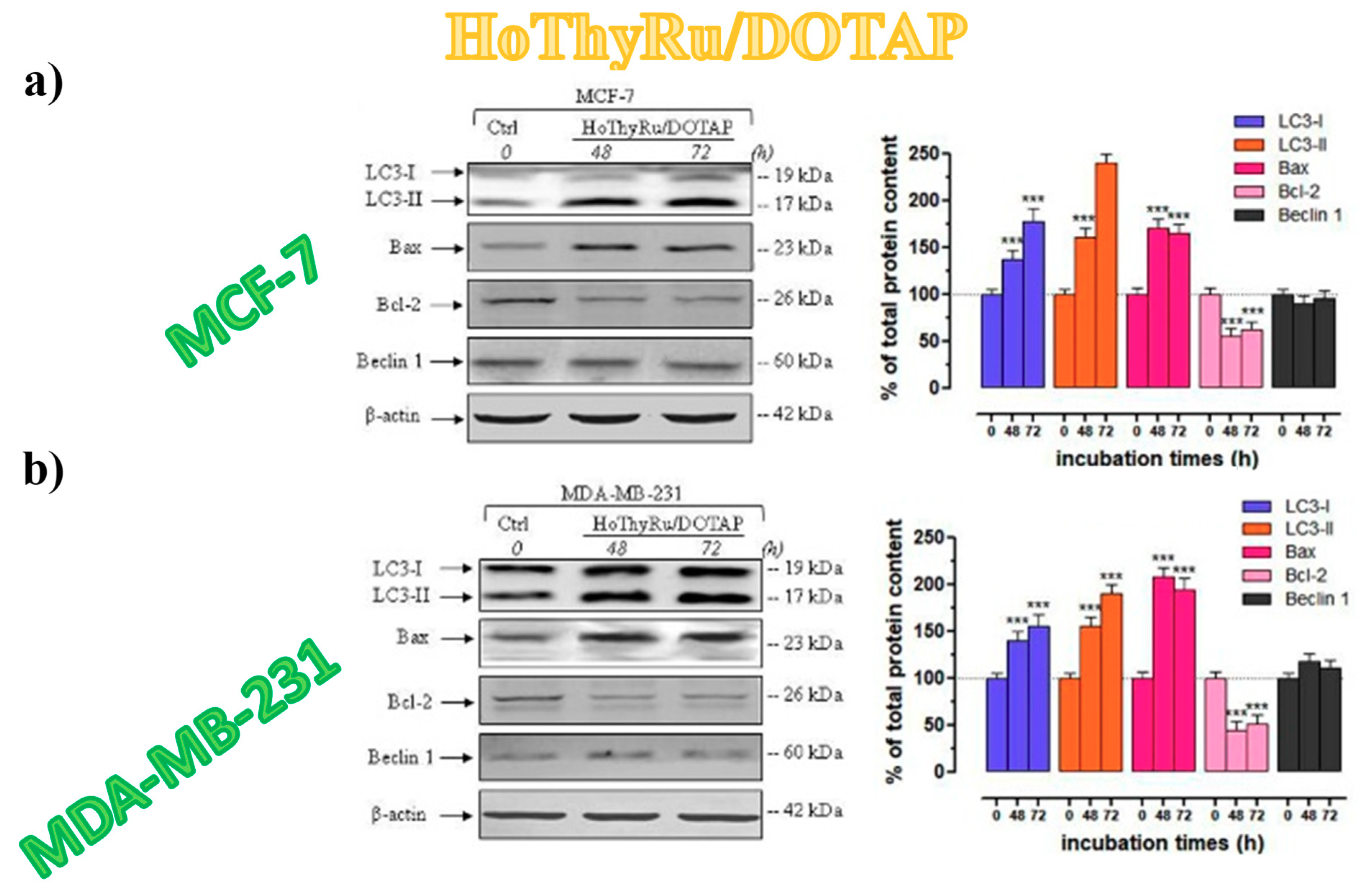
3.5.5. Autophagy-Related Proteins Expression in Breast Cancer Cells
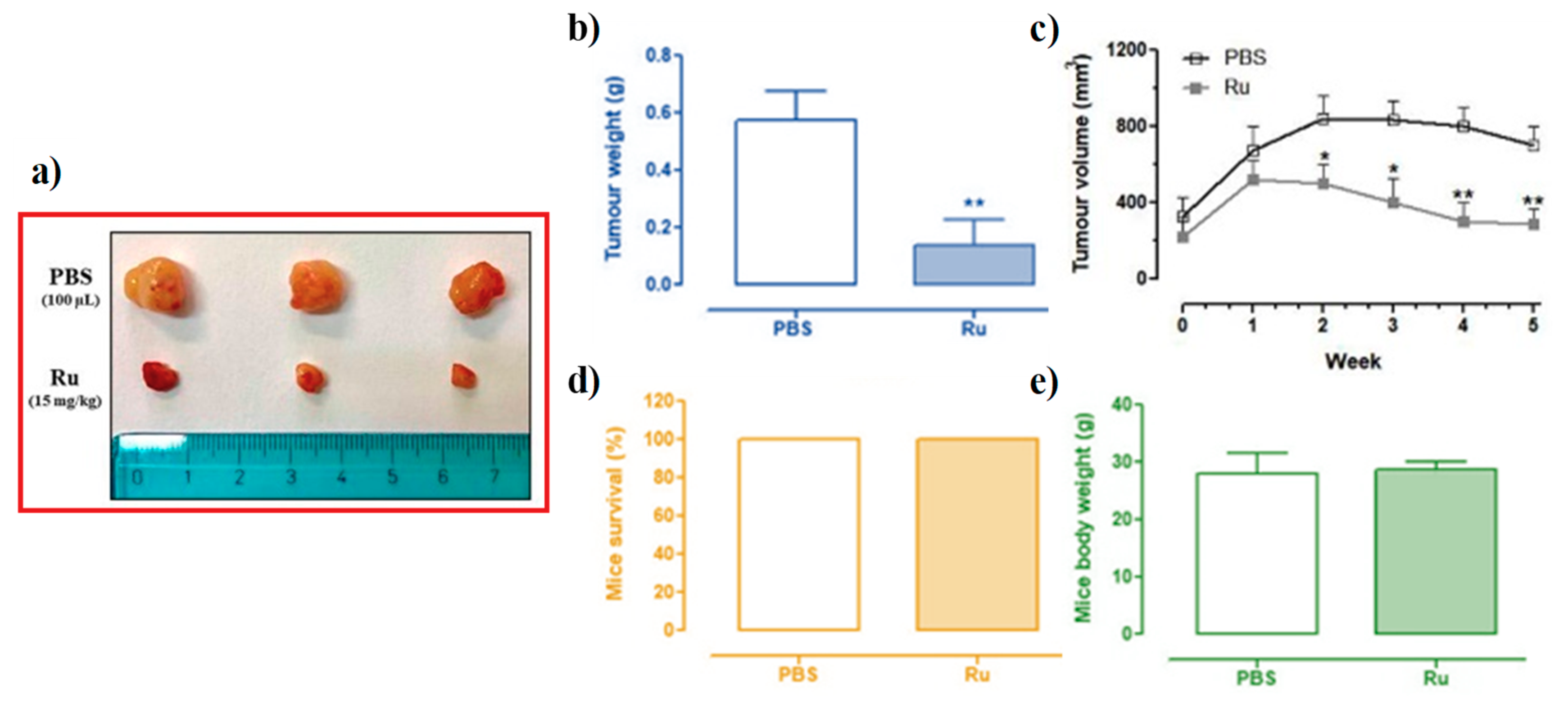
3.6. In Vivo Anticancer Efficacy of Ru(III)-Containing Liposomes in Mice BCC Xenografts
4. Improved Ru(III)-Containing Nanosystems: Introduction of Targeting and/or Diagnostic Agents for Theranostic Applications
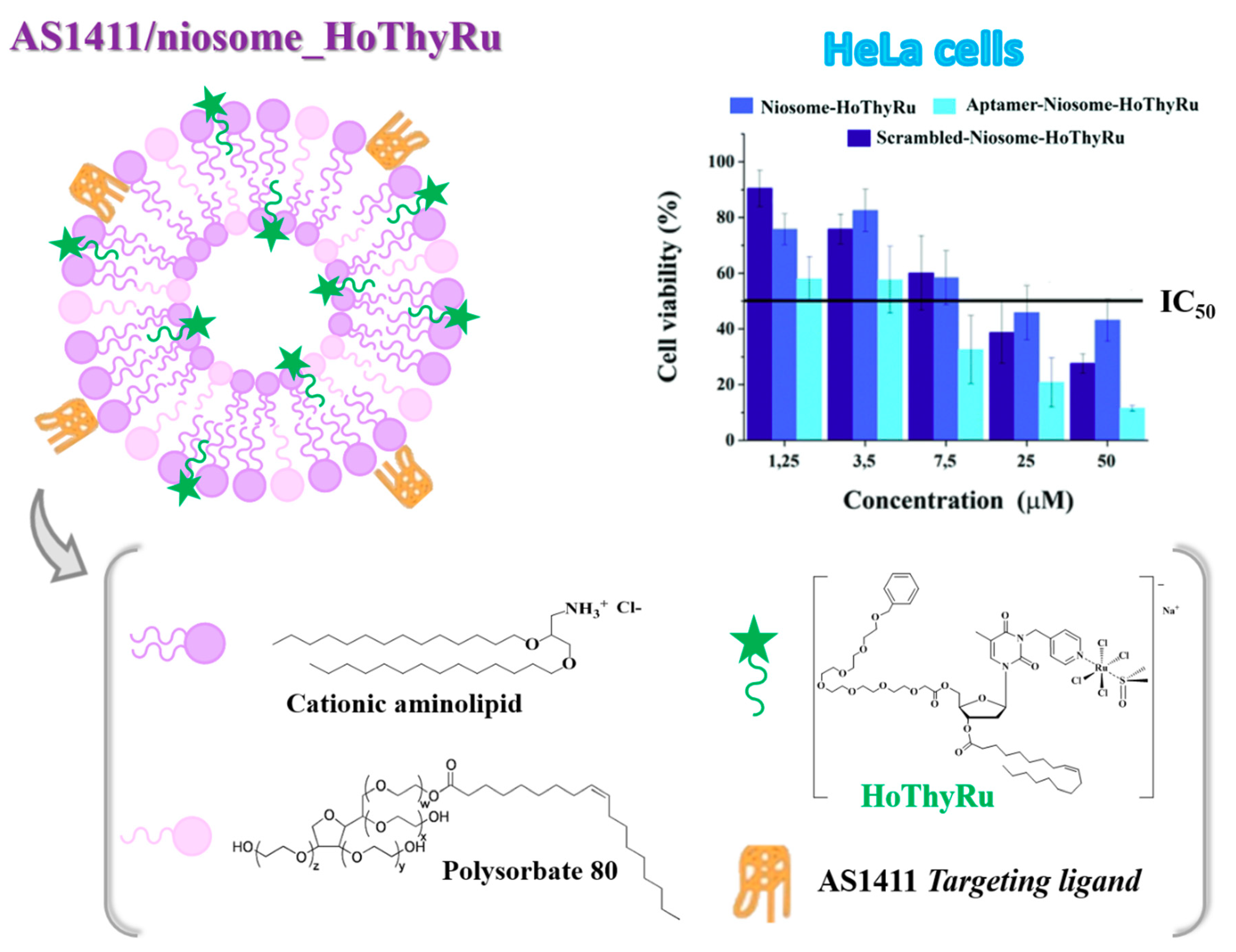
4.1. Niosome-Based Systems Containing Nucleolipid-Based Ru(III) Complexes
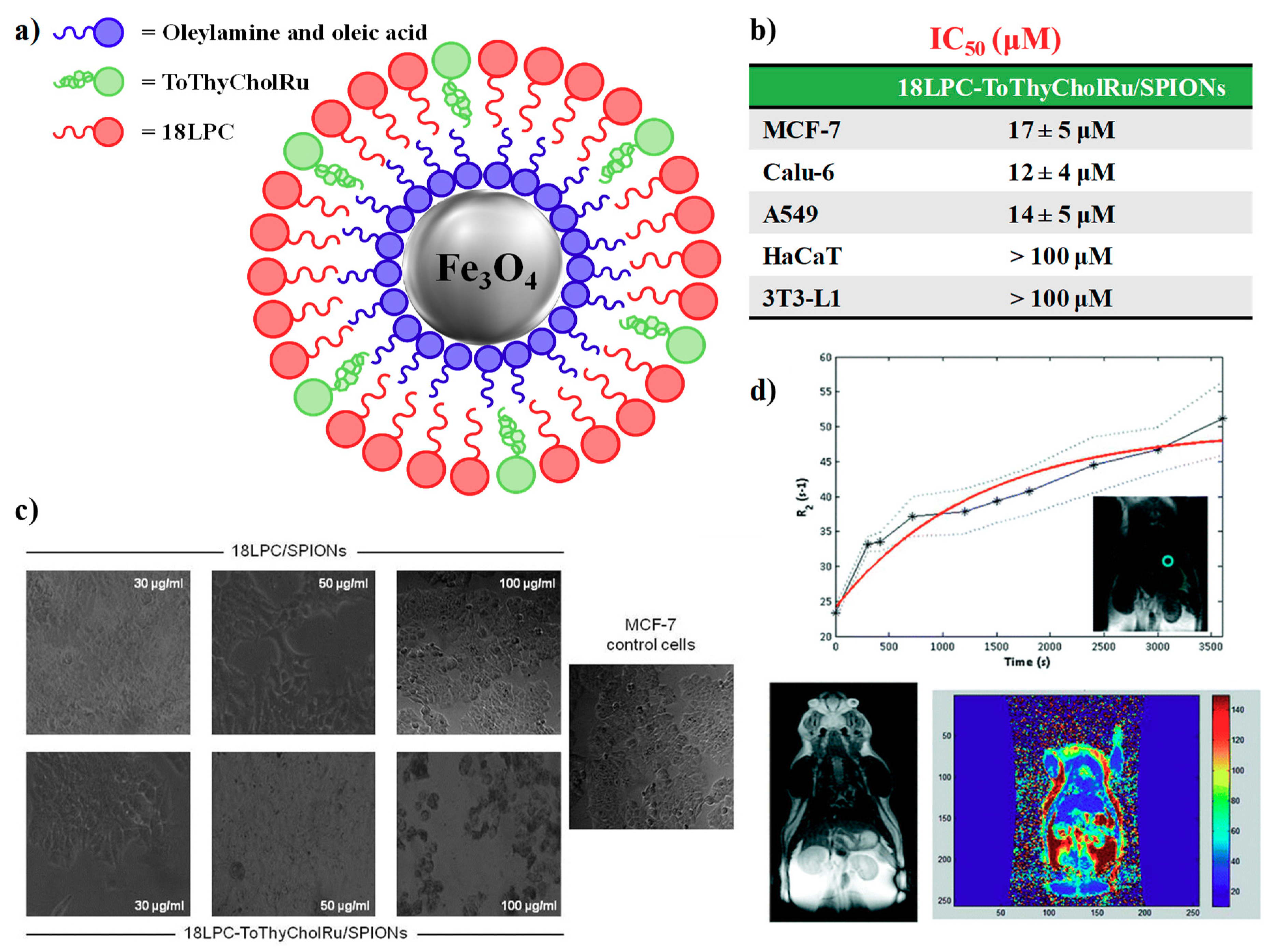
4.2. Nanoparticle-Based Systems Containing Nucleolipid-Based Ru(III) Complexes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of cell division in escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; van Camp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.S.; Berners-Price, S.J.; Hambley, T.W. Slowing of cisplatin aquation in the presence of DNA but not in the presence of phosphate: Improved understanding of sequence selectivity and the roles of monoaquated and diaquated species in the binding of cisplatin to DNA. Inorg. Chem. 2000, 39, 5603–5613. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, V.; Fuertes, M.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, J. Platinum anticancer coordination compounds: Study of DNA binding inspires new drug design. Eur. J. Inorg. Chem. 2009, 10, 1303–1312. [Google Scholar] [CrossRef]
- Todd, R.C.; Lippard, S.J. Structure of duplex DNA containing the cisplatin 1,2-{Pt(NH3)2}2+-d(GpG) cross-link at 1.77Å resolution. J. Inorg. Biochem. 2010, 104, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, N. Platinum resistance in breast and ovarian cancer cell lines. J. Exp. Clin. Cancer Res. 2011, 30, 91. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalt. Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Harrap, K.R. Preclinical studies identifying carboplatin as a viable cisplatin alternative. Cancer Treat. Rev. 1985, 12, 21–33. [Google Scholar] [CrossRef]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]
- Galanski, M.; Jakupec, M.A.; Keppler, B.K. Update of the preclinical situation of anticancer platinum complexes: Novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.A.; Wani, W.; Saleem, K.; Haque, A. Platinum compounds: A hope for future cancer chemotherapy. Anticancer Agents Med. Chem. 2013, 13, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.; Gust, R. Non platinum metal complexes as anti-cancer drugs. Arch. Pharm. 2007, 340, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Bergamo, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef]
- Bergamo, A.; Gaiddon, C.; Schellens, J.H.; Beijnen, J.H.; Sava, G. Approaching tumour therapy beyond platinum drugs: Status of the art and perspectives of ruthenium drug candidates. J. Inorg. Biochem. 2012, 106, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy – an update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.X.; Zhong, H.J.; Yang, G.; Vellaisamy, K.; Ma, D.L.; Leung, C.H. Recent development of transition metal complexes with in vivo antitumor activity. J. Inorg. Biochem. 2017, 177, 276–286. [Google Scholar] [CrossRef]
- Lazarević, T.; Rilak, A.; Bugarčić, Ž.D. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef]
- Hanif, M.; Hartinger, C.G. Anticancer metallodrugs: Where is the next cisplatin? Future Med. Chem. 2018, 10, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Bratsos, I.; Jedner, S.; Gianferrara, T.; Alessio, E. Ruthenium anticancer compounds: Challenges and expectations. Chim. Int. J. Chem. 2007, 61, 692–697. [Google Scholar] [CrossRef]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Emadi, A. Ruthenium-based chemotherapeutics: Are they ready for prime time? Cancer Chemother. Pharm. 2010, 66, 1–9. [Google Scholar] [CrossRef]
- Bergamo, A.; Sava, G. Ruthenium anticancer compounds: Myths and realities of the emerging metal-based drugs. Dalton Trans. 2011, 40, 7817–7823. [Google Scholar] [CrossRef] [PubMed]
- Blunden, B.M.; Stenzel, M.H. Incorporating ruthenium into advanced drug delivery carriers—An innovative generation of chemotherapeutics. J. Chem. Technol. Biotechnol. 2015, 90, 1177–1195. [Google Scholar] [CrossRef]
- Alessio, E. Thirty years of the drug candidate NAMI-A and the myths in the field of ruthenium anticancer compounds: A personal perspective. Eur. J. Inorg. Chem. 2017, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Zheng, K.; Wu, Q.; Wang, C.; Tan, W.; Mei, W. Ruthenium (II) complexes as potential apoptosis inducers in chemotherapy. Anticancer Agents Med. Chem. 2017, 17, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru (II) compounds: Next-generation anticancer metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure-activity relationships for ruthenium and osmium anticancer agents-towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing ruthenium anticancer drugs: What have we learnt from the key drug candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef]
- Wani, W.A.; Prashar, S.; Shreaz, S.; Gómez-Ruiz, S. Nanostructured materials functionalized with metal complexes: In search of alternatives for administering anticancer metallodrugs. Coord. Chem. Rev. 2016, 312, 67–98. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Irace, C.; Paduano, L.; Montesarchio, D. Ru (III) complexes for anticancer therapy: The importance of being nucleolipidic. Eur. J. Org. Chem. 2017, 2017, 1100–1119. [Google Scholar] [CrossRef]
- Sarkar, A. Novel platinum compounds and nanoparticles as anticancer agents. Pharm. Pat. Anal. 2018, 7, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Poursharifi, M.; Wlodarczyk, M.T.; Mieszawska, A.J. Nano-based systems and biomacromolecules as carriers for metallodrugs in anticancer therapy. Inorganics 2019, 7, 2. [Google Scholar] [CrossRef]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: Nanosized vehicles for drug delivery in cancer. Trends Pharm. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Pillai, G. Nanomedicines for cancer therapy: An update of FDA approved and those under various stages of development. SOJ Pharm. Pharm. Sci. 2014. [Google Scholar] [CrossRef]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in nanomedicine: Approved and investigational nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Villemin, E.; Ong, Y.C.; Thomas, C.M.; Gasser, G. Polymer encapsulation of ruthenium complexes for biological and medicinal applications. Nat. Rev. Chem. 2019, 3, 261–282. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, P.; Viswanath, B.; Kim, S. Recent developments in the nanostructured materials functionalized with ruthenium complexes for targeted drug delivery to tumors. Int. J. Nanomed. 2017, 12, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, S.; Pongratz, M.; Jakupec, M.A.; Heffeter, P.; Berger, W.; Lackinger, L.; Keppler, B.K.; Marian, B. Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells. J. Cancer Res. Clin. Oncol. 2005, 131, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Jakupec, M.A.; Arion, V.B.; Kapitza, S.; Reisner, E.; Eichinger, A.; Pongratz, M.; Marian, B.; von Keyserlingk Graf, N.; Keppler, B.K. KP1019 (FFC14A) from bench to bedside: Preclinical and early clinical development- an overview. Int. J. Clin. Pharm. Ther. 2005, 43, 595–596. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent-preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Azad, G.K.; Mandal, P.; Reddy, M.A.; Tomar, R.S. Anti-cancer drug KP1019 modulates epigenetics and induces DNA damage response in Saccharomyces cerevisiae. FEBS Lett. 2014, 588, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339—A clinically investigated ruthenium-based drug—Involves ER- and ROS-related effects in colon carcinoma cell lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase i study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [PubMed]
- Lizardo, M.M.; Morrow, J.J.; Miller, T.E.; Hong, E.S.; Ren, L.; Mendoza, A.; Halsey, C.H.; Scacheri, P.C.; Helman, L.J.; Khanna, C. Upregulation of glucose-regulated protein 78 in metastatic cancer cells is necessary for lung metastasis progression. Neoplasia 2016, 18, 699–710. [Google Scholar] [CrossRef]
- Schoenhacker-Alte, B.; Mohr, T.; Pirker, C.; Kryeziu, K.; Kuhn, P.S.; Buck, A.; Hofmann, T.; Gerner, C.; Hermann, G.; Koellensperger, G.; et al. Sensitivity towards the GRP78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption of ER homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Blazevic, A.; Hummer, A.A.; Heffeter, P.; Berger, W.; Filipits, M.; Cibin, G.; Keppler, B.K.; Rompel, A. Electronic state of sodium trans-[tetrachloridobis (1H-indazole) ruthenate (III)] (NKP-1339) in tumor, liver and kidney tissue of a SW480-bearing mouse. Sci. Rep. 2017, 7, 40966. [Google Scholar] [CrossRef]
- Alessio, E.; Messori, L. Anticancer drug candidates face-to-face: A case story in medicinal inorganic chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef]
- Messori, L.; Orioli, P.; Vullo, D.; Alessio, E.; Iengo, E. A spectroscopic study of the reaction of NAMI, a novel ruthenium(III)anti-neoplastic complex, with bovine serum albumin. Eur. J. Biochem. 2000, 267, 1206–1213. [Google Scholar] [CrossRef]
- Bytzek, A.K.; Boeck, K.; Hermann, G.; Hann, S.; Keppler, B.K.; Hartinger, C.G.; Koellensperger, G. LC- and CZE-ICP-MS approaches for the in vivo analysis of the anticancer drug candidate sodium trans-[tetrachloridobis (1H-indazole) ruthenate (III)] (KP1339) in mouse plasma. Metallomics 2011, 3, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Dömötör, O.; Hartinger, C.G.; Bytzek, A.K.; Kiss, T.; Keppler, B.K.; Enyedy, E.A. Characterization of the binding sites of the anticancer ruthenium (III) complexes KP1019 and KP1339 on human serum albumin via competition studies. J. Biol. Inorg. Chem. 2013, 18, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.I.; Walsby, C.J. Albumin binding and ligand-exchange processes of the Ru (III) anticancer agent NAMI-A and its bis-DMSO analogue determined by ENDOR spectroscopy. Dalton Trans. 2015, 44, 17482–17493. [Google Scholar] [CrossRef] [PubMed]
- Bijelic, A.; Theiner, S.; Keppler, B.K.; Rompel, A. X-ray structure analysis of indazolium trans-[tetrachlorobis (1H-indazole) ruthenate (III)] (KP1019) bound to human serum albumin reveals two ruthenium binding sites and provides insights into the drug binding mechanism. J. Med. Chem. 2016, 59, 5894–5903. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Kratz, F.; Alessio, E. The interaction of the antitumor aomplexes Na[trans-RuCl4(DMSO) (Im)] and Na[trans-RuCl4(DMSO)(Ind)] with apotransferrin: A spectroscopic study. Met. Based Drugs 1996, 3, 1–9. [Google Scholar] [CrossRef]
- Pongratz, M.; Schluga, P.; Jakupec, M.A.; Arion, V.B.; Hartinger, C.G.; Allmaier, G.; Keppler, B.K. Transferrin binding and transferrin-mediated cellular uptake of the ruthenium coordination compound KP1019, studied by means of AAS, ESI-MS and CD spectroscopy. J. Anal. Atomic Spectrom. 2004, 19, 46–51. [Google Scholar] [CrossRef]
- Mazuryk, O.; Kurpiewska, K.; Lewinski, K.; Stochel, G.; Brindell, M. Interaction of apo-transferrin with anticancer ruthenium complexes NAMI-A and its reduced form. J. Inorg. Biochem. 2012, 116, 11–18. [Google Scholar] [CrossRef]
- Spiewak, K.; Brindell, M. Impact of low- and high-molecular-mass components of human serum on NAMI-A binding to transferrin. J. Biol. Inorg. Chem. 2015, 20, 695–703. [Google Scholar] [CrossRef]
- Ciambellotti, S.; Pratesi, A.; Severi, M.; Ferraro, G.; Alessio, E.; Merlino, A.; Messori, L. The NAMI A-human ferritin system: A biophysical characterization. Dalton Trans. 2018, 47, 11429–11437. [Google Scholar] [CrossRef]
- Pizarro, A.M.; Sadler, P.J. Unusual DNA binding modes for metal anticancer complexes. Biochimie 2009, 91, 1198–1211. [Google Scholar] [CrossRef]
- Alessio, E.; Balducci, G.; Lutman, A.; Mestroni, G.; Calligaris, M.; Attia, W.M. Synthesis and characterization of two new classes of ruthenium (III)-sulfoxide complexes with nitrogen donor ligands (L): Na[trans-RuCl4(R2SO)(L)] and mer, cis-RuCl3(R2SO)(R2SO)(L). The crystal structure of Na[trans-RuCl4(DMSO)(NH3)] 2DMSO, Na[trans-RuCl4(DMSO)(Im)] H2O, Me2CO (Im = imidazole) and mer, cis-RuCl3(DMSO)(DMSO)(NH3). Inorg. Chim. Acta 1993, 203, 205–217. [Google Scholar] [CrossRef]
- Groessl, M.; Zava, O.; Dyson, P.J. Cellular uptake and subcellular distribution of ruthenium-based metallodrugs under clinical investigation versus cisplatin. Metallomics 2011, 3, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Stevens, S.K.; Strehle, A.P.; Miller, R.L.; Gammons, S.H.; Hoffman, K.J.; McCarty, J.T.; Miller, M.E.; Stultz, L.K.; Hanson, P.K. The anticancer ruthenium complex KP1019 induces DNA damage, leading to cell cycle delay and cell death in saccharomyces cerevisiae. Mol. Pharm. 2013, 83, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Aitken, J.B.; Antony, S.; Weekley, C.M.; Lai, B.; Spiccia, L.; Harris, H.H. Distinct cellular fates for KP1019 and NAMI-A determined by X-ray fluorescence imaging of single cells. Metallomics 2012, 4, 1051. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.I.; Chard, R.A.; Al-Jobory, Y.M.; Jones, M.R.; Wong, E.W.Y.; Walsby, C.J. Pyridine analogs of the antimetastatic Ru(III) complex NAMI-A targeting non-covalent interactions with albumin. Inorg. Chem. 2012, 51, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Chang, S.W.; Prosser, K.E.; Leung, A.W.Y.; Santacruz, S.; Jang, T.; Thompson, J.R.; Yapp, D.T.T.; Warren, J.J.; Bally, M.B.; et al. Induction of cytotoxicity in pyridine analogues of the anti-metastatic Ru(III) complex NAMI-A by ferrocene functionalization. Inorg. Chem. 2016, 55, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Mangiapia, G.; D’Errico, G.; Simeone, L.; Irace, C.; Radulescu, A.; Di Pascale, A.; Colonna, A.; Montesarchio, D.; Paduano, L. Ruthenium-based complex nanocarriers for cancer therapy. Biomaterials 2012, 33, 3770–3782. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Wu, S.; Lai, S.; Wang, M.; Chen, Y.; Zhou, L.; Zhu, Y.; Lian, W.; Peng, W.; Ji, L.; et al. Synthesis, structures, cellular uptake and apoptosis-inducing properties of highly cytotoxic ruthenium-norharman complexes. Dalton Trans. 2011, 40, 8611–8621. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, G.; Luchini, A.; D’Errico, G.; Santamaria, R.; Capuozzo, A.; Irace, C.; Montesarchio, D.; Paduano, L. Cationic liposomes as efficient nanocarriers for the drug delivery of an anticancer cholesterol-based ruthenium complex. J. Mater. Chem. B 2015, 3, 3011–3023. [Google Scholar] [CrossRef]
- Łakomska, I.; Fandzloch, M.; Muzioł, T.; Lis, T.; Jezierska, J. Synthesis, characterization and antitumor properties of two highly cytotoxic ruthenium(III) complexes with bulky triazolopyrimidine ligands. Dalton Trans. 2013, 42, 6219. [Google Scholar] [CrossRef]
- Webb, M.I.; Wu, B.; Jang, T.; Chard, R.A.; Wong, E.W.Y.; Wong, M.Q.; Yapp, D.T.T.; Walsby, C.J. Increasing the bioavailability of RuIII anticancer complexes through hydrophobic albumin interactions. Chem. Eur. J. 2013, 19, 17031–17042. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W.; Lewis, A.R.; Prosser, K.E.; Thompson, J.R.; Gladkikh, M.; Bally, M.B.; Warren, J.J.; Walsby, C.J. CF3 derivatives of the anticancer Ru(III) complexes KP1019, NKP-1339, and their imidazole and pyridine analogues show enhanced lipophilicity, albumin interactions, and cytotoxicity. Inorg. Chem. 2016, 55, 4850–4863. [Google Scholar] [CrossRef] [PubMed]
- Vergara, A.; D’Errico, G.; Montesarchio, D.; Paduano, L.; Merlino, A. Interaction of anticancer ruthenium compounds with proteins high-resolution X-ray structures and raman microscopy studies of the adduct between hen egg white lysozyme and AziRu. Inorg. Chem. 2013, 52, 4157–4159. [Google Scholar] [CrossRef]
- Vergara, A.; Russo Krauss, I.; Montesarchio, D.; Paduano, L.; Merlino, A. Investigating the ruthenium metalation of proteins: X-ray structure and Raman microspectroscopy of the complex between RNase A and AziRu. Inorg. Chem. 2013, 52, 10714–10716. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Rozza, L.; Merlino, A.; Paduano, L.; Marzo, T.; Massai, L.; Messori, L.; Montesarchio, D. Interaction of anticancer Ru(III) complexes with single stranded and duplex DNA model systems. Dalton Trans. 2015, 44, 13914–13925. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Herrmann, M.; Merlino, A.; Riccardi, C.; Montesarchio, D.; Mroginski, M.A.; Musumeci, D.; Ruffo, F.; Paduano, L.; Hildebrandt, P.; et al. On the pH-modulated Ru-based prodrug activation mechanism. Inorg. Chem. 2019, 58, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Merlino, A. Ruthenium metalation of proteins: The X-ray structure of the complex formed between NAMI-A and hen egg white lysozyme. Dalton Trans. 2014, 43, 6128–6131. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Baracco, S.; Cassino, C.; Zanello, P.; Osella, D. Appraisal of the redox behaviour of the antimetastatic ruthenium(III) complex [ImH][RuCl4(DMSO)(Im)], NAMI-A. Dalton Trans. 2004, 2347–2351. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Hotze, A.C.G.; van der Schilden, K.; Haasnoot, J.G.; Pacor, S.; Alessio, E.; Sava, G.; Reedijk, J. The hydrolysis of the anti-cancer ruthenium complex NAMI-A affects its DNA binding and antimetastatic activity: An NMR evaluation. J. Inorg. Biochem. 2004, 98, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Timerbaev, A.R.; Rudnev, A.V.; Semenova, O.; Hartinger, C.G.; Keppler, B.K. Comparative binding of antitumor indazolium [trans-tetrachlorobis (1H- indazole) ruthenate(III)] to serum transport proteins assayed by capillary zone electrophoresis. Anal. Biochem. 2005, 341, 326–333. [Google Scholar] [CrossRef]
- Chen, J.; Chen, L.; Liao, S.; Zheng, K.; Ji, L. A theoretical study on the hydrolysis process of the antimetastatic ruthenium(III) complex NAMI-A. J. Phys. Chem. B 2007, 111, 7862–7869. [Google Scholar] [CrossRef] [PubMed]
- Besker, N.; Coletti, C.; Marrone, A.; Re, N. Aquation of the ruthenium-based anticancer drug NAMI-A: A density functional study. J. Phys. Chem. B 2008, 112, 3871–3875. [Google Scholar] [CrossRef]
- Vargiu, A.V.; Robertazzi, A.; Magistrato, A.; Ruggerone, P.; Carloni, P. The hydrolysis mechanism of the anticancer ruthenium drugs NAMI-A and ICR investigated by DFT-PCM calculations. J. Phys. Chem. B 2008, 112, 4401–4409. [Google Scholar] [CrossRef] [PubMed]
- Pashkunova-Martic, I.; Losantos, B.C.; Kandler, N.; Keppler, B. Studies of KP46 and KP1019 and the hydrolysis product of KP1019 in lipiodol emulsions: Preparation and initial characterizations as potential theragnostic agents. Curr. Drug Deliv. 2018, 15, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Nandi, U.; Mukherjee, D. Detailed account on activation mechanisms of ruthenium coordination complexes and their role as antineoplastic agents. Eur. J. Med. Chem. 2018, 150, 419–445. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, G.; Alessio, E.; Sava, G.; Pacor, S.; Coluccia, M.; Boccarelli, A. Water-soluble ruthenium(III)-dimethyl sulfoxide complexes: Chemical behaviour and pharmaceutical properties. Met. Based Drugs 1994, 1, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Bouma, M.; Nuijen, B.; Jansen, M.T.; Sava, G.; Flaibani, A.; Bult, A.; Beijnen, J.H. A kinetic study of the chemical stability of the antimetastatic ruthenium complex NAMI-A. Int. J. Pharm. 2002, 248, 239–246. [Google Scholar] [CrossRef]
- Sava, G.; Bergamo, A.; Zorzet, S.; Gava, B.; Casarsa, C.; Cocchietto, M.; Furlani, A.; Scarcia, V.; Serli, B.; Iengo, E.; et al. Influence of chemical stability on the activity of the antimetastasis ruthenium compound NAMI-A. Eur. J. Cancer 2002, 38, 427–435. [Google Scholar] [CrossRef]
- Galanski, M.; Keppler, B.K. Searching for the magic bullet: Anticancer platinum drugs which can be accumulated or activated in the tumor tissue. Anticancer Agents Med. Chem. 2007, 7, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, M.; Misso, G.; Ferraro, M.G.; Riccardi, C.; Capuozzo, A.; Zarone, M.R.; Maione, F.; Trifuoggi, M.; Stiuso, P.; D’Errico, G.; et al. Exploring cellular uptake, accumulation and mechanism of action of a cationic Ru-based nanosystem in human preclinical models of breast cancer. Sci. Rep. 2019, 9, 7006. [Google Scholar] [CrossRef]
- Baillet, J.; Desvergnes, V.; Hamoud, A.; Latxague, L.; Barthélémy, P. Lipid and nucleic acid chemistries: Combining the best of both worlds to construct advanced biomaterials. Adv. Mater. 2018, 30, 1705078. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, M.; Del Litto, R.; Mangiapia, G.; Carnerup, A.M.; D’Errico, G.; Ruffo, F.; Paduano, L. Lipid based nanovectors containing ruthenium complexes: A potential route in cancer therapy. Chem. Commun. 2009, 1404–1406. [Google Scholar] [CrossRef] [PubMed]
- Simeone, L.; Mangiapia, G.; Irace, C.; Di Pascale, A.; Colonna, A.; Ortona, O.; De Napoli, L.; Montesarchio, D.; Paduano, L. Nucleolipid nanovectors as molecular carriers for potential applications in drug delivery. Mol. Biosyst. 2011, 7, 3075–3086. [Google Scholar] [CrossRef]
- Simeone, L.; Irace, C.; Di Pascale, A.; Ciccarelli, D.; D’Errico, G.; Montesarchio, D. Synthesis, self-aggregation and bioactivity properties of a cationic aminoacyl surfactant, based on a new class of highly functionalized nucleolipids. Eur. J. Med. Chem. 2012, 57, 429–440. [Google Scholar] [CrossRef]
- Simeone, L.; Mangiapia, G.; Vitiello, G.; Irace, C.; Colonna, A.; Ortona, O.; Montesarchio, D.; Paduano, L. Cholesterol-based nucleolipid-ruthenium complex stabilized by lipid aggregates for antineoplastic therapy. Bioconj. Chem. 2012, 23, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Mangiapia, G.; Vitiello, G.; Irace, C.; Santamaria, R.; Colonna, A.; Angelico, R.; Radulescu, A.; D’Errico, G.; Montesarchio, D.; Paduano, L. Anticancer cationic ruthenium nanovectors: From rational molecular design to cellular uptake and bioactivity. Biomacromolecules 2013, 14, 2549–2560. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Montesarchio, D.; Mangiapia, G.; Vitiello, G.; Musumeci, D.; Irace, C.; Santamaria, R.; D’Errico, G.; Paduano, L. A new design for nucleolipid-based Ru(III) complexes as anticancer agents. Dalton Trans. 2013, 42, 16697–16708. [Google Scholar] [CrossRef]
- Luchini, A.; Irace, C.; Santamaria, R.; Montesarchio, D.; Heenan, R.K.; Szekely, N.; Flori, A.; Menichetti, L.; Paduano, L. Phosphocholine-decorated superparamagnetic iron oxide nanoparticles: Defining the structure and probing in vivo applications. Nanoscale 2016, 8, 10078–10086. [Google Scholar] [CrossRef]
- Riccardi, C.; Fàbrega, C.; Grijalvo, S.; Vitiello, G.; D’Errico, G.; Eritja, R.; Montesarchio, D. AS1411-decorated niosomes as effective nanocarriers for Ru(III)-based drugs in anticancer strategies. J. Mater. Chem. B 2018, 6, 5368–5384. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Capuozzo, A.; Irace, C.; King, S.; Russo Krauss, I.; Paduano, L.; Montesarchio, D. “Dressing up” an old drug: An aminoacyl lipid for the functionalization of Ru(III)-based anticancer agents. ACS Biomater. Sci. Eng. 2018, 4, 163–174. [Google Scholar] [CrossRef]
- Fischer, B.; Heffeter, P.; Kryeziu, K.; Gille, L.; Meier, S.M.; Berger, W.; Kowol, C.R.; Keppler, B.K. Poly (lactic acid) nanoparticles of the lead anticancer ruthenium compound KP1019 and its surfactant-mediated activation. Dalton Trans. 2014, 43, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, T.; Riabtseva, A.; Kowol, C.R.; Jungwith, U.; Heffeter, P.; Yanchuk, I.; Zaichenko, A.; Körner, W.; Senkiv, Y.; Berger, W.; et al. Nanoformulation improves activity of the (pre)clinical anticancer ruthenium complex KP1019. J. Biomed. Nanotechnol. 2014, 10, 877–884. [Google Scholar] [CrossRef]
- Xu, W.; Ling, P.; Zhang, T. Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. J. Drug Deliv. 2013, 2013, 340315. [Google Scholar] [CrossRef] [PubMed]
- Tanbour, R.M.; Martins, A.G.; Pitt, W.A.; Husseini, G. Drug delivery systems based on polymeric micelles and ultrasound: A review. Curr. Pharm. Des. 2016, 22, 2796–27807. [Google Scholar] [CrossRef] [PubMed]
- Blunden, B.M.; Rawal, A.; Lu, H.; Stenzel, M.H. Superior chemotherapeutic benefits from the ruthenium-based anti-metastatic drug NAMI-A through conjugation to polymeric micelles. Macromolecules 2014, 47, 1646–1655. [Google Scholar] [CrossRef]
- D’Amora, A.; Cucciolito, M.E.; Iannitti, R.; Morelli, G.; Palumbo, R.; Ruffo, F.; Tesauro, D. Pyridine ruthenium (III) complexes entrapped in liposomes with enhanced cytotoxic properties in PC-3 prostate cancer cells. J. Drug Deliv. Sci. Technol. 2019, 51, 552–558. [Google Scholar] [CrossRef]
- Scintilla, S.; Brustolin, L.; Gambalunga, A.; Chiara, F.; Trevisan, A.; Nardon, C.; Fregona, D. Ru(III) anticancer agents with aromatic and non-aromatic dithiocarbamates as ligands: Loading into nanocarriers and preliminary biological studies. J. Inorg. Biochem. 2016, 165, 159–169. [Google Scholar] [CrossRef]
- Escobar-Chávez, J.J.; López-Cervantes, M.; Naïk, A.; Kalia, Y.N.; Quintanar-Guerrero, D.; Ganem-Quintanar, A. Applications of thermo-reversible pluronic F-127 gels in pharmaceutical formulations. J. Pharm. Pharm. Sci. 2006, 9, 339–358. [Google Scholar] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharm. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.; Karimi, N.; Safaei, M. Application of various types of liposomes in drug delivery systems. Adv. Pharm. Bull. 2017, 7, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Yingchoncharoen, P.; Kalinowski, D.S.; Richardson, D.R. Lipid-based drug delivery systems in cancer therapy: What is available and what is yet to come. Pharm. Rev. 2016, 68, 701–787. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Jani, R.K.; Gohil, K.M.; Kaushalkumar Jani, R. Liposomal formulations in cancer therapy: Basic concepts to advanced strategies. Int. J. Pharm. Sci. Drug Res. 2018, 10, 386–393. [Google Scholar] [CrossRef]
- Felice, B.; Prabhakaran, M.P.; Rodríguez, A.P.; Ramakrishna, S. Drug delivery vehicles on a nano-engineering perspective. Mater. Sci. Eng. C 2014, 41, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Fanciullino, R.; Ciccolini, J. Liposome-encapsulated anticancer drugs: Still waiting for the magic bullet? Curr. Med. Chem. 2009, 16, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Hang, Z.; Cooper, M.A.; Ziora, Z.M. Platinum-based anticancer drugs encapsulated liposome and polymeric micelle formulation in clinical trials. Biochem. Compd. 2016, 4, 1. [Google Scholar] [CrossRef]
- Stoyanova, E.; Petrov, P.; Karadjova, I.; Momekov, G.; Koseva, N. Cisplatin delivery vehicles based on stabilized polymeric aggregates comprising poly (acrylic acid) chains. Polym. J. 2017, 49, 607–615. [Google Scholar] [CrossRef][Green Version]
- Caracciolo, G. Clinically approved liposomal nanomedicines: Lessons learned from the biomolecular corona. Nanoscale 2018, 10, 4167–4172. [Google Scholar] [CrossRef]
- Stathopoulos, G.P.; Boulikas, T. Lipoplatin formulation review article. J. Drug Deliv. 2012, 2012, 581363. [Google Scholar] [CrossRef]
- Qi, P.; Cao, M.; Song, L.; Chen, C.; Liu, M.; Li, N.; Wu, D.; Peng, J.; Hu, G.; Zhao, J. The biological activity of cationic liposomes in drug delivery and toxicity test in animal models. Environ. Toxicol. Pharm. 2016, 47, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, J.K.; Weaver, E.M.; Hummon, A.B. Analyzing liposomal drug delivery systems in three-dimensional cell culture models using MALDI imaging mass spectrometry. Anal. Chem. 2017, 89, 8453–8458. [Google Scholar] [CrossRef] [PubMed]
- Walde, P.; Ichikawa, S. Enzymes inside lipid vesicles: Preparation, reactivity and applications. Biomol. Eng. 2001, 18, 143–177. [Google Scholar] [CrossRef]
- Irace, C.; Misso, G.; Capuozzo, A.; Piccolo, M.; Riccardi, C.; Luchini, A.; Caraglia, M.; Paduano, L.; Montesarchio, D.; Santamaria, R. Antiproliferative effects of ruthenium-based nucleolipidic nanoaggregates in human models of breast cancer in vitro: Insights into their mode of action. Sci. Rep. 2017, 7, 45236–45249. [Google Scholar] [CrossRef] [PubMed]
- John, R. Masters HeLa cells 50 years on: The good, the bad and the ugly. Nat. Rev. Cancer 2002, 2, 315–319. [Google Scholar] [CrossRef]
- Grobben, B.; De Deyn, P.P.; Slegers, H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 2002, 310, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Miniaci, M.C.; Irace, C.; Capuozzo, A.; Piccolo, M.; Di Pascale, A.; Russo, A.; Lippiello, P.; Lepre, F.; Russo, G.; Santamaria, R. Cysteine prevents the reduction in keratin synthesis induced by iron deficiency in human keratinocytes. J. Cell. Biochem. 2016, 117, 402–412. [Google Scholar] [CrossRef]
- Ji, L.; Zheng, W.; Lin, Y.; Wang, X.; Lü, S.; Hao, X.; Luo, Q.; Li, X.; Yang, L.; Wang, F. Novel ruthenium complexes ligated with 4-anilinoquinazoline derivatives: Synthesis, characterisation and preliminary evaluation of biological activity. Eur. J. Med. Chem. 2014, 77, 110–120. [Google Scholar] [CrossRef]
- Nikolova, A.; Momekov, G.; Bakalova, A.; Nikolova, K.; Ivanov, D. Novel Ru(III) complexes with some benzothiazole derivatives: Synthesis, physicochemical and pharmacological investigations. Drug Res. 2015, 65, 317–322. [Google Scholar] [CrossRef]
- Dömötör, O.; de Almeida, R.F.M.; Côrte-Real, L.; Matos, C.P.; Marques, F.; Matos, A.; Real, C.; Kiss, T.; Enyedy, É.A.; Helena Garcia, M.; et al. Studies on the mechanism of action of antitumor bis(aminophenolate) ruthenium(III) complexes. J. Inorg. Biochem. 2017, 168, 27–37. [Google Scholar] [CrossRef][Green Version]
- Sahyon, H.A.; El-Bindary, A.A.; Shoair, A.F.; Abdellatif, A.A. Synthesis and characterization of ruthenium(III) complex containing 2-aminomethyl benzimidazole, and its anticancer activity of in vitro and in vivo models. J. Mol. Liq. 2018, 255, 122–134. [Google Scholar] [CrossRef]
- Hutchinson, L. Breast cancer: Challenges, controversies, breakthroughs. Nat. Rev. Clin. Oncol. 2010, 7, 669–670. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef]
- Lacroix, M.; Leclercq, G. Relevance of breast cancer cell lines as models for breast tumours: An update. Breast Cancer Res. Treat. 2004, 83, 249–289. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Brabec, V.; Novàkovà, O. DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resist. Update 2006, 9, 111–122. [Google Scholar] [CrossRef]
- Bergamo, A.; Sava, G. Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans. 2007, 36, 1267–1272. [Google Scholar] [CrossRef]
- Coppola, C.; Paciello, A.; Mangiapia, G.; Licen, S.; Boccalon, M.; De Napoli, L.; Paduano, L.; Tecilla, P.; Montesarchio, D. Design, synthesis and characterisation of a fluorescently labelled CyPLOS ionophore. Chemistry 2010, 16, 13757–13772. [Google Scholar] [CrossRef]
- De Tito, S.; Morvan, F.; Meyer, A.; Vasseur, J.J.; Cummaro, A.; Petraccone, L.; Pagano, B.; Novellino, E.; Randazzo, A.; Giancola, C.; et al. Fluorescence enhancement upon G-quadruplex folding: Synthesis, structure, and biophysical characterization of a dansyl/cyclodextrin-tagged thrombin binding aptamer. Bioconj. Chem. 2013, 24, 1917–1927. [Google Scholar] [CrossRef]
- Riccardi, C.; Russo Krauss, I.; Musumeci, D.; Morvan, F.; Meyer, A.; Vasseur, J.J.; Paduano, L.; Montesarchio, D. Fluorescent thrombin binding aptamer-tagged nanoparticles for an efficient and reversible control of thrombin activity. ACS Appl. Mater. Interfaces 2017, 9, 35574–35587. [Google Scholar] [CrossRef]
- Li, C.; Ip, K.-W.; Man, W.-L.; Song, D.; He, M.-L.; Yiu, S.-M.; Lau, T.-C.; Zhu, G. Cytotoxic (salen) ruthenium(III) anticancer complexes exhibit different modes of cell death directed by axial ligands. Chem. Sci. 2017, 8, 6865–6870. [Google Scholar] [CrossRef]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A.; Hartinger, C.G.; Dyson, P.J. The ruthenium(II)-arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53-JNK pathways. J. Biol. Inorg. Chem. 2008, 1, 1149–1155. [Google Scholar] [CrossRef]
- De Lima, A.P.; Pereira, F.D.C.; Vilanova-Costa, C.A.S.T.; Soares, J.R.; Pereira, L.C.G.; Porto, H.K.P.; Pavanin, L.A.; Dos Santos, W.B.; Silveira-Lacerda, E.D.P. Induction of cell cycle arrest and apoptosis by ruthenium complex cis-(dichloro)tetramineruthenium(III) chloride in human lung carcinoma cells A549. Biol. Trace Elem. Res. 2012, 147, 8–15. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, Q.; Qin, X.; Sun, D.; Zhang, J.; Liu, J. Studies of ruthenium(II)-2,2′-bisimidazole complexes on binding to G-quadruplex DNA and inducing apoptosis in HeLa cells. New J. Chem. 2013, 37, 3706–3715. [Google Scholar] [CrossRef]
- Vilanova-Costa, C.A.S.T.; Porto, H.K.P.; Pereira, F.D.C.; De Lima, A.P.; Dos Santos, W.B.; Silveira-Lacerda, E.D.P. The ruthenium complexes cis-(dichloro)tetramineruthenium(III) chloride and cis-tetraammine(oxalato)ruthenium(III) dithionate overcome resistance inducing apoptosis on human lung carcinoma cells (A549). BioMetals 2014, 27, 459–469. [Google Scholar] [CrossRef]
- Berndsen, R.H.; Weiss, A.; Abdul, U.K.; Wong, T.J.; Meraldi, P.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. Combination of ruthenium(II)-arene complex [Ru(η6-p-cymene)Cl2 (pta)] (RAPTA-C) and the epidermal growth factor receptor inhibitor erlotinib results in efficient angiostatic and antitumor activity. Sci. Rep. 2017, 7, 43005. [Google Scholar] [CrossRef]
- Popolin, C.P.; Reis, J.P.B.; Becceneri, A.B.; Graminha, A.E.; Almeida, M.A.P.; Corrêa, R.S.; Colina-Vegas, L.A.; Ellena, J.; Batista, A.A.; Cominetti, M.R. Cytotoxicity and anti-tumor effects of new ruthenium complexes on triple negative breast cancer cells. PLoS ONE 2017, 12, e0183275. [Google Scholar] [CrossRef]
- Popolin, C.P.; Cominetti, M.R. A review of ruthenium complexes activities on breast cancer cells. Mini-Rev. Med. Chem. 2017, 17, 1435–1441. [Google Scholar] [CrossRef]
- Misso, G.; Giuberti, G.; Lombardi, A.; Grimaldi, A.; Ricciardiello, F.; Giordano, A.; Tagliaferri, P.; Abbruzzese, A.; Caraglia, M. Pharmacological inhibition of HSP90 and ras activity as a new strategy in the treatment of HNSCC. J. Cell. Physiol. 2013, 228, 130–141. [Google Scholar] [CrossRef]
- Okamura, M.; Hashimoto, K.; Shimada, J.; Sakagami, H. Apoptosis-inducing activity of cisplatin (CDDP) against human hepatoma and oral squamous cell carcinoma cell lines. Anticancer Res. 2004, 24, 655–661. [Google Scholar]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef]
- Devarajan, E.; Sahin, A.A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef]
- Oberhammer, F.; Wilson, J.W.; Dive, C.; Morris, I.D.; Hickman, J.A.; Wakeling, A.E.; Walker, P.R.; Sikorska, M. Apoptotic death in epithelial cells: Cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 1993, 12, 3679–3684. [Google Scholar] [CrossRef]
- Liang, Y.; Yan, C.; Schor, N.F. Apoptosis in the absence of caspase 3. Oncogene 2001, 20, 6570–6578. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Zheng, J.H.; Viacava Follis, A.; Kriwacki, R.W.; Moldoveanu, T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J. 2016, 283, 2690–2700. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef]
- Sturm, I.; Papadopoulos, S.; Hillebrand, T.; Benter, T.; Lück, H.J.; Wolff, G.; Dörken, B.; Daniel, P.T. Impaired BAX protein expression in breast cancer: Mutational analysis of the BAX and the p53 gene. Int. J. Cancer 2000, 27, 517–521. [Google Scholar] [CrossRef]
- Merino, D.; Lok, S.W.; Visvader, J.E.; Lindeman, G.J. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene 2016, 35, 1877–1887. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumor Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef]
- Qian, C.; Wang, J.Q.; Song, C.L.; Wang, L.L.; Ji, L.N.; Chao, H. The induction of mitochondria-mediated apoptosis in cancer cells by ruthenium(II) asymmetric complexes. Metallomics 2013, 5, 844–854. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Lima, A.P.; Pereira, F.C.; Almeida, M.A.P.; Mello, F.M.S.; Pires, W.C.; Pinto, T.M.; Delella, F.K.; Felisbino, S.L.; Moreno, V.; Batista, A.A.; et al. Cytoxicity and apoptotic mechanism of ruthenium(II) amino acid complexes in sarcoma-180 tumor cells. PLoS ONE 2014, 9, e105865. [Google Scholar] [CrossRef]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Maiuri, M.C.; Madeo, F.; Kroemer, G. Autophagy induction for the treatment of cancer. Autophagy 2016, 12, 1962–1964. [Google Scholar] [CrossRef]
- Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting autophagy to overcome human diseases. Int. J. Mol. Sci. 2019, 20, E725. [Google Scholar] [CrossRef]
- Ryter, S.W.; Mizumura, K.; Choi, A.M.K. The impact of autophagy on cell death modalities. Int. J. Cell Biol. 2014, 2014, 502676. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of selective autophagy in normal physiology and cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Ozpolat, B.; Benbrook, D.M. Targeting autophagy in cancer management-strategies and developments. Cancer Manag. Res. 2015, 7, 291–299. [Google Scholar] [CrossRef]
- Lippai, M.; Szatmári, Z. Autophagy-from molecular mechanisms to clinical relevance. Cell Biol. Toxicol. 2017, 33, 145–168. [Google Scholar] [CrossRef]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy modulation in cancer: Current knowledge on action and therapy. Oxid. Med. Cell. Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol Cell Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. Recent insights into cell death and autophagy. FEBS J. 2015, 282, 4279–4288. [Google Scholar] [CrossRef]
- Sambi, M.; Haq, S.; Samuel, V.; Qorri, B.; Haxho, F.; Hill, K.; Harless, W.; Szewczuk, M.R. Alternative therapies for metastatic breast cancer: Multimodal approach targeting tumor cell heterogeneity. Breast Cancer Targets Ther. 2017, 9, 85–93. [Google Scholar] [CrossRef]
- Lim, B.; Hortobagyi, G.N. Current challenges of metastatic breast cancer. Cancer Metastasis Rev. 2016, 35, 495–514. [Google Scholar] [CrossRef]
- Turashvili, G.; Brogi, E. Tumor heterogeneity in breast cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 2010, 5, e9996. [Google Scholar] [CrossRef]
- Ojha, R.; Ishaq, M.; Singh, S. Caspase-mediated crosstalk between autophagy and apoptosis: Mutual adjustment or matter of dominance. J. Cancer Res. Ther. 2015, 11, 514–524. [Google Scholar] [CrossRef]
- Ueno, T.; Masuda, N.; Kamigaki, S.; Morimoto, T.; Saji, S.; Imoto, S.; Sasano, H.; Toi, M. Differential involvement of autophagy and apoptosis in response to chemoendocrine and endocrine therapy in breast cancer: JBCRG-07TR. Int. J. Mol. Sci. 2019, 20, E984. [Google Scholar] [CrossRef]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem. Pharm. 2015, 94, 1–11. [Google Scholar] [CrossRef]
- Yuan, J.; Lei, Z.; Wang, X.; Zhu, F.; Chen, D. Ruthenium complex Λ-WH0402 induces hepatocellular carcinoma LM6 (HCCLM6) cell death by triggering the Beclin-1-dependent autophagy pathway. Metallomics 2015, 7, 896–907. [Google Scholar] [CrossRef]
- Chen, L.; Li, G.; Peng, F.; Jie, X.; Dongye, G.; Cai, K.; Feng, R.; Li, B.; Zeng, Q.; Lun, K.; et al. The induction of autophagy against mitochondria-mediated apoptosis in lung cancer cells by a ruthenium (II) imidazole complex. Oncotarget 2016, 7, 80716–80734. [Google Scholar] [CrossRef]
- Maier, K.E.; Levy, M. From selection hits to clinical leads: Progress in aptamer discovery. Mol. Ther. Methods Clin. Dev. 2016, 5, 16014–16023. [Google Scholar] [CrossRef]
- Grimaldi, A.; Santini, D.; Zappavigna, S.; Lombardi, A.; Misso, G.; Boccellino, M.; Desiderio, V.; Vitiello, P.P.; Di Lorenzo, G.; Zoccoli, A.; et al. Antagonistic effects of chloroquine on autophagy occurrence potentiate the anticancer effects of everolimus on renal cancer cells. Cancer Biol. Ther. 2015, 16, 567–579. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef]
- Fujii, T.; Yajima, R.; Tatsuki, H.; Oosone, K.; Kuwano, H. Anticancer effect of rapamycin on MCF-7 via downregulation of VEGF expression. Vitr. Cell. Dev. Biol. Anim. 2016, 52, 45–48. [Google Scholar] [CrossRef]
- Tanida, I.; Waguri, S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 2010, 648, 193–214. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef]
- Fu, L.L.; Cheng, Y.; Liu, B. Beclin-1: Autophagic regulator and therapeutic target in cancer. Int. J. Biochem. Cell Biol. 2013, 45, 921–924. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Lee, Y.K.; Koo, J.S. The potential of Beclin 1 as a therapeutic target for the treatment of breast cancer. Expert Opin. Ther. Targets 2016, 20, 167–178. [Google Scholar] [CrossRef]
- Zarzynska, J.M. The importance of autophagy regulation in breast cancer development and treatment. Biomed. Res. Int. 2014, 2014, 710345. [Google Scholar] [CrossRef]
- Zhang, X.D.; Wang, Y.; Wu, J.C.; Lin, F.; Han, R.; Han, F.; Fukunaga, K.; Qin, Z.H. Down-regulation of Bcl-2 enhances autophagy activation and cell death induced by mitochondrial dysfunction in rat striatum. J. Neurosci. Res. 2009, 87, 3600–3610. [Google Scholar] [CrossRef]
- Oh, S.; Xiaofei, E.; Ni, D.; Pirooz, S.D.; Lee, J.Y.; Lee, D.; Zhao, Z.; Lee, S.; Lee, H.; Ku, B.; et al. Downregulation of autophagy by Bcl-2 promotes MCF7 breast cancer cell growth independent of its inhibition of apoptosis. Cell Death Differ. 2011, 18, 452–464. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Uchegbu, I.F.; Florence, A.T. Non-ionic surfactant vesicles (niosomes): Physical and pharmaceutical chemistry. Adv. Colloid Interface Sci. 1995, 58, 1–55. [Google Scholar] [CrossRef]
- Uchegbu, I.F.; Vyas, S.P. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 1998, 172, 33–70. [Google Scholar] [CrossRef]
- Marianecci, C.; Di Marzio, L.; Rinaldi, F.; Celia, C.; Paolino, D.; Alhaique, F.; Esposito, S.; Carafa, M. Niosomes from 80 s to present: The state of the art. Adv. Colloid Interface Sci. 2014, 205, 187–206. [Google Scholar] [CrossRef]
- Grimaldi, N.; Andrade, F.; Segovia, N.; Ferrer-Tasies, L.; Sala, S.; Veciana, J.; Ventosa, N. Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 2016, 45, 6520–6545. [Google Scholar] [CrossRef]
- Khan, R.; Irchhaiya, R. Niosomes: A potential tool for novel drug delivery. J. Pharm. Investig. 2016, 46, 195–204. [Google Scholar] [CrossRef]
- Ag Seleci, D.; Seleci, M.; Walter, J.G.; Stahl, F.; Scheper, T. Niosomes as nanoparticular drug carriers: Fundamentals and recent applications. J. Nanomater. 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Grijalvo, S.; Puras, G.; Zárate, J.; Sainz-Ramos, M.; Qtaish, N.A.L.; López, T.; Mashal, M.; Attia, N.; Díaz, D.; Pons, R.; et al. Cationic niosomes as non-viral vehicles for nucleic acids: Challenges and opportunities in gene delivery. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef]
- Moghassemi, S.; Hadjizadeh, A. Nano-niosomes as nanoscale drug delivery systems: An illustrated review. J. Control. Release 2014, 185, 22–36. [Google Scholar] [CrossRef]
- Hua, S. Lipid-based nano-delivery systems for skin delivery of drugs and bioactives. Front. Pharm. 2015, 6, 219–224. [Google Scholar] [CrossRef]
- Marianecci, C.; Paolino, D.; Celia, C.; Fresta, M.; Carafa, M.; Alhaique, F. Non-ionic surfactant vesicles in pulmonary glucocorticoid delivery: Characterization and interaction with human lung fibroblasts. J. Control. Release 2010, 147, 127–135. [Google Scholar] [CrossRef]
- Abdelkader, H.; Alani, A.W.G.; Alany, R.G. Recent advances in non-ionic surfactant vesicles (niosomes): Self-assembly, fabrication, characterization, drug delivery applications and limitations. Drug Deliv. 2014, 21, 87–100. [Google Scholar] [CrossRef]
- Asthana, G.S.; Sharma, P.K.; Asthana, A. In vitro and in vivo evaluation of niosomal formulation for controlled delivery of clarithromycin. Scientifica 2016, 2016, 6492953. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Teeranachaideekul, V.; Supaperm, T. Effect of charged and non-ionic membrane additives on physicochemical properties and stability of niosomes. AAPS PharmSciTech 2008, 9, 851–859. [Google Scholar] [CrossRef]
- Redondo-Morata, L.; Giannotti, M.I.; Sanz, F. Influence of cholesterol on the phase transition of lipid bilayers: A temperature-controlled force spectroscopy study. Langmuir 2012, 28, 12851–12860. [Google Scholar] [CrossRef]
- Bates, P.J.; Reyes-reyes, E.M.; Malik, M.T.; Murphy, E.M.; Toole, M.G.O.; Trent, J.O. G-quadruplex oligonucleotide AS1411 as a cancer-targeting agent: Uses and mechanisms. BBA Gen. Subj. 2017, 1861, 1414–1428. [Google Scholar] [CrossRef]
- Guo, J.; Gao, X.; Su, L.; Xia, H.; Gu, G.; Pang, Z.; Jiang, X.; Yao, L.; Chen, J.; Chen, H. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 2011, 32, 8010–8020. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, R.; Chen, F.; Chen, M.; Wang, Y. Nucleolin targeting AS1411 aptamer modified pH-sensitive micelles: A dual-functional strategy for paclitaxel delivery. J. Control. Release 2015, 213, e137–e138. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, R.; Fang, X.; Chen, F.; Wang, Y.; Chen, M. Nucleolin targeting AS1411 aptamer modified pH-sensitive micelles for enhanced delivery and antitumor efficacy of paclitaxel. Nano Res. 2015, 8, 201–218. [Google Scholar] [CrossRef]
- Li, F.; Lu, J.; Liu, J.; Liang, C.; Wang, M.; Wang, L.; Li, D.; Yao, H.; Zhang, Q.; Wen, J.; et al. A water-soluble nucleolin aptamer-paclitaxel conjugate for tumor-specific targeting in ovarian cancer. Nat. Commun. 2017, 8, 1390. [Google Scholar] [CrossRef]
- Gao, H.; Qian, J.; Cao, S.; Yang, Z.; Pang, Z.; Pan, S.; Fan, L.; Xi, Z.; Jiang, X.; Zhang, Q. Precise glioma targeting of and penetration by aptamer and peptide dual-functioned nanoparticles. Biomaterials 2012, 33, 5115–5123. [Google Scholar] [CrossRef]
- Tao, W.; Zeng, X.; Wu, J.; Zhu, X.; Yu, X.; Zhang, X.; Zhang, J.; Liu, G.; Mei, L. Polydopamine-based surface modification of novel nanoparticle-aptamer bioconjugates for in vivo breast cancer targeting and enhanced therapeutic effects. Theranostics 2016, 6, 470–484. [Google Scholar] [CrossRef]
- Xu, G.; Yu, X.; Zhang, J.; Sheng, Y.; Liu, G.; Tao, W.; Mei, L. Robust aptamer-polydopamine-functionalized M-PLGA-TPGS nanoparticles for targeted delivery of docetaxel and enhanced cervical cancer therapy. Int. J. Nanomed. 2016, 11, 2953–2965. [Google Scholar] [CrossRef]
- Chen, D.; Li, B.; Cai, S.; Wang, P.; Peng, S.; Sheng, Y.; He, Y.; Gu, Y.; Chen, H. Dual targeting luminescent gold nanoclusters for tumor imaging and deep tissue therapy. Biomaterials 2016, 100, 1–16. [Google Scholar] [CrossRef]
- Xing, H.; Tang, L.; Yang, X.; Hwang, K.; Wang, W.; Yin, Q.; Wong, N.Y.; Dobrucki, L.W.; Yasui, N.; Katzenellenbogen, J.A.; et al. Selective delivery of an anticancer drug with aptamer-functionalized liposomes to breast cancer cells in vitro and in vivo. J. Mater. Chem. B 2013, 1, 5288–5297. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, L.; Jiao, X.; Yandan, J.; Zhu, X.; Hongji, L.; Chen, X.; Ren, J.; Xia, Y.; Zhang, Z. In vitro and in vivo evaluation of antitumor drug -loaded aptamer targeted single-walled carbon nanotubes system. Curr. Pharm. Biotechnol. 2014, 14, 1105–1117. [Google Scholar] [CrossRef]
- Zhang, B.; Luo, Z.; Liu, J.; Ding, X.; Li, J.; Cai, K. Cytochrome c end-capped mesoporous silica nanoparticles as redox-responsive drug delivery vehicles for liver tumor-targeted triplex therapy in vitro and in vivo. J. Control. Release 2014, 192, 192–201. [Google Scholar] [CrossRef]
- Li, X.; Yu, Y.; Ji, Q.; Qiu, L. Targeted delivery of anticancer drugs by aptamer AS1411 mediated Pluronic F127/cyclodextrin-linked polymer composite micelles. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 175–184. [Google Scholar] [CrossRef]
- Peng, L.H.; Zhang, Y.H.; Han, L.J.; Zhang, C.Z.; Wu, J.H.; Wang, X.R.; Gao, J.Q.; Mao, Z.W. Cell membrane capsules for encapsulation of chemotherapeutic and cancer cell targeting in vivo. ACS Appl. Mater. Interfaces 2015, 7, 18628–18637. [Google Scholar] [CrossRef]
- Trinh, T.L.; Zhu, G.; Xiao, X.; Puszyk, W.; Sefah, K.; Wu, Q.; Tan, W.; Liu, C. A synthetic aptamer-drug adduct for targeted liver cancer therapy. PLoS ONE 2015, 10, e0136673. [Google Scholar] [CrossRef]
- Liao, Z.X.; Chuang, E.Y.; Lin, C.C.; Ho, Y.C.; Lin, K.J.; Cheng, P.Y.; Chen, K.J.; Wei, H.J.; Sung, H.W. An AS1411 aptamer-conjugated liposomal system containing a bubble-generating agent for tumor-specific chemotherapy that overcomes multidrug resistance. J. Control. Release 2015, 208, 42–51. [Google Scholar] [CrossRef]
- Li, X.; Zhu, X.; Qiu, L. Constructing aptamer anchored nanovesicles for enhanced tumor penetration and cellular uptake of water soluble chemotherapeutics. Acta Biomater. 2016, 35, 269–279. [Google Scholar] [CrossRef]
- Liu, X.; Wu, L.; Wang, L.; Jiang, W. A dual-targeting DNA tetrahedron nanocarrier for breast cancer cell imaging and drug delivery. Talanta 2018, 179, 356–363. [Google Scholar] [CrossRef]
- Taghdisi, S.M.; Danesh, N.M.; Ramezani, M.; Lavaee, P.; Jalalian, S.H.; Robati, R.Y.; Abnous, K. Double targeting and aptamer-assisted controlled release delivery of epirubicin to cancer cells by aptamers-based dendrimer in vitro and in vivo. Eur. J. Pharm. Biopharm. 2016, 102, 152–158. [Google Scholar] [CrossRef]
- Velikyan, I. Molecular imaging and radiotherapy: Theranostics for personalized patient management. Theranostics 2012, 2, 424–426. [Google Scholar] [CrossRef]
- Bao, G.; Mitragotri, S.; Tong, S. Multifunctional nanoparticles for drug delivery and molecular imaging. Annu. Rev. Biomed. Eng. 2013, 15, 253–282. [Google Scholar] [CrossRef]
- Ochoa, G.P.; Sesma, J.Z.; Díez, M.A.; Díaz-Tahoces, A.; Avilés-Trigeros, M.; Grijalvo, S.; Eritja, R.; Fernández, E.; Pedraz, J.L. A novel formulation based on 2,3-di(tetradecyloxy)propan-1-amine cationic lipid combined with polysorbate 80 for efficient gene delivery to the retina. Pharm. Res. 2014, 31, 1665–1675. [Google Scholar] [CrossRef]
- Grijalvo, S.; Alagia, A.; Puras, G.; Zárate, J.; Pedraz, J.L.; Eritja, R. Cationic vesicles based on non-ionic surfactant and synthetic aminolipids mediate delivery of antisense oligonucleotides into mammalian cells. Colloids Surf. B Biointerfaces 2014, 119, 30–37. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Arango, D.; Shi, Q.; Wilson, A.J.; Corner, G.A.; Nicholas, C.; Aranes, M.J.; Lesser, M.; Schwartz, E.L.; Augenlicht, L.H. Gene expression profiling-based prediction of response of colon carcinoma cells to 5-fluorouracil and camptothecin. Cancer Res. 2003, 63, 8791–8812. [Google Scholar]
- Jorge, A.F.; Aviñó, A.; Pais, A.A.C.C.; Eritja, R.; Fàbrega, C. DNA-based nanoscaffolds as vehicles for 5-fluoro-2′-deoxyuridine oligomers in colorectal cancer therapy. Nanoscale 2018, 10, 7238–7249. [Google Scholar] [CrossRef]
- Ojeda, E.; Puras, G.; Agirre, M.; Zárate, J.; Grijalvo, S.; Pons, R.; Eritja, R.; Fernández, E.; Pedraz, J.L. Niosomes based on synthetic cationic lipids for gene delivery: The influence of polar head-groups on the transfection efficiency in HEK-293, ARPE-19 and MSC-D1 cells. Org. Biomol. Chem. 2015, 13, 1068–1081. [Google Scholar] [CrossRef]
- Luchini, A.; Vitiello, G.; Rossi, F.; Ruiz De Ballesteros, O.; Radulescu, A.; D’Errico, G.; Montesarchio, D.; de Julián Fernández, C.; Paduano, L. Developing functionalized Fe3O4–Au nanoparticles: A physico-chemical insight. Phys. Chem. Chem. Phys. 2015, 17, 6087–6097. [Google Scholar] [CrossRef]
- Luchini, A.; Vitiello, G. Understanding the nano-bio interfaces: Lipid-coatings for inorganic nanoparticles as promising strategy for biomedical applications. Front. Chem. 2019, 7, 1–16. [Google Scholar] [CrossRef]
- Estelrich, J.; Sánchez-Martín, M.J.; Busquets, M.A. Nanoparticles in magnetic resonance imaging: From simple to dual contrast agents. Int. J. Nanomed. 2015, 10, 1727–1741. [Google Scholar] [CrossRef]
- Alvares, R.D.A.; Szulc, D.A.; Cheng, H.L.M. A scale to measure MRI contrast agent sensitivity. Sci. Rep. 2017, 7, 15493. [Google Scholar] [CrossRef]
- Boni, A.; Bardi, G.; Bertero, A.; Cappello, V.; Emdin, M.; Flori, A.; Gemmi, M.; Innocenti, C.; Menichetti, L.; Sangregorio, C.; et al. Design and optimization of lipid-modified poly (amidoamine) dendrimer coated iron oxide nanoparticles as probes for biomedical applications. Nanoscale 2015, 7, 7307–7317. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Russo Krauss, I.; Piccolo, M.; Irace, C.; Paduano, L.; Montesarchio, D. Exploring the conformational behaviour and aggregation properties of lipid-conjugated AS1411 aptamers. Int. J. Biol. Macromol. 2018, 118, 1384–1399. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | ||||||
|---|---|---|---|---|---|---|
| MCF-7 | WiDr | C6 | HeLa | L6 | HaCaT | |
| NAMI-A [78] | 620 ± 30 | - | - | 626 ± 45 | - | - |
| AziRu [77,79] | 305 ± 16 [77] | 441 ± 20 [77] | 318 ± 12 [77] | 382 ± 19 [79] | > 500 [79] | > 500 [79] |
| POPC-based formulations | ||||||
| ToThyRu/POPC [77,134] | 27.8 ± 0.1 [134] | 75 ± 4 [77] | 36 ± 8 [77] | - | - | - |
| HoThyRu/POPC [77] | 7 ± 4 | 40 ± 5 | 81 ± 7 | - | - | - |
| DoHuRu/POPC [77,134] | 18.9 ± 0.1 [134] | 99 ± 5 [77] | 24 ± 5 [77] | - | - | - |
| ToThyCholRu/POPC [79,105] | 70 ± 12 [105] | 165 ± 10 [105] | - | - | >500 [79] | >500 [79] |
| HoUrRu/POPC [107] | 14 ± 7 | 20 ± 8 | - | - | - | - |
| DOTAP-based formulations | ||||||
| ToThyRu/DOTAP [106,134] | 10.1 ± 0.1 [134] | 50 ± 11 [106] | 54 ± 8 [106] | - | - | - |
| HoThyRu/DOTAP [100,106] | 13 ± 4 [100] | 65 ± 8 [106] | 43 ± 11 [106] | - | - | - |
| DoHuRu/DOTAP [106,134] | 10.3 ± 0.2 [134] | 41 ± 10 [106] | 34 ± 9 [106] | - | - | - |
| ToThyCholRu/DOTAP [79] | 13 ± 2 | 23 ± 8 | - | 34 ± 4 | 187 ± 1 | 377 ± 3 |
| HoUrRu/DOTAP [107] | 8 ± 5 | 12 ± 5 | - | - | - | - |
| I/DOTAP [110] | 31.2 ± 2.7 | - | 35.4 ± 2.7 | 45.6 ± 3 | - | >150 |
| The Composite Sustainability Indicator (CSI) | Average Score① | Growth Rate② | ||||||
|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | |||
| Beijing | 0.565 | 0.59 | 0.609 | 0.605 | 0.588 | 0.634 | 0.598 | 2.44% |
| Tianjin | 0.562 | 0.555 | 0.553 | 0.513 | 0.522 | 0.515 | 0.537 | −1.66% |
| Hebei | 0.403 | 0.401 | 0.414 | 0.426 | 0.467 | 0.503 | 0.436 | 4.93% |
| Shanxi | 0.407 | 0.419 | 0.442 | 0.434 | 0.437 | 0.482 | 0.437 | 3.70% |
| Inner Mongolia | 0.522 | 0.523 | 0.532 | 0.556 | 0.558 | 0.598 | 0.548 | 2.89% |
| Liaoning | 0.474 | 0.486 | 0.515 | 0.509 | 0.509 | 0.453 | 0.491 | −0.91% |
| Jilin | 0.406 | 0.442 | 0.455 | 0.466 | 0.467 | 0.517 | 0.459 | 5.46% |
| Heilongjiang | 0.393 | 0.397 | 0.432 | 0.431 | 0.432 | 0.46 | 0.424 | 3.42% |
| Shanghai | 0.45 | 0.455 | 0.449 | 0.477 | 0.496 | 0.556 | 0.48 | 4.69% |
| Jiangsu | 0.53 | 0.517 | 0.555 | 0.547 | 0.566 | 0.586 | 0.55 | 2.11% |
| Zhejiang | 0.534 | 0.545 | 0.576 | 0.588 | 0.594 | 0.613 | 0.575 | 2.96% |
| Anhui | 0.412 | 0.43 | 0.503 | 0.483 | 0.51 | 0.582 | 0.486 | 8.25% |
| Fujian | 0.487 | 0.525 | 0.537 | 0.547 | 0.574 | 0.586 | 0.543 | 4.03% |
| Jiangxi | 0.403 | 0.42 | 0.45 | 0.423 | 0.45 | 0.507 | 0.442 | 5.20% |
| Shandong | 0.537 | 0.537 | 0.572 | 0.574 | 0.561 | 0.588 | 0.561 | 1.90% |
| Henan | 0.33 | 0.337 | 0.351 | 0.382 | 0.402 | 0.484 | 0.381 | 9.30% |
| Hubei | 0.421 | 0.446 | 0.458 | 0.501 | 0.518 | 0.574 | 0.486 | 7.27% |
| Hunan | 0.398 | 0.431 | 0.433 | 0.464 | 0.508 | 0.551 | 0.464 | 7.70% |
| Guangdong | 0.474 | 0.451 | 0.482 | 0.471 | 0.514 | 0.534 | 0.488 | 2.54% |
| Guangxi | 0.415 | 0.431 | 0.436 | 0.443 | 0.477 | 0.522 | 0.454 | 5.18% |
| Hainan | 0.547 | 0.596 | 0.595 | 0.585 | 0.569 | 0.598 | 0.582 | 1.87% |
| Chongqing | 0.496 | 0.531 | 0.541 | 0.571 | 0.572 | 0.604 | 0.553 | 4.36% |
| Sichuan | 0.388 | 0.401 | 0.407 | 0.417 | 0.46 | 0.512 | 0.431 | 6.41% |
| Guizhou | 0.393 | 0.427 | 0.422 | 0.452 | 0.473 | 0.518 | 0.448 | 6.35% |
| Yunnan | 0.427 | 0.432 | 0.446 | 0.457 | 0.449 | 0.504 | 0.452 | 3.58% |
| Tibet | 0.534 | 0.55 | 0.56 | 0.582 | 0.582 | 0.558 | 0.561 | 0.92% |
| Shaanxi | 0.483 | 0.521 | 0.532 | 0.526 | 0.545 | 0.568 | 0.529 | 3.53% |
| Gansu | 0.421 | 0.429 | 0.442 | 0.442 | 0.43 | 0.474 | 0.44 | 2.49% |
| Qinghai | 0.476 | 0.485 | 0.49 | 0.504 | 0.502 | 0.549 | 0.501 | 3.06% |
| Ningxia | 0.457 | 0.481 | 0.501 | 0.512 | 0.503 | 0.551 | 0.501 | 4.15% |
| Xinjiang | 0.5 | 0.494 | 0.498 | 0.515 | 0.546 | 0.567 | 0.52 | 2.68% |
| East China③ | 0.509 | 0.517 | 0.534 | 0.533 | 0.545 | 0.571 | 0.535 | 2.45% |
| Middle China | 0.395 | 0.414 | 0.439 | 0.448 | 0.471 | 0.53 | 0.449 | 6.83% |
| West China | 0.459 | 0.475 | 0.484 | 0.498 | 0.508 | 0.544 | 0.495 | 3.68% |
| Northeastern China | 0.424 | 0.442 | 0.467 | 0.469 | 0.469 | 0.477 | 0.458 | 2.46% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riccardi, C.; Musumeci, D.; Trifuoggi, M.; Irace, C.; Paduano, L.; Montesarchio, D. Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity. Pharmaceuticals 2019, 12, 146. https://doi.org/10.3390/ph12040146
Riccardi C, Musumeci D, Trifuoggi M, Irace C, Paduano L, Montesarchio D. Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity. Pharmaceuticals. 2019; 12(4):146. https://doi.org/10.3390/ph12040146
Chicago/Turabian StyleRiccardi, Claudia, Domenica Musumeci, Marco Trifuoggi, Carlo Irace, Luigi Paduano, and Daniela Montesarchio. 2019. "Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity" Pharmaceuticals 12, no. 4: 146. https://doi.org/10.3390/ph12040146
APA StyleRiccardi, C., Musumeci, D., Trifuoggi, M., Irace, C., Paduano, L., & Montesarchio, D. (2019). Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity. Pharmaceuticals, 12(4), 146. https://doi.org/10.3390/ph12040146