Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells



Abstract
1. Introduction
2. Results and Discussion
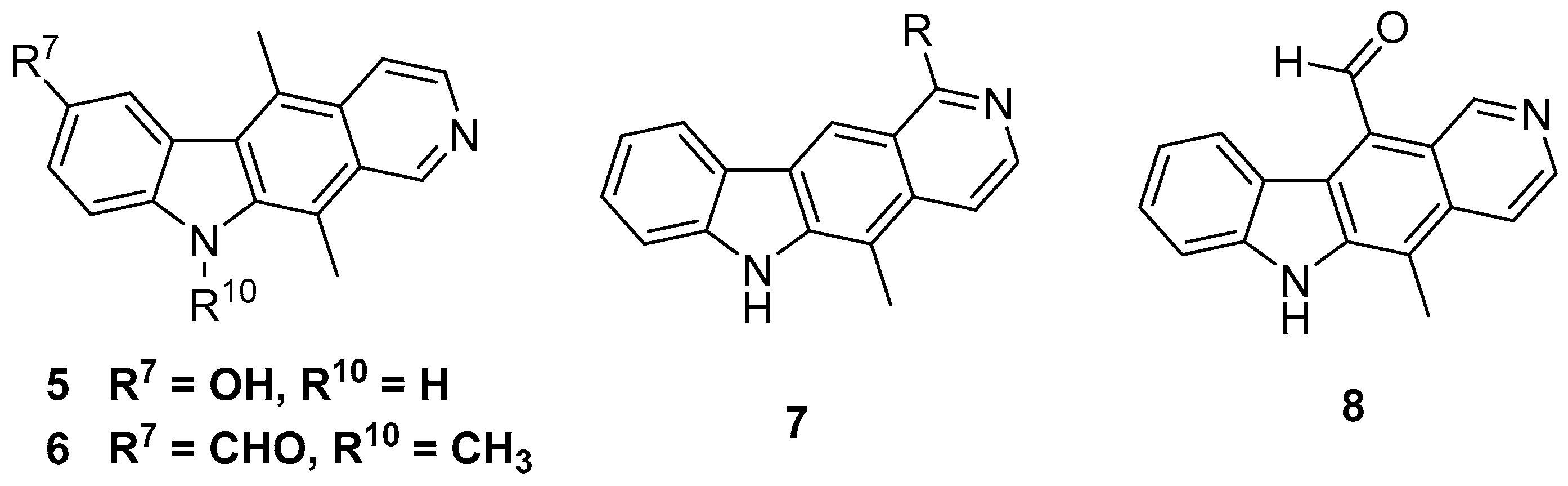
2.1. Synthesis of 11-Substituted Ellipticine Derivatives
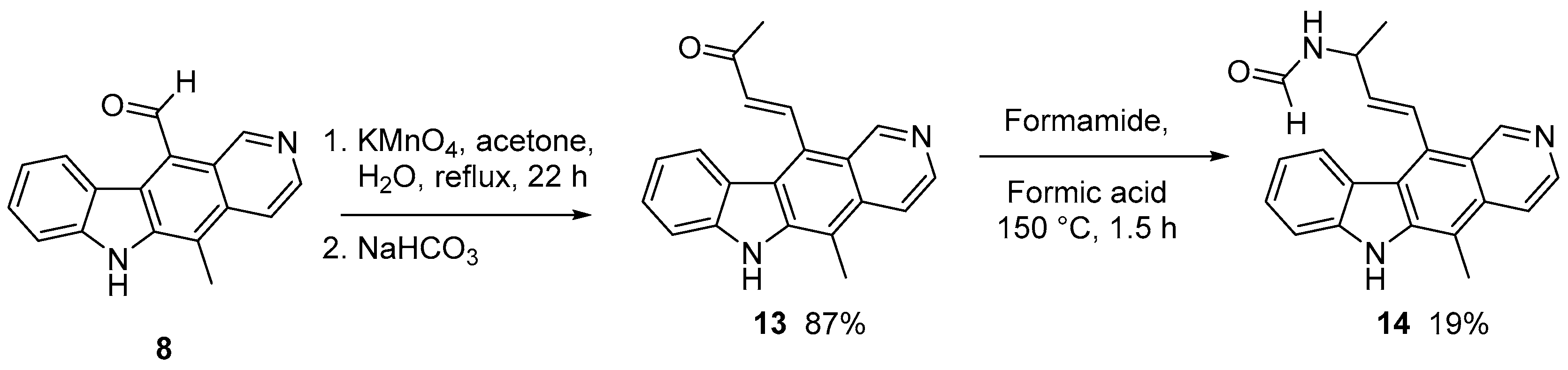
2.1.1. Modification of 11-formylellipticine
2.1.2. Modification of the 9-position of 11-substituted Ellipticines
2.2. Biological Evaluation of 11-Substituted Ellipticines
2.2.1. Inhibition of Topoisomerase II
2.2.2. Inhibition of Cancer Cell Growth
Inhibition of Cancer Cell Growth—One Dose Assay
Inhibition of Cancer Cell Growth—Five Dose Assay
2.3. COMPARE Analysis of Compounds 11 and 13
3. Materials and Methods
- [(Ti−Tz)/(C−Tz)] × 100 for concentrations for which Ti ≥ Tz
- [(Ti−Tz)/Tz] × 100 for concentrations for which Ti < Tz.
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein Kinase B |
| CK2 | Casein Kinase 2 |
| GI50 | Growth Inhibition 50% |
| LC50 | Lethal concentration 50% |
| NCI | National Cancer Institute |
| NSC | numeric identifier for substances submitted to the National Cancer Institute (NCI) |
| Topo | Topoisomerase |
| SAR | Structure Activity Relationship |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; Smith, A.F.; Horning, E.C. Alkaloids of Ochrosia elliptica Labill. J. Am. Chem. Soc. 1959, 81, 1903–1908. [Google Scholar] [CrossRef]
- Miller, C.M.; McCarthy, F.O. Isolation, biological activity and synthesis of the natural product ellipticine and related pyridocarbazoles. R. Soc. Chem. Adv. 2012, 2, 8883–8918. [Google Scholar] [CrossRef]
- O’Sullivan, E.C.; Miller, C.M.; Deane, F.M.; McCarthy, F.O. Emerging Targets in the Bioactivity of Ellipticines and Derivatives. Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2013; pp. 189–232. [Google Scholar]
- Paoletti, C.; Le Pecq, J.B.; Dat-Xuong, N.; Juret, P.; Garnier, H.; Amiel, J.L.; Rouesse, J. Antitumor activity, pharmacology, and toxicity of ellipticines, ellipticinium, and 9-hydroxy derivatives: preliminary clinical trials of 2-methyl-9-hydroxy ellipticinium (NSC 264-137). Recent Results Cancer Res. 1980, 74, 107–123. [Google Scholar] [PubMed]
- Ohashi, M.; Oki, T. Ellipticine and related anticancer agents. Expert Opin. Ther. Pat. 1996, 6, 1285–1294. [Google Scholar] [CrossRef]
- Rouesse, J.; Spielmann, M.; Turpin, F.; Le Chevalier, T.; Azab, M.; Mondesir, J.M. Phase II study of elliptinium acetate salvage treatment of advanced breast cancer. Eur. J. Cancer 1993, 6, 856–859. [Google Scholar] [CrossRef]
- Monnot, M.; Mauffret, O.; Simon, V.; Lescot, E.; Psaume, B.; Saucier, J.M.; Charra, M.; Belehradek, J., Jr.; Fermandjian, S. DNA-drug recognition and effects on topoisomerase II-mediated cytotoxicity. A three-mode binding model for ellipticine derivatives. J. Biol. Chem. 1991, 266, 1820–1829. [Google Scholar]
- Froelich-Ammon, S.J.; Patchan, M.W.; Osheroff, N.; Thompson, R.B. Topoisomerase II binds to ellipticine in the absence or presence of DNA. Characterization of enzyme-drug interactions by fluorescence spectroscopy. J. Biol. Chem. 1995, 270, 14998–15004. [Google Scholar] [CrossRef]
- Fossé, P.; René, B.; Charra, M.; Paoletti, C.; Saucier, J.M. Stimulation of topoisomerase II-mediated DNA cleavage by ellipticine derivatives: structure-activity relationship. Mol. Pharmacol. 1992, 42, 590–595. [Google Scholar]
- Poljakova, J.; Eckschlager, T.; Hrabeta, J.; Hrebackova, J.; Smutny, S.; Frei, E.; Martinek, V.; Kizek, R.; Stiborova, M. The mechanism of cytotoxicity and DNA adduct formation by the anticancer drug ellipticine in human neuroblastoma cells. Biochem. Pharmacol. 2009, 77, 1466–1479. [Google Scholar] [CrossRef]
- Vendôme, J.; Letard, S.; Martin, F.; Svinarchuk, F.; Dubreuil, P.; Auclair, C.; Le Bret, M. Molecular modelling of wild-type and D816V c-kit inhibition based on ATP-competitive binding of ellipticine derivatives to tyrosine kinases. J. Med. Chem. 2005, 48, 6194–6201. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Vassal-Stermann, E.; Nguyen, C.-H.; Pillet, C.; Martinez, A.; Prunier, C.; Barette, C.; Soleilhac, E.; Filhol, O.; Beghin, A. Pharmacological inhibition of LIM kinase stabilizes microtubules and inhibits neoplastic growth. Cancer Res. 2012, 72, 4429–4439. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wang, W.; El-Deiry, W.S. Non-genotoxic anti-neoplastic effects of ellipticine derivative NSC176327 in p53-deficient human colon carcinoma cells involve stimulation of p73. Cancer Biol. Ther. 2008, 7, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Andrews, W.J.; Panova, T.; Normand, C.; Gadal, O.; Tikhonova, I.G.; Panov, K.I. Old drug, new target: ellipticines selectively inhibit RNA polymerase I transcription. J. Biol. Chem. 2013, 288, 4567–4582. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.V.; Wang, T.; Chappeta, V.R.; Wu, G.; Onel, B.; Chawla, R.; Quijada, H.; Camp, S.M.; Chiang, E.T.; Lassiter, Q.R.; et al. The Consequences of Overlapping G-Quadruplexes and i-Motifs in the Platelet-Derived Growth Factor Receptor β Core Promoter Nuclease Hypersensitive Element Can Explain the Unexpected Effects of Mutations and Provide Opportunities for Selective Targeting of Both Structures by Small Molecules To Downregulate Gene Expression. J. Am. Chem. Soc. 2017, 139, 7456–7475. [Google Scholar] [PubMed]
- Miller, C.M.; O’Sullivan, E.C.; Devine, K.J.; McCarthy, F.O. Synthesis and biological evaluation of novel isoellipticine derivatives and salts. Org. Biomol. Chem. 2012, 10, 7912–7921. [Google Scholar] [CrossRef]
- Deane, F.M.; O’Sullivan, E.C.; Maguire, A.R.; Gilbert, J.; Sakoff, J.A.; McCluskey, A.; McCarthy, F.O. Synthesis and evaluation of novel ellipticines as potential anti-cancer agents. Org. Biomol. Chem. 2013, 11, 1334–1344. [Google Scholar] [CrossRef]
- Russell, E.G.; O’Sullivan, E.C.; Miller, C.M.; Stanicka, J.; McCarthy, F.O.; Cotter, T.G. Ellipticine derivative induces potent cytostatic effect in acute myeloid leukaemia cells. Investig. New Drugs 2014, 32, 1113–1122. [Google Scholar] [CrossRef]
- Russell, E.G.; Guo, J.; O’Sullivan, E.C.; O’Driscoll, C.M.; McCarthy, F.O.; Cotter, T.G. 7-formyl-10-methylisoellipticine, a novel ellipticine derivative, induces mitochondrial reactive oxygen species (ROS) and shows anti-leukaemic activity in mice. Investig. New Drugs 2016, 34, 15–23. [Google Scholar] [CrossRef]
- Kutney, J.P.; Noda, M.; Lewis, N.G.; Monteiro, B.; Mostowicz, D.; Worth, B.R. Dihydropyridines in synthesis and biosynthesis. V. Synthesis of pyridocarbazole alkaloids: Olivacine and (±)-guantambuine. Can. J. Chem. 1982, 60, 2426–2430. [Google Scholar]
- Mosher, C.W.; Crews, O.P.; Acton, E.M.; Goodman, L. Preparation and antitumor activity of olivacine and some new analogs. J. Med. Chem. 1966, 9, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Theumer, G.; Jäger, A.; Kataeva, O.; Wan, B.; Franzblau, S.G.; Knölker, H.-J. Synthesis and Activity against Mycobacterium tuberculosis of Olivacine and Oxygenated Derivatives. Molecules 2018, 23, 1402. [Google Scholar] [CrossRef] [PubMed]
- Awada, A.; Giacchetti, S.; Gerard, B.; Eftekhary, P.; Lucas, C.; de Valeriola, D.; Poullain, M.G.; Soudon, J.; Dosquet, C.; Brillanceau, M.-H. Clinical phase I and pharmacokinetic study of S 16020, a new olivacine derivative: report on three infusion schedules. Ann. Oncol. 2002, 13, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Miller, C.M.; McCarthy, F.O. Computer Simulations Reveal a Novel Nucleotide-type Binding Orientation for Ellipticine-based Anticancer c-kit Kinase Inhibitors. Biochemistry 2008, 47, 10333–10344. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W.; Saulnier, M.G.; Obaza-Nutaitis, J.A.; Ketcha, D.M. A versatile and efficient construction of the 6H-pyrido[4,3-b]carbazole ring system. Syntheses of the antitumor alkaloids ellipticine, 9-methoxyellipticine, and olivacine, and their analogs. J. Org. Chem. 1992, 57, 5891–5899. [Google Scholar] [CrossRef]
- Saulnier, M.G.; Gribble, G.W. An efficient synthesis of ellipticine. J. Org. Chem. 1982, 47, 2810–2812. [Google Scholar] [CrossRef]
- Saulnier, M.G.; Gribble, G.W. Efficient construction of the 10H-pyrido[3,4-b]carbazole ring system. Syntheses of isoellipticine and 7-methoxyisoellipticine. J. Org. Chem. 1983, 48, 2690–2695. [Google Scholar] [CrossRef]
- Gribble, G.W.; Fletcher, G.L.; Ketcha, D.M.; Rajopadhye, M. Metalated heterocycles in the synthesis of ellipticine analogs. A new route to the 10H-pyrido[2,3-b]carbazole ring system. J. Org. Chem. 1989, 54, 3264–3269. [Google Scholar] [CrossRef]
- Modi, S.P.; Carey, J.J.; Archer, S. Synthesis of 5-methyl-6h-pyrido[4,3-b]carbazole-11-methanol. Tetrahedron Lett. 1990, 31, 5845–5848. [Google Scholar] [CrossRef]
- Modi, S.P.; Michael, M.A.; Archer, S.; Carey, J.J. An efficient synthesis of C-11 substituted 6H-pyrido[4,3-b]carbazoles. Tetrahedron 1991, 47, 6539–6548. [Google Scholar] [CrossRef]
- Plug, J.P.M.; Koomen, G.-J.; Pandit, U.K. An Expedient Synthesis of 9-Hydroxyellipticine. Synthesis 1992, 1992, 1221–1222. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tagat, J.R.; Kerekes, A.D.; Doll, R.J.; Zhang, Y.; Xiao, Y.; Esposite, S.; Belanger, D.B.; Curran, P.J.; Mandal, A.K.; et al. Discovery of a Potent, Injectable Inhibitor of Aurora Kinases Based on the Imidazo-[1,2-a]-Pyrazine Core. ACS Med. Chem. Lett. 2010, 1, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Developmental Therapeutics Program. NCI-60 Human Tumour Cell Lines Screen. Available online: https://dtp.cancer.gov/discovery_development/nci-60/default.htm (accessed on 14 May 2019).
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and Analysis of Patterns of Differential Activity of Drugs Against Human Tumor Cell Lines: Development of Mean Graph and COMPARE Algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
R1 R2 R2 | Topo II Inhibition a | NSC No | NCI Mean Growth % | ||
|---|---|---|---|---|---|
| 8 | CHO | H | − | Not tested | |
| 9 | COOH | H | − | 762124 | 99.92 |
| 11 | CONHCH2Ph | H | − | 762144 | 21.22 |
| 13 | CH=CH-C(O)Me | H | + | 762123 | 17.83 |
| 15 | COOH | CHO | − | 762141 | 95.56 |
| 16 | COO− +NH3CH2Ph | CH=NCH2Ph | + | 762142 | 106.19 |
| 17 | COOH | CH2NHCH2Ph | − | 762143 | 101.72 |
| Cell Line | Cancer Subtype | 11 | 13 | ||
|---|---|---|---|---|---|
| GI50 | LC50 | GI50 | LC50 | ||
| HOP-62 | Lung | 2.15 | >100 | 1.77 | 26.0 |
| SW-620 | Colon | 2.86 | >100 | 1.65 | 44.0 |
| SNB-75 | CNS | 2.05 | >100 | 2.65 | 34.8 |
| OVCAR-3 | Ovarian | 2.33 | <10 | 2.53 | 28.0 |
| OVCAR-4 | Ovarian | 1.71 | 6.19 | 2.88 | 41.5 |
| 786-0 | Renal | 2.79 | 72.0 | 2.79 | 29.8 |
| A498 | Renal | 50.5 | >100 | 0.386 | 7.48 |
| UO-31 | Renal | 2.73 | >100 | 1.25 | 33.8 |
| MCF7 | Breast | 2.71 | >100 | 1.74 | 52.3 |
| MDA-MB-231/ATCC | Breast | 2.43 | >100 | 1.74 | 41.3 |
| HS578T | Breast | 2.61 | >100 | 1.96 | 48.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, C.M.; O’Sullivan, E.C.; McCarthy, F.O. Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells. Pharmaceuticals 2019, 12, 90. https://doi.org/10.3390/ph12020090
Miller CM, O’Sullivan EC, McCarthy FO. Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells. Pharmaceuticals. 2019; 12(2):90. https://doi.org/10.3390/ph12020090
Chicago/Turabian StyleMiller, Charlotte M., Elaine C. O’Sullivan, and Florence O. McCarthy. 2019. "Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells" Pharmaceuticals 12, no. 2: 90. https://doi.org/10.3390/ph12020090
APA StyleMiller, C. M., O’Sullivan, E. C., & McCarthy, F. O. (2019). Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells. Pharmaceuticals, 12(2), 90. https://doi.org/10.3390/ph12020090