Novel Treatment Targets Based on Insights in the Etiology of Depression: Role of IL-6 Trans-Signaling and Stress-Induced Elevation of Glutamate and ATP

Abstract
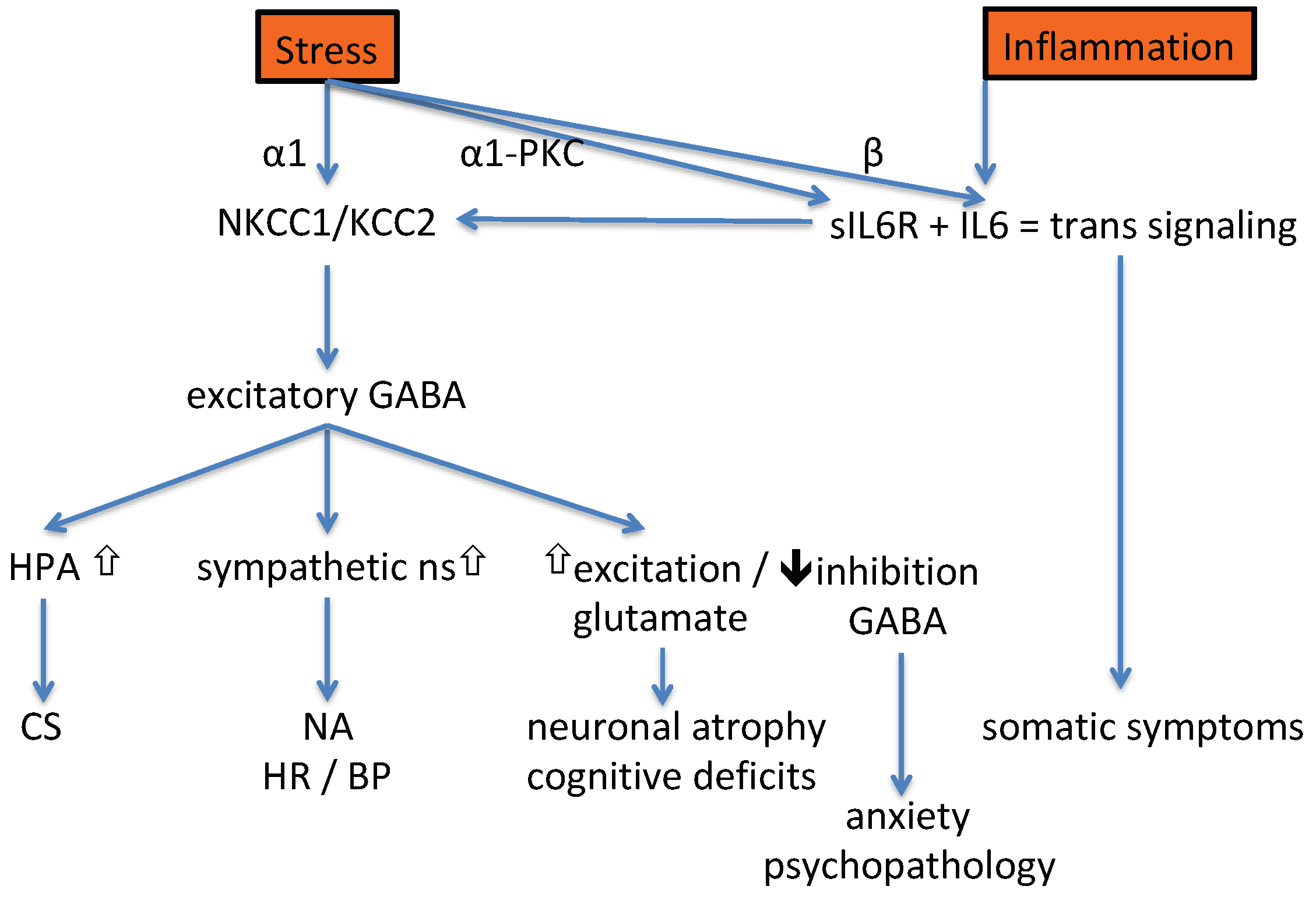
1. Introduction
2. Stress as a Risk Factor for Depression and Suicide
2.1. Stress Modifies KCC2 and NKCC1 Activity
2.2. Hyperactivation of the HPA Axis
2.3. Stress Induces Increased Central Glutamate Signaling
2.4. Stress Increases Levels of Interleukin-6
3. Inflammation as a Risk Factor for Depression and Suicide
3.1. IL-6 Trans-Signaling in Depression
3.2. IL-6 Trans-Signaling in the Brain
4. Potential for Therapeutic Intervention
4.1. Intervening in the Sequence of Events Provoked by Stress
4.2. Intervening in the Sequence of Events Provoked by Inflammation
4.3. Common Pathways Activated by Stress and Inflammation
5. Biomarkers
5.1. Biomarker for IL-6 Trans-Signaling
5.2. Biomarkers for Stress-Induced Effects
5.3. Biomarkers for Inflammation-Induced Effects
6. General Remarks
Funding
Conflicts of Interest
References
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Wittchen, H.U.; Jacobi, F.; Rehm, J.; Gustavsson, A.; Svensson, M.; Jonsson, B.; Olesen, J.; Allgulander, C.; Alonso, J.; Faravelli, C.; et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 655–679. [Google Scholar] [CrossRef] [PubMed]
- Monteggia, L.M.; Malenka, R.C.; Deisseroth, K. Depression: The best way forward. Nature 2014, 515, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Tian, L.; Zhang, S. Molecular networks in drug discovery. Crit. Rev. Biomed. Eng. 2010, 38, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Beumer, W.; Gibney, S.M.; Drexhage, R.C.; Pont-Lezica, L.; Doorduin, J.; Klein, H.C.; Steiner, J.; Connor, T.J.; Harkin, A.; Versnel, M.A.; et al. The immune theory of psychiatric diseases: A key role for activated microglia and circulating monocytes. J. Leukoc. Biol. 2012, 92, 959–975. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and psychiatric illness. J. Neuroinflamm. 2013, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.; Mondelli, V.; Pariante, C.M. Genetic Contributions of Inflammation to Depression. Neuropsychopharmacology 2017, 42, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef]
- Bartolomucci, A.; Leopardi, R. Stress and depression: Preclinical research and clinical implications. PLoS ONE 2009, 4, e4265. [Google Scholar] [CrossRef]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef]
- Kendler, K.S.; Karkowski, L.M.; Prescott, C.A. Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry 1999, 156, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C. The effects of stressful life events on depression. Annu. Rev. Psychol. 1997, 48, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Littrell, J.L. Taking the Perspective that a Depressive State Reflects Inflammation: Implications for the Use of Antidepressants. Front. Psychol. 2012, 3, 297. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Saricicek, A. GABAergic contributions to the pathophysiology of depression and the mechanism of antidepressant action. CNS Neurol. Disord. Drug. Targets 2007, 6, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Slavich, G.M.; Irwin, M.R. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull. 2014, 140, 774–815. [Google Scholar] [CrossRef]
- Charney, D.S.; Manji, H.K. Life stress, genes, and depression: Multiple pathways lead to increased risk and new opportunities for intervention. Sci. STKE 2004, 2004, re5. [Google Scholar] [CrossRef] [PubMed]
- Nock, M.K.; Borges, G.; Bromet, E.J.; Cha, C.B.; Kessler, R.C.; Lee, S. Suicide and suicidal behavior. Epidemiol. Rev. 2008, 30, 133–154. [Google Scholar] [CrossRef]
- Ambelas, A. Psychologically stressful events in the precipitation of manic episodes. Br. J. Psychiatry 1979, 135, 15–21. [Google Scholar] [CrossRef]
- Malkoff-Schwartz, S.; Frank, E.; Anderson, B.P.; Hlastala, S.A.; Luther, J.F.; Sherrill, J.T.; Houck, P.R.; Kupfer, D.J. Social rhythm disruption and stressful life events in the onset of bipolar and unipolar episodes. Psychol. Med. 2000, 30, 1005–1016. [Google Scholar] [CrossRef]
- Kirschbaum, C.; Pirke, K.M.; Hellhammer, D.H. The Trier Social Stress Test—A tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology 1993, 28, 76–81. [Google Scholar] [CrossRef]
- Bierhaus, A.; Wolf, J.; Andrassy, M.; Rohleder, N.; Humpert, P.M.; Petrov, D.; Ferstl, R.; von Eynatten, M.; Wendt, T.; Rudofsky, G.; et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc. Natl. Acad. Sci. USA 2003, 100, 1920–1925. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.L.; Gawuga, C.E.; Tyrka, A.R.; Lee, J.K.; Anderson, G.M.; Price, L.H. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology 2010, 35, 2617–2623. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, S.S.; Gable, S.L.; Irwin, M.R.; Aziz, N.; Kemeny, M.E. Social-evaluative threat and proinflammatory cytokine regulation: An experimental laboratory investigation. Psychol. Sci. 2009, 20, 1237–1244. [Google Scholar] [CrossRef]
- Pace, T.W.; Mletzko, T.C.; Alagbe, O.; Musselman, D.L.; Nemeroff, C.B.; Miller, A.H.; Heim, C.M. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am. J. Psychiatry 2006, 163, 1630–1633. [Google Scholar] [CrossRef] [PubMed]
- Blandino, P., Jr.; Barnum, C.J.; Deak, T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J. Neuroimmunol. 2006, 173, 87–95. [Google Scholar] [CrossRef]
- Powell, N.D.; Sloan, E.K.; Bailey, M.T.; Arevalo, J.M.; Miller, G.E.; Chen, E.; Kobor, M.S.; Reader, B.F.; Sheridan, J.F.; Cole, S.W. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc. Natl. Acad. Sci. USA 2013, 110, 16574–16579. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Woster, A.P.; Dahlman, J.; Sauter, E.R.; Combs, C.K.; Porter, J.E. Alpha1-adrenergic receptors positively regulate Toll-like receptor cytokine production from human monocytes and macrophages. J. Pharmacol. Exp. Ther. 2011, 338, 648–657. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. Beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef]
- Bailey, M.T.; Engler, H.; Powell, N.D.; Padgett, D.A.; Sheridan, J.F. Repeated social defeat increases the bactericidal activity of splenic macrophages through a Toll-like receptor-dependent pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1180–R1190. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Baratta, M.V.; Sprunger, D.B.; Watkins, L.R.; Maier, S.F. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav. Immun. 2007, 21, 47–59. [Google Scholar] [CrossRef]
- Hinwood, M.; Morandini, J.; Day, T.A.; Walker, F.R. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb. Cortex 2012, 22, 1442–1454. [Google Scholar] [CrossRef]
- Jankord, R.; Zhang, R.; Flak, J.N.; Solomon, M.B.; Albertz, J.; Herman, J.P. Stress activation of IL-6 neurons in the hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R343–R351. [Google Scholar] [CrossRef]
- Audet, M.C.; Anisman, H. Interplay between pro-inflammatory cytokines and growth factors in depressive illnesses. Front. Cell Neurosci. 2013, 7, 68. [Google Scholar] [CrossRef]
- Kaufmann, F.N.; Costa, A.P.; Ghisleni, G.; Diaz, A.P.; Rodrigues, A.L.S.; Peluffo, H.; Kaster, M.P. NLRP3 inflammasome-driven pathways in depression: Clinical and preclinical findings. Brain Behav. Immun. 2017, 64, 367–383. [Google Scholar] [CrossRef]
- Tynan, R.J.; Naicker, S.; Hinwood, M.; Nalivaiko, E.; Buller, K.M.; Pow, D.V.; Day, T.A.; Walker, F.R. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav. Immun. 2010, 24, 1058–1068. [Google Scholar] [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a microglial disease. Trends Neurosci. 2015, 38, 637–658. [Google Scholar] [CrossRef]
- Spitzer, N.C. How GABA generates depolarization. J. Physiol. 2010, 588, 757–758. [Google Scholar] [CrossRef]
- Levy, B.H.; Tasker, J.G. Synaptic regulation of the hypothalamic-pituitary-adrenal axis and its modulation by glucocorticoids and stress. Front. Cell Neurosci. 2012, 6, 24. [Google Scholar] [CrossRef]
- Zhou, J.J.; Gao, Y.; Zhang, X.; Kosten, T.A.; Li, D.P. Enhanced Hypothalamic NMDA Receptor Activity Contributes to Hyperactivity of HPA Axis in Chronic Stress in Male Rats. Endocrinology 2018, 159, 1537–1546. [Google Scholar] [CrossRef]
- Hewitt, S.A.; Wamsteeker, J.I.; Kurz, E.U.; Bains, J.S. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 2009, 12, 438–443. [Google Scholar] [CrossRef]
- Sarkar, J.; Wakefield, S.; MacKenzie, G.; Moss, S.J.; Maguire, J. Neurosteroidogenesis is required for the physiological response to stress: Role of neurosteroid-sensitive GABAA receptors. J. Neurosci. 2011, 31, 18198–18210. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, J.J.; Zhu, Y.; Kosten, T.; Li, D.P. Chronic Unpredictable Mild Stress Induces Loss of GABA Inhibition in Corticotrophin-Releasing Hormone-Expressing Neurons through NKCC1 Upregulation. Neuroendocrinology 2017, 104, 194–208. [Google Scholar] [CrossRef]
- Dai, S.; Ma, Z. BDNF-trkB-KCC2-GABA pathway may be related to chronic stress-induced hyperalgesia at both the spinal and supraspinal level. Med. Hypotheses 2014, 83, 772–774. [Google Scholar] [CrossRef]
- Ferrini, F.; De Koninck, Y. Microglia control neuronal network excitability via BDNF signalling. Neural. Plast. 2013, 2013, 429815. [Google Scholar] [CrossRef]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef]
- Ding, B.; Frisina, R.D.; Zhu, X.; Sakai, Y.; Sokolowski, B.; Walton, J.P. Direct control of Na(+)-K(+)-2Cl(-)-cotransport protein (NKCC1) expression with aldosterone. Am. J. Physiol. Cell Physiol. 2014, 306, C66–C75. [Google Scholar] [CrossRef]
- Gold, P.W.; Chrousos, G.P. Organization of the stress system and its dysregulation in melancholic and atypical depression: High vs. low CRH/NE states. Mol. Psychiatry 2002, 7, 254–275. [Google Scholar] [CrossRef]
- Wahbeh, H.; Oken, B.S. Salivary cortisol lower in posttraumatic stress disorder. J. Trauma. Stress 2013, 26, 241–248. [Google Scholar] [CrossRef]
- Melhem, N.M.; Keilp, J.G.; Porta, G.; Oquendo, M.A.; Burke, A.; Stanley, B.; Cooper, T.B.; Mann, J.J.; Brent, D.A. Blunted HPA Axis Activity in Suicide Attempters Compared to those at High Risk for Suicidal Behavior. Neuropsychopharmacology 2016, 41, 1447–1456. [Google Scholar] [CrossRef]
- Nair, A.; Bonneau, R.H. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J. Neuroimmunol. 2006, 171, 72–85. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Deak, T.; Owens, S.M.; Kohno, T.; Fleshner, M.; Watkins, L.R.; Maier, S.F. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J. Neurosci. 1998, 18, 2239–2246. [Google Scholar] [CrossRef]
- Danese, A.; Moffitt, T.E.; Pariante, C.M.; Ambler, A.; Poulton, R.; Caspi, A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch. Gen. Psychiatry 2008, 65, 409–415. [Google Scholar] [CrossRef]
- Lowy, M.T.; Gault, L.; Yamamoto, B.K. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J. Neurochem. 1993, 61, 1957–1960. [Google Scholar] [CrossRef]
- Moghaddam, B.; Bolinao, M.L.; Stein-Behrens, B.; Sapolsky, R. Glucocorticoids mediate the stress-induced extracellular accumulation of glutamate. Brain Res. 1994, 655, 251–254. [Google Scholar] [CrossRef]
- Munhoz, C.D.; Lepsch, L.B.; Kawamoto, E.M.; Malta, M.B.; Lima Lde, S.; Avellar, M.C.; Sapolsky, R.M.; Scavone, C. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-kappaB in the frontal cortex and hippocampus via glucocorticoid secretion. J. Neurosci. 2006, 26, 3813–3820. [Google Scholar] [CrossRef]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Sanacora, G.; Duman, R.S.; Krystal, J.H. The neurobiology of depression, ketamine and rapid-acting antidepressants: Is it glutamate inhibition or activation? Pharmacol. Ther. 2018, 190, 148–158. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Li, X.Y.; Sakaue, F.; Li, N.; Dutheil, S.; Banasr, M.; Duric, V.; Yamanashi, T.; Kaneko, K.; et al. Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol. Psychiatry 2016, 80, 12–22. [Google Scholar] [CrossRef]
- Banasr, M.; Duman, R.S. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 2008, 64, 863–870. [Google Scholar] [CrossRef]
- Czeh, B.; Simon, M.; Schmelting, B.; Hiemke, C.; Fuchs, E. Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 2006, 31, 1616–1626. [Google Scholar] [CrossRef]
- Frank, M.G.; Thompson, B.M.; Watkins, L.R.; Maier, S.F. Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav. Immun. 2012, 26, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F. Stress weakens prefrontal networks: Molecular insults to higher cognition. Nat. Neurosci. 2015, 18, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; Miller, M.M.; Goldwater, D.S.; Radley, J.J.; Rocher, A.B.; Hof, P.R.; Morrison, J.H.; McEwen, B.S. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006, 26, 7870–7874. [Google Scholar] [CrossRef] [PubMed]
- Radley, J.J.; Rocher, A.B.; Miller, M.; Janssen, W.G.; Liston, C.; Hof, P.R.; McEwen, B.S.; Morrison, J.H. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb. Cortex 2006, 16, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Henter, I.D.; Manji, H.K. A role for PKC in mediating stress-induced prefrontal cortical structural plasticity and cognitive function. Proc. Natl. Acad. Sci. USA 2009, 106, 17613–17614. [Google Scholar] [CrossRef] [PubMed]
- Hains, A.B.; Vu, M.A.; Maciejewski, P.K.; van Dyck, C.H.; Gottron, M.; Arnsten, A.F. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc. Natl. Acad. Sci. USA 2009, 106, 17957–17962. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Garcia-Oscos, F.; Mendez, J.A. Role of IL-6 in the etiology of hyperexcitable neuropsychiatric conditions: Experimental evidence and therapeutic implications. Future Med. Chem. 2012, 4, 2177–2192. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Steptoe, A.; Hamer, M.; Chida, Y. The effects of acute psychological stress on circulating inflammatory factors in humans: A review and meta-analysis. Brain Behav. Immun. 2007, 21, 901–912. [Google Scholar] [CrossRef]
- Gouin, J.P.; Glaser, R.; Malarkey, W.B.; Beversdorf, D.; Kiecolt-Glaser, J.K. Childhood abuse and inflammatory responses to daily stressors. Ann. Behav. Med. 2012, 44, 287–292. [Google Scholar] [CrossRef]
- Wang, N.; Yu, H.Y.; Shen, X.F.; Gao, Z.Q.; Yang, C.; Yang, J.J.; Zhang, G.F. The rapid antidepressant effect of ketamine in rats is associated with down-regulation of pro-inflammatory cytokines in the hippocampus. Ups. J. Med. Sci. 2015, 120, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.; Donegan, J.J.; Morilak, D.A. Influence of hypothalamic IL-6/gp130 receptor signaling on the HPA axis response to chronic stress. Psychoneuroendocrinology 2013, 38, 1158–1169. [Google Scholar] [CrossRef] [PubMed]
- Sukoff Rizzo, S.J.; Neal, S.J.; Hughes, Z.A.; Beyna, M.; Rosenzweig-Lipson, S.; Moss, S.J.; Brandon, N.J. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Transl. Psychiatry 2012, 2, e199. [Google Scholar] [CrossRef] [PubMed]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Erta, M.; Lim, S.L.; Frausto, R.; May, U.; Rose-John, S.; Scheller, J.; Hidalgo, J. Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J. Neurosci. 2014, 34, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef]
- Wan, J.; Fu, A.K.; Ip, F.C.; Ng, H.K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, J.; Kwak, Y.; Park, S.K. Depression research: Where are we now? Mol. Brain 2010, 3, 8. [Google Scholar] [CrossRef]
- Dumitrescu, A.L. Depression and Inflammatory Periodontal Disease Considerations-An Interdisciplinary Approach. Front. Psychol. 2016, 7, 347. [Google Scholar] [CrossRef]
- Jiang, M.; Qin, P.; Yang, X. Comorbidity between depression and asthma via immune-inflammatory pathways: A meta-analysis. J. Affect. Disord. 2014, 166, 22–29. [Google Scholar] [CrossRef]
- Siegert, R.J.; Abernethy, D.A. Depression in multiple sclerosis: A review. J. Neurol. Neurosurg. Psychiatry 2005, 76, 469–475. [Google Scholar] [CrossRef]
- Lu, M.C.; Guo, H.R.; Lin, M.C.; Livneh, H.; Lai, N.S.; Tsai, T.Y. Bidirectional associations between rheumatoid arthritis and depression: A nationwide longitudinal study. Sci Rep. 2016, 6, 20647. [Google Scholar] [CrossRef]
- Matcham, F.; Rayner, L.; Steer, S.; Hotopf, M. The prevalence of depression in rheumatoid arthritis: A systematic review and meta-analysis. Rheumatology 2013, 52, 2136–2148. [Google Scholar] [CrossRef]
- Nerurkar, L.; Siebert, S.; McInnes, I.B.; Cavanagh, J. Rheumatoid arthritis and depression: An inflammatory perspective. Lancet Psychiatry 2019, 6, 164–173. [Google Scholar] [CrossRef]
- Aarsland, D.; Pahlhagen, S.; Ballard, C.G.; Ehrt, U.; Svenningsson, P. Depression in Parkinson disease—Epidemiology, mechanisms and management. Nat. Rev. Neurol. 2011, 8, 35–47. [Google Scholar] [CrossRef]
- Jacob, E.L.; Gatto, N.M.; Thompson, A.; Bordelon, Y.; Ritz, B. Occurrence of depression and anxiety prior to Parkinson’s disease. Parkinsonism. Relat. Disord. 2010, 16, 576–581. [Google Scholar] [CrossRef]
- Benoit, M.; Berrut, G.; Doussaint, J.; Bakchine, S.; Bonin-Guillaume, S.; Fremont, P.; Gallarda, T.; Krolak-Salmon, P.; Marquet, T.; Mekies, C.; et al. Apathy and depression in mild Alzheimer’s disease: A cross-sectional study using diagnostic criteria. J. Alzheimers Dis. 2012, 31, 325–334. [Google Scholar] [CrossRef]
- Diniz, B.S.; Butters, M.A.; Albert, S.M.; Dew, M.A.; Reynolds, C.F., 3rd. Late-life depression and risk of vascular dementia and Alzheimer’s disease: Systematic review and meta-analysis of community-based cohort studies. Br. J. Psychiatry 2013, 202, 329–335. [Google Scholar] [CrossRef]
- Rutledge, T.; Reis, V.A.; Linke, S.E.; Greenberg, B.H.; Mills, P.J. Depression in heart failure a meta-analytic review of prevalence, intervention effects, and associations with clinical outcomes. J. Am. Coll. Cardiol. 2006, 48, 1527–1537. [Google Scholar] [CrossRef]
- Mezuk, B.; Eaton, W.W.; Albrecht, S.; Golden, S.H. Depression and type 2 diabetes over the lifespan: A meta-analysis. Diabetes Care 2008, 31, 2383–2390. [Google Scholar] [CrossRef]
- De Wit, L.; Luppino, F.; van Straten, A.; Penninx, B.; Zitman, F.; Cuijpers, P. Depression and obesity: A meta-analysis of community-based studies. Psychiatry Res. 2010, 178, 230–235. [Google Scholar] [CrossRef]
- Dunn, A.J.; Swiergiel, A.H.; de Beaurepaire, R. Cytokines as mediators of depression: What can we learn from animal studies? Neurosci. Biobehav. Rev. 2005, 29, 891–909. [Google Scholar] [CrossRef]
- Kopschina Feltes, P.; Doorduin, J.; Klein, H.C.; Juarez-Orozco, L.E.; Dierckx, R.A.; Moriguchi-Jeckel, C.M.; de Vries, E.F. Anti-inflammatory treatment for major depressive disorder: Implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J. Psychopharmacol. 2017, 31, 1149–1165. [Google Scholar] [CrossRef]
- Silic, A.; Karlovic, D.; Serretti, A. Increased inflammation and lower platelet 5-HT in depression with metabolic syndrome. J. Affect. Disord. 2012, 141, 72–78. [Google Scholar] [CrossRef]
- Rudolf, S.; Greggersen, W.; Kahl, K.G.; Huppe, M.; Schweiger, U. Elevated IL-6 levels in patients with atypical depression but not in patients with typical depression. Psychiatry Res. 2014, 217, 34–38. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Haapakoski, R.; Mathieu, J.; Ebmeier, K.P.; Alenius, H.; Kivimaki, M. Cumulative meta-analysis of interleukins 6 and 1beta, tumour necrosis factor alpha and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 2015, 49, 206–215. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Zhao, G.; Wang, F.; Fang, Y. Identification of IL6 as a susceptibility gene for major depressive disorder. Sci. Rep. 2016, 6, 31264. [Google Scholar] [CrossRef]
- Maes, M.; Ombelet, W.; De Jongh, R.; Kenis, G.; Bosmans, E. The inflammatory response following delivery is amplified in women who previously suffered from major depression, suggesting that major depression is accompanied by a sensitization of the inflammatory response system. J. Affect. Disord. 2001, 63, 85–92. [Google Scholar] [CrossRef]
- Modabbernia, A.; Taslimi, S.; Brietzke, E.; Ashrafi, M. Cytokine alterations in bipolar disorder: A meta-analysis of 30 studies. Biol. Psychiatry 2013, 74, 15–25. [Google Scholar] [CrossRef]
- Munkholm, K.; Brauner, J.V.; Kessing, L.V.; Vinberg, M. Cytokines in bipolar disorder vs. healthy control subjects: A systematic review and meta-analysis. J. Psychiatr. Res. 2013, 47, 1119–1133. [Google Scholar] [CrossRef]
- Michalopoulou, M.; Nikolaou, C.; Tavernarakis, A.; Alexandri, N.M.; Rentzos, M.; Chatzipanagiotou, S.; Cambouri, C.; Vassilopoulos, D. Soluble interleukin-6 receptor (sIL-6R) in cerebrospinal fluid of patients with inflammatory and non inflammatory neurological diseases. Immunol. Lett. 2004, 94, 183–189. [Google Scholar] [CrossRef]
- Gananca, L.; Oquendo, M.A.; Tyrka, A.R.; Cisneros-Trujillo, S.; Mann, J.J.; Sublette, M.E. The role of cytokines in the pathophysiology of suicidal behavior. Psychoneuroendocrinology 2016, 63, 296–310. [Google Scholar] [CrossRef]
- Isung, J.; Aeinehband, S.; Mobarrez, F.; Nordstrom, P.; Runeson, B.; Asberg, M.; Piehl, F.; Jokinen, J. High interleukin-6 and impulsivity: Determining the role of endophenotypes in attempted suicide. Transl. Psychiatry 2014, 4, e470. [Google Scholar] [CrossRef]
- Lindqvist, D.; Janelidze, S.; Hagell, P.; Erhardt, S.; Samuelsson, M.; Minthon, L.; Hansson, O.; Bjorkqvist, M.; Traskman-Bendz, L.; Brundin, L. Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol. Psychiatry 2009, 66, 287–292. [Google Scholar] [CrossRef]
- Sasayama, D.; Hattori, K.; Wakabayashi, C.; Teraishi, T.; Hori, H.; Ota, M.; Yoshida, S.; Arima, K.; Higuchi, T.; Amano, N.; et al. Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. J. Psychiatr. Res. 2013, 47, 401–406. [Google Scholar] [CrossRef]
- O’Donovan, A.; Rush, G.; Hoatam, G.; Hughes, B.M.; McCrohan, A.; Kelleher, C.; O’Farrelly, C.; Malone, K.M. Suicidal ideation is associated with elevated inflammation in patients with major depressive disorder. Depress. Anxiety 2013, 30, 307–314. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattei, D.; Westrin, A.; Traskman-Bendz, L.; Brundin, L. Cytokine levels in the blood may distinguish suicide attempters from depressed patients. Brain Behav. Immun. 2011, 25, 335–339. [Google Scholar] [CrossRef]
- Bergmans, R.S.; Kelly, K.M.; Mezuk, B. Inflammation as a unique marker of suicide ideation distinct from depression syndrome among U.S. adults. J. Affect. Disord 2019, 245, 1052–1060. [Google Scholar] [CrossRef]
- Brundin, L.; Erhardt, S.; Bryleva, E.Y.; Achtyes, E.D.; Postolache, T.T. The role of inflammation in suicidal behaviour. Acta Psychiatr. Scand. 2015, 132, 192–203. [Google Scholar] [CrossRef]
- Marsland, A.L.; Prather, A.A.; Petersen, K.L.; Cohen, S.; Manuck, S.B. Antagonistic characteristics are positively associated with inflammatory markers independently of trait negative emotionality. Brain Behav. Immun. 2008, 22, 753–761. [Google Scholar] [CrossRef]
- Marini, S.; Vellante, F.; Matarazzo, I.; De Berardis, D.; Serroni, N.; Gianfelice, D.; Olivieri, L.; Di Renzo, F.; Di Marco, A.; Fornaro, M.; et al. Inflammatory markers and suicidal attempts in depressed patients: A review. Int. J. Immunopathol. Pharmacol. 2016, 29, 583–594. [Google Scholar] [CrossRef]
- Kubera, M.; Obuchowicz, E.; Goehler, L.; Brzeszcz, J.; Maes, M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 744–759. [Google Scholar] [CrossRef]
- Baran, P.; Hansen, S.; Waetzig, G.H.; Akbarzadeh, M.; Lamertz, L.; Huber, H.J.; Ahmadian, M.R.; Moll, J.M.; Scheller, J. The balance of interleukin (IL)-6, IL-6.soluble IL-6 receptor (sIL-6R), and IL-6.sIL-6R.sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 2018, 293, 6762–6775. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef]
- Sohn, S.J.; Barrett, K.; Van Abbema, A.; Chang, C.; Kohli, P.B.; Kanda, H.; Smith, J.; Lai, Y.; Zhou, A.; Zhang, B.; et al. A restricted role for TYK2 catalytic activity in human cytokine responses revealed by novel TYK2-selective inhibitors. J. Immunol. 2013, 191, 2205–2216. [Google Scholar] [CrossRef]
- Briso, E.M.; Dienz, O.; Rincon, M. Cutting edge: Soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J. Immunol. 2008, 180, 7102–7106. [Google Scholar] [CrossRef]
- Maes, M.; Anderson, G.; Kubera, M.; Berk, M. Targeting classical IL-6 signalling or IL-6 trans-signalling in depression? Expert Opin. Ther. Targets 2014, 18, 495–512. [Google Scholar] [CrossRef]
- Burton, M.D.; Sparkman, N.L.; Johnson, R.W. Inhibition of interleukin-6 trans-signaling in the brain facilitates recovery from lipopolysaccharide-induced sickness behavior. J. Neuroinflamm. 2011, 8, 54. [Google Scholar] [CrossRef]
- Hsu, M.P.; Frausto, R.; Rose-John, S.; Campbell, I.L. Analysis of IL-6/gp130 family receptor expression reveals that in contrast to astroglia, microglia lack the oncostatin M receptor and functional responses to oncostatin M. Glia 2015, 63, 132–141. [Google Scholar] [CrossRef]
- Riethmueller, S.; Somasundaram, P.; Ehlers, J.C.; Hung, C.W.; Flynn, C.M.; Lokau, J.; Agthe, M.; Dusterhoft, S.; Zhu, Y.; Grotzinger, J.; et al. Proteolytic Origin of the Soluble Human IL-6R In Vivo and a Decisive Role of N-Glycosylation. PLoS Biol. 2017, 15, e2000080. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- Jones, S.A.; Novick, D.; Horiuchi, S.; Yamamoto, N.; Szalai, A.J.; Fuller, G.M. C-reactive protein: A physiological activator of interleukin 6 receptor shedding. J. Exp. Med. 1999, 189, 599–604. [Google Scholar] [CrossRef]
- Sluzewska, A.; Rybakowski, J.K.; Laciak, M.; Mackiewicz, A.; Sobieska, M.; Wiktorowicz, K. Interleukin-6 serum levels in depressed patients before and after treatment with fluoxetine. Ann. NY Acad. Sci. 1995, 762, 474–476. [Google Scholar] [CrossRef]
- Patel, A.; Zhu, Y.; Kuzhikandathil, E.V.; Banks, W.A.; Siegel, A.; Zalcman, S.S. Soluble interleukin-6 receptor induces motor stereotypies and co-localizes with gp130 in regions linked to cortico-striato-thalamo-cortical circuits. PLoS ONE 2012, 7, e41623. [Google Scholar] [CrossRef]
- Raber, J.; O’Shea, R.D.; Bloom, F.E.; Campbell, I.L. Modulation of hypothalamic-pituitary-adrenal function by transgenic expression of interleukin-6 in the CNS of mice. J. Neurosci. 1997, 17, 9473–9480. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, G.; Weber, J.S.; Magiakou, M.A.; Gunn, H.; Chrousos, G.P. Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: Potential implications for the syndrome of inappropriate vasopressin secretion. J. Clin. Endocrinol. Metab. 1994, 79, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Watanabe, M.; Moorhouse, A.J.; Kanematsu, T.; Horibe, S.; Matsukawa, N.; Asai, K.; Ojika, K.; Hirata, M.; Nabekura, J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 2007, 27, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Pieraut, S.; Lucas, O.; Sangari, S.; Sar, C.; Boudes, M.; Bouffi, C.; Noel, D.; Scamps, F. An autocrine neuronal interleukin-6 loop mediates chloride accumulation and NKCC1 phosphorylation in axotomized sensory neurons. J. Neurosci. 2011, 31, 13516–13526. [Google Scholar] [CrossRef] [PubMed]
- Garza, J.C.; Guo, M.; Zhang, W.; Lu, X.Y. Leptin restores adult hippocampal neurogenesis in a chronic unpredictable stress model of depression and reverses glucocorticoid-induced inhibition of GSK-3beta/beta-catenin signaling. Mol. Psychiatry 2012, 17, 790–808. [Google Scholar] [CrossRef] [PubMed]
- Hodes, G.E.; Menard, C.; Russo, S.J. Integrating Interleukin-6 into depression diagnosis and treatment. Neurobiol. Stress 2016, 4, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Marz, P.; Cheng, J.G.; Gadient, R.A.; Patterson, P.H.; Stoyan, T.; Otten, U.; Rose-John, S. Sympathetic neurons can produce and respond to interleukin 6. Proc. Natl. Acad. Sci. USA 1998, 95, 3251–3256. [Google Scholar] [CrossRef]
- Helwig, B.G.; Craig, R.A.; Fels, R.J.; Blecha, F.; Kenney, M.J. Central nervous system administration of interleukin-6 produces splenic sympathoexcitation. Auton. Neurosci. 2008, 141, 104–111. [Google Scholar] [CrossRef]
- Palsson, J.; Ricksten, S.E.; Delle, M.; Lundin, S. Changes in renal sympathetic nerve activity during experimental septic and endotoxin shock in conscious rats. Circ. Shock 1988, 24, 133–141. [Google Scholar]
- Rudiger, A.; Jeger, V.; Arrigo, M.; Schaer, C.A.; Hildenbrand, F.F.; Arras, M.; Seifert, B.; Singer, M.; Schoedon, G.; Spahn, D.R.; et al. Heart rate elevations during early sepsis predict death in fluid-resuscitated rats with fecal peritonitis. Intensive Care Med. Exp. 2018, 6, 28. [Google Scholar] [CrossRef]
- Franke, H.; Verkhratsky, A.; Burnstock, G.; Illes, P. Pathophysiology of astroglial purinergic signalling. Purinergic Signal. 2012, 8, 629–657. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Lee, W.H.; Suk, K. Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 2016, 103, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Pariante, C.M. The glucocorticoid receptor: Part of the solution or part of the problem? J. Psychopharmacol. 2006, 20, 79–84. [Google Scholar] [CrossRef]
- Lotrich, F.E. Inflammatory cytokine-associated depression. Brain Res. 2015, 1617, 113–125. [Google Scholar] [CrossRef]
- Boku, S.; Nakagawa, S.; Masuda, T.; Nishikawa, H.; Kato, A.; Kitaichi, Y.; Inoue, T.; Koyama, T. Glucocorticoids and lithium reciprocally regulate the proliferation of adult dentate gyrus-derived neural precursor cells through GSK-3beta and beta-catenin/TCF pathway. Neuropsychopharmacology 2009, 34, 805–815. [Google Scholar] [CrossRef][Green Version]
- Nitta, A.; Zheng, W.H.; Quirion, R. Insulin-like growth factor 1 prevents neuronal cell death induced by corticosterone through activation of the PI3k/Akt pathway. J. Neurosci. Res. 2004, 76, 98–103. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Y.; Wu, W.; Xu, T.; Yin, Y.; Zhang, J.; Huang, D.; Li, W. Chronic glucocorticoid exposure activates BK-NLRP1 signal involving in hippocampal neuron damage. J. Neuroinflamm. 2017, 14, 139. [Google Scholar] [CrossRef]
- Sanacora, G.; Schatzberg, A.F. Ketamine: Promising path or false prophecy in the development of novel therapeutics for mood disorders? Neuropsychopharmacology 2015, 40, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Gould, T.D. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Bjorkholm, C.; Monteggia, L.M. BDNF—A key transducer of antidepressant effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Li, H.; Thomas-Crusells, J.; Lahtinen, H.; Viitanen, T.; Nanobashvili, A.; Kokaia, Z.; Airaksinen, M.S.; Voipio, J.; Kaila, K.; et al. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J. Cell Biol. 2002, 159, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Liu, C.Y.; Zhang, F.; Duan, X.; Wen, Z.; Song, J.; Feighery, E.; Lu, B.; Rujescu, D.; St Clair, D.; et al. Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 2012, 148, 1051–1064. [Google Scholar] [CrossRef]
- North, R.A.; Surprenant, A. Pharmacology of cloned P2X receptors. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 563–580. [Google Scholar] [CrossRef]
- Ase, A.R.; Honson, N.S.; Zaghdane, H.; Pfeifer, T.A.; Seguela, P. Identification and characterization of a selective allosteric antagonist of human P2X4 receptor channels. Mol. Pharmacol. 2015, 87, 606–616. [Google Scholar] [CrossRef]
- Matsumura, Y.; Yamashita, T.; Sasaki, A.; Nakata, E.; Kohno, K.; Masuda, T.; Tozaki-Saitoh, H.; Imai, T.; Kuraishi, Y.; Tsuda, M.; et al. A novel P2X4 receptor-selective antagonist produces anti-allodynic effect in a mouse model of herpetic pain. Sci. Rep. 2016, 6, 32461. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Leitner, N.R.; Witalisz-Siepracka, A.; Strobl, B.; Muller, M. Tyrosine kinase 2—Surveillant of tumours and bona fide oncogene. Cytokine 2017, 89, 209–218. [Google Scholar] [CrossRef]
- He, X.; Chen, X.; Zhang, H.; Xie, T.; Ye, X.Y. Selective Tyk2 inhibitors as potential therapeutic agents: A patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 137–149. [Google Scholar] [CrossRef]
- Menet, C.J. Toward selective TYK2 inhibitors as therapeutic agents for the treatment of inflammatory diseases. Pharm. Pat. Anal. 2014, 3, 449–466. [Google Scholar] [CrossRef]
- Tracey, K.J. Reflex control of immunity. Nat. Rev. Immunol. 2009, 9, 418–428. [Google Scholar] [CrossRef]
- Bencherif, M.; Lippiello, P.M.; Lucas, R.; Marrero, M.B. Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell Mol. Life Sci. 2011, 68, 931–949. [Google Scholar] [CrossRef]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; Leon, R.; Lopez, M.G. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- De Jonge, W.J.; van der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.R.; Uematsu, S.; Akira, S.; van den Wijngaard, R.M.; et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef]
- Bajbouj, M.; Merkl, A.; Schlaepfer, T.E.; Frick, C.; Zobel, A.; Maier, W.; O’Keane, V.; Corcoran, C.; Adolfsson, R.; Trimble, M.; et al. Two-year outcome of vagus nerve stimulation in treatment-resistant depression. J. Clin. Psychopharmacol. 2010, 30, 273–281. [Google Scholar] [CrossRef]
- Berry, S.M.; Broglio, K.; Bunker, M.; Jayewardene, A.; Olin, B.; Rush, A.J. A patient-level meta-analysis of studies evaluating vagus nerve stimulation therapy for treatment-resistant depression. Med. Devices 2013, 6, 17–35. [Google Scholar] [CrossRef]
- Kalkman, H.O.; Feuerbach, D. Modulatory effects of alpha7 nAChRs on the immune system and its relevance for CNS disorders. Cell Mol. Life Sci. 2016, 73, 2511–2530. [Google Scholar] [CrossRef]
- Cui, W.Y.; Zhao, S.; Polanowska-Grabowska, R.; Wang, J.; Wei, J.; Dash, B.; Chang, S.L.; Saucerman, J.J.; Gu, J.; Li, M.D. Identification and characterization of poly(I:C)-induced molecular responses attenuated by nicotine in mouse macrophages. Mol. Pharmacol. 2013, 83, 61–72. [Google Scholar] [CrossRef]
- Kalkman, H.O.; Feuerbach, D. Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacol. Ther. 2016, 163, 82–93. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Goldstein, R.S.; Gallowitsch-Puerta, M.; Yang, L.; Valdes-Ferrer, S.I.; Patel, N.B.; Chavan, S.; Al-Abed, Y.; Yang, H.; Tracey, K.J. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol. Med. 2009, 15, 195–202. [Google Scholar] [CrossRef]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef]
- Liu, Z.; Neff, R.A.; Berg, D.K. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science 2006, 314, 1610–1613. [Google Scholar] [CrossRef]
- Sinkus, M.L.; Graw, S.; Freedman, R.; Ross, R.G.; Lester, H.A.; Leonard, S. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation, and function. Neuropharmacology 2015, 96, 274–288. [Google Scholar] [CrossRef]
- Van Zwieten, P.A.; Thoolen, M.J.; Timmermans, P.B. The hypotensive activity and side effects of methyldopa, clonidine, and guanfacine. Hypertension 1984, 6, II28–II33. [Google Scholar] [CrossRef]
- Chobanyan-Jurgens, K.; Jordan, J. Autonomic nervous system activity and inflammation: Good ideas, good treatments, or both? Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1999–H2001. [Google Scholar] [CrossRef]
- Lomasney, J.W.; Lorenz, W.; Allen, L.F.; King, K.; Regan, J.W.; Yang-Feng, T.L.; Caron, M.G.; Lefkowitz, R.J. Expansion of the alpha 2-adrenergic receptor family: Cloning and characterization of a human alpha 2-adrenergic receptor subtype, the gene for which is located on chromosome 2. Proc. Natl. Acad. Sci. USA 1990, 87, 5094–5098. [Google Scholar] [CrossRef]
- De Quervain, D.J.; Kolassa, I.T.; Ertl, V.; Onyut, P.L.; Neuner, F.; Elbert, T.; Papassotiropoulos, A. A deletion variant of the alpha2b-adrenoceptor is related to emotional memory in Europeans and Africans. Nat. Neurosci. 2007, 10, 1137–1139. [Google Scholar] [CrossRef]
- Rasch, B.; Spalek, K.; Buholzer, S.; Luechinger, R.; Boesiger, P.; Papassotiropoulos, A.; de Quervain, D.J. A genetic variation of the noradrenergic system is related to differential amygdala activation during encoding of emotional memories. Proc. Natl. Acad. Sci. USA 2009, 106, 19191–19196. [Google Scholar] [CrossRef]
- Xie, W.; Cappiello, M.; Meng, M.; Rosenthal, R.; Zhang, W. ADRA2B deletion variant and enhanced cognitive processing of emotional information: A meta-analytical review. Neurosci. Biobehav. Rev. 2018, 92, 402–416. [Google Scholar] [CrossRef]
- Small, K.M.; Brown, K.M.; Forbes, S.L.; Liggett, S.B. Polymorphic deletion of three intracellular acidic residues of the alpha 2B-adrenergic receptor decreases G protein-coupled receptor kinase-mediated phosphorylation and desensitization. J. Biol. Chem. 2001, 276, 4917–4922. [Google Scholar] [CrossRef]
- Suzuki, N.; Matsunaga, T.; Nagasumi, K.; Yamamura, T.; Shihara, N.; Moritani, T.; Ue, H.; Fukushima, M.; Tamon, A.; Seino, Y.; et al. Alpha(2B)-adrenergic receptor deletion polymorphism associates with autonomic nervous system activity in young healthy Japanese. J. Clin. Endocrinol. Metab. 2003, 88, 1184–1187. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Damier, P.; Lemonnier, E. Failure of the Nemo Trial: Bumetanide Is a Promising Agent to Treat Many Brain Disorders but Not Newborn Seizures. Front. Cell Neurosci. 2016, 10, 90. [Google Scholar] [CrossRef]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Cytokines and psychopathology: Lessons from interferon-alpha. Biol. Psychiatry 2004, 56, 819–824. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Cattaneo, A.; Gennarelli, M.; Uher, R.; Breen, G.; Farmer, A.; Aitchison, K.J.; Craig, I.W.; Anacker, C.; Zunsztain, P.A.; McGuffin, P.; et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: Differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 2013, 38, 377–385. [Google Scholar] [CrossRef]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef]
- Lanquillon, S.; Krieg, J.C.; Bening-Abu-Shach, U.; Vedder, H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology 2000, 22, 370–379. [Google Scholar] [CrossRef]
- O’Brien, S.M.; Scully, P.; Fitzgerald, P.; Scott, L.V.; Dinan, T.G. Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J. Psychiatr. Res. 2007, 41, 326–331. [Google Scholar] [CrossRef]
- Yang, J.J.; Wang, N.; Yang, C.; Shi, J.Y.; Yu, H.Y.; Hashimoto, K. Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biol. Psychiatry 2015, 77, e19–e20. [Google Scholar] [CrossRef]
- Maruyama, Y.; Kawano, A.; Okamoto, S.; Ando, T.; Ishitobi, Y.; Tanaka, Y.; Inoue, A.; Imanaga, J.; Kanehisa, M.; Higuma, H.; et al. Differences in salivary alpha-amylase and cortisol responsiveness following exposure to electrical stimulation versus the Trier Social Stress Tests. PLoS ONE 2012, 7, e39375. [Google Scholar] [CrossRef]
- Schumacher, S.; Kirschbaum, C.; Fydrich, T.; Strohle, A. Is salivary alpha-amylase an indicator of autonomic nervous system dysregulations in mental disorders?—A review of preliminary findings and the interactions with cortisol. Psychoneuroendocrinology 2013, 38, 729–743. [Google Scholar] [CrossRef]
- McGirr, A.; Diaconu, G.; Berlim, M.T.; Pruessner, J.C.; Sable, R.; Cabot, S.; Turecki, G. Dysregulation of the sympathetic nervous system, hypothalamic-pituitary-adrenal axis and executive function in individuals at risk for suicide. J. Psychiatry Neurosci. 2010, 35, 399–408. [Google Scholar] [CrossRef]
- Harwood, A.J. Lithium and bipolar mood disorder: The inositol-depletion hypothesis revisited. Mol. Psychiatry 2005, 10, 117–126. [Google Scholar] [CrossRef]
- Manji, H.K.; Bebchuk, J.M.; Moore, G.J.; Glitz, D.; Hasanat, K.A.; Chen, G. Modulation of CNS signal transduction pathways and gene expression by mood-stabilizing agents: Therapeutic implications. J. Clin. Psychiatry 1999, 60 (Suppl. 2), 27–39; 0–21, 113–116. [Google Scholar]
- Le-Niculescu, H.; Levey, D.F.; Ayalew, M.; Palmer, L.; Gavrin, L.M.; Jain, N.; Winiger, E.; Bhosrekar, S.; Shankar, G.; Radel, M.; et al. Discovery and validation of blood biomarkers for suicidality. Mol. Psychiatry 2013, 18, 1249–1264. [Google Scholar] [CrossRef]
- Kim, H.G.; Cheon, E.J.; Bai, D.S.; Lee, Y.H.; Koo, B.H. Stress and Heart Rate Variability: A Meta-Analysis and Review of the Literature. Psychiatry Investig. 2018, 15, 235–245. [Google Scholar] [CrossRef]
- Hartmann, R.; Schmidt, F.M.; Sander, C.; Hegerl, U. Heart Rate Variability as Indicator of Clinical State in Depression. Front. Psychiatry 2018, 9, 735. [Google Scholar] [CrossRef]
- Kemp, A.H.; Quintana, D.S.; Gray, M.A.; Felmingham, K.L.; Brown, K.; Gatt, J.M. Impact of depression and antidepressant treatment on heart rate variability: A review and meta-analysis. Biol. Psychiatry 2010, 67, 1067–1074. [Google Scholar] [CrossRef]
- Kemp, A.H.; Quintana, D.S.; Quinn, C.R.; Hopkinson, P.; Harris, A.W. Major depressive disorder with melancholia displays robust alterations in resting state heart rate and its variability: Implications for future morbidity and mortality. Front. Psychol. 2014, 5, 1387. [Google Scholar] [CrossRef]
- Wilson, S.T.; Chesin, M.; Fertuck, E.; Keilp, J.; Brodsky, B.; Mann, J.J.; Sonmez, C.C.; Benjamin-Phillips, C.; Stanley, B. Heart rate variability and suicidal behavior. Psychiatry Res. 2016, 240, 241–247. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Pariante, C.M.; Lightman, S.L. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci. 2008, 31, 464–468. [Google Scholar] [CrossRef]
- Thase, M.E. Using biomarkers to predict treatment response in major depressive disorder: Evidence from past and present studies. Dialogues Clin. Neurosci. 2014, 16, 539–544. [Google Scholar]
- Coryell, W.; Schlesser, M. The dexamethasone suppression test and suicide prediction. Am. J. Psychiatry 2001, 158, 748–753. [Google Scholar] [CrossRef]
- Jokinen, J.; Nordstrom, P. HPA axis hyperactivity as suicide predictor in elderly mood disorder inpatients. Psychoneuroendocrinology 2008, 33, 1387–1393. [Google Scholar] [CrossRef]
- Kamali, M.; Saunders, E.F.; Prossin, A.R.; Brucksch, C.B.; Harrington, G.J.; Langenecker, S.A.; McInnis, M.G. Associations between suicide attempts and elevated bedtime salivary cortisol levels in bipolar disorder. J. Affect. Disord 2012, 136, 350–358. [Google Scholar] [CrossRef]
- Pfennig, A.; Kunzel, H.E.; Kern, N.; Ising, M.; Majer, M.; Fuchs, B.; Ernst, G.; Holsboer, F.; Binder, E.B. Hypothalamus-pituitary-adrenal system regulation and suicidal behavior in depression. Biol. Psychiatry 2005, 57, 336–342. [Google Scholar] [CrossRef]
- Woo, J.M.; Gibbons, R.D.; Qin, P.; Komarow, H.; Kim, J.B.; Rogers, C.A.; Mann, J.J.; Postolache, T.T. Suicide and prescription rates of intranasal corticosteroids and nonsedating antihistamines for allergic rhinitis: An ecological study. J. Clin. Psychiatry 2011, 72, 1423–1428. [Google Scholar] [CrossRef]
- Kalkman, H.O. Circumstantial evidence for a role of glutamine-synthetase in suicide. Med. Hypotheses 2011, 76, 905–907. [Google Scholar] [CrossRef]
- Zou, J.; Wang, Y.X.; Dou, F.F.; Lu, H.Z.; Ma, Z.W.; Lu, P.H.; Xu, X.M. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem. Int. 2010, 56, 577–584. [Google Scholar] [CrossRef]
- Hansson, E. Regulation of glutamine synthetase synthesis and activity by glucocorticoids and adrenoceptor activation in astroglial cells. Neurochem. Res. 1989, 14, 585–587. [Google Scholar] [CrossRef]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef]
- Marcus, S.R.; Nadiger, H.A.; Chandrakala, M.V.; Rao, T.I.; Sadasivudu, B. Acute and short-term effects of lithium on glutamate metabolism in rat brain. Biochem. Pharmacol. 1986, 35, 365–369. [Google Scholar] [CrossRef]
- Ren, X.; Rizavi, H.S.; Khan, M.A.; Dwivedi, Y.; Pandey, G.N. Altered Wnt signalling in the teenage suicide brain: Focus on glycogen synthase kinase-3beta and beta-catenin. Int. J. Neuropsychopharmacol. 2013, 16, 945–955. [Google Scholar] [CrossRef]
- Baldessarini, R.J.; Tondo, L.; Hennen, J. Lithium treatment and suicide risk in major affective disorders: Update and new findings. J. Clin. Psychiatry 2003, 64 (Suppl. 5), 44–52. [Google Scholar]
- Beurel, E.; Jope, R.S. Inflammation and lithium: Clues to mechanisms contributing to suicide-linked traits. Transl. Psychiatry 2014, 4, e488. [Google Scholar] [CrossRef]
- Lamers, F.; de Jonge, P.; Nolen, W.A.; Smit, J.H.; Zitman, F.G.; Beekman, A.T.; Penninx, B.W. Identifying depressive subtypes in a large cohort study: Results from the Netherlands Study of Depression and Anxiety (NESDA). J. Clin. Psychiatry 2010, 71, 1582–1589. [Google Scholar] [CrossRef]
- Arling, T.A.; Yolken, R.H.; Lapidus, M.; Langenberg, P.; Dickerson, F.B.; Zimmerman, S.A.; Balis, T.; Cabassa, J.A.; Scrandis, D.A.; Tonelli, L.H.; et al. Toxoplasma gondii antibody titers and history of suicide attempts in patients with recurrent mood disorders. J. Nerv. Ment. Dis. 2009, 197, 905–908. [Google Scholar] [CrossRef]
- Lamers, F.; Bot, M.; Jansen, R.; Chan, M.K.; Cooper, J.D.; Bahn, S.; Penninx, B.W. Serum proteomic profiles of depressive subtypes. Transl. Psychiatry 2016, 6, e851. [Google Scholar] [CrossRef]
- Okusaga, O.; Langenberg, P.; Sleemi, A.; Vaswani, D.; Giegling, I.; Hartmann, A.M.; Konte, B.; Friedl, M.; Groer, M.W.; Yolken, R.H.; et al. Toxoplasma gondii antibody titers and history of suicide attempts in patients with schizophrenia. Schizophr. Res. 2011, 133, 150–155. [Google Scholar] [CrossRef]
- Rapaport, M.H.; Nierenberg, A.A.; Schettler, P.J.; Kinkead, B.; Cardoos, A.; Walker, R.; Mischoulon, D. Inflammation as a predictive biomarker for response to omega-3 fatty acids in major depressive disorder: A proof-of-concept study. Mol. Psychiatry 2016, 21, 71–79. [Google Scholar] [CrossRef]
- MacKenzie, E.M.; Odontiadis, J.; Le Melledo, J.M.; Prior, T.I.; Baker, G.B. The relevance of neuroactive steroids in schizophrenia, depression, and anxiety disorders. Cell Mol. Neurobiol. 2007, 27, 541–574. [Google Scholar] [CrossRef]
- Rethorst, C.D.; Bernstein, I.; Trivedi, M.H. Inflammation, obesity, and metabolic syndrome in depression: Analysis of the 2009-2010 National Health and Nutrition Examination Survey (NHANES). J. Clin. Psychiatry 2014, 75, e1428–e1432. [Google Scholar] [CrossRef]
- Capuron, L.; Lasselin, J.; Castanon, N. Role of Adiposity-Driven Inflammation in Depressive Morbidity. Neuropsychopharmacology 2017, 42, 115–128. [Google Scholar] [CrossRef]
- Perez-Cornago, A.; de la Iglesia, R.; Lopez-Legarrea, P.; Abete, I.; Navas-Carretero, S.; Lacunza, C.I.; Lahortiga, F.; Martinez-Gonzalez, M.A.; Martinez, J.A.; Zulet, M.A. A decline in inflammation is associated with less depressive symptoms after a dietary intervention in metabolic syndrome patients: A longitudinal study. Nutr. J. 2014, 13, 36. [Google Scholar] [CrossRef]
- Sublette, M.E.; Galfalvy, H.C.; Fuchs, D.; Lapidus, M.; Grunebaum, M.F.; Oquendo, M.A.; Mann, J.J.; Postolache, T.T. Plasma kynurenine levels are elevated in suicide attempters with major depressive disorder. Brain Behav. Immun. 2011, 25, 1272–1278. [Google Scholar] [CrossRef]
- Erhardt, S.; Lim, C.K.; Linderholm, K.R.; Janelidze, S.; Lindqvist, D.; Samuelsson, M.; Lundberg, K.; Postolache, T.T.; Traskman-Bendz, L.; Guillemin, G.J.; et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 2013, 38, 743–752. [Google Scholar] [CrossRef]
- Sadkowski, M.; Dennis, B.; Clayden, R.C.; Elsheikh, W.; Rangarajan, S.; Dejesus, J.; Samaan, Z. The role of the serotonergic system in suicidal behavior. Neuropsychiatr. Dis. Treat. 2013, 9, 1699–1716. [Google Scholar] [CrossRef][Green Version]
- Sandyk, R.; Awerbuch, G.I. Nocturnal melatonin secretion in multiple sclerosis patients with affective disorders. Int. J. Neurosci. 1993, 68, 227–240. [Google Scholar] [CrossRef]
- Stanley, M.; Brown, G.M. Melatonin levels are reduced in the pineal glands of suicide victims. Psychopharmacol. Bull. 1988, 24, 484–488. [Google Scholar]
- Teraishi, T.; Hori, H.; Sasayama, D.; Matsuo, J.; Ogawa, S.; Ota, M.; Hattori, K.; Kajiwara, M.; Higuchi, T.; Kunugi, H. (13)C-tryptophan breath test detects increased catabolic turnover of tryptophan along the kynurenine pathway in patients with major depressive disorder. Sci. Rep. 2015, 5, 15994. [Google Scholar] [CrossRef]
- Kraus, C.; Kadriu, B.; Lanzenberger, R.; Zarate, C.A., Jr.; Kasper, S. Prognosis and improved outcomes in major depression: A review. Transl. Psychiatry 2019, 9, 127. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Comorbidity | Citation |
|---|---|
| Viral infection (e.g., HIV*) | [36,81] |
| Bacterial infection (e.g., periodontitis) | [68,82] |
| Allergic inflammation (e.g., asthma) | [83] |
| Autoimmune disease (MS) | [84] |
| Autoimmune disease (RA) | [85,86,87] |
| Neurological disorder (Parkinson’s) | [88,89] |
| Neurological disorder (Alzheimer’s) | [90,91] |
| Cardiovascular disease (heart failure) | [92] |
| Diabetes | [93] |
| Obesity | [94] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalkman, H.O. Novel Treatment Targets Based on Insights in the Etiology of Depression: Role of IL-6 Trans-Signaling and Stress-Induced Elevation of Glutamate and ATP. Pharmaceuticals 2019, 12, 113. https://doi.org/10.3390/ph12030113
Kalkman HO. Novel Treatment Targets Based on Insights in the Etiology of Depression: Role of IL-6 Trans-Signaling and Stress-Induced Elevation of Glutamate and ATP. Pharmaceuticals. 2019; 12(3):113. https://doi.org/10.3390/ph12030113
Chicago/Turabian StyleKalkman, Hans O. 2019. "Novel Treatment Targets Based on Insights in the Etiology of Depression: Role of IL-6 Trans-Signaling and Stress-Induced Elevation of Glutamate and ATP" Pharmaceuticals 12, no. 3: 113. https://doi.org/10.3390/ph12030113
APA StyleKalkman, H. O. (2019). Novel Treatment Targets Based on Insights in the Etiology of Depression: Role of IL-6 Trans-Signaling and Stress-Induced Elevation of Glutamate and ATP. Pharmaceuticals, 12(3), 113. https://doi.org/10.3390/ph12030113