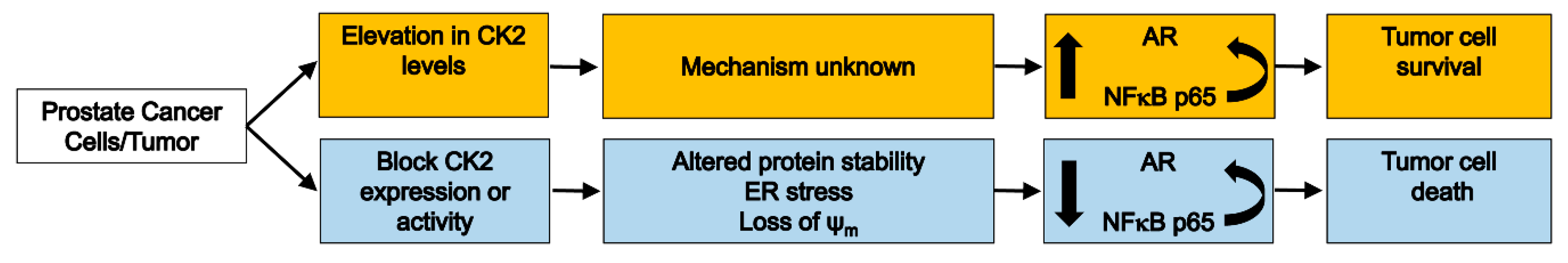
CK2 Pro-Survival Role in Prostate Cancer Is Mediated via Maintenance and Promotion of Androgen Receptor and NFκB p65 Expression

 ,
, 

Abstract
1. Introduction
2. Results
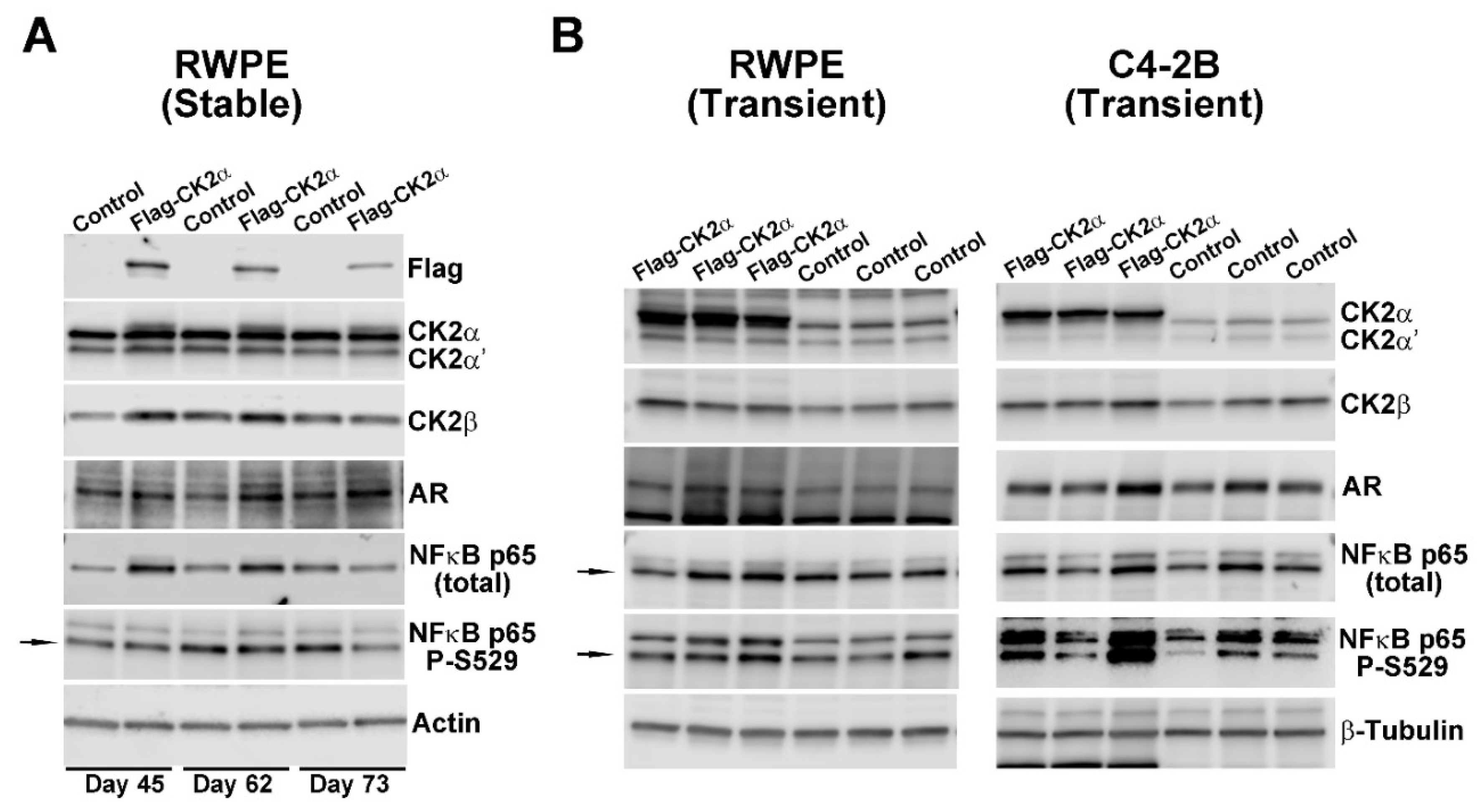
2.1. Effect of Increased CK2 Level on AR and NFκB p65 Expression in Benign and Malignant Prostate Cells
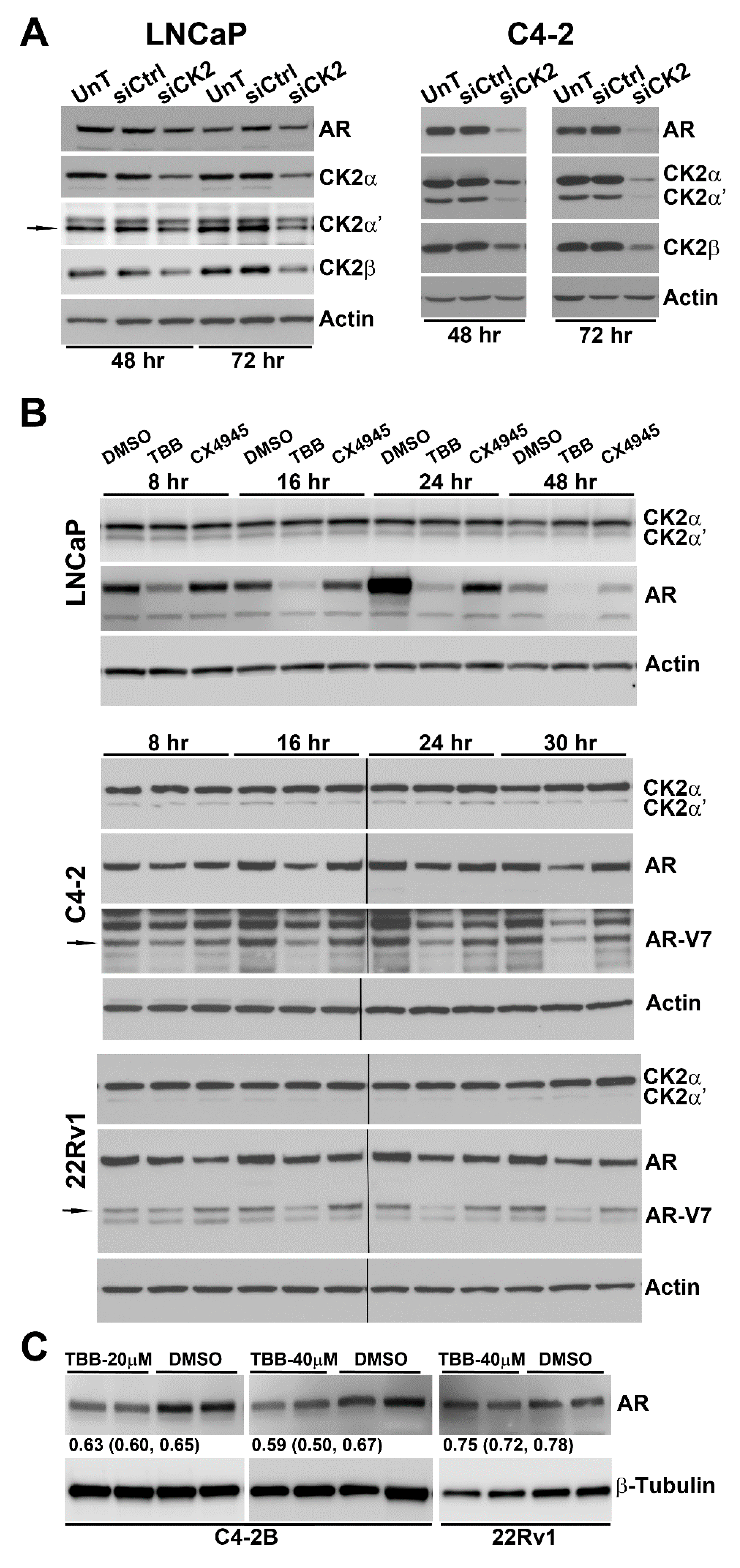
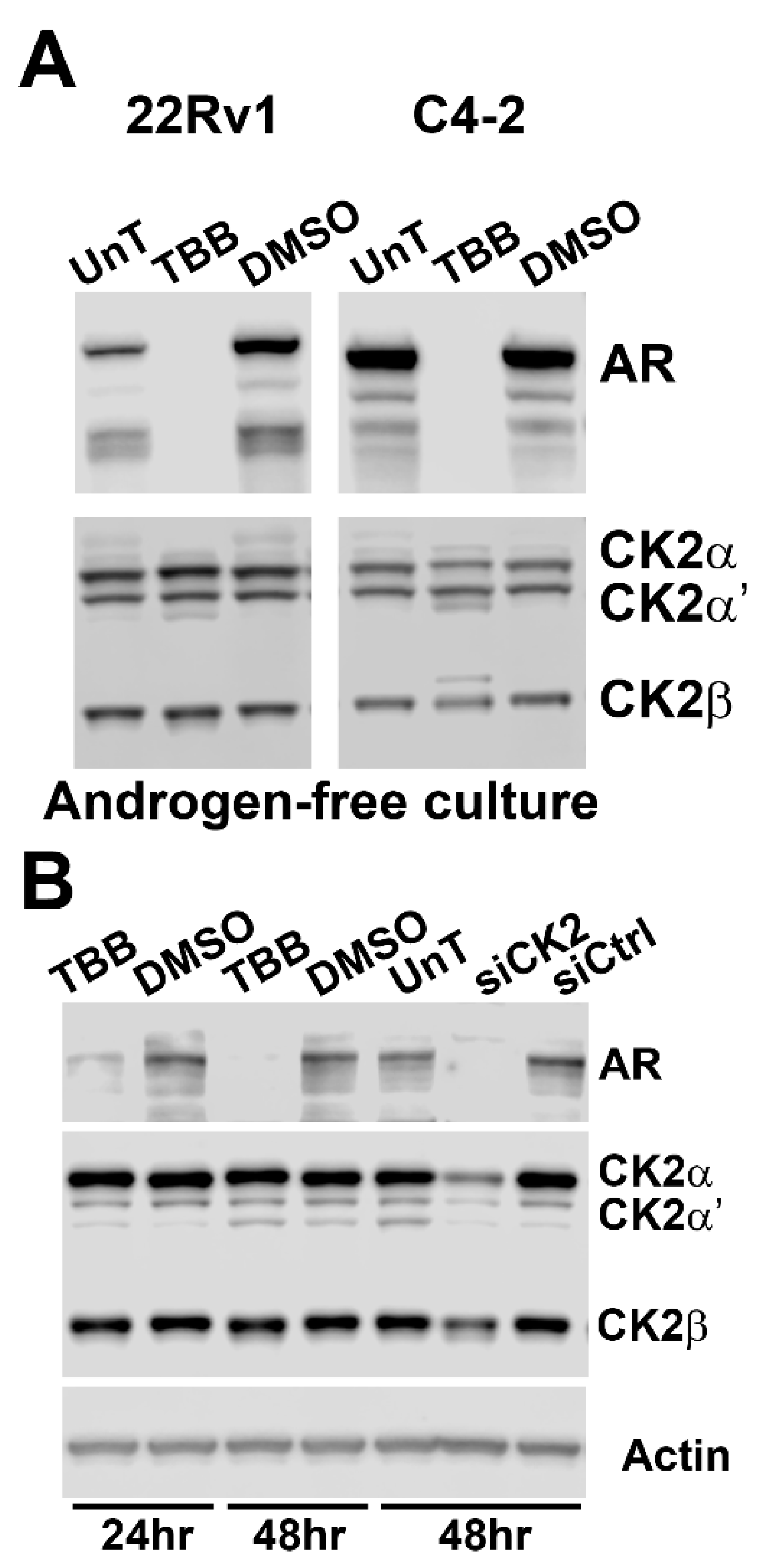
2.2. Effect of Reducing CK2 Level or Activity on AR Protein Levels in Prostate Cancer Cells
2.3. Effect of TBB or siRNA-mediated CK2 Inhibition on AR mRNA Levels in Prostate Cancer Cells
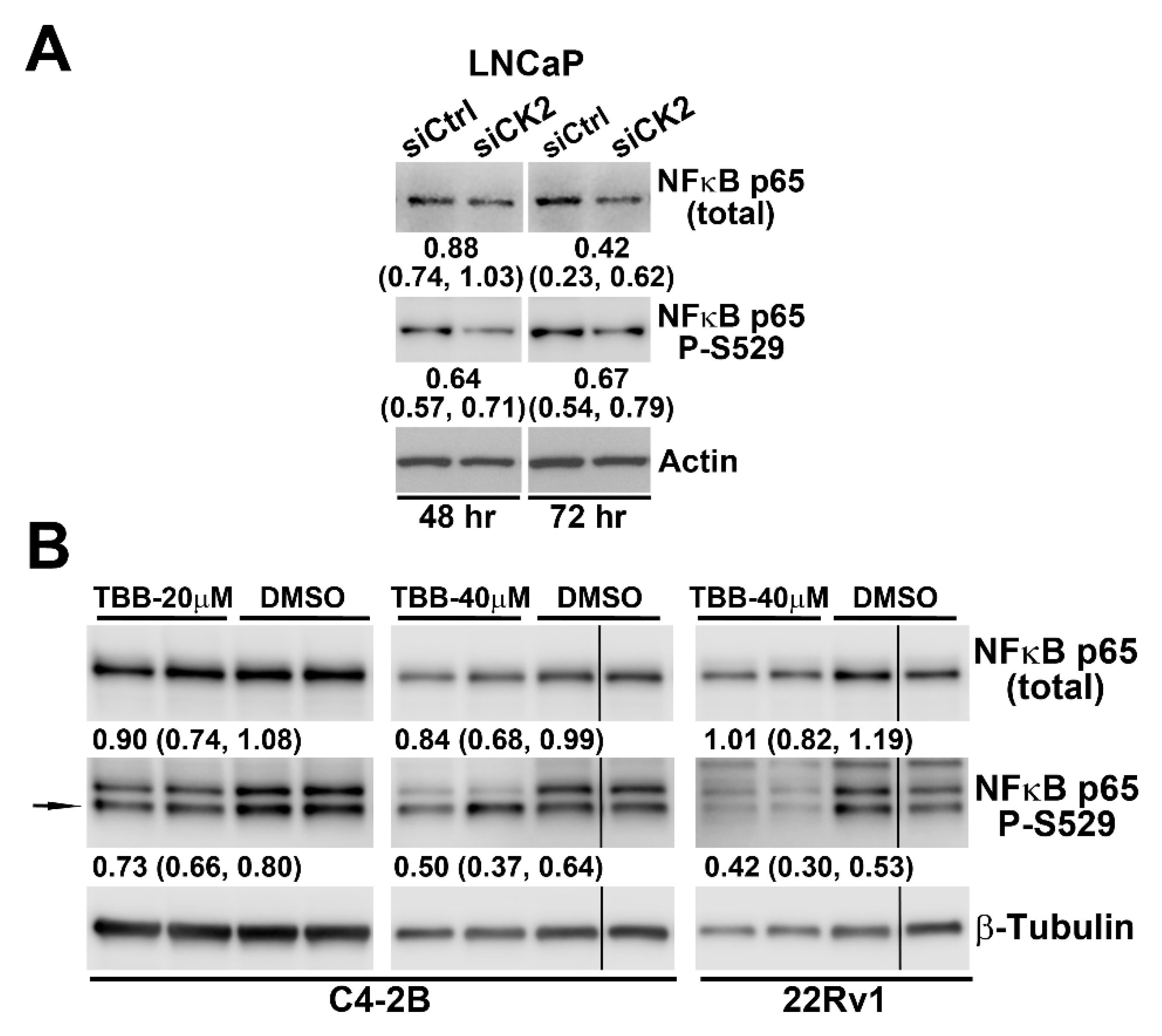
2.4. Effect of Reducing CK2 Level or Activity on NFκB p65 Protein Levels and Activation in Prostate Cancer Cells
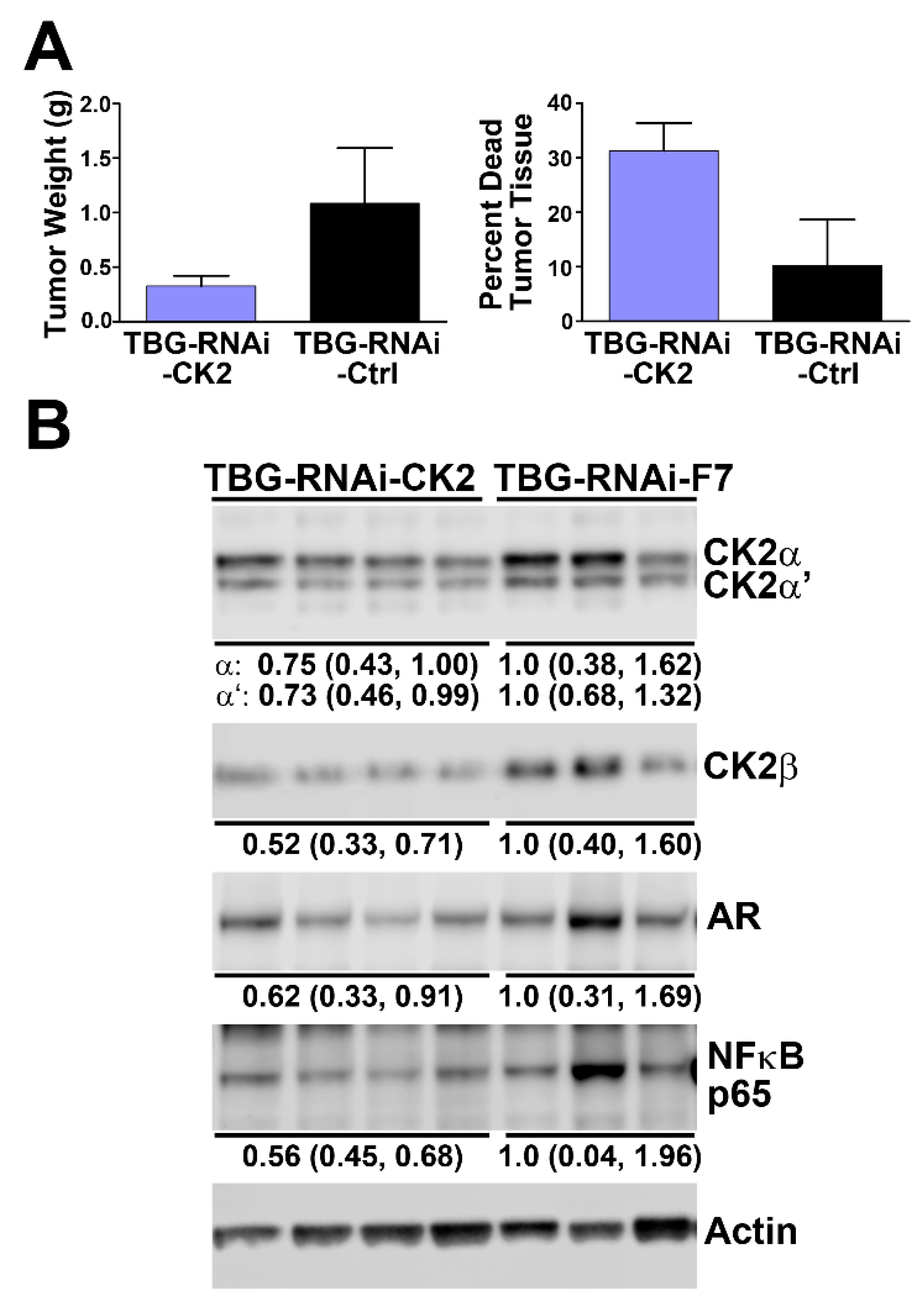
2.5. Effect of Reducing CK2 Level in vivo on AR and NFκB p65 Protein Levels in Xenograft Prostate Cancer Tumors
2.6. Androgen is Not Required for Effects on AR Status by CK2 Modulation
2.7. AR Loss is Equivalent in Non-malignant RWPE-1 Cells upon CK2 Inhibition or Loss
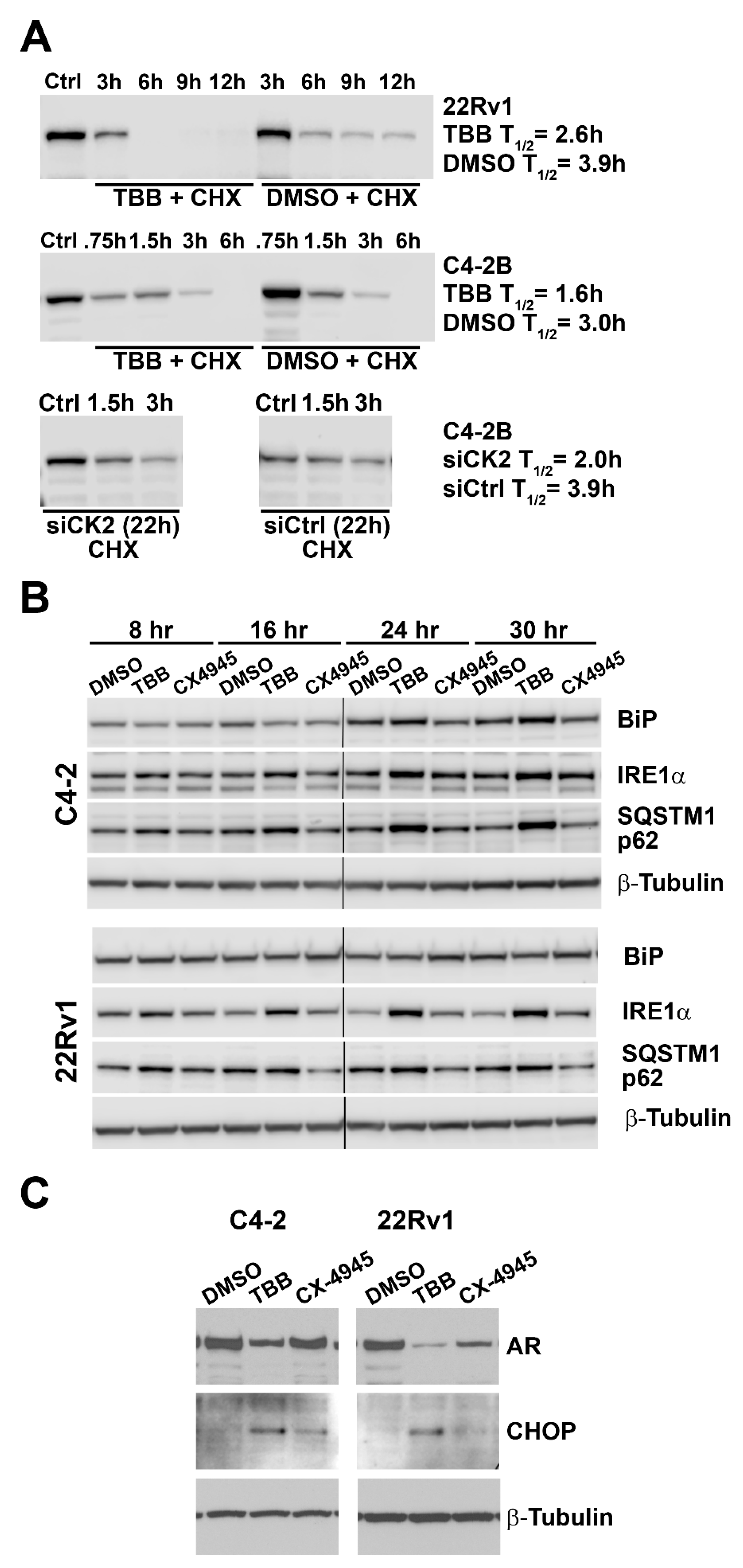
2.8. Effect of Altered CK2 Expression and Activity on AR Protein Half-life and ER Stress
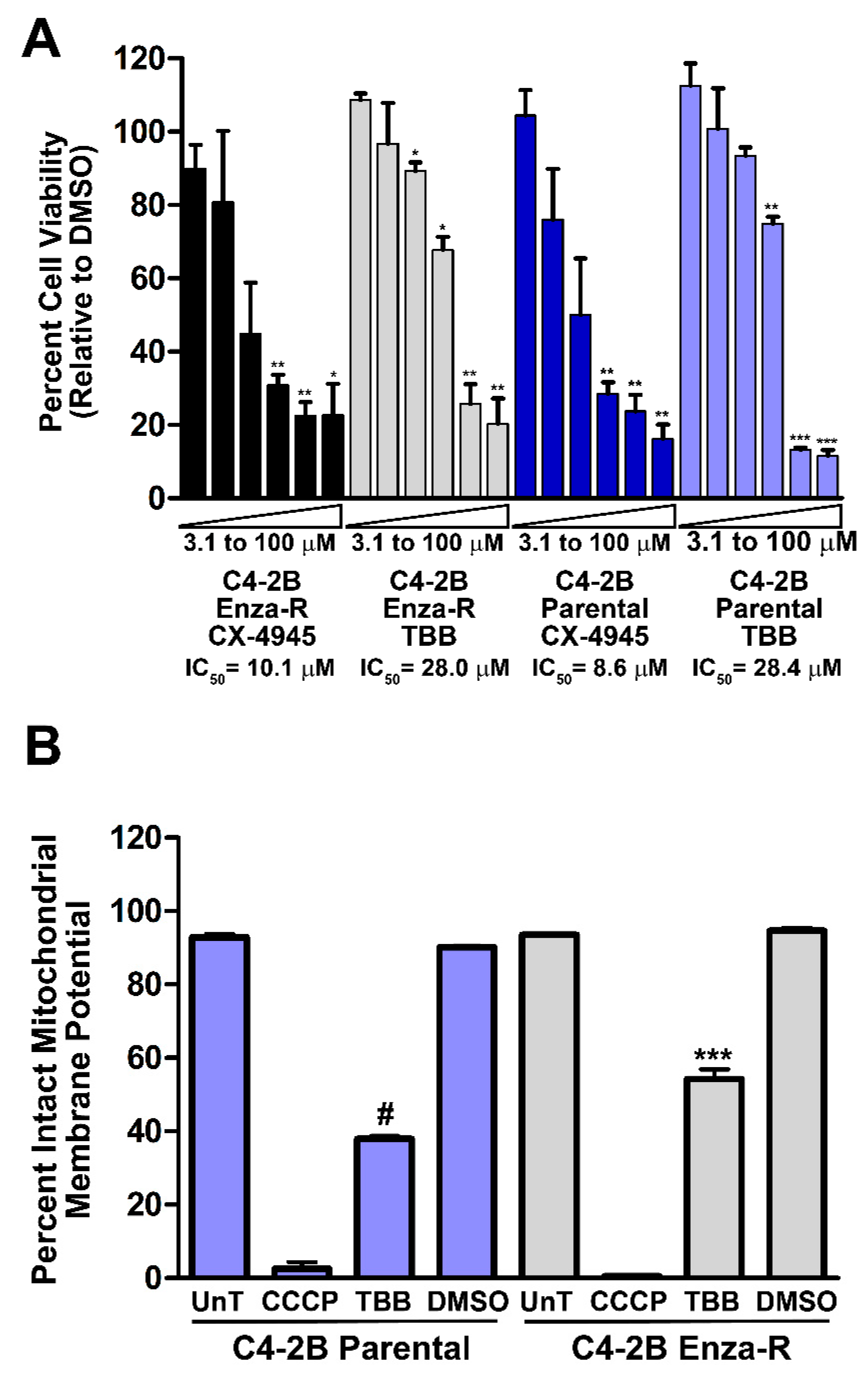
2.9. Effect of Inhibition of CK2 on Viability of Enzalutamide-resistant C4-2B Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. siRNA and Plasmid Transfections and Stable Cell Line Production
4.3. Cell Viability Assays
4.4. Immunoblot Analysis
4.5. Quantitative Real-Time RT-PCR Analysis
4.6. AR Protein Half Life Experiments
4.7. JC-1 FACS Analysis of Cultured Cells
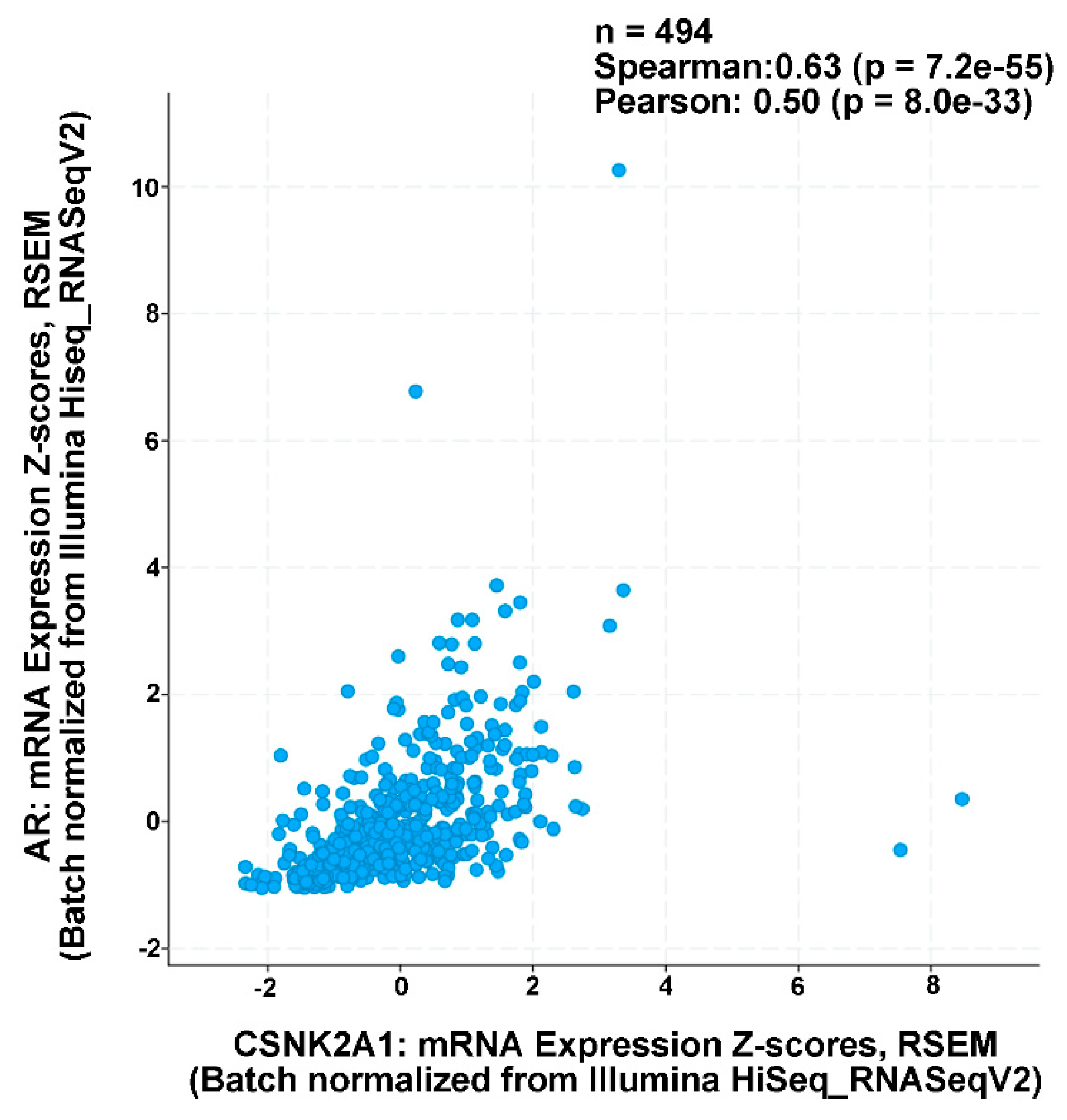
4.8. Human Prostate Cancer TCGA Data Analysis
4.9. Prostate Cancer Xenograft Experiment
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Dehm, S.M.; Tindall, D.J. Androgen receptor structural and functional elements: Role and regulation in prostate cancer. Mol. Endocrinol. 2007, 21, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Coffey, D.S.; Isaacs, J.T. Control of prostate growth. Urology 1981, 17, 17–24. [Google Scholar] [PubMed]
- Ahmed, K. Phosphoprotein metabolism in primary and accessory sex tissues. Adv. Sex Horm. Res. 1975, 1, 129–165. [Google Scholar] [PubMed]
- Ahmed, K.; Ishida, H. Effect of testosterone on nuclear phosphoproteins of rat ventral prostate. Mol. Pharmacol. 1971, 7, 323–327. [Google Scholar]
- Ahmed, K.; Wilson, M.J. Chromatin-associated protein phosphokinases of rat ventral prostate. Characteristics and effects of androgenic status. J. Biol. Chem. 1975, 250, 2370–2375. [Google Scholar]
- Ahmed, K.; Wilson, M.J.; Goueli, S.A. Biochemistry of protein kinase reactions in the prostate in relation to androgen action. Prog. Clin. Biol. Res. 1981, 75A, 55–74. [Google Scholar]
- Ahmed, K.; Yenice, S.; Davis, A.; Goueli, S.A. Association of casein kinase 2 with nuclear chromatin in relation to androgenic regulation of rat prostate. Proc. Natl. Acad. Sci. USA 1993, 90, 4426–4430. [Google Scholar] [CrossRef]
- Tawfic, S.; Yu, S.; Wang, H.; Faust, R.; Davis, A.; Ahmed, K. Protein kinase CK-2 signal in neoplasia. Histol. Histopathol. 2001, 16, 573–582. [Google Scholar]
- Yenice, S.; Davis, A.T.; Goueli, S.A.; Akdas, A.; Limas, C.; Ahmed, K. Nuclear casein kinase 2 (CK2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 1994, 24, 11–16. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: Ck2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.-G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Nunez de Villavicencio-Diaz, T.; Rabalski, A.J.; Litchfield, D.W. Protein kinase CK2: Intricate relationships within regulatory cellular networks. Pharmaceuticals 2017, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.Y.; Dominguez, I.; Toselli, P.; Landesman-Bollag, E.; O’Brien, C.; Seldin, D.C. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol. Cell. Biol. 2008, 28, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Buchou, T.; Vernet, M.; Blond, O.; Jensen, H.H.; Pointu, H.; Olsen, B.B.; Cochet, C.; Issinger, O.-G.; Boldyreff, B. Disruption of the regulatory b subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol. Cell. Biol. 2003, 23, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yu, S.; Davis, A.T.; Wang, H.; Green, J.E.; Ahmed, K. A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J. Biol. Chem. 2001, 276, 5992–5999. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2—A key suppressor of apoptosis. Adv. Enzyme Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef]
- Ahmed, K.; Gerber, D.A.; Cochet, C. Joining the cell survival squad: An emerging role for protein kinase CK2. Trends Cell Biol. 2002, 12, 226–230. [Google Scholar] [CrossRef]
- Trembley, J.H.; Qaiser, F.; Kren, B.T.; Ahmed, K. CK2—A global regulator of cell death. In Protein Kinase CK2 Cellular Function in Normal and Disease States; Ahmed, K., Issinger, O.-G., Szyszka, R., Eds.; Springer International Publishing: Cham, Switzerland, 2015; Volume 12, pp. 159–181. [Google Scholar]
- Goueli, S.A.; Holtzman, J.L.; Ahmed, K. Phosphorylation of the androgen receptor by a nuclear camp-independent protein kinase. Biochem. Biophys. Res. Commun. 1984, 123, 778–784. [Google Scholar] [CrossRef]
- Gotz, C.; Bachmann, C.; Montenarh, M. Inhibition of protein kinase CK2 leads to a modulation of androgen receptor dependent transcription in prostate cancer cells. Prostate 2007, 67, 125–134. [Google Scholar] [CrossRef]
- Ryu, B.J.; Baek, S.-H.; Kim, J.; Bae, S.J.; Chang, S.-Y.; Heo, J.-N.; Lee, H.; Lee, S.Y.; Kim, S.H. Anti-androgen receptor activity of apoptotic CK2 inhibitor cx4945 in human prostate cancer lncap cells. Bioorg. Med. Chem. Lett. 2012, 22, 5470–5474. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Youn, H.; Gao, X.; Huang, B.; Zhou, F.; Li, B.; Han, H. Casein kinase 2 inhibition attenuates androgen receptor function and cell proliferation in prostate cancer cells. Prostate 2012, 72, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Chen, J.; Guo, S.; Wang, Y.; Zhou, Q.; Li, Z.; Yang, X.; Yu, X.; Zhang, Z.; Zhou, F.; et al. Cx4945 suppresses the growth of castration-resistant prostate cancer cells by reducing AR-v7 expression. World J. Urol. 2017, 35, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Davis, A.; Yu, S.; Ahmed, K. Response of cancer cells to molecular interruption of the CK2 signal. Mol. Cell. Biochem. 2001, 227, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Unger, G.; Ahmad, K.A.; Slaton, J.W.; Ahmed, K. Downregulation of CK2 induces apoptosis in cancer cells—A potential approach to cancer therapy. Mol. Cell. Biochem. 2005, 274, 77–84. [Google Scholar] [CrossRef]
- Trembley, J.H.; Kren, B.T.; Abedin, M.J.; Vogel, R.I.; Cannon, C.M.; Unger, G.M.; Ahmed, K. CK2 molecular targeting-tumor cell-specific delivery of rnai in various models of cancer. Pharmaceuticals 2017, 10, 25. [Google Scholar] [CrossRef]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef]
- Chua, M.M.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in cancer: Cellular and biochemical mechanisms and potential therapeutic target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Wilding, G. The importance of steroid hormones in prostate cancer. Cancer Surv. 1992, 14, 113–130. [Google Scholar]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2018, 22, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Maughan, B.L.; Antonarakis, E.S. Androgen pathway resistance in prostate cancer and therapeutic implications. Expert Opin. Pharmacother. 2015, 16, 1521–1537. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Regulation of androgen receptor signaling in prostate cancer. Expert Rev. Anticancer Ther. 2005, 5, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S. Tumor necrosis factor α-induced phosphorylation of rela/p65 on ser529 is controlled by casein kinase ii. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Li, J.Y.; Yadav, S.S.; Tewari, A.K. Recent insights into NF-kappa B signalling pathways and the link between inflammation and prostate cancer. BJU Int. 2014, 114, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Diallo, O.T.; Hu, M.; Ehsanian, R.; Yang, X.; Arun, P.; Lu, H.; Korman, V.; Unger, G.; Ahmed, K.; et al. CK2 modulation of NF-κB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin. Cancer Res. 2010, 16, 2295–2307. [Google Scholar] [CrossRef]
- Brown, M.; Cohen, J.; Arun, P.; Chen, Z.; Van Waes, C. NF-kappaB in carcinoma therapy and prevention. Expert Opin. Ther. Targets 2008, 12, 1109–1122. [Google Scholar] [CrossRef]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. Protein kinase CK2 in health and disease: CK2 and its role in wnt and NF-kappaB signaling: Linking development and cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Qaiser, F.; Trembley, J.H.; Sadiq, S.; Muhammad, I.; Younis, R.; Hashmi, S.N.; Murtaza, B.; Rector, T.S.; Naveed, A.K.; Ahmed, K.; et al. Examination of CK2alpha and NF-kappaB p65 expression in human benign prostatic hyperplasia and prostate cancer tissues. Mol. Cell. Biochem. 2016, 420, 43–51. [Google Scholar] [CrossRef]
- Trembley, J.H.; Unger, G.M.; Korman, V.L.; Abedin, M.J.; Nacusi, L.P.; Vogel, R.I.; Slaton, J.W.; Kren, B.T.; Ahmed, K. Tenfibgen ligand nanoencapsulation delivers bi-functional anti-CK2 RNAi oligomer to key sites for prostate cancer targeting using human xenograft tumors in mice. PLoS ONE 2014, 9, e109970. [Google Scholar] [CrossRef] [PubMed]
- Unger, G.M.; Kren, B.T.; Korman, V.L.; Kimbrough, T.G.; Vogel, R.I.; Ondrey, F.G.; Trembley, J.H.; Ahmed, K. Mechanism and efficacy of sub-50-nm tenfibgen nanocapsules for cancer cell-directed delivery of anti-CK2 RNAi to primary and metastatic squamous cell carcinoma. Mol. Cancer Ther. 2014, 13, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Kren, B.T.; Abedin, M.J.; Vogel, R.I.; Shaughnessy, D.P.; Nacusi, L.; Korman, V.L.; Li, Y.; Dehm, S.M.; Zimmerman, C.L.; et al. CK2 targeted RNAi therapeutic delivered via malignant cell-directed tenfibgen nanocapsule: Dose and molecular mechanisms of response in xenograft prostate tumors. Oncotarget 2016, 7, 61789–61805. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Unger, G.M.; Tobolt, D.K.; Korman, V.L.; Wang, G.; Ahmad, K.A.; Slaton, J.W.; Kren, B.T.; Ahmed, K. Systemic administration of antisense oligonucleotides simultaneously targeting CK2alpha and alpha’ subunits reduces orthotopic xenograft prostate tumors in mice. Mol. Cell. Biochem. 2011, 356, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioportal. Sci. Signal 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Brancalion, A.; Tubi, L.Q.; Colpo, A.; Pavan, L.; Cabrelle, A.; Ave, E.; Zaffino, F.; Di Maira, G.; Ruzzene, M.; et al. Protein kinase CK2 protects multiple myeloma cells from ER stress–induced apoptosis and from the cytotoxic effect of hsp90 inhibition through regulation of the unfolded protein response. Clin. Cancer Res. 2012, 18, 1888–1900. [Google Scholar] [CrossRef]
- Hessenauer, A.; Schneider, C.C.; Gotz, C.; Montenarh, M. CK2 inhibition induces apoptosis via the er stress response. Cell. Signal. 2011, 23, 145–151. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; et al. Cytotoxic activity of the casein kinase 2 inhibitor cx-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2alpha) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Wang, G.; Slaton, J.; Unger, G.; Ahmed, K. Targeting CK2 for cancer therapy. Anticancer Drugs 2005, 16, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Pinna, L.A.; Allende, J.E. Protein kinase CK2 in health and disease: Protein kinase ck2: An ugly duckling in the kinome pond. Cell. Mol. Life Sci. 2009, 66, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. Mek-Erk signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Asim, M.; Hafeez, B.B.; Adhami, V.M.; Tarapore, R.S.; Mukhtar, H. Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. FASEB J. 2011, 25, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Harris, N.H.; Johnson, A.D.; Lindvall, H.C.; Wang, G.; Ahmed, K. Protein kinase CK2 modulates apoptosis induced by resveratrol and epigallocatechin-3-gallate in prostate cancer cells. Mol. Cancer Ther. 2007, 6, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.C.; Ampofo, E.; Montenarh, M. CK2 regulates atf4 and chop transcription within the cellular stress response signalling pathway. Cell. Signal. 2012, 24, 1797–1802. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Shourideh, M.; Goodrich, D.W.; Koochekpour, S. Riluzole induces AR degradation via endoplasmic reticulum stress pathway in androgen-dependent and castration-resistant prostate cancer cells. Prostate 2018, 79, 140–150. [Google Scholar] [CrossRef]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef]
- Zhang, L.; Altuwaijri, S.; Deng, F.; Chen, L.; Lal, P.; Bhanot, U.K.; Korets, R.; Wenske, S.; Lilja, H.G.; Chang, C.; et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am. J. Pathol. 2009, 175, 489–499. [Google Scholar] [CrossRef]
- Chappell, W.H.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Martelli, A.M.; Cocco, L.; Rakus, D.; Gizak, A.; Terrian, D.; Steelman, L.S.; et al. Novel roles of androgen receptor, epidermal growth factor receptor, TP53, regulatory RNAs, NF-kappa-B, chromosomal translocations, neutrophil associated gelatinase, and matrix metalloproteinase-9 in prostate cancer and prostate cancer stem cells. Adv. Biol. Regul. 2016, 60, 64–87. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yamashita, H.; Yu, X.; Wang, J.; Franco, O.E.; Wang, Y.; Hayward, S.W.; Matusik, R.J. Inhibition of NF-kappa B signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene 2015, 34, 3700–3710. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Unger, G.M.; Gomez, O.C.; Abedin, J.; Korman, V.L.; Vogel, R.I.; Niehans, G.; Kren, B.T.; Ahmed, K. Tenfibgen-DMAT nanocapsule delivers CK2 inhibitor DMAT to prostate cancer xenograft tumors causing inhibition of cell proliferation. Mol. Cell. Pharmacol. 2014, 6, 15–25. [Google Scholar] [PubMed]
- Qaiser, F.; Trembley, J.H.; Kren, B.T.; Wu, J.J.; Naveed, A.K.; Ahmed, K. Protein kinase CK2 inhibition induces cell death via early impact on mitochondrial function. J. Cell. Biochem. 2014, 115, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Slaton, J.W.; Unger, G.M.; Sloper, D.T.; Davis, A.T.; Ahmed, K. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol. Cancer Res. 2004, 2, 712–721. [Google Scholar] [PubMed]
- Trembley, J.H.; Unger, G.M.; Korman, V.L.; Tobolt, D.K.; Kazimierczuk, Z.; Pinna, L.A.; Kren, B.T.; Ahmed, K. Nanoencapsulated anti-CK2 small molecule drug or siRNA specifically targets malignant cancer but not benign cells. Cancer Lett. 2012, 315, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Wang, T.Y.; Chang, Y.T.; Chu, C.Y.; Lee, C.L.; Hsu, H.W.; Zhou, T.A.; Wu, Z.; Kim, R.H.; Desai, S.J.; et al. Autophagy pathway is required for IL-6 induced neuroendocrine differentiation and chemoresistance of prostate cancer lncap cells. PLoS ONE 2014, 9, e88556. [Google Scholar] [CrossRef]
- Ahmed, K.; Unger, G.M.; Kren, B.T.; Trembley, J.H. Targeting ck2 for cancer therapy using a nanomedicine approach. In Protein Kinase CK2 in Cellular Function in Normal and Disease States; Ahmed, K., Issinger, O.-G., Szyszka, R., Eds.; Springer International Publishing: Cham, Switzerland, 2015; Volume 12, pp. 299–315. [Google Scholar]
- Ahmed, R.L.; Shaughnessy, D.P.; Knutson, T.P.; Vogel, R.I.; Ahmed, K.; Kren, B.T.; Trembley, J.H. Cdk11 loss induces cell cycle dysfunction and death of BRAF and NRAS melanoma cells. Pharmaceuticals 2019, 12, 50. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Protein Detected | ||||||
|---|---|---|---|---|---|---|---|
| CK2α | CK2α′ | CK2β | AR | NFκB p65 | NFκB p65 P-S529 | Cyclin D1 | |
| RWPE 1 (stable) | 1.5 (1.1, 1.9) | 1.2 (1.0, 1.3) | 1.6 (0.4, 2.8) | 1.5 (0.9, 2.0) | 1.8 (0.9, 2.8) | 0.9 (0.6, 1.2) | 0.6 (0.5, 0.7) |
| RWPE 2 (transient) | 2.9 (2.2, 3.5) | 1.9 (1.7, 2.2) | 1.6 (1.3, 1.9) | 1.4 (1.2, 1.9) | 1.3 (0.9, 1.6) | 1.2 (0.6, 1.7) | 1.0 (0.9, 1.1) |
| C4-2B 2 (transient) | 4.4 (4.1, 4.7) | 1.2 (1.0, 1.4) | 1.4 (0.9, 1.8) | 1.2 (0.8, 1.7) | 1.0 (0.8, 1.2) | 3.3 (0.2, 6.4) | 1.1 (0.9, 1.3) |
| LNCAP 3 (transient) | 3.6 (2.5, 4.6) | 1.7 (1.6, 1.7) | 1.2 (1.2, 1.2) | 1.4 (1.3, 1.5) | 1.0 (0.8, 1.1) | 1.9 (1.7, 2.2) | ND |
| Cell Line | Time Point (h) | Protein Detected | |||
|---|---|---|---|---|---|
| CK2α | CK2α′ | CK2β | AR | ||
| LNCaP | 48 | 0.60 (0.48,0.72) | 0.68 (0.59,0.77) | 0.67 (0.59,0.76) | 0.79 (0.63,0.96) |
| 72 | 0.54 (0.36,0.73) | 0.43 (0.19,0.68) | 0.49 (0.35,0.62) | 0.70 (0.53,0.87) | |
| C4-2 | 48 | 0.56 (0.47,0.65) | 0.20 (0.10,0.30) | 0.58 (0.53,0.63) | 0.27 (0.18,0.36) |
| 72 | 0.21 (0.12,0.30) | 0.10 (0.08,0.12) | 0.50 (0.35,0.65) | 0.15 (0.13,017) | |
| Cell Line | Time Point (h) | TBB | CX-4945 |
|---|---|---|---|
| LNCaP | 8 | 0.71 (0.42, 1.00) | 1.13 (0.68, 1.58) |
| 16 | 0.31 (0.11, 0.51) | 0.96 (0.84, 1.08) | |
| 24 | 0.36 (−0.25, 0.97) | 0.62 (0.06, 1.18) | |
| 48 | 0.17 (−0.06, 0.40) | 0.56 (0.10, 1.01) | |
| C4-2 | 8 | 0.87 (0.74, 1.00) | 0.97 (0.73, 1.21) |
| 16 | 0.69 (0.44, 0.94) | 0.89 (0.88, 0.90) | |
| 24 | 0.62 (0.46, 0.78) | 0.67 (0.30, 1.04) | |
| 30 | 0.33 (−0.19, 0.85) | 1.92 (0.16, 3.68) | |
| 22Rv1 | 8 | 0.84 (0.81, 0.87) | 0.70 (0.49, 0.91) |
| 16 | 0.79 (0.73, 0.85) | 0.75 (0.58, 0.92) | |
| 24 | 0.77 (0.76, 0.78) | 0.75 (0.48, 1.02) | |
| 30 | 0.65 (0.62, 0.68) | 0.74 (0.68, 0.80) |
| Cell Line | Time Point (h) | TBB | siCK2 |
|---|---|---|---|
| 22Rv1 | 8 | 0.86 ± 0.07 | ND |
| 16 | 0.75 ± 0.06 | ND | |
| 24 | 0.60 ± 0.10 | ND | |
| 48 | 0.29 ± 0.23 | ND | |
| C4-2 | 8 | 0.71 ± 0.04 | ND |
| 16 | 0.36 ± 0.01 | ND | |
| 24 | 0.35 ± 0.03 | ND | |
| 48 | 0.48 ± 0.02 | 1.25 ± 0.09 | |
| 72 | ND | 1.80 ± 0.19 | |
| LNCaP | 48 | 0.12 ± 0.02 | ND |
| 72 | ND | 0.82 ± 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trembley, J.H.; Kren, B.T.; Abedin, M.J.; Shaughnessy, D.P.; Li, Y.; Dehm, S.M.; Ahmed, K. CK2 Pro-Survival Role in Prostate Cancer Is Mediated via Maintenance and Promotion of Androgen Receptor and NFκB p65 Expression. Pharmaceuticals 2019, 12, 89. https://doi.org/10.3390/ph12020089
Trembley JH, Kren BT, Abedin MJ, Shaughnessy DP, Li Y, Dehm SM, Ahmed K. CK2 Pro-Survival Role in Prostate Cancer Is Mediated via Maintenance and Promotion of Androgen Receptor and NFκB p65 Expression. Pharmaceuticals. 2019; 12(2):89. https://doi.org/10.3390/ph12020089
Chicago/Turabian StyleTrembley, Janeen H., Betsy T. Kren, Md. J. Abedin, Daniel P. Shaughnessy, Yingming Li, Scott M. Dehm, and Khalil Ahmed. 2019. "CK2 Pro-Survival Role in Prostate Cancer Is Mediated via Maintenance and Promotion of Androgen Receptor and NFκB p65 Expression" Pharmaceuticals 12, no. 2: 89. https://doi.org/10.3390/ph12020089
APA StyleTrembley, J. H., Kren, B. T., Abedin, M. J., Shaughnessy, D. P., Li, Y., Dehm, S. M., & Ahmed, K. (2019). CK2 Pro-Survival Role in Prostate Cancer Is Mediated via Maintenance and Promotion of Androgen Receptor and NFκB p65 Expression. Pharmaceuticals, 12(2), 89. https://doi.org/10.3390/ph12020089