Therapeutic Potential of Kappa Opioid Agonists


Abstract

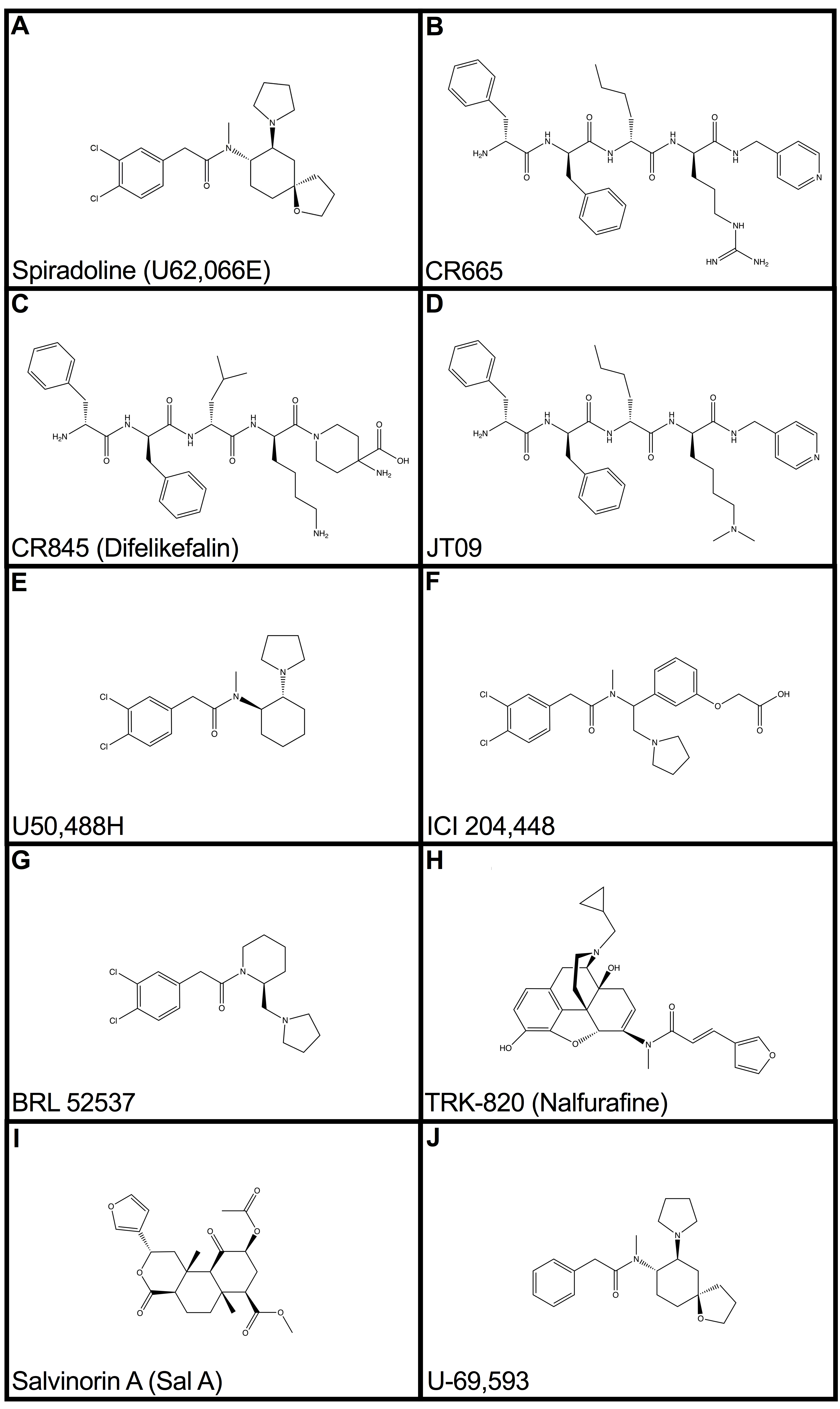{kind=link}
{kind=link}
1. Introduction
2. Indications
2.1. Chronic Pain
2.2. Myocardial Infarction
2.3. Anti-Pruritic
2.4. Anti-Inflammatory and Anti-Edema
2.5. HIV-Induced Neuroinflammation
2.6. Anti-Emetic
2.7. Spinal Anesthesia
2.8. Stroke and Neuroprotection
2.9. Hypoxic Pulmonary Hypertension
2.10. Multiple Sclerosis
2.11. Addiction
2.12. Post-Traumatic Cartilage Regeneration
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Annual Surveillance Report of Drug-Related Risks and Outcomes—United States, 2017. Surveillance Special Report: Centers for Disease Control and Prevention, U.S. Department of Health and Human Services. Available online: https://www.cdc.gov/drugoverdose/pdf/pubs/2017-cdc-drug-surveillance-report.pdf (accessed on 22 February 2019).
- Opioid Overdose. Available online: https://www.cdc.gov/drugoverdose/data/prescribing.html (accessed on 19 February 2019).
- Bree, D. The Kappa Opioid Receptor Takes Shape. Available online: https://www.painresearchforum.org/news/93114-kappa-opioid-receptor-takes-shape (accessed on 19 February 2019).
- DeHaven-Hudkins, D.L.; Dolle, R.E. Peripheral restricted opioid agonists and novel analgesic agents. Curr. Pharm. Des. 2004, 10, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Opioid Overdose: Understanding the Epidemic. Available online: https://www.cdc.gov/drugoverdose/epidemic/index.html (accessed on 19 February 2019).
- Beck, T.C.; Reichel, C.M.; Helke, K.L.; Bhadsavle, S.S.; Dix, T.A. Non-addictive orally-active kappa opioid agonist for the treatment of peripheral pain. Eur. J. Pharmacol. 2019, 856, 172396. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.C.; Clark, D.J.; Oh, U.; Vasko, M.R.; Wilcox, G.L.; Overland, A.C.; Vanderah, T.W.; Spencer, R.H. Peripheral mechanisms of pain and analgesia. Brain Res. Rev. 2008, 60, 90. [Google Scholar] [CrossRef] [PubMed]
- Rech, R.H.; Mokler, D.J.; Briggs, S.L. Effects of Combined Opioids on Pain and Mood in Mammals. Pain Res. Treat. 2012, 2012, 145965. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Arico, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, P. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. [Google Scholar] [CrossRef]
- Hughes, F.M.; Shaner, B.E.; Brower, J.O.; Woods, R.J.; Dix, T.A. Development of a Peptide-derived orally-active kappa-opioid receptor agonist targeting peripheral pain. Open Med. Chem. J. 2013, 7, 16–22. [Google Scholar] [CrossRef]
- Pharmaceutical Product Development for Pruritus & Pain. Available online: https://www.caratherapeutics.com/ (accessed on 22 February 2019).
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics—2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Turpie, A.G. Burden of Disease: Medical and Economic Impact of Acute Coronary Syndromes. Am. J. Manag. Care 2006, 12, S430–434. [Google Scholar]
- Zafari, M. Myocardial Infarction Treatment & Management. Available online: https://emedicine.medscape.com/article/155919-treatment (accessed on 10 March 2019).
- Gerber, Y.; Weston, S.A.; Enriquez-Sarano, M.; Manemann, S.M.; Chamberlain, A.M.; Jiang, R.; Roger, V.L. Atherosclerotic Burden and Heart Failure After Myocardial Infarction. JAMA Cardiol. 2016, 1, 152–162. [Google Scholar] [CrossRef]
- Sobanski, P.; Krajnik, M.; Shaqura, M.; Bloch-Boguslawska, E.; Schäfer, M.; Mousa, S.A. The presence of mu-, delta- and kappa-opioid receptors in human heart tissue. Heart Vessels 2014, 29, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kersten, J.R.; Riess, M.L. Opioid-Induced Cardioprotection. Curr. Pharm. Des. 2014, 20, 5696–5705. [Google Scholar] [CrossRef] [PubMed]
- Peart, J.N.; Gross, E.R.; Gross, G.J. Effect of Exogenous Kappa-Opioid Receptor Activation in Rat Model of Myocardial Infarction. J. Cardiovasc. Pharmacol. 2004, 43, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.; Zhang, B.; Zhou, X.; Zhao, J.; Sun, Z.; Tao, Y.; Pei, J.; Zhang, W. Kappa-Opioid Agonist U50,488-Mediated Protection Against Heart Failure Following Ischemia/Reperfusion: Dual Roles of Heme Oxygenase. Cell. Physiol. Biochem. 2016, 39, 2158–2172. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Wang, H.; Li, J.; Wang, Q.; Zhang, S.; Feng, N.; Fan, R.; Pei, J. κ-Opioid Receptor Stimulation Modulates TLR4/NF-κB signaling in the Rat Heart Subjected to Ischemia-Reperfusion. Cytokine 2013, 61, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, W.; Bian, J.; Wong, T.M. Pro- and Anti-Arrhythmic Effects of a κ-Opioid Receptor Agonist: A Model for the Biphasic Action of a Local Hormone in the Heart. Clin. Exp. Pharmacol. Physiol. 1999, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Wadenberg, M.L. A review of the properties of spiradoline: A potent and selective kappa-opioid receptor agonist. CNS Drug Rev. 2003, 9, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Reeder, G.S.; Kennedy, H.L. Acute myocardial infarction: Role of beta blocker therapy. UpToDate 2019. Available online: https://www.uptodate.com/contents/acute-myocardial-infarction-role-of-beta-blocker-therapy (accessed on 10 June 2019).
- Tivoli, Y.A.; Rubenstein, R.M. Pruritus: An Updated Look on an Outdated Problem. J. Clin. Aesthet. Dermatol. 2009, 2, 30–36. [Google Scholar]
- Bigliardi, P.L.; Stammer, H.; Jost, G.; Rufli, T.; Büchner, S.; Bigliardi-Qi, M. Treatment of Pruritus with Topically Applied Opiate Receptor Anatagonist. J. Am. Acad. Dermatol. 2007, 56, 979–988. [Google Scholar] [CrossRef]
- Burden of Disease: Chronic Inflammation and Inflammatory Disease. Available online: https://www.pfizer.com/sites/default/files/health/VOM_Chronic_Inflammation_and_Inflammatory_Diseases.pdf (accessed on 11 April 2019).
- Walker, J.S. Anti-Inflammatory Effects of Opioids. Adv. Exp. Med. Biol. 2003, 521, 148–160. [Google Scholar] [PubMed]
- Chavez-Valdez, R.; Kovell, L.; Ahlawat, R.; McLemore, G.L.; Wills-Karp, M.; Gauda, E.B. Opioids and clonidine modulate cytokine production and opioid receptor expression in neonatal immune cells. J. Perinatol. 2013, 33, 374–382. [Google Scholar] [CrossRef]
- Zhang, L.; Rogers, T.J. κ-Opioid Regulation of Thymocyte IL-7 Receptor and C-C Chemokine Receptor 2 Expression. J. Immunol. 2000, 164, 5088–5093. [Google Scholar] [CrossRef] [PubMed]
- Flaishon, L.; Hart, G.; Zelman, E.; Moussion, C.; Grabovsky, V.; Lapidot Tal, G.; Feigelson, S.; Margalit, R.; Harmelin, A.; Avin-Wittenberg, T.; et al. Anti-inflammatory effects of an inflammatory chemokine: CCL2 inhibits lymphocyte homing by modulation of CCL21-triggered integrin-mediated adhesions. Blood 2008, 112, 5016–5025. [Google Scholar] [CrossRef] [PubMed]
- Joris, J.; Costello, A.; Dubner, R.; Hargreaves, K.M. Opiates suppress carrageenan-induced edema and hyperthermia at doses that inhibit hyperalgesia. Pain 1990, 43, 95–103. [Google Scholar] [CrossRef]
- Nash, B.; Meucci, O. Functions of the Chemokine Receptor CXCR4 in the Central Nervous System and Its Regulation by u-Opioid Receptors. Neuroimmune Signal. Drug Actions Addict. Int. Rev. Neurobiol. 2014, 118, 105–128. [Google Scholar] [CrossRef]
- Finley, M.J.; Happel, C.M.; Kaminsky, D.E.; Rogers, T.J. Opioid and Nociceptin Receptors Regulate Cytokine and Cytokine Receptor Expression. Cell. Immunol. 2008, 252, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Hasler, W.L.; Chey, W.D. Nausea and vomiting. Gastroenterology 2003, 125, 1860–1867. [Google Scholar] [CrossRef]
- Blancquaert, J.-P.; Lefebvre, R.A.; Willems, J.L. Emetic and antiemetic effects of opioids in the dog. Eur. J. Pharmacol. 1986, 128, 143–150. [Google Scholar] [CrossRef]
- Johnston, K.D. The potential for mu-opioid receptor agonists to be anti-emetic in humans: A review of clinical data. Acta Anaesthesiol. Scand. 2010, 54, 132–140. [Google Scholar] [CrossRef]
- Porreca, F.; Ossipov, M.H. Nausea and Vomiting Side Effects with Opioid Analgesics during Treatment of Chronic Pain: Mechanisms, Implications, and Management Options. Pain Med. 2009, 10, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.D.; Eriksson, L.I.; Fleisher, L.A.; Wiener-Kronish, J.P.; Young, W.L. Miller’s Anesthesia, 7th ed.; Churchill Livingstone: Philadelphia, PA, USA, 2009. [Google Scholar] [CrossRef]
- Kuusniemi, K.S.; Pihlajamäki, K.K.; Pitkänen, M.T.; Helenius, H.Y.; Kirvelä, O.A. The Use of Bupivicaine and Fentanyl for Spinal Anesthesia for Urologic Surgery. Anesth. Analg. 2000, 91, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Singh, S.I. Neuraxial Opioid-Induced Pruritus: An Update. J. Anaesthesiol. Clin. Pharmacol. 2013, 29, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Bumgarner, G.A.; Kaye, A.D. Species Variaion in effects of Intrathecal K-Opioid Receptor Agonist on Morphine-Induced Itch and Antinociception. Reg. Anesth. Pain Med. 2016, 41, 414. [Google Scholar] [CrossRef] [PubMed]
- Gouardères, C.; Kopp, N.; Cros, J.; Quirion, R. Kappa opioid receptors in human lumbo-sacral spinal cord. Brain Res. Bull. 1986, 16, 355–361. [Google Scholar] [CrossRef]
- Harris, J.A.; Chang, P.C.; Drake, C.T. Kappa opioid receptors in rat spinal cord: Sex-linked distribution differences. Neuroscience 2004, 124, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Castillo, R.; Kissin, I.; Bradley, E.L. Selective kappa opioid agonist for spinal analgesia without the risk of respiratory depression. Anesth. Analg. 1986, 65, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Dosaka-Akita, K.; Tortella, F.C.; Holaday, J.W.; Long, J.B. The Kappa Opioid Agonist U-50,488H Antagonizes Respiratory Effects of Mu Opioid Receptor Agonists in Conscious Rats. J. Pharmacol. Exp. Ther. 1993, 264, 631–637. [Google Scholar]
- The Internet Stroke Center. Available online: http://www.strokecenter.org/patients/about-stroke/stroke-statistics/ (accessed on 7 April 2019).
- Stroke Treatment. Available online: http://www.strokeassociation.org/STROKEORG/AboutStroke/Treatment/Stroke-Treatment_UCM_492017_SubHomePage.jsp (accessed on 7 April 2019).
- Goyagi, T.; Toung, T.J.; Kirsch, J.R.; Traystman, R.J.; Koehler, R.C.; Hurn, P.D.; Bhardwaj, A. Neuroprotective kappa-opioid receptor agonist BRL 52537 attenuates ischemia-evoked nitric oxide production in vivo in rats. Stroke 2003, 34, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Silvia, R.C.; Slizgi, G.R.; Luden, J.H.; Tang, A.H. Protection from ischemia-induced cerebral edema in the rat by U-50488H, a kappa opioid receptor agonist. Brain Res. 1987, 403, 52–57. [Google Scholar] [CrossRef]
- Chen, T.Y.; Goyagi, T.; Toung, T.J.; Kirsch, J.R.; Hurn, P.D.; Koehler, R.C.; Bhardwaj, A. Prolonged opportunity for ischemic neuroprotection with selective kappa-opioid receptor agonist in rats. Stroke 2004, 35, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Shah, K.; Karamyan, V.T.; Abbruscato, T.J. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011, 1383, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tian, X.; Wang, W.; Wang, Y.; Fan, R.; Wang, Y.; Feng, N.; Zhang, S.; Guo, H.; Gu, X.; et al. Quaternary ammonium salt of U50,488H elicits protective effects against hypoxic pulmonary hypertension. Eur. J. Pharmacol. 2018, 832, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Multiple Sclerosis FAQs. Available online: https://www.nationalmssociety.org/What-is-MS/MS-FAQ-s - question-What-is-multiple-sclerosis (accessed on 10 May 2019).
- MS Facts and Data. Available online: https://multiplesclerosis.net/what-is-ms/statistics/ (accessed on 10 May 2019).
- Mei, F.; Mayoral, S.R.; Nobuta, H.; Wang, F.; Desponts, C.; Lorrain, D.S.; Xiao, L.; Green, A.J.; Rowitch, D.; Whistler, J.; et al. Identification of the Kappa-Opioid Receptor as a Therapeutic Target for Oligodendrocyte Remyelination. J. Neurosci. 2016, 36, 7925–7935. [Google Scholar] [CrossRef] [PubMed]
- Treatment Approaches for Drug Addiction: National Institute on Drug Abuse. Available online: https://www.drugabuse.gov/publications/drugfacts/treatment-approaches-drug-addiction (accessed on 11 May 2019).
- Maisonneuve, I.M.; Archer, S.; Glick, S.D. U50,488, a κ opioid receptor agonist, attenuates cocaine-induced increases in extracellular dopamine in the nucleus accumbens of rats. Neurosci. Lett. 1994, 181, 57–60. [Google Scholar] [CrossRef]
- Svingos, M.L.; Chavkin, C.; Colago, E.E.; Pickel, V.M. Major co-expression of kappa-opioid receptors and the dopamine transporter in nucleus accumbens axonal profiles. Synapse 2001, 42, 185–192. [Google Scholar] [CrossRef]
- Thompson, A.C.; Zapata, A.; Justice, J.B.; Vaughan, R.A.; Sharpe, L.G.; Shippenberg, T.S. Kappa-opioid receptor activation modifies dopamine uptake in the nucleus accumbens and opposes the effects of cocaine. J. Neurosci. 2000, 20, 9333–9340. [Google Scholar] [CrossRef] [PubMed]
- Kivell, B.M.; Ewald, A.W.M.; Prisinzano, T.E. Salvinorin A analogs and other kappa opioid receptor compounds as treatments for cocaine abuse. Adv. Pharmacol. 2015, 69, 481–511. [Google Scholar] [CrossRef]
- Osteoarthritis. Available online: https://www.mayoclinic.org/diseases-conditions/osteoarthritis/symptoms-causes/syc-20351925 (accessed on 13 May 2019).
- Wu, L.; Zhang, S.; Shkhyan, R.; Lee, S.; Gullo, F.; Eliasberg, C.D.; Petrigliano, F.A.; Ba, K.; Wang, J.; Lin, Y.; et al. Kappa opioid receptor signaling protects cartilage tissue against posttraumatic degeneration. JCI Insight 2017, 2, e88553. [Google Scholar] [CrossRef] [PubMed]
- Binder, W.; Machelska, H.; Mousa, S.; Schmitt, T.; Rivière, P.J.; Junien, J.L.; Stein, C.; Schäfer, M. Analgesic and Antiinflammatory Effects of Two Novel κ-Opioid Peptides. Anesthesiology 2001, 94, 1034–1044. [Google Scholar] [CrossRef]
- Sobczak, M.; Salaga, M.; Storr, M.A.; Fichna, J. Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: Current concepts and future perspectives. J. Gastroenterol. 2014, 49, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Barron, B.A. Opioid peptides and the heart. Cardiovasc. Res. 1999, 43, 13–16. [Google Scholar] [CrossRef]
- Bolte, C.; Newman, G.; Shultz, J.E.J. Kappa and delta opioid receptor signaling is augmented in the failing heart. J. Mol. Cell. Cardiol. 2009, 47, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Hileman, C.O.; Funderburg, N.T. Inflammation, Immune Activation, and Antiretroviral Therapy in HIV. Curr. HIV/AIDS Rep. 2017, 14, 93–100. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beck, T.C.; Hapstack, M.A.; Beck, K.R.; Dix, T.A. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals 2019, 12, 95. https://doi.org/10.3390/ph12020095
Beck TC, Hapstack MA, Beck KR, Dix TA. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals. 2019; 12(2):95. https://doi.org/10.3390/ph12020095
Chicago/Turabian StyleBeck, Tyler C., Matthew A. Hapstack, Kyle R. Beck, and Thomas A. Dix. 2019. "Therapeutic Potential of Kappa Opioid Agonists" Pharmaceuticals 12, no. 2: 95. https://doi.org/10.3390/ph12020095
APA StyleBeck, T. C., Hapstack, M. A., Beck, K. R., & Dix, T. A. (2019). Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals, 12(2), 95. https://doi.org/10.3390/ph12020095