Hierarchical Virtual Screening of Potential Insectides Inhibitors of Acetylcholinesterase and Juvenile Hormone from Temephos

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Empirical Force Field Methods (Molecular Mechanics–MM)
2.2. Hierarchical Virtual Screening
2.2.1. Rapid Overlay of Chemical Structures (Using ROCS)
2.2.2. Electrostatic Similarity (Using EON)
2.2.3. Pharmacokinetic and Toxicological Properties
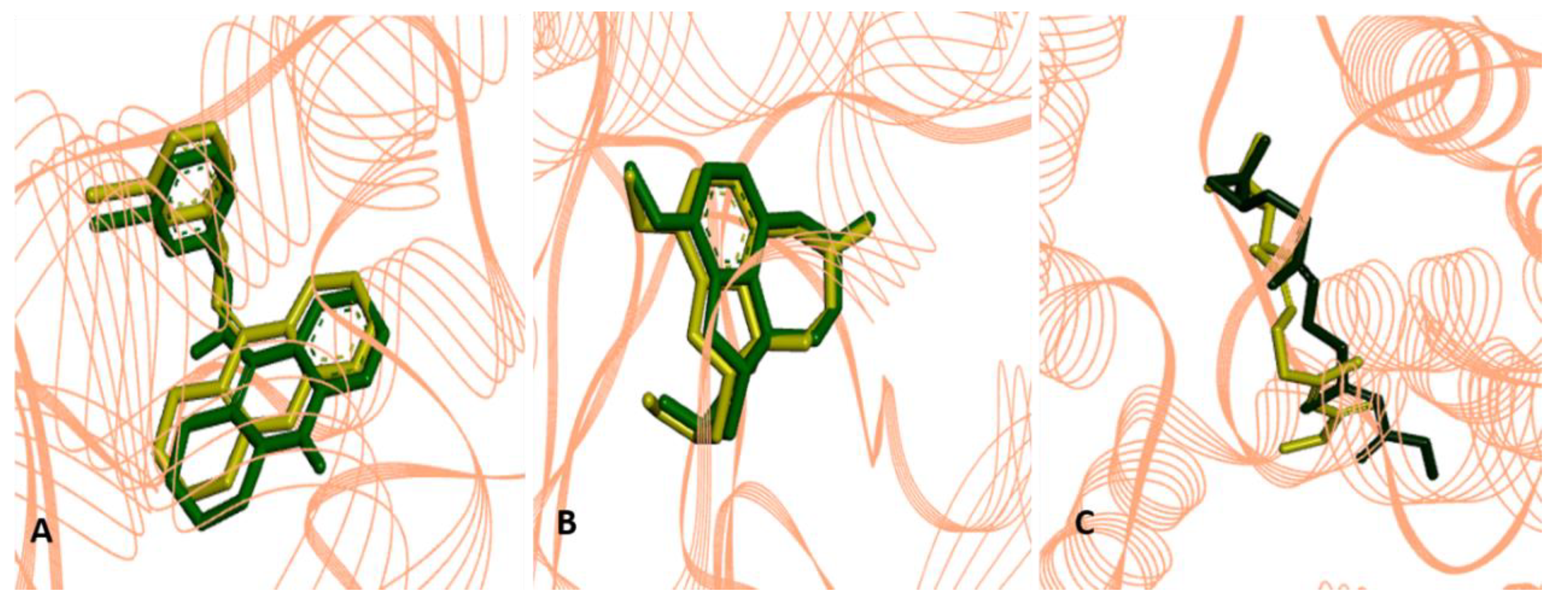
2.2.4. Molecular Docking Procedures
2.3. Structure-Activity Relationship of the Promising Molecule
3. Materials and Methods
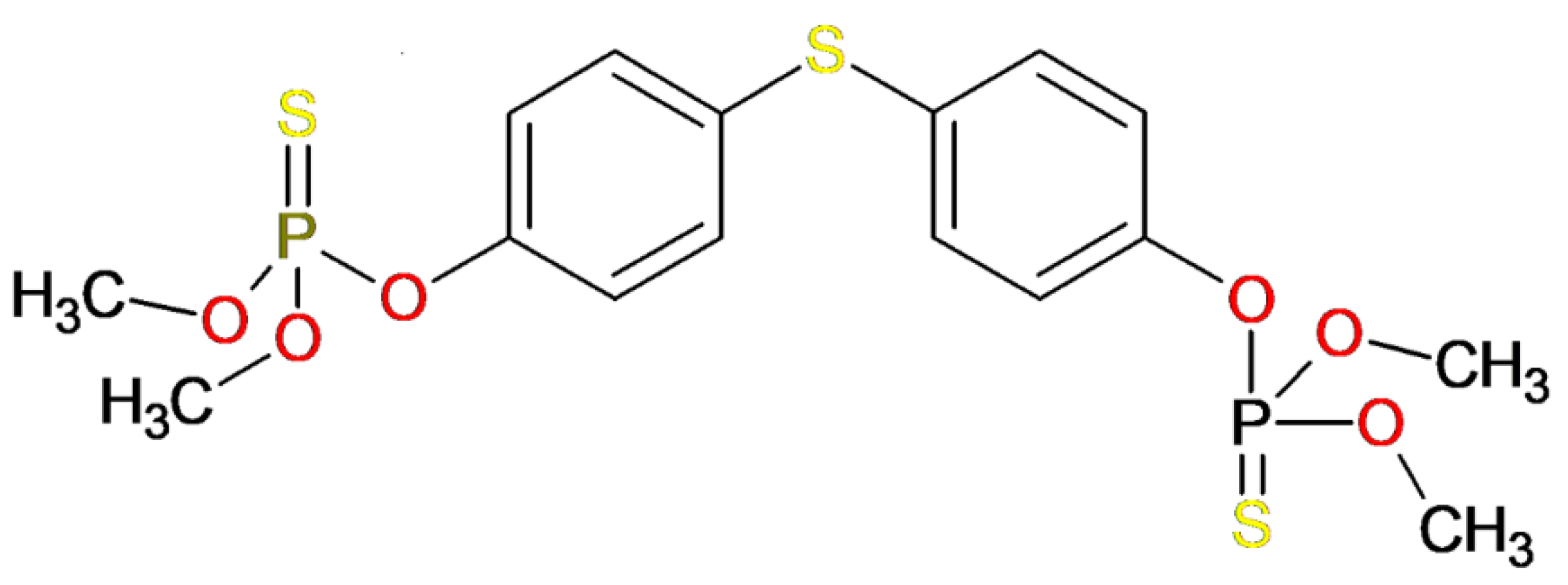
3.1. Template Compound
3.2. Generation of Conformers Library in Database
3.3. Virtual Screening Procedures
3.3.1. ROCS (for Shape Similarity)
3.3.2. EON (for Electrostatic Similarity)
3.3.3. In Silico Pharmacokinetic and Toxicological Properties
3.3.4. Molecular Docking Simulations
Selection of the Proteins and Ligands Structures
Docking Study with AutoDock 4.2/Vina 1.1.2 via Graphical Interface Pyrx (Version 0.8.30)
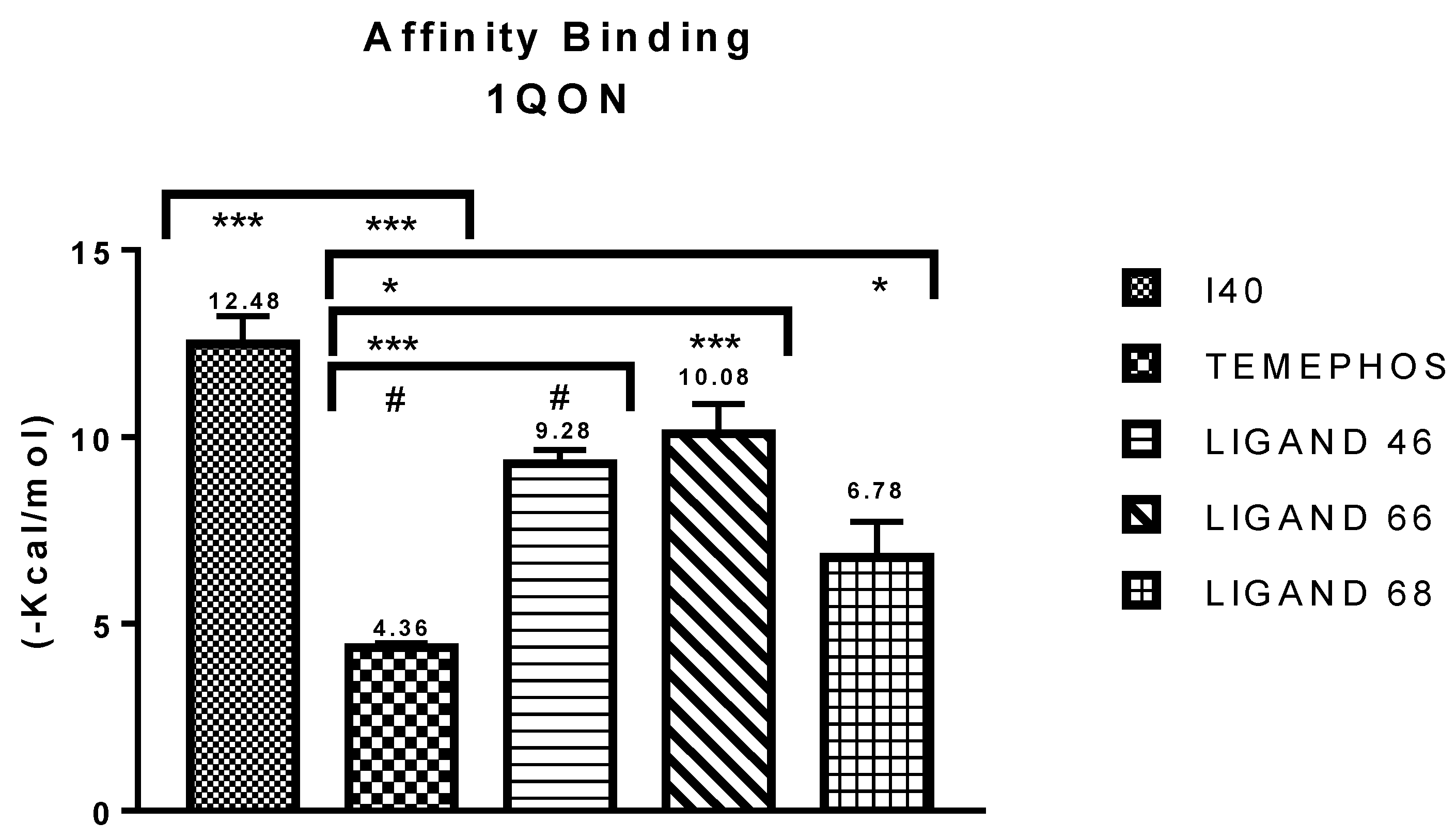
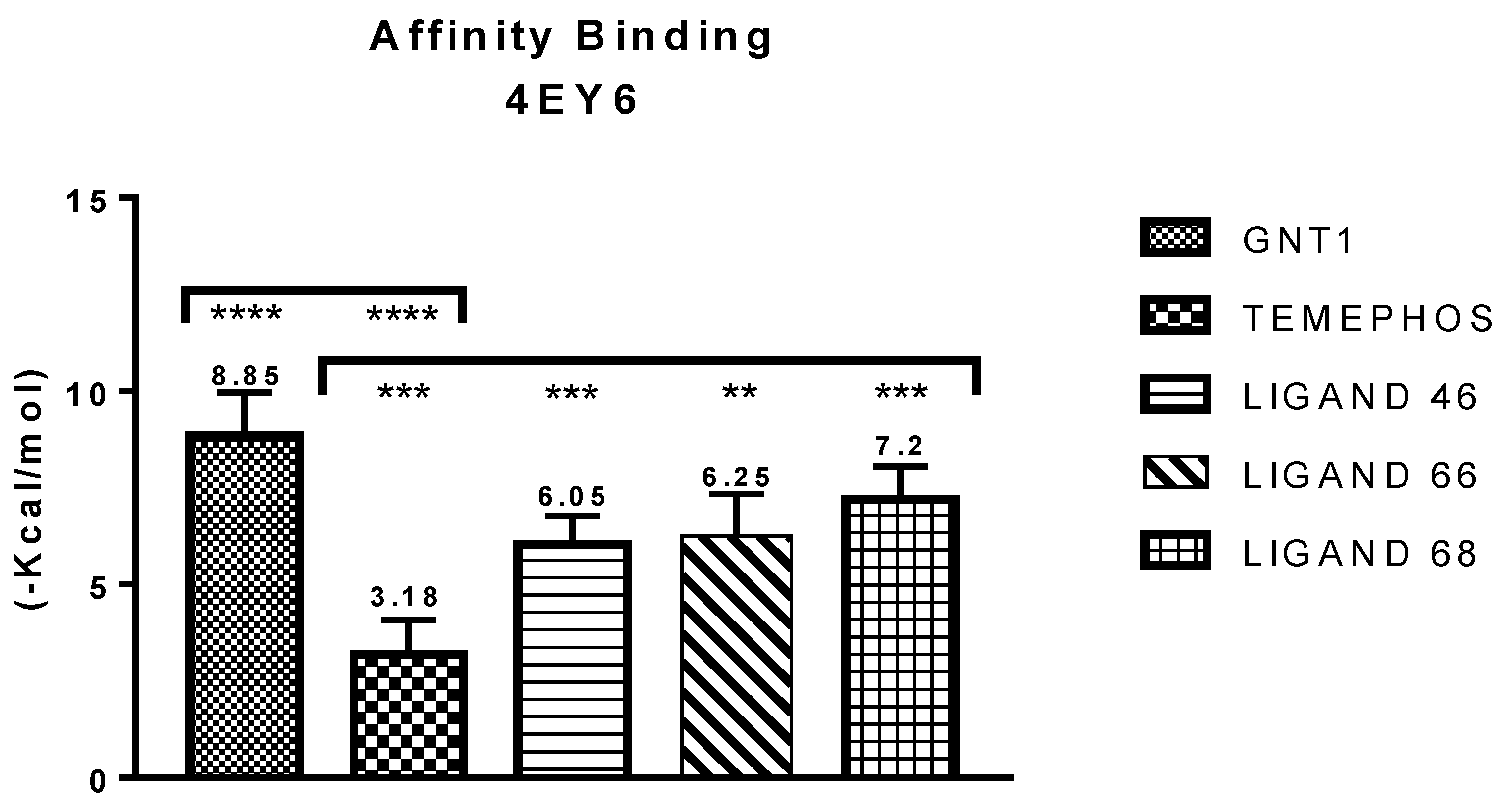
Molecular Binding Affinity Statistics
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hay, S.I.; George, D.B.; Wint, G.R.W.; Hoen, A.G.; Brownstein, J.S.; Bhatt, S.; Drake, J.M.; Myers, M.F.; Farlow, A.W.; Simmons, C.P.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar]
- Poupardin, R.; Srisukontarat, W.; Yunta, C.; Ranson, H. Identification of Carboxylesterase Genes Implicated in Temephos Resistance in the Dengue Vector Aedes aegypti. PLoS Negl. Trop. Dis. 2014, 8, e2743. [Google Scholar] [CrossRef]
- Kobayashi, H.; Suzuki, T.; Akahori, F.; Satoh, T. Receptors: Brain Regional Heterogeneity. In: Tetsuo Satoh Ramesh C. Gupta, editor. Anticholinesterase Pestic. Metab. Neurotox. Epidemiol. 2011, 3, 3–18. [Google Scholar]
- Loza-Mejía, M.A.; Salazar, J.R.; Sánchez-Tejeda, J.F. In Silico Studies on Compounds Derived from Calceolaria: Phenylethanoid Glycosides as Potential Multitarget Inhibitors for the Development of Pesticides. Biomolecules 2018, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Winteringham, F.P.; Barnes, J.M. Comparative response of insects and mammals to certain halogenated hydrocarbons used as insecticides. Physiol. Rev. 1955, 35, 701–739. [Google Scholar] [CrossRef] [PubMed]
- Areiza, M. Regulation of Juvenile Hormone Synthesis by 20-Hydroxyecdysone in the Yellow-fever Mosquito, Aedes aegypti. Ph.D. Thesis, Florida International University, Miami, FL, USA, 2018. [Google Scholar]
- Nakagawa, Y.; Hormann, R.E.; Smagghe, G.; Fitzpatrick, G.E.; Dowell, R.V.; Entomology, V.; Ferreira-Pereira, A.; Sorgine, M.H.F.; Moreira, M.F.; Franco, T.A.; et al. Reproductive toxicity of organophosphate and carbamate pesticides. Toxicol. Organophosphate Carbamate Compd. 2006, 1, 447–462. [Google Scholar]
- Hemingway, J.; Ranson, H. Insecticide Resistance in Insect Vectors of Human Disease. Annu. Rev. Entomol. 2000, 45, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Gholivand, K.; Ebrahimi Valmoozi, A.A.; Bonsaii, M. Synthesis and crystal structure of new temephos analogues as cholinesterase inhibitor: Molecular docking, qsar study, and hydrogen bonding analysis of solid state. J. Agric. Food Chem. 2014, 62, 5761–5771. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Stillh, C. MacroModel—An Integrated Software System for Modeling Organic and Bioorganic Molecules Using Molecular Mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Emerson, C.L.; Computation, S. Optimization Scheme of Equilibrium Structures and Transition States FELIU MASERAS IMOMM: A New Integrated Ab Initio +. J. Comput. Chem. 1995, 16, 1170–1179. [Google Scholar]
- Hochreiter, S.; Klambauer, G.; Rarey, M. Machine Learning in Drug Discovery. J. Chem. Inf. Model. 2018, 58, 1723–1724. [Google Scholar] [CrossRef]
- Poli, G.; Lapillo, M.; Jha, V.; Mouawad, N.; Caligiuri, I.; Macchia, M.; Minutolo, F.; Rizzolio, F.; Tuccinardi, T.; Granchi, C. Computationally driven discovery of phenyl(piperazin-1-yl)methanone derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 589–596. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Z.; Yu, D.J.; Zhang, Y. LS-align: An atom-level, flexible ligand structural alignment algorithm for high-throughput virtual screening. Bioinformatics 2018, 34, 2209–2218. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 11, 572–584. [Google Scholar] [CrossRef]
- Borges, N.M.; Sartori, G.R.; Ribeiro, J.F.R.; Rocha, J.R.; Martins, J.B.L.; Montanari, C.A.; Gargano, R. Similarity search combined with docking and molecular dynamics for novel hAChE inhibitor scaffolds. J. Mol. Model. 2018, 24, 41. [Google Scholar] [CrossRef]
- Burt, B.P.E. Biophysical aspects of nervous activity in relation to studies on the mode of action of insecticides. Pest. Sci. 1970, 1, 88–92. [Google Scholar] [CrossRef]
- Webb, J.E.; Green, R.A. On the penetration of insecticides through the insect cuticle. J. Exp. Biol. 1945, 22, 8–20. [Google Scholar]
- Gerolt, P. The mode of entry of contact insecticides. Pestic. Sci. 1970, 1, 209–212. [Google Scholar] [CrossRef]
- Hurst, H. Principles of insecticidal action as a guide to drug reactivity-phase distribution relationships. Trans. Faraday Soc. 1943, 39, 390–411. [Google Scholar] [CrossRef]
- Lauger, P.; Martin, H.; Muller, P. Über Konstitution und toxische Wirkung von natürlichen und neuen synthetischen insektentötenden Stoffen. Helv. Chim. Acta 1944, 27, 893–934. [Google Scholar]
- Matthews, B.J.; McBride, C.S.; DeGennaro, M.; Despo, O.; Vosshall, L.B. The neurotranscriptome of the Aedes aegypti mosquito. BMC Genom. 2016, 17, 1–20. [Google Scholar] [CrossRef]
- Onguéné, P.A.; Ntie-Kang, F.; Mbah, J.A.; Lifongo, L.L.; Ndom, J.C.; Sippl, W.; Mbaze, L.M. The potential of anti-malarial compounds derived from African medicinal plants, part III: An in silico evaluation of drug metabolism and pharmacokinetics profiling. Org. Med. Chem. Lett. 2014, 4, 6. [Google Scholar] [CrossRef]
- Chauhan, N.; Vidyarthi, A.S.; Poddar, R. Comparative Analysis of Different DNA-Binding Drugs for Leishmaniasis Cure: A Pharmacoinformatics Approach. Chem. Biol. Drug Des. 2012, 80, 54–63. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef]
- Print, I.; Online, I.; S, H.U.; Ravi, T.K.; George, S. Drug design and synthesis of 1,3,4-oxadiazolo cinnoline analogs as potent antitubercular agents. World J. Pharm. Sci. 2015, 3, 2273–2279. [Google Scholar]
- Ramos, R.; Costa, J.; Macêdo, W.; Rodrigues, A.; Silva, C.; Santos, C.; da Costa, G.; Taft, C.; Silva, R.; Souto, R.; et al. Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals 2019, 12, 20. [Google Scholar] [CrossRef]
- WHO. Temephos in Drinking-Water: Use for Vector Control in Drinking-water Sources and Containers; Background Document for Development of WHO Guidelines for Drinking-Water Quality; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Voutchkova, A.M.; Ferris, L.A.; Zimmerman, J.B.; Anastas, P.T. Toward molecular design for hazard reduction-fundamental relationships between chemical properties and toxicity. Tetrahedron 2010, 66, 1031–1039. [Google Scholar] [CrossRef]
- Regina, K.J.; Brown, S.M.; Kharasch, E.D.; Crafford, A.; Holtzman, M.J.; Campbell, S.D. P-Glycoprotein Is a Major Determinant of Norbuprenorphine Brain Exposure and Antinociception. J. Pharmacol. Exp. Ther. 2012, 343, 53–61. [Google Scholar]
- Lipinski, A.C.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Aiub, C.A.F.; Coelho, E.C.A.; Sodré, E.; Pinto, L.F.R.; Felzenszwalb, I. Genotoxic evaluation of the organophosphorous pesticide temephos. Genet. Mol. Res. 2002, 1, 159–166. [Google Scholar]
- Lee, B.M.; Scott, G.I. Acute toxicity of temephos, fenoxycarb, diflubenzuron, and methoprene and Bacillus thuringiensis var. israelensis to the mummichog (Fundulus heteroclitus). Bull. Environ. Contam. Toxicol. 1989, 43, 827–832. [Google Scholar] [CrossRef]
- Fitzpatrick, G.E.; Dowell, R.V. Survival and Emergence of Citrus Blackfly Parasitoids Mter Exposure to Insecticides. Environ. Entomol. 1981, 10, 728–731. [Google Scholar] [CrossRef]
- Printes, L.B.; Callaghan, A. A comparative study on the relationship between acetylcholinesterase activity and acute toxicity in Daphnia magna exposed to anticholinesterase insecticides. Environ. Toxicol. Chem. 2004, 23, 1241–1247. [Google Scholar] [CrossRef]
- Sparling, D.W.; Lowe, T.P.; Pinkney, A.E. Toxicity of Abate® to green frog tadpoles. Bull. Environ. Contam. Toxicol. 1997, 58, 475–481. [Google Scholar] [CrossRef]
- Junges, C.M.; Maglianese, M.I.; Lajmanovich, R.C.; Peltzer, P.M.; Attademo, A.M. Acute Toxicity and Etho-toxicity of Three Insecticides Used for Mosquito Control on Amphibian Tadpoles. Water Air Soil Pollut. 2017, 228, 143. [Google Scholar] [CrossRef]
- Anadu, D.I.; Anaso, H.U.; Onyeka, O.N.D. Acute toxicity of the insect larvicide Abate® (temephos) on the fish Tilapia melanopleura and the dragonfly larvae Neurocordelia virginiensis. J. Environ. Sci. Heal. Part B Pestic. Food Contam. Agric. Wastes 1996, 31, 1363–1375. [Google Scholar] [CrossRef]
- Zaheer-ul-haq; Wellenzohn, B.; Liedl, K.R.; Rode, B.M. Molecular Docking Studies of Natural Cholinesterase-Inhibiting Steroidal Alkaloids from Sarcococca s aligna. J. Med. Chem. 2007, 46, 5087–5090. [Google Scholar] [CrossRef]
- Yusuf, D.; Davis, A.M.; Kleywegt, G.J.; Schmitt, S. An alternative method for the evaluation of docking performance: RSR vs RMSD. J. Chem. Inf. Model. 2008, 48, 1411–1422. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Harel, M.; Kryger, G.; Rosenberry, T.L.; Mallender, W.D.; Lewis, T.; Fletcher, R.J.; Guss, J.M.; Silman, I.; Sussman, J.L. Three-dimensional structures of Drosophila melanogaster acetylcholinesterase and of its complexes with two potent inhibitors. Protein Sci. 2000, 9, 1063–1072. [Google Scholar] [CrossRef]
- Kua, J.; Zhang, Y.; McCammon, J.A. Studying enzyme binding specificity in acetylcholinesterase using a combined molecular dynamics and multiple docking approach. J. Am. Chem. Soc. 2002, 124, 8260–8267. [Google Scholar] [CrossRef]
- Atanasova, M.; Stavrakov, G.; Philipova, I.; Zheleva, D.; Yordanov, N.; Doytchinova, I. Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorg. Med. Chem. 2015, 23, 5382–5389. [Google Scholar] [CrossRef]
- Han, F.; Qian, M.; Wang, S.; Zhang, H.; Wan, Y.; Huang, H.; Guan, S. Structural Basis of Fullerene Derivatives as Novel Potent Inhibitors of Protein Acetylcholinesterase without Catalytic Active Site Interaction: Insight into the Inhibitory Mechanism through Molecular Modeling Studies. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef]
- Srivastava, P.; Tripathi, P.N.; Sharma, P.; Rai, S.N.; Singh, S.P.; Srivastava, R.K.; Shankar, S.; Shrivastava, S.K. Design and development of some phenyl benzoxazole derivatives as a potent acetylcholinesterase inhibitor with antioxidant property to enhance learning and memory. Eur. J. Med. Chem. 2018, 163, 116–135. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Massoulié, J.; Pezzementi, L.; Bon, S.; Krejci, E.; Vallette, F.M. Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 1993, 41, 31–91. [Google Scholar] [CrossRef]
- Guo, J.X.; Wu, J.J.Q.; Wright, J.B.; Lushington, G.H. Mechanistic insight into acetylcholinesterase inhibition and acute toxicity of organophosphorus compounds: A molecular modeling study. Chem. Res. Toxicol. 2006, 19, 209–216. [Google Scholar] [CrossRef]
- Kim, I.H.; Pham, V.; Jablonka, W.; Goodman, W.G.; Ribeiro, J.M.C.; Andersen, J.F. A mosquito hemolymph odorant-binding protein family member specifically binds juvenile hormone. J. Biol. Chem. 2017, 292, 15329–15339. [Google Scholar] [CrossRef]
- Noriega, F.G.; Ribeiro, J.M.C.; Koener, J.F.; Valenzuela, J.G.; Hernandez-Martinez, S.; Pham, V.M.; Feyereisen, R. Comparative genomics of insect juvenile hormone biosynthesis. Insect Biochem. Mol. Biol. 2006, 36, 366–374. [Google Scholar] [CrossRef]
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.Z.; Zhou, B.; Zhou, C.X.; Ding, X.L.; Liu, Y.X. Fluorescence study on the interaction of bovine serum albumin with P-aminoazobenzene. J. Fluoresc. 2008, 18, 109–118. [Google Scholar]
- Perozzo, R.; Folkers, G.; Scapozza, L. Thermodynamics of protein-ligand interactions: History, presence, and future aspects. J. Recept. Signal Transduct. 2004, 24, 1–52. [Google Scholar] [CrossRef]
- Mofidifar, S.; Sohraby, F.; Bagheri, M.; Aryapour, H. Repurposing existing drugs for new AMPK activators as a strategy to extend lifespan: A computer-aided drug discovery study. Biogerontology 2018, 19, 133–143. [Google Scholar] [CrossRef]
- Milan, D.; Peal, D. National Center for Biotechnology Information. U.S. Patent 2009005354, 2013. [Google Scholar] [CrossRef]
- Maienfisch, P. Selective Feeding Blockers: Pymetrozine, Flonicamid, and Pyrifluquinazon. Mod. Crop Prot. Compd. 2019, 1501–1526. [Google Scholar]
- Ausborn, J. The insecticide pymetrozine selectively affects chordotonal mechanoreceptors. J. Exp. Biol. 2005, 208, 4451–4466. [Google Scholar] [CrossRef]
- Sechser, B.; Reber, B.; Bourgeois, F. Pymetrozine: Selectivity spectrum to beneficial arthropods and fitness for integrated pest management. Anzeiger fur Schadlingskd. 2002, 75, 72–77. [Google Scholar] [CrossRef]
- Horowitz, A.R.; Ishaaya, I. Insect Pest Management: Field and Protected Crops; Springer Science & Business Media: New York, NY, USA, 2004. [Google Scholar]
- Duvall, L.B.; Ramos-espiritu, L.; Barsoum, K.E.; Fraser, J.; Vosshall, L.B. Novel small molecule agonists of an Aedes aegypti neuropeptide Y receptor block mosquito biting behavior. bioRxiv 2018. [Google Scholar] [CrossRef]
- Duvall, L.B.; Ramos-Espiritu, L.; Barsoum, K.E.; Glickman, J.F.; Vosshall, L.B. Small-Molecule Agonists of Ae. aegypti Neuropeptide Y Receptor Block Mosquito Biting. Cell 2019, 176, 687–701. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Lee, A.S.Y.; Berger, J.M.; Doudna, J.A. Structure of Human cGAS Reveals a Conserved Family of Second-Messenger Enzymes in Innate Immunity. Cell Rep. 2013, 3, 1362–1368. [Google Scholar] [CrossRef]
- Ascano, M.; Imaeda, T.; Vincent, J.; Adura, C.; Asano, Y.; Reasoner, S.; Patel, D.J.; Gao, P.; Aso, K.; Lama, L.; et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 2017, 8, 750. [Google Scholar]
- Trosset, J.-Y.; Scheraga, H.A. Reaching the global minimum in docking simulations: A Monte Carlo energy minimization approach using Bezier splines. Proc. Natl. Acad. Sci. USA 2002, 95, 8011–8015. [Google Scholar] [CrossRef]
- Wiggers, P.; Mertens, B.; Rothkrantz, L. Dynamic Bayesian networks for situational awareness in the presence of noisy. In Proceedings of the 12th International Conference on Computer Systems and Technologies, Vienna, Austria, 16–17 June 2011; pp. 411–416. [Google Scholar]
- Cannalire, R.; Tarantino, D.; Astolfi, A.; Barreca, M.L.; Sabatini, S.; Massari, S.; Tabarrini, O.; Milani, M.; Querat, G.; Mastrangelo, E.; et al. Functionalized 2,1-benzothiazine 2,2-dioxides as new inhibitors of Dengue NS5 RNA-dependent RNA polymerase. Eur. J. Med. Chem. 2018, 143, 1667–1676. [Google Scholar] [CrossRef]
- Cabarcas-Montalvo, M.; Maldonado-Rojas, W.; Montes-Grajales, D.; Bertel-Sevilla, A.; Wagner-Döbler, I.; Sztajer, H.; Reck, M.; Flechas-Alarcon, M.; Ocazionez, R.; Olivero-Verbel, J. Discovery of antiviral molecules for dengue: In silico search and biological evaluation. Eur. J. Med. Chem. 2016, 110, 87–97. [Google Scholar] [CrossRef]
- Bureau, R.; Rault, S.; Dulin, F.; Lozano, S.; Halm-Lemeille, M.-P.; Lepailleur, A.; Sopkova-de Oliveira Santos, J. Interpretation of honeybees contact toxicity associated to acetylcholinesterase inhibitors. Ecotoxicol. Environ. Saf. 2012, 79, 13–21. [Google Scholar]
- Silva, R.; Brasil, D.; Poiani, J.; Ramos, R.; Costa, J.; Santos, C.; Silva, C. Ligand- and structure-based virtual screening from 16-(N,N-diisobutylaminomethyl)-6α-hydroxyivouacapan-7β,17β-lactone compound with potential anti-prostate cancer activity. J. Serbian Chem. Soc. 2019, 84, 153–174. [Google Scholar] [CrossRef]
- Cruz, J.; Do Socorro Barros Brasil, D.; da Silva Lopes Costa, K.; Silva, L.B.; da Cruz Macedo, W.J.; dos Santos, C.B.R.; da Silva Ramos, R.; da Silva Costa, J.V.; de Paula da Silva, C.H.T. Virtual Screening and Statistical Analysis in the Design of New Caffeine Analogues Molecules with Potential Epithelial Anticancer Activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | ID | Star a | CNS b | MW c | ClogP/w d | log (BB) e | MDCK (nm/s) f | HBD g | HBA h | R5 i |
|---|---|---|---|---|---|---|---|---|---|---|
| Normal range | - | 0–5 | −2 to +2 | 130–725 | −2.0 to 6.5 | −3.0 to 1.2 | <25 poor, >500 great | 0–6 | 2–20 | Max. 4 |
| TRI (toxic) compounds l | - | - | - | - | −2.1 to 6.99 | −1.67 to 0.794 | 25.65 to 104 | 0–3 | 0–9.53 | - |
| Temephos | - | 5 | 0 | 466.458 | 7.333 | −0.491 | 104 | 0 | 3.00 | 1 |
| omega_22273_1_1_66 | L66 | 0 | 0 | 345.406 | 4.251 | −0.430 | 947.552 | 1 | 5.00 | 0 |
| omega_23792_1_1_68 | L68 | 0 | 0 | 350.425 | 3.507 | −0.656 | 563.417 | 1 | 6.50 | 0 |
| omega_32859_1_1_12 | L12 | 0 | 1 | 315.429 | 4.163 | 0.269 | 3898.807 | 0 | 3.75 | 0 |
| omega_5087_1_1_46 | L46 | 0 | 1 | 287.401 | 2.931 | 0.079 | 3128.836 | 0 | 4.70 | 0 |
| omega_36855_1_1_257 | L257 | 0 | 0 | 372.441 | 3.550 | −0.423 | 1578.497 | 0 | 7.20 | 0 |
| omega_31303_1_1_291 | L291 | 0 | 0 | 348.404 | 3.560 | −0.644 | 538.073 | 1 | 6.25 | 0 |
| omega_20865_1_1_102 | L102 | 0 | 1 | 282.401 | 2.380 | −0.049 | 3166.685 | 0 | 4.70 | 0 |
| omega_12589_1_1_18 | L18 | 0 | 0 | 381.474 | 3.147 | −0.425 | 696.935 | 0 | 7.50 | 0 |
| omega_45720_1_1_22 | L22 | 0 | 0 | 339,317 | 3.894 | −0.052 | 3333.563 | 1 | 5.70 | 0 |
| omega_28697_1_1_35 | L35 | 0 | 0 | 354.451 | 2.310 | −0.581 | 620.602 | 0 | 7.50 | 0 |
| omega_34836_1_1_50 | L50 | 0 | 0 | 341.452 | 2.661 | −0.581 | 648.835 | 1 | 6.50 | 0 |
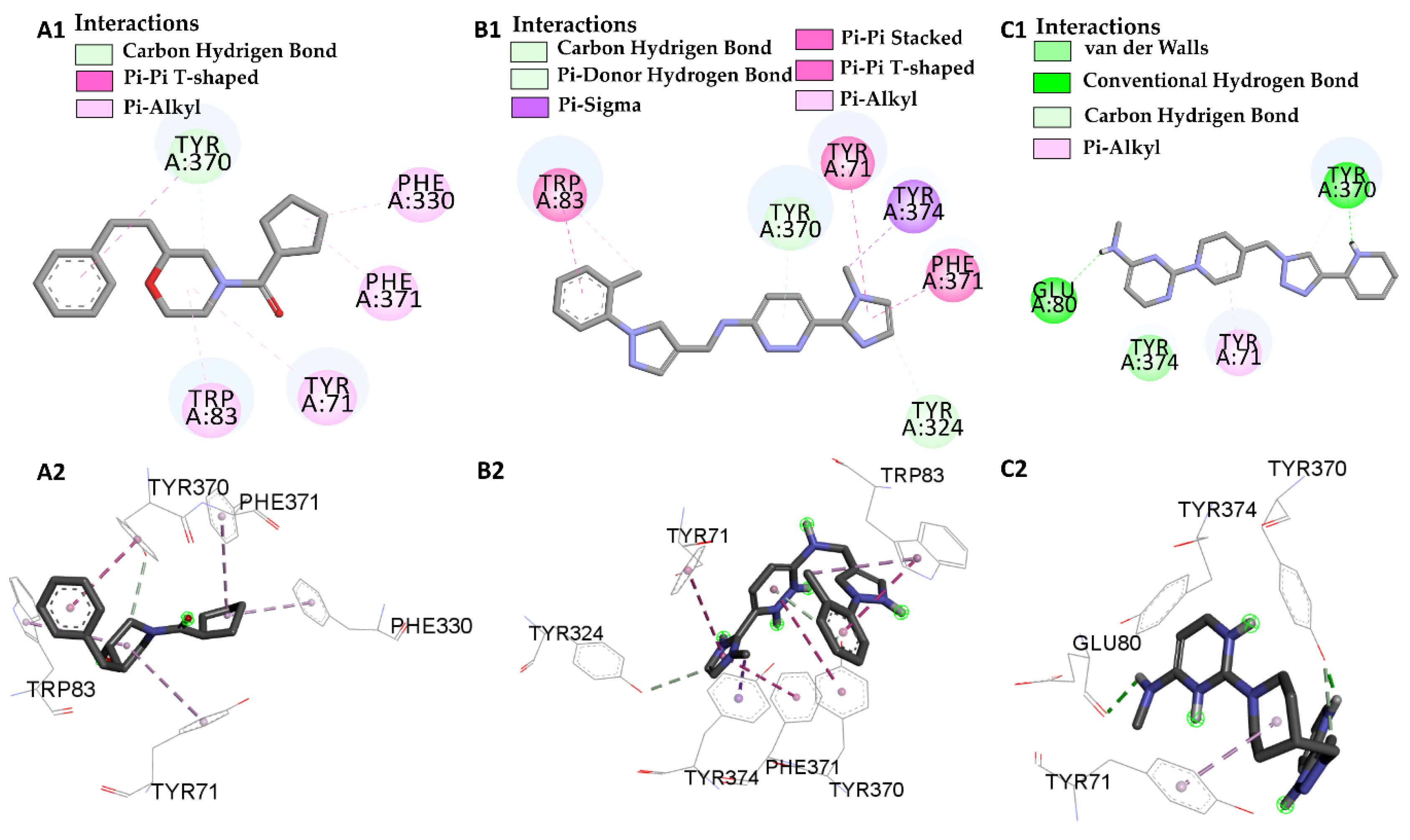
| Molecular Docking | Residues | Distance (Å) | Type | ΔG (kcal/mol) |
|---|---|---|---|---|
| I40 vs PDB ID 1QON | Trp-83 | 4.1/4.497 | Pi-Alkyl | −12.66 |
| Trp-83 | 3.66/5.66 | Pi-Pi Stacked | ||
| His-480 | 2.75 | Conventional Hydrogen Bond | ||
| Phe-371 | 4.88 | Pi-Alkyl | ||
| Try-370 | 3.79/4.18 | Pi-Pi Stacked | ||
| Try-370 | 2.99 | Pi-donor | ||
| Try-374 | 4.34 | Pi-Alkyl | ||
| Try-71 | 4.49 | Pi-Pi Stacked | ||
| Try-71 | 3.98 | Pi-donor | ||
| L46 vs PDB ID 1QON | Trp-83 | 5.00 | Pi-Alkyl | −6.94 |
| Try-370 | 4.37 | Pi-T-Stacked | ||
| Try-370 | 3.26 | Carbon Hydrogen Bond | ||
| Try-71 | 5.41 | Pi-Alkyl | ||
| Phe-330 | 4.39 | Pi-Alkyl | ||
| Phe-371 | 5.41 | Pi-Alkyl | ||
| L66 vs PDB ID 1QON | Trp-83 | 4.41 | Pi-Pi-Stacked | −9.55 |
| Trp-83 | 4.65 | Pi-Alkyl | ||
| Try-370 | 5.98 | Pi-Pi-Stacked | ||
| Try-371 | 4.01 | Pi-Alkyl | ||
| Try-324 | 3.68 | Carbon Hydrogen Bond | ||
| Try-374 | 3.89 | Pi-Sigma | ||
| Phe-371 | 4.55 | Pi-T-Stacked | ||
| L68 vs PDB ID 1QON | Try-370 | 1.73 | Convent. Hydrogen Bond | −1.33 |
| Try-370 | 3.32 | Carbon Hydrogen Bond | ||
| Try-374 | 3.98 | Pi-Sigma | ||
| Try-371 | 4.01 | Pi-Alkyl | ||
| Glu-80 | 2.80 | Convent. Hydrogen Bond |
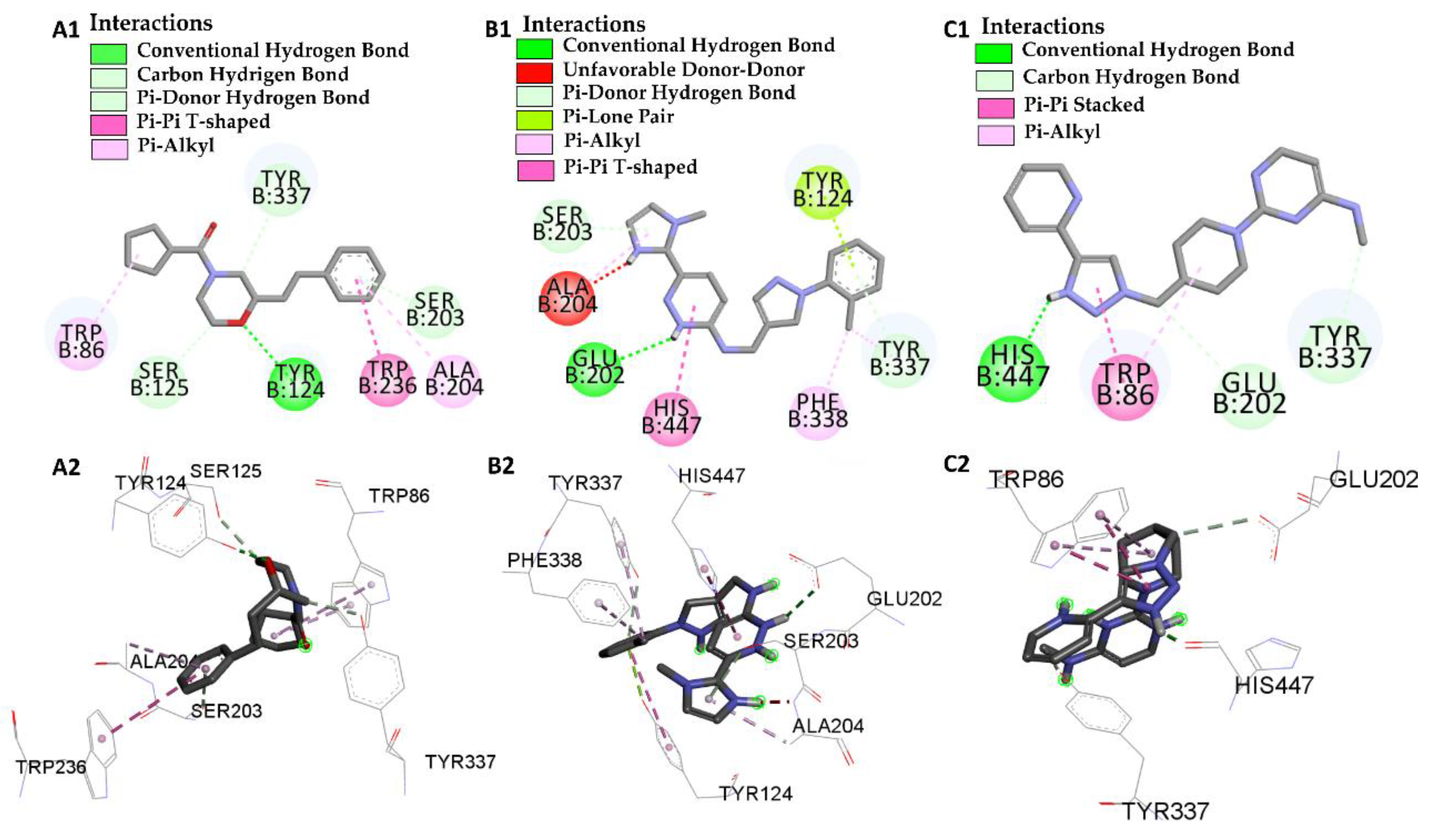
| Molecular Docking | Residues | Distance (Å) | Type | ΔG (kcal/mol) |
|---|---|---|---|---|
| GNT vs PDB ID 4EY6 | Trp-86 | 5.08/4.54/4.14 | Pi-Alkyl | −9.72 |
| Ser-203 | 2.61 | Conventional Hydrogen Bond | ||
| His-447 | 3.54 | Carbon Hydrogen Bond | ||
| Glu-202 | 2.31 | Conventional Hydrogen Bond | ||
| Tyr-337 | 5.45 | Pi-Alkyl | ||
| Tyr-124 | 3.73 | Carbon Hydrogen Bond | ||
| L46 vs PDB ID 4EY6 | Trp-86 | 4.30 | Pi-Alkyl | −7.64 |
| Trp-86 | 5.31 | Pi-Alkyl | ||
| Ser-125 | 2.78 | Carbon Hydrogen Bond | ||
| Tyr-124 | 2.65 | Conventional Hydrogen Bond | ||
| Trp-286 | 5.64 | Pi-Pi-T-Shaped | ||
| Ala-204 | 5.18 | Pi-Alkyl | ||
| Ser-203 | 2.78 | Pi-donor Hydrogen Bond | ||
| Tyr-337 | 2.66 | Carbon Hydrogen Bond | ||
| L66 vs PDB ID 4EY6 | Ser-203 | 2.93 | Pi-Donor | −3.88 |
| His-447 | 5.54 | Pi-T-Stacked | ||
| Tyr-124 | 2.91 | Pi-T-Stacked | ||
| Tyr-124 | 5.63 | Carbon Hydrogen Bond | ||
| Tyr-337 | 4.73 | Pi-Alkyl | ||
| Tyr-337 | 3.43 | Pi-Donor | ||
| Glu-202 | 2.72 | Conventional Hydrogen Bond | ||
| Phe-338 | 4.88 | Pi-Alkyl | ||
| Ala-204 | 4.09 | Pi-Alkyl | ||
| Ala-204 | 2.08 | Donor-Donor | ||
| L68 vs PDB ID 4EY6 | His-447 | 1.74 | Conventional Hydrogen Bond | −5.31 |
| Trp-86 | 4.80/5.28 | Pi-Alkyl | ||
| Trp-86 | 3.42/3.90 | Pi-T-Stacked | ||
| Try-337 | 2.97 | Carbon Hydrogen Bond | ||
| Glu-202 | 3.74 | Conventional Hydrogen Bond |
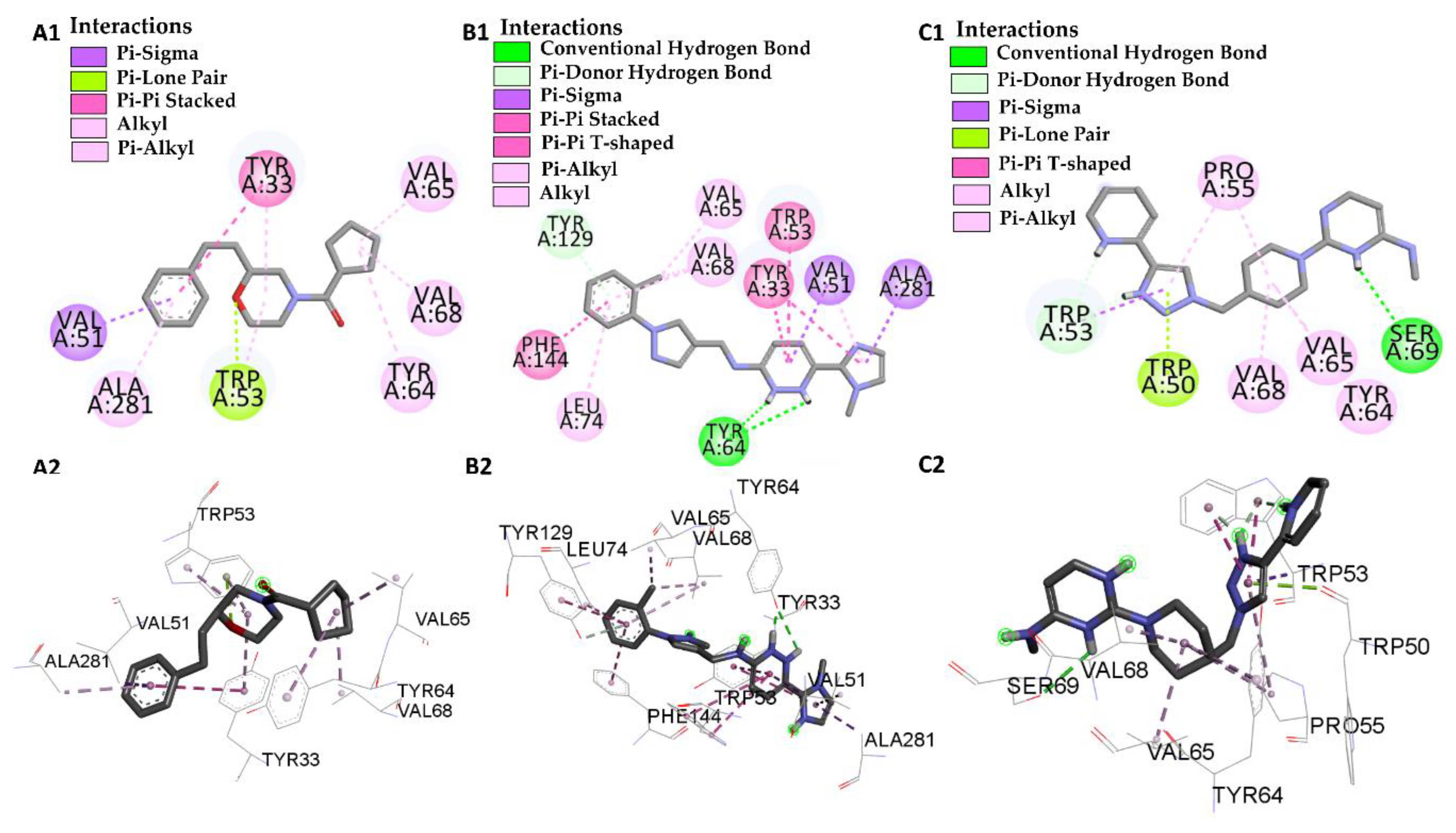
| Molecular Docking | Residues | Distance (Å) | Type | ΔG (Kcal·mol−1) |
|---|---|---|---|---|
| JHIII vs PDB ID 5V13 | Trp-53 | 4.53/4.95/5.02 | Pi-Alkyl | −5.61 |
| Val-51 | 4.74 | Alkyl | ||
| Leu-74 | 4.98 | Alkyl | ||
| Val-68 | 4.89 | Alkyl | ||
| Tyr-129 | 5.28/5.80 | Pi-Alkyl | ||
| Tyr-133 | 5.28/5.29 | Pi-Alkyl | ||
| Tyr-64 | 5.01 | Pi-Alkyl | ||
| Ile-140 | 4.98 | Alkyl | ||
| Phe-144 | 5.00 | Pi-Alkyl | ||
| L46 vs PDB ID 5V13 | Trp 53 | 4.12 | Pi-Alkyl | −8.46 |
| Trp 53 | 2.98 | Pi-Lone-Pair | ||
| Val-51 | 3.94 | Pi-Sigma | ||
| Val-68 | 3.93 | Alkyl | ||
| Val-65 | 3.65 | Alkyl | ||
| Ala-281 | 4.24 | Pi-Alkyl | ||
| Tyr-64 | 5.25 | Pi-Alkyl | ||
| Tyr-33 | 5.07 | Pi-Pi-Stacked | ||
| Tyr-33 | 4.40 | Pi-Alkyl | ||
| L66 vs PDB ID 5V13 | Trp-53 | 4.69/2.40 | Pi-T-Stacked | −10.56 |
| Val-51 | 4.94 | Pi-Alkyl | ||
| Val-51 | 3.72 | Pi-Sigma | ||
| Val-68 | 3.81/5.32 | Pi-Alkyl | ||
| Val-65 | 3.90 | Pi-Alkyl | ||
| Phe-144 | 5.24 | Pi-T-Stacked | ||
| Tyr-129 | 4.95 | Pi-T-Stacked | ||
| Tyr-129 | 3.23 | Pi-donor | ||
| Tyr-64 | 2.80/1.90 | Conventional Hydrogen Bond | ||
| Tyr-33 | 5.69/4.40 | Pi-Pi-Stacked | ||
| Ala-281 | 3.76 | Pi-Sigma | ||
| Leu-74 | 5.30 | Pi-Alkyl | ||
| L68 vs PDB ID 5V13 | Trp-53 | 3.83 | Pi-donor Hydrogen Bond | −8.70 |
| Pro-55 | 5.34 | Pi-Alkyl | ||
| Trp-50 | 2.99 | Pi-lone Pair | ||
| Val-68 | 3.93 | Alkyl | ||
| Val-65 | 4.35 | Alkyl | ||
| Tyr-64 | 4.67 | Alkyl | ||
| Ser-69 | 2.75 | Conventional Hydrogen Bond |
| Receptor | Ligand | Ligand Coordinates of the Grid Center | Grid Size (Points) |
|---|---|---|---|
| AChE (PDB ID 1QON) | 9-(3-Iodobenzylamino)-1,2,3,4-tetrahydroacridine | X = 33.8687 Y = 67.6341 Z = 10.1163 | 74 x 33 y 18 z |
| AChE (PDB ID 4EY6) | (–)-Galantamine | X = 8.2053 Y = −60.5991 Z = −24.2265 | 74 x 32 y 18 z |
| Juvenile hormone (PDB ID 5V13) | Methyl(2E,6E)-9-[(2R)-3,3-dimethyloxiran-2-yl]-3,7-dimethylnona-2,6-dienoate | X = -251.7312 Y = 9.0631 Z = 353.4562 | 48 x 29 y 20 z |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
V. da Costa, G.; Ferreira, E.F.B.; da S. Ramos, R.; B. da Silva, L.; M. F. de Sá, E.; K. P. da Silva, A.; M. Lobato, C.; N. P. Souto, R.; T. de P. da Silva, C.H.; B. Federico, L.; et al. Hierarchical Virtual Screening of Potential Insectides Inhibitors of Acetylcholinesterase and Juvenile Hormone from Temephos. Pharmaceuticals 2019, 12, 61. https://doi.org/10.3390/ph12020061
V. da Costa G, Ferreira EFB, da S. Ramos R, B. da Silva L, M. F. de Sá E, K. P. da Silva A, M. Lobato C, N. P. Souto R, T. de P. da Silva CH, B. Federico L, et al. Hierarchical Virtual Screening of Potential Insectides Inhibitors of Acetylcholinesterase and Juvenile Hormone from Temephos. Pharmaceuticals. 2019; 12(2):61. https://doi.org/10.3390/ph12020061
Chicago/Turabian StyleV. da Costa, Glauber, Elenilze F. B. Ferreira, Ryan da S. Ramos, Luciane B. da Silva, Ester M. F. de Sá, Alicia K. P. da Silva, Cássio M. Lobato, Raimundo N. P. Souto, Carlos Henrique T. de P. da Silva, Leonardo B. Federico, and et al. 2019. "Hierarchical Virtual Screening of Potential Insectides Inhibitors of Acetylcholinesterase and Juvenile Hormone from Temephos" Pharmaceuticals 12, no. 2: 61. https://doi.org/10.3390/ph12020061
APA StyleV. da Costa, G., Ferreira, E. F. B., da S. Ramos, R., B. da Silva, L., M. F. de Sá, E., K. P. da Silva, A., M. Lobato, C., N. P. Souto, R., T. de P. da Silva, C. H., B. Federico, L., M. C. Rosa, J., & B. R. dos Santos, C. (2019). Hierarchical Virtual Screening of Potential Insectides Inhibitors of Acetylcholinesterase and Juvenile Hormone from Temephos. Pharmaceuticals, 12(2), 61. https://doi.org/10.3390/ph12020061