Ironing out Macrophage Immunometabolism


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
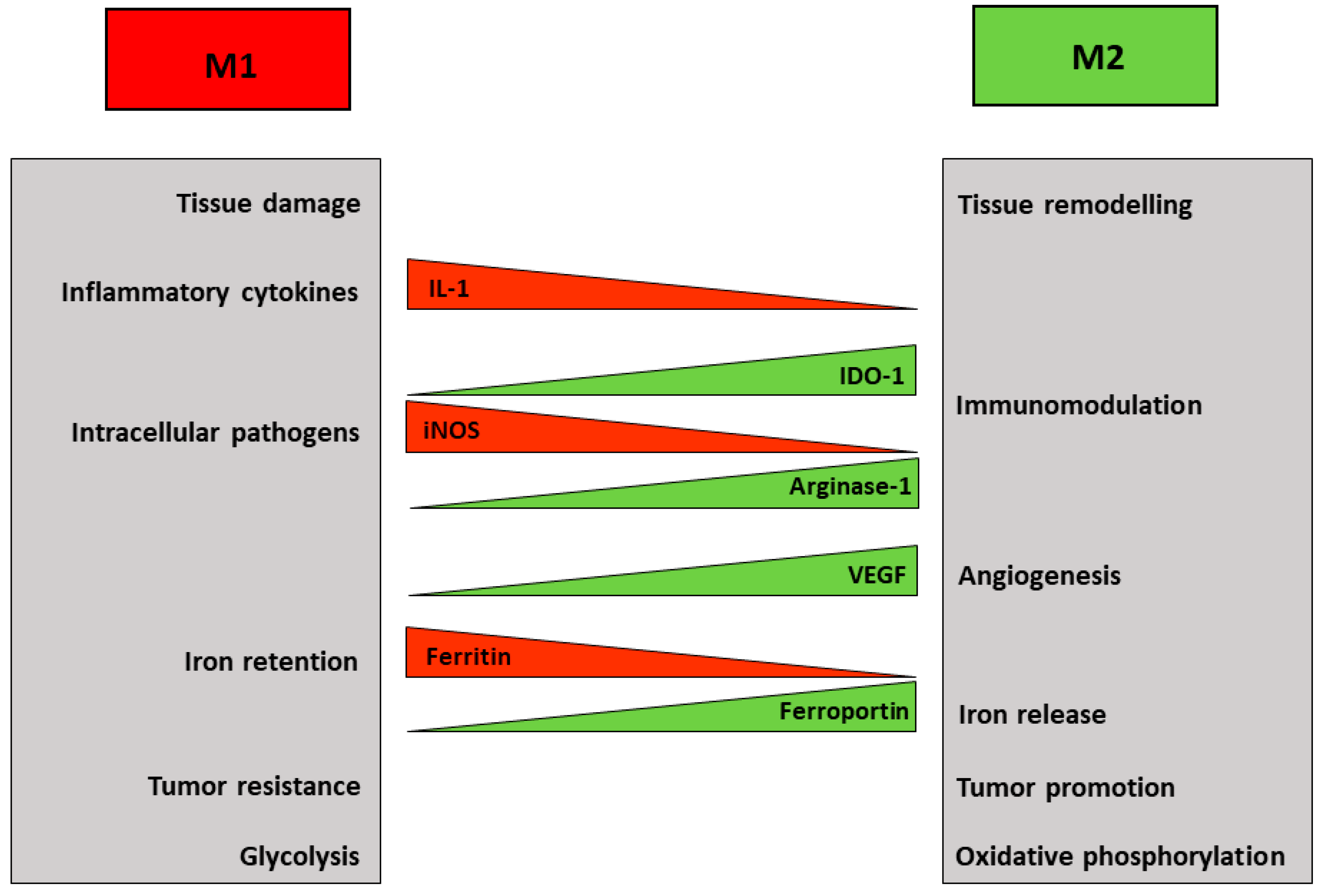
2. Macrophage Polarization
3. Macrophage Diversity and Metabolism
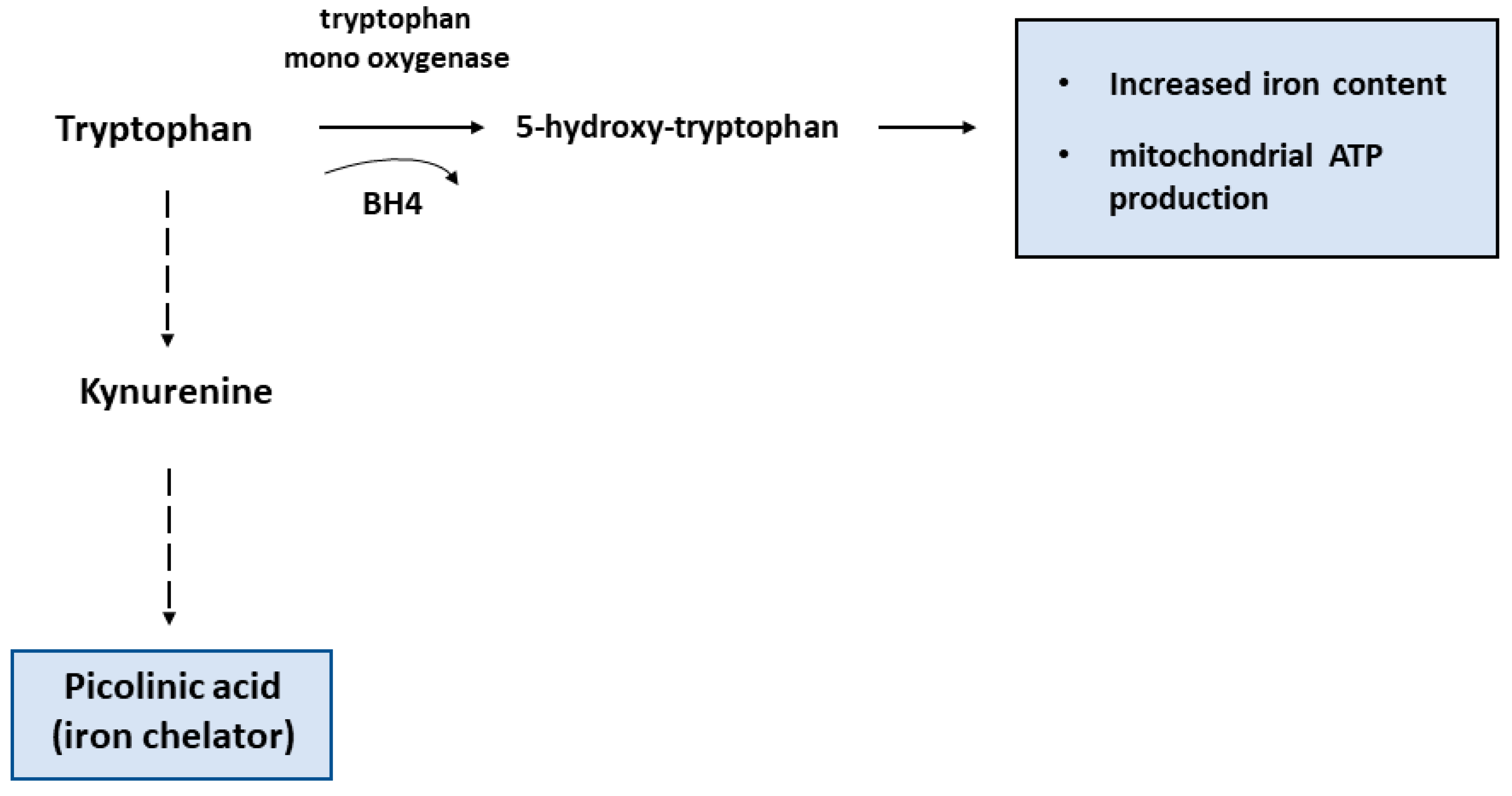
3.1. Amino Acid Metabolism
3.2. Glucose Metabolism
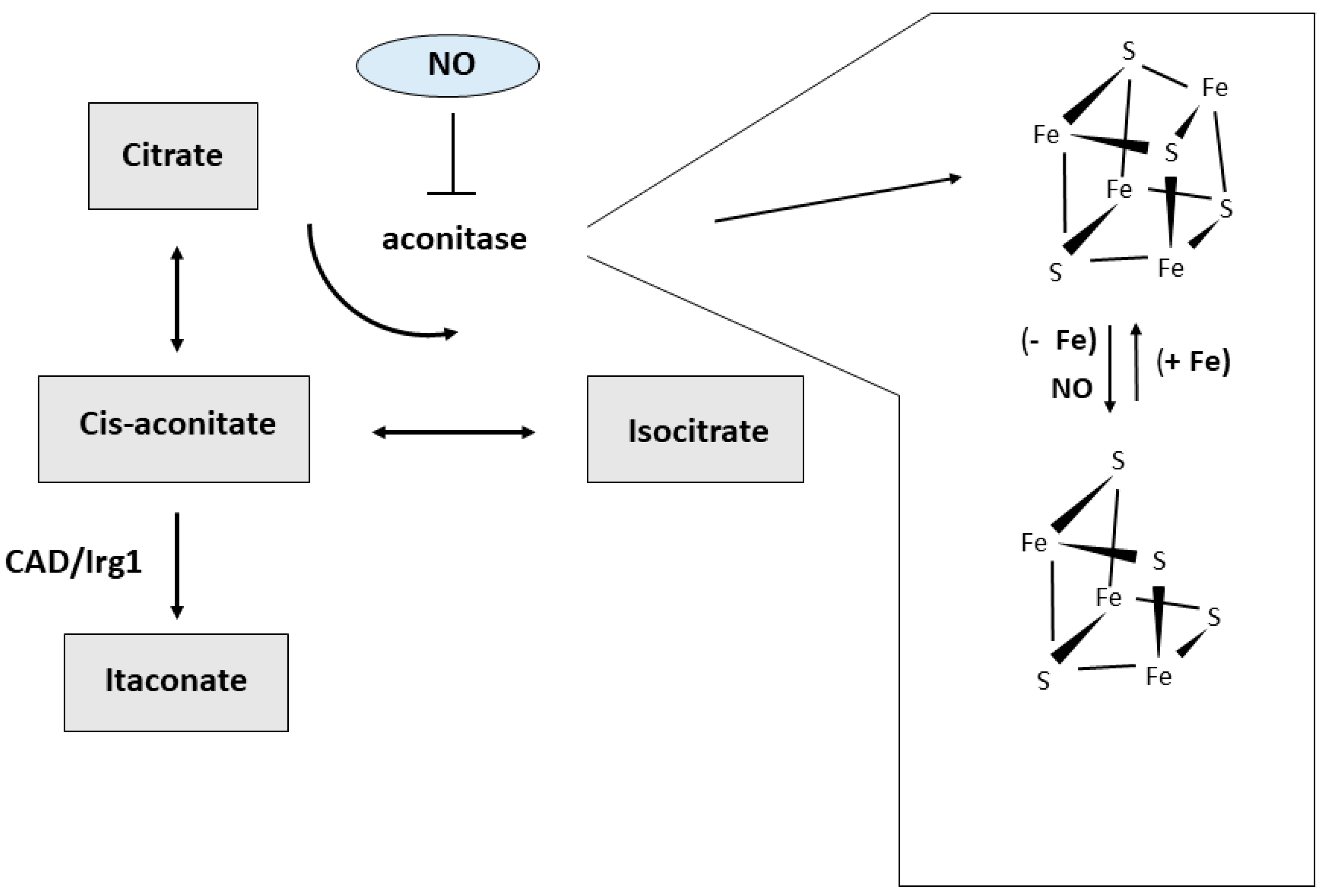
4. Macrophages and Iron Metabolism
5. Macrophage Iron and Immunometabolism
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jiang, S.; Yan, W. T-cell immunometabolism against cancer. Cancer Lett. 2016, 382, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e413. [Google Scholar] [CrossRef] [PubMed]
- El-Zaatari, M.; Kao, J.Y. Role of Dietary Metabolites in Regulating the Host Immune Response in Gastrointestinal Disease. Front. Immunol. 2017, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Rani, R.; Jordan, M.B.; Divanovic, S.; Herbert, D.R. IFN-γ-driven IDO production from macrophages protects IL-4Rα-deficient mice against lethality during Schistosoma mansoni infection. Am. J. Pathol. 2012, 180, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Memari, B.; Bouttier, M.; Dimitrov, V.; Ouellette, M.; Behr, M.A.; Fritz, J.H.; White, J.H. Engagement of the Aryl Hydrocarbon Receptor in Mycobacterium tuberculosis-Infected Macrophages Has Pleiotropic Effects on Innate Immune Signaling. J. Immunol. 2015, 195, 4479–4491. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.J.F.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L.; et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 2018, 563, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Langston, P.K.; Shibata, M.; Horng, T. Metabolism Supports Macrophage Activation. Front. Immunol. 2017, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Luan, H.H.; Medzhitov, R. Food Fight: Role of Itaconate and Other Metabolites in Antimicrobial Defense. Cell Metab. 2016, 24, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Cordes, T.; Michelucci, A.; Hiller, K. Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite. Annu. Rev. Nutr. 2015, 35, 451–473. [Google Scholar] [CrossRef] [PubMed]
- Papathanassiu, A.E.; Ko, J.H.; Imprialou, M.; Bagnati, M.; Srivastava, P.K.; Vu, H.A.; Cucchi, D.; McAdoo, S.P.; Ananieva, E.A.; Mauro, C.; et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat. Commun. 2017, 8, 16040. [Google Scholar] [CrossRef]
- Sica, A.; Strauss, L.; Consonni, F.M.; Travelli, C.; Genazzani, A.; Porta, C. Metabolic regulation of suppressive myeloid cells in cancer. Cytokine Growth Factor Rev. 2017, 35, 27–35. [Google Scholar] [CrossRef]
- Huang, S.C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef]
- Ouimet, M.; Ediriweera, H.N.; Gundra, U.M.; Sheedy, F.J.; Ramkhelawon, B.; Hutchison, S.B.; Rinehold, K.; van Solingen, C.; Fullerton, M.D.; Cecchini, K.; et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Investig. 2015, 125, 4334–4348. [Google Scholar] [CrossRef] [PubMed]
- Morioka, S.; Perry, J.S.A.; Raymond, M.H.; Medina, C.B.; Zhu, Y.; Zhao, L.; Serbulea, V.; Onengut-Gumuscu, S.; Leitinger, N.; Kucenas, S.; et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 2018, 563, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456.e445. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Sergushichev, A.; Schilling, J.D. Integrating immunometabolism and macrophage diversity. Semin. Immunol. 2016, 28, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ganta, V.C.; Choi, M.H.; Kutateladze, A.; Fox, T.E.; Farber, C.R.; Annex, B.H. A MicroRNA93-Interferon Regulatory Factor-9-Immunoresponsive Gene-1-Itaconic Acid Pathway Modulates M2-Like Macrophage Polarization to Revascularize Ischemic Muscle. Circulation 2017, 135, 2403–2425. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Cairo, G. Systemic and cellular consequences of macrophage control of iron metabolism. Semin. Immunol. 2012, 24, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Hamza, I. Macrophages and Iron Metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Zhou, Y.; Que, K.T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.P.; Liu, Z.J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018, 7, 4012–4022. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ye, S.; Fujiwara, T.; Manolagas, S.C.; Zhao, H. Steap4 plays a critical role in osteoclastogenesis in vitro by regulating cellular iron/reactive oxygen species (ROS) levels and cAMP response element-binding protein (CREB) activation. J. Biol. Chem. 2013, 288, 30064–30074. [Google Scholar] [CrossRef]
- Alkhateeb, A.A.; Han, B.; Connor, J.R. Ferritin stimulates breast cancer cells through an iron-independent mechanism and is localized within tumor-associated macrophages. Breast Cancer Res. Treat. 2013, 137, 733–744. [Google Scholar] [CrossRef]
- Recalcati, S.; Gammella, E.; Buratti, P.; Doni, A.; Anselmo, A.; Locati, M.; Cairo, G. Macrophage ferroportin is essential for stromal cell proliferation in wound healing. Haematologica 2019, 104, 47–58. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Passos Dos Santos, R.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S. Iron-regulatory proteins: molecular biology and pathophysiological implications. Expert Rev. Mol. Med. 2007, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H.; Kennedy, M.C. Aconitase, a two-faced protein: enzyme and iron regulatory factor. FASEB J. 1993, 7, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Ronchi, R.; Recalcati, S.; Campanella, A.; Minotti, G. Nitric oxide and peroxynitrite activate the iron regulatory protein-1 of J774A.1 macrophages by direct disassembly of the Fe-S cluster of cytoplasmic aconitase. Biochemistry 2002, 41, 7435–7442. [Google Scholar] [CrossRef]
- Tong, W.H.; Maio, N.; Zhang, D.L.; Palmieri, E.M.; Ollivierre, H.; Ghosh, M.C.; McVicar, D.W.; Rouault, T.A. TLR-activated repression of Fe-S cluster biogenesis drives a metabolic shift and alters histone and tubulin acetylation. Blood Adv. 2018, 2, 1146–1156. [Google Scholar] [CrossRef]
- Pastorino, S.; Carta, L.; Puppo, M.; Melillo, G.; Bosco, M.C.; Varesio, L. Picolinic acid- or desferrioxamine-inducible autocrine activation of macrophages engineered to produce IFNgamma: An approach for gene therapy. Gene Ther. 2004, 11, 560–568. [Google Scholar] [CrossRef]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef]
- Prodinger, J.; Loacker, L.J.; Schmidt, R.L.; Ratzinger, F.; Greiner, G.; Witzeneder, N.; Hoermann, G.; Jutz, S.; Pickl, W.F.; Steinberger, P.; et al. The tryptophan metabolite picolinic acid suppresses proliferation and metabolic activity of CD4+ T cells and inhibits c-Myc activation. J. Leukoc. Biol. 2016, 99, 583–594. [Google Scholar] [CrossRef]
- Melillo, G.; Cox, G.W.; Biragyn, A.; Sheffler, L.A.; Varesio, L. Regulation of nitric-oxide synthase mRNA expression by interferon-gamma and picolinic acid. J. Biol. Chem. 1994, 269, 8128–8133. [Google Scholar]
- Recalcati, S.; Gammella, E.; Cairo, G. New perspectives on the molecular basis of the interaction between oxygen homeostasis and iron metabolism. Hypoxia 2015, 2015, 93–103. [Google Scholar]
- Buqué, A.; Bloy, N.; Aranda, F.; Cremer, I.; Eggermont, A.; Fridman, W.H.; Fucikova, J.; Galon, J.; Spisek, R.; Tartour, E.; et al. Trial Watch-Small molecules targeting the immunological tumor microenvironment for cancer therapy. Oncoimmunology 2016, 5, e1149674. [Google Scholar] [CrossRef] [PubMed]
- Bayeva, M.; Khechaduri, A.; Puig, S.; Chang, H.C.; Patial, S.; Blackshear, P.J.; Ardehali, H. mTOR regulates cellular iron homeostasis through tristetraprolin. Cell Metab. 2012, 16, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Orr, J.S.; Kennedy, A.; Anderson-Baucum, E.K.; Webb, C.D.; Fordahl, S.C.; Erikson, K.M.; Zhang, Y.; Etzerodt, A.; Moestrup, S.K.; Hasty, A.H. Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes 2014, 63, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Sakamoto, A.; Cornelissen, A.; Hong, C.C.; Finn, A.V. Ironing-Out the Role of Hepcidin in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 303–305. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Recalcati, S.; Gammella, E.; Cairo, G. Ironing out Macrophage Immunometabolism. Pharmaceuticals 2019, 12, 94. https://doi.org/10.3390/ph12020094
Recalcati S, Gammella E, Cairo G. Ironing out Macrophage Immunometabolism. Pharmaceuticals. 2019; 12(2):94. https://doi.org/10.3390/ph12020094
Chicago/Turabian StyleRecalcati, Stefania, Elena Gammella, and Gaetano Cairo. 2019. "Ironing out Macrophage Immunometabolism" Pharmaceuticals 12, no. 2: 94. https://doi.org/10.3390/ph12020094
APA StyleRecalcati, S., Gammella, E., & Cairo, G. (2019). Ironing out Macrophage Immunometabolism. Pharmaceuticals, 12(2), 94. https://doi.org/10.3390/ph12020094