Strategies for the Development of Glycomimetic Drug Candidates


Abstract
1. Introduction
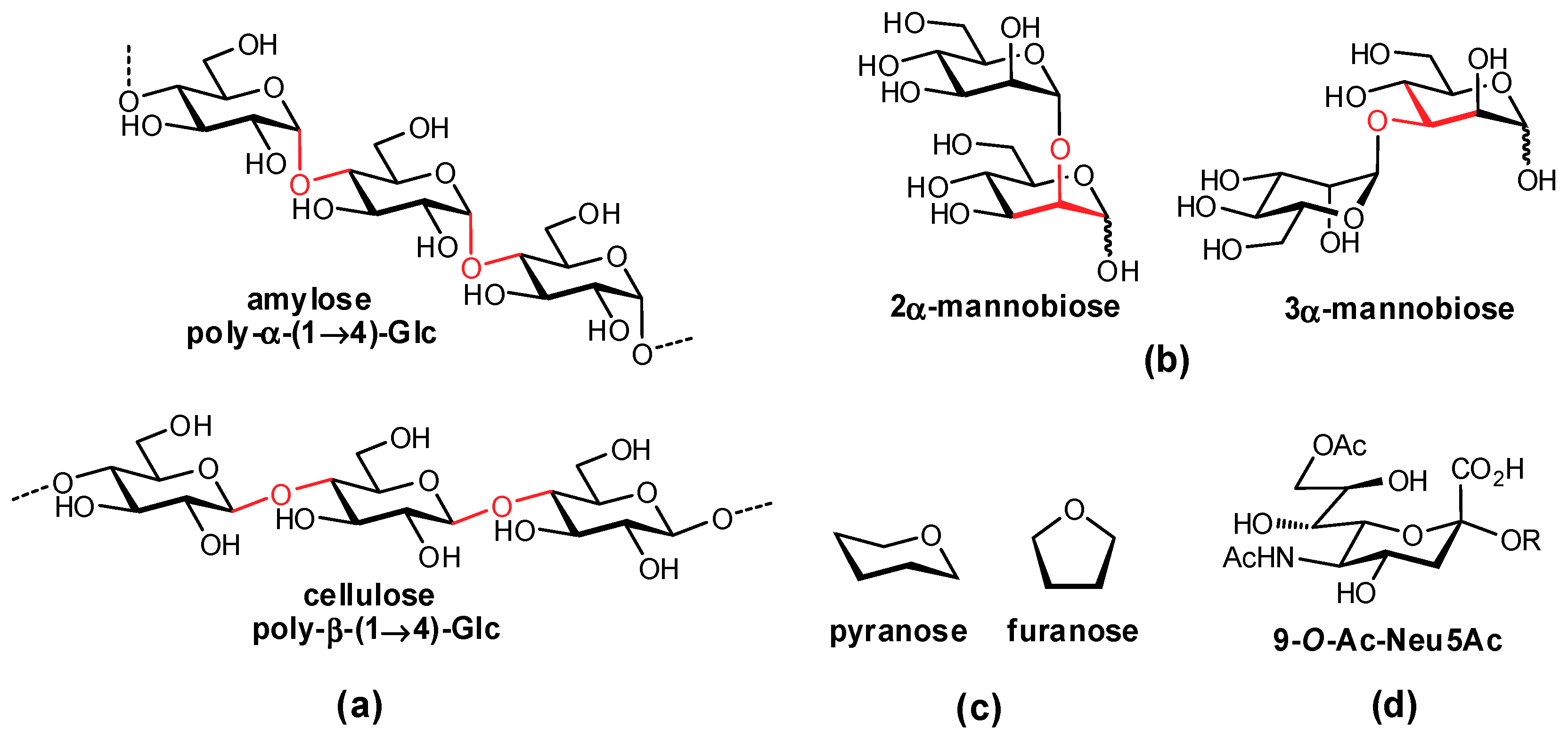
1.1. Carbohydrates in Disease
1.2. Native Carbohydrates as Pharmaceutical Agents
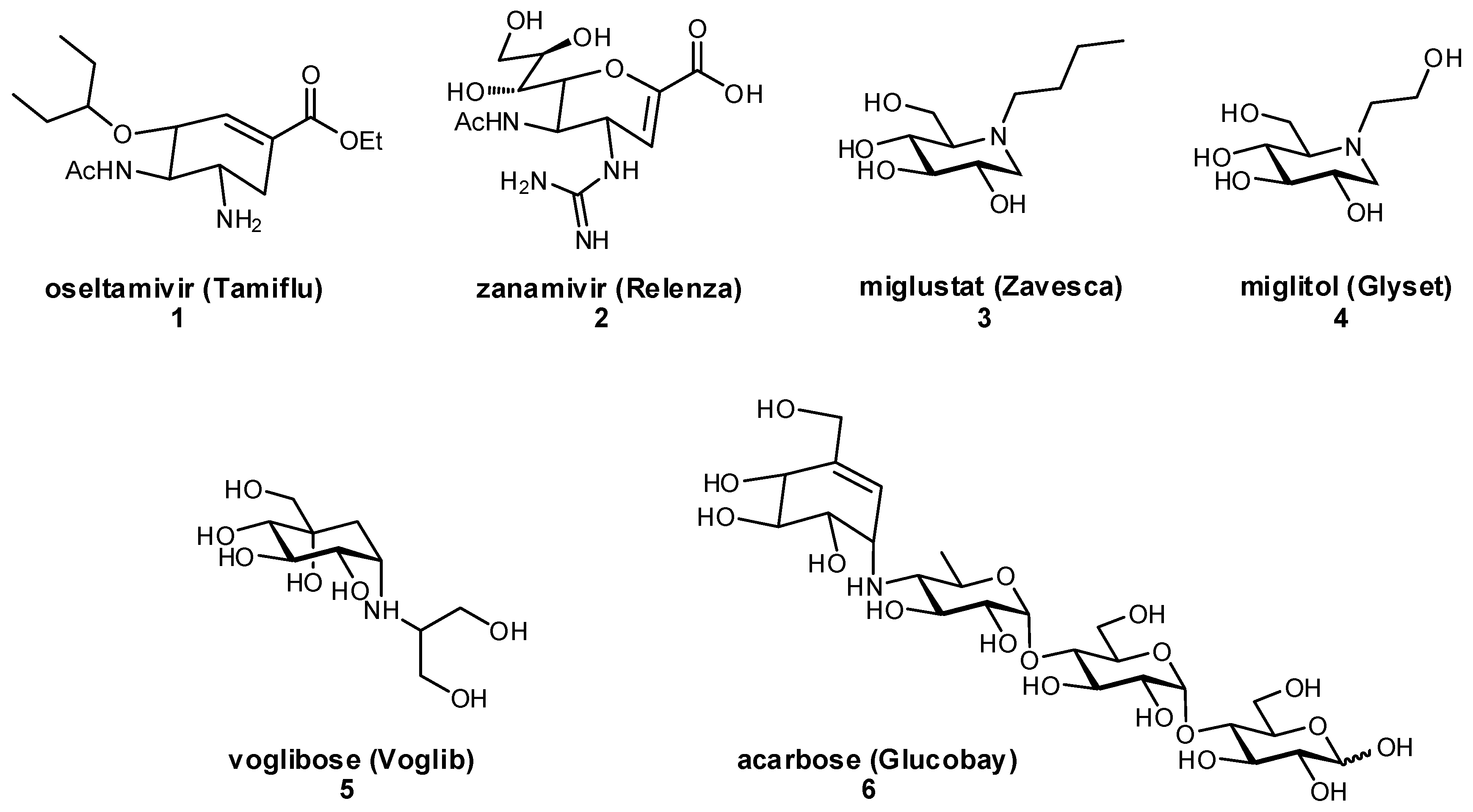
1.3. Development of Glycomimetic Drug Candidates
2. Glycomimetic Design – Strategies to Improve Binding Affinities
2.1. Deoxygenation
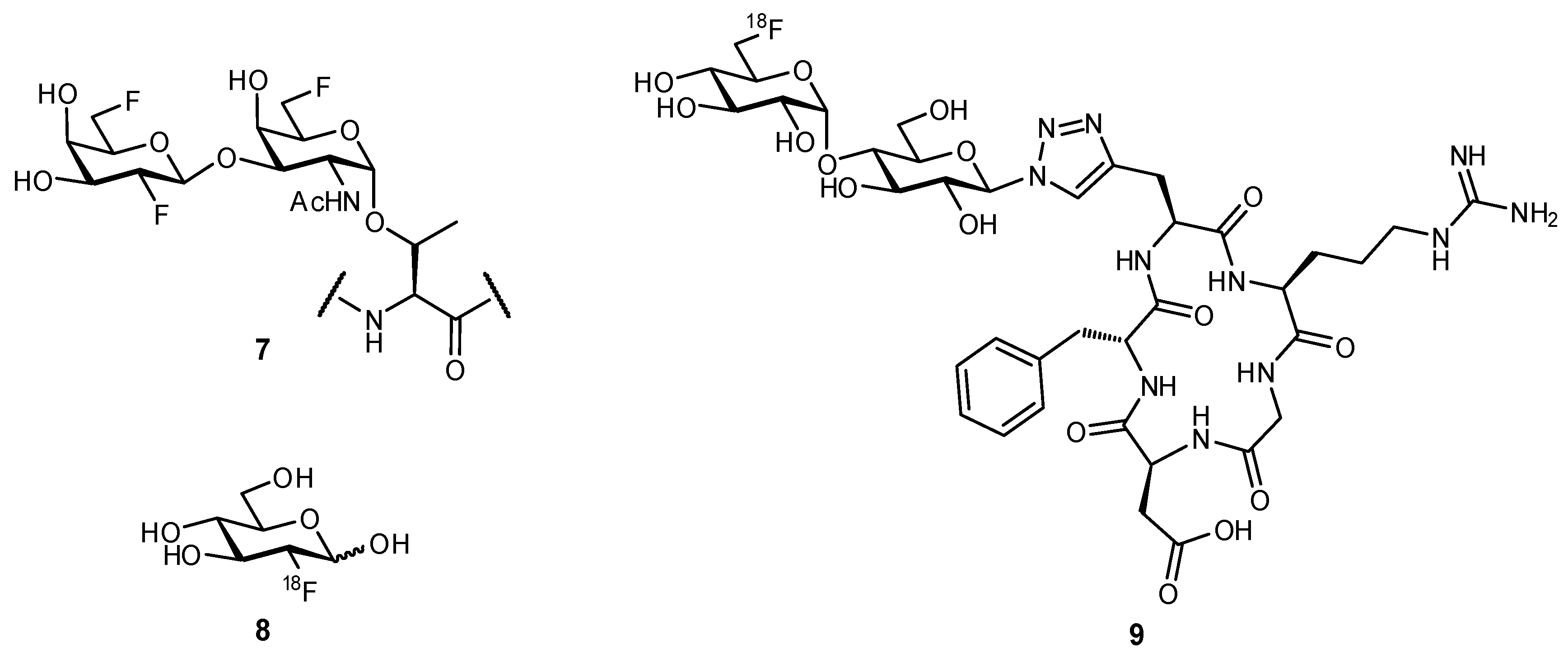
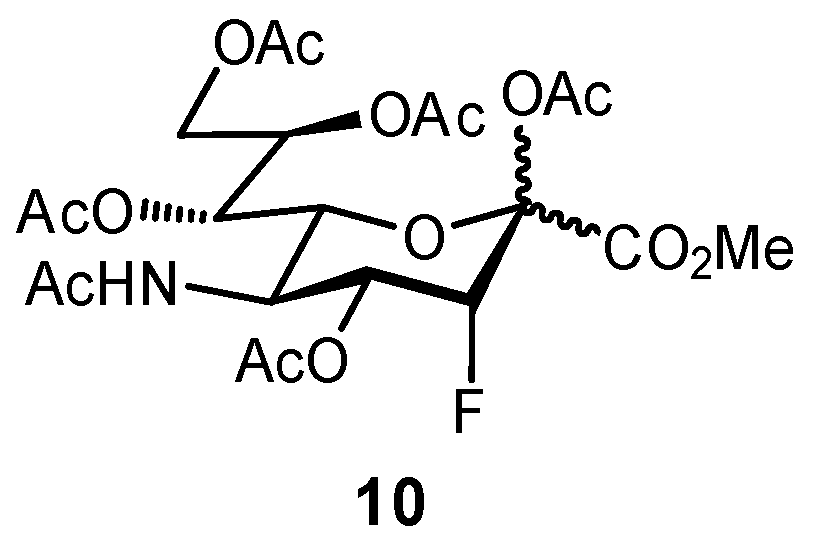
2.2. Biomimetic Replacement of Functional Groups
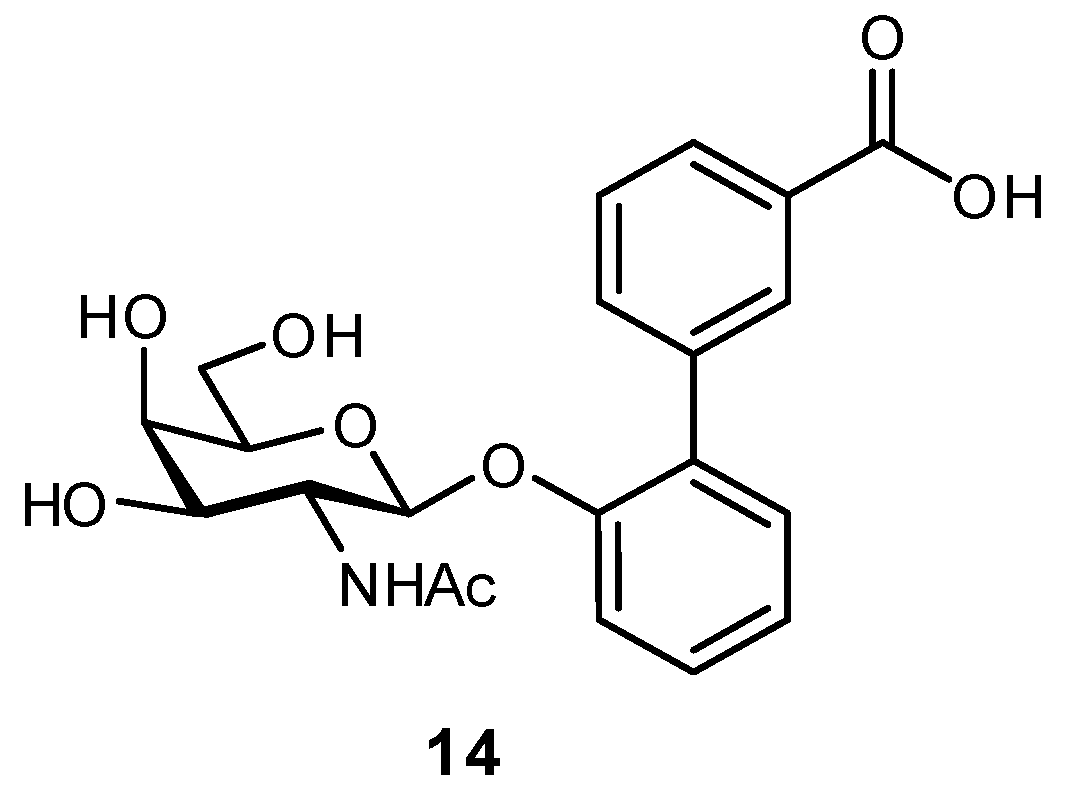
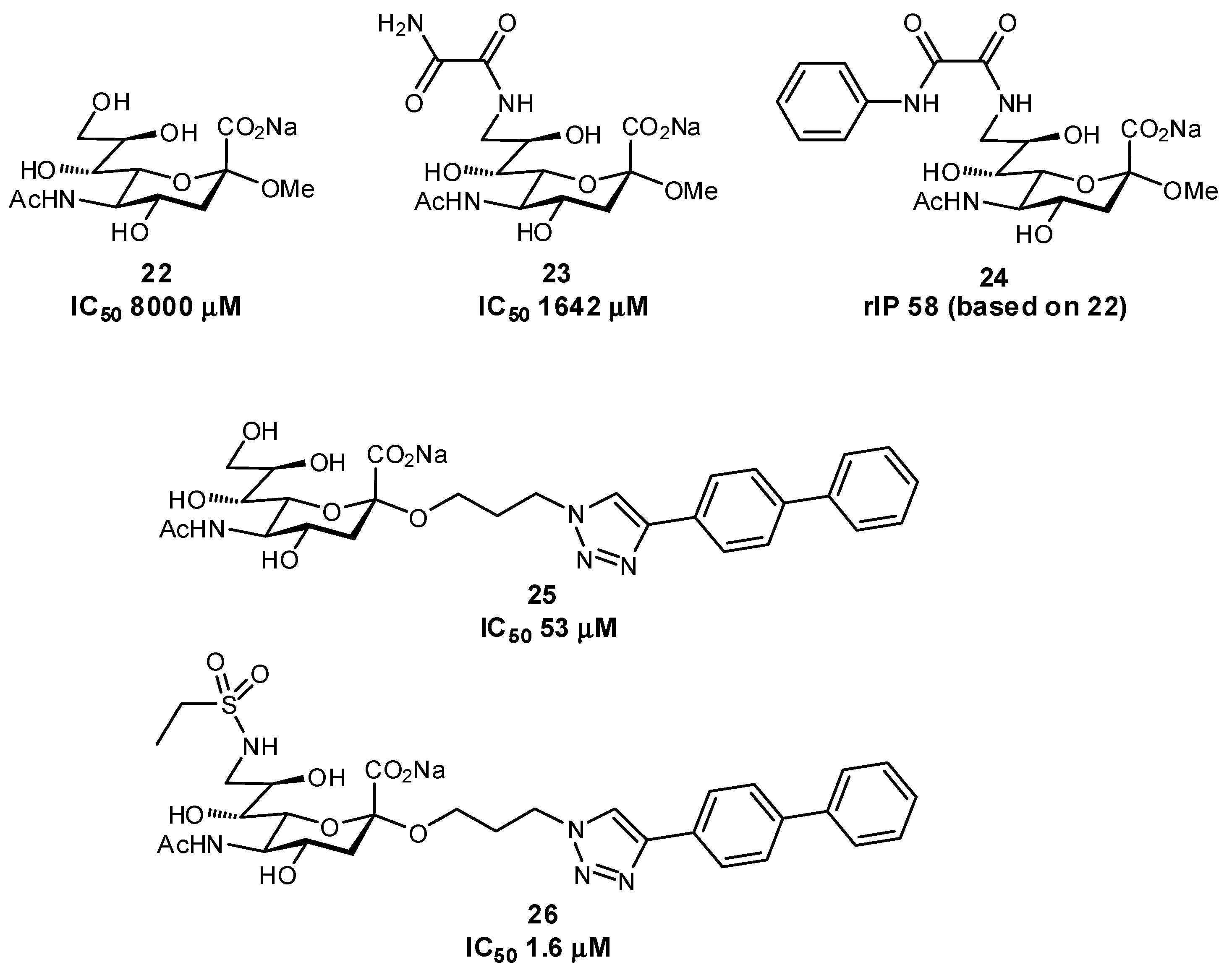
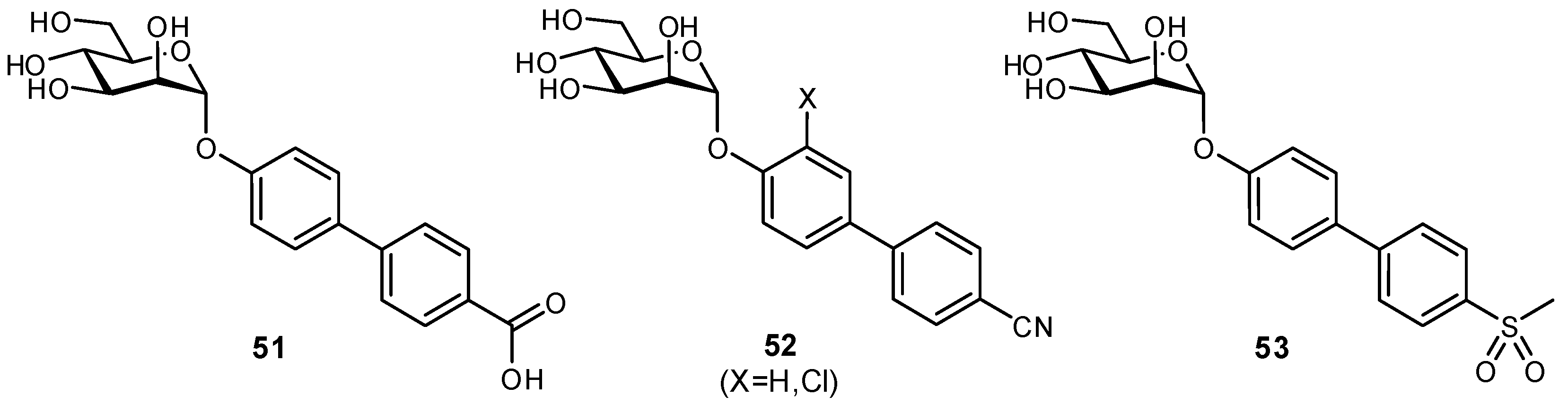
2.3. Targeting Neighboring Regions of the Binding Site
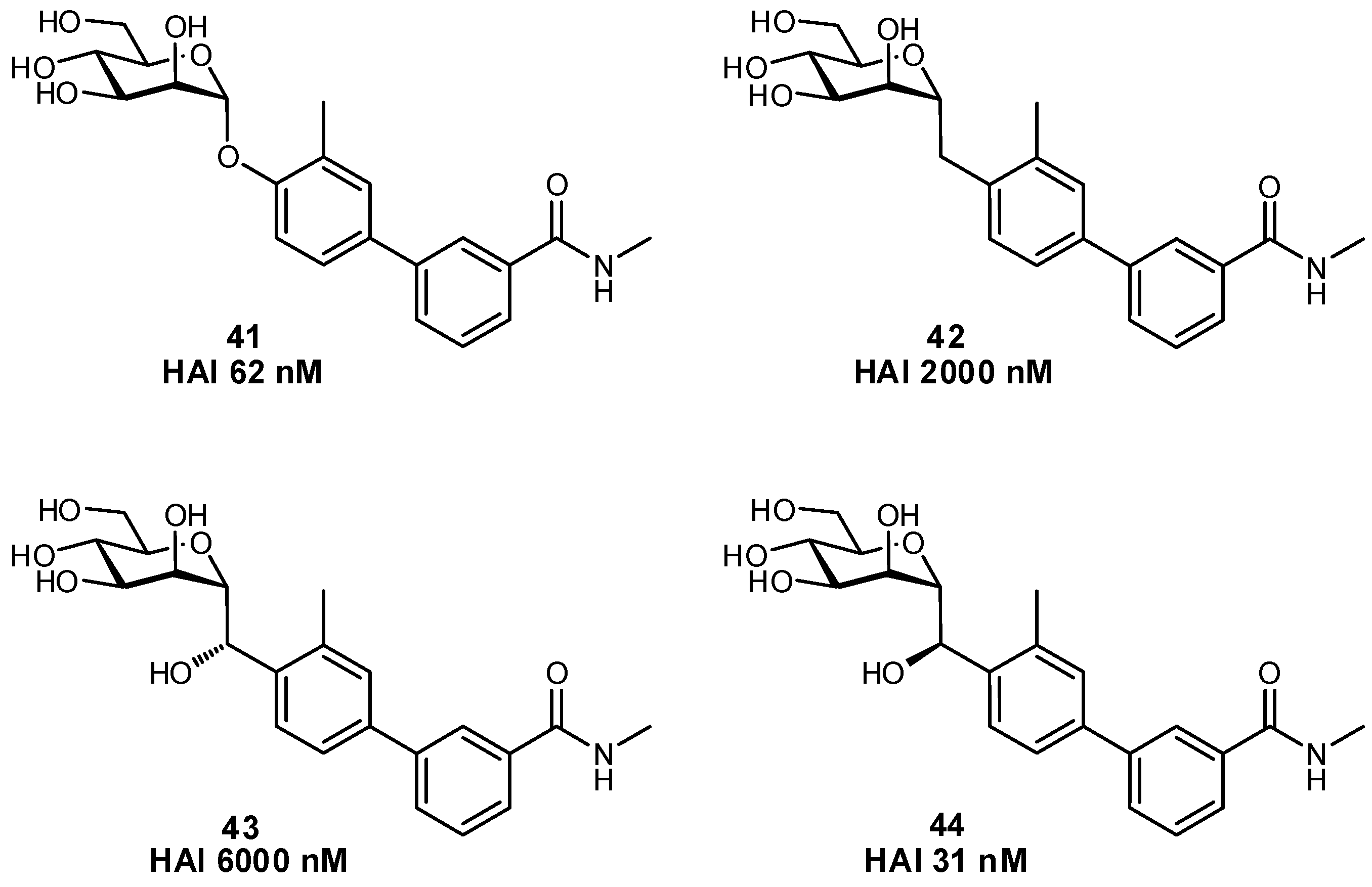
2.4. Conformational Pre-organization
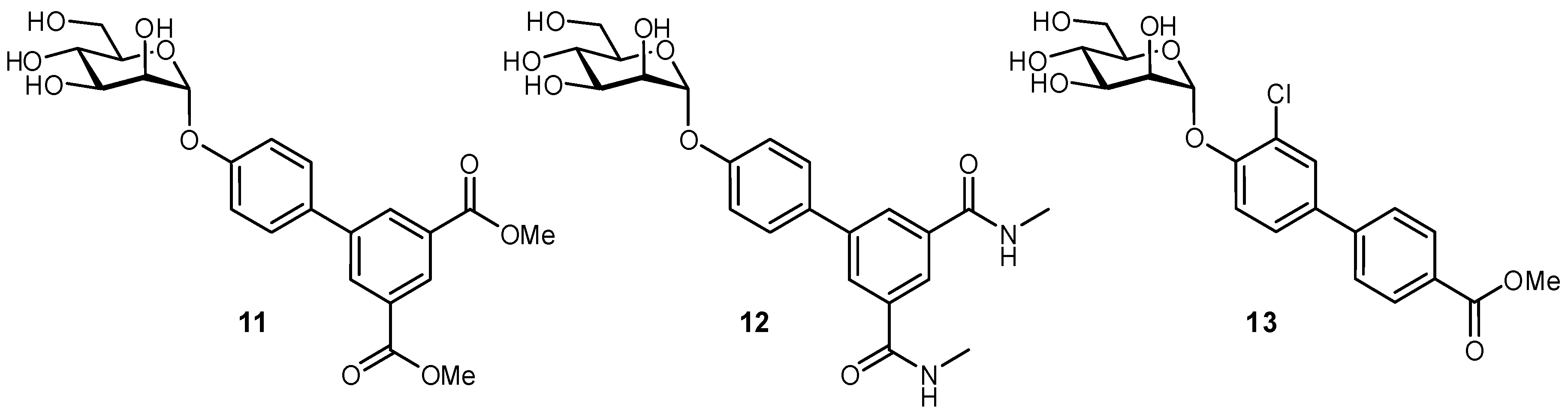
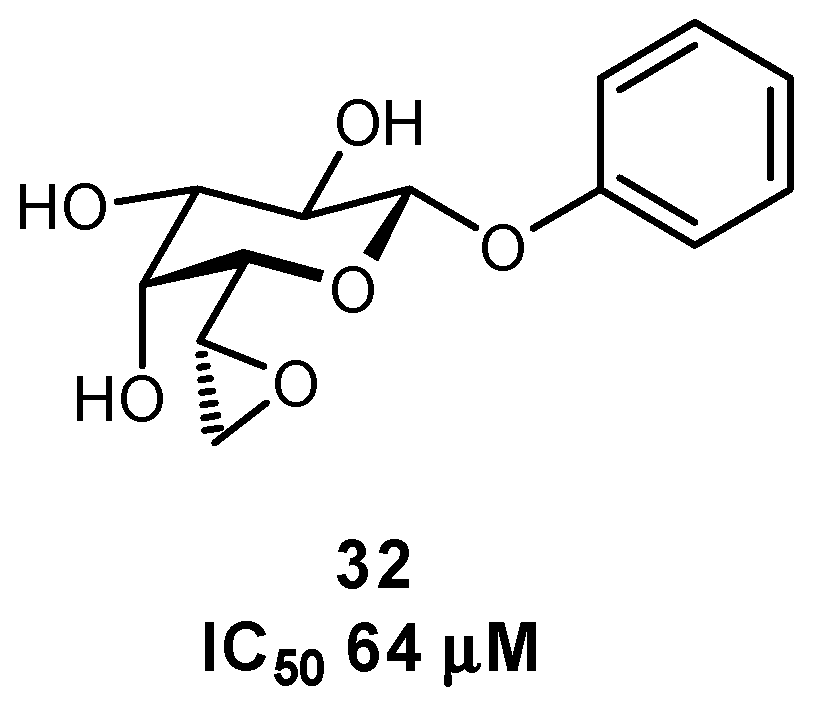
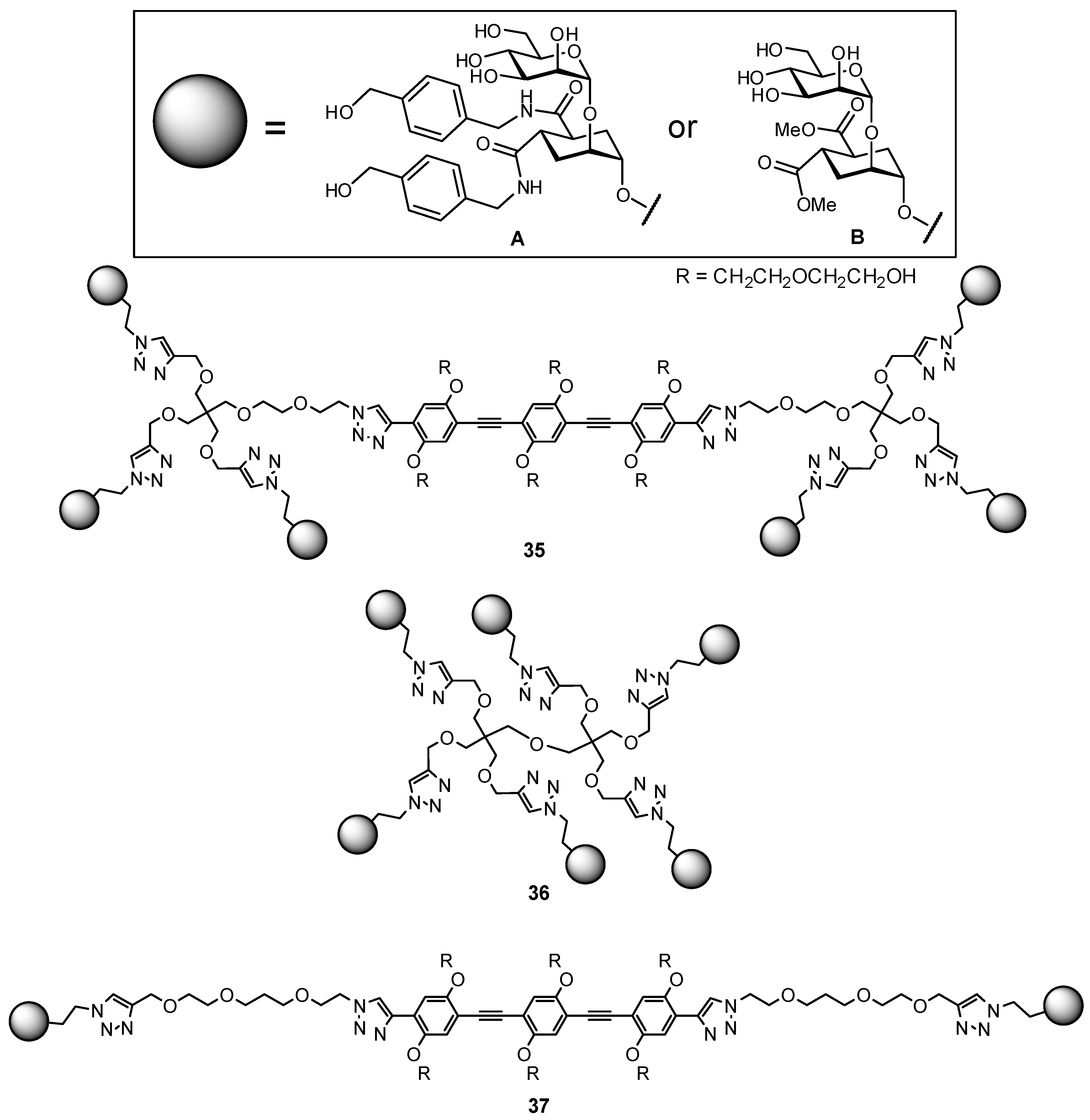
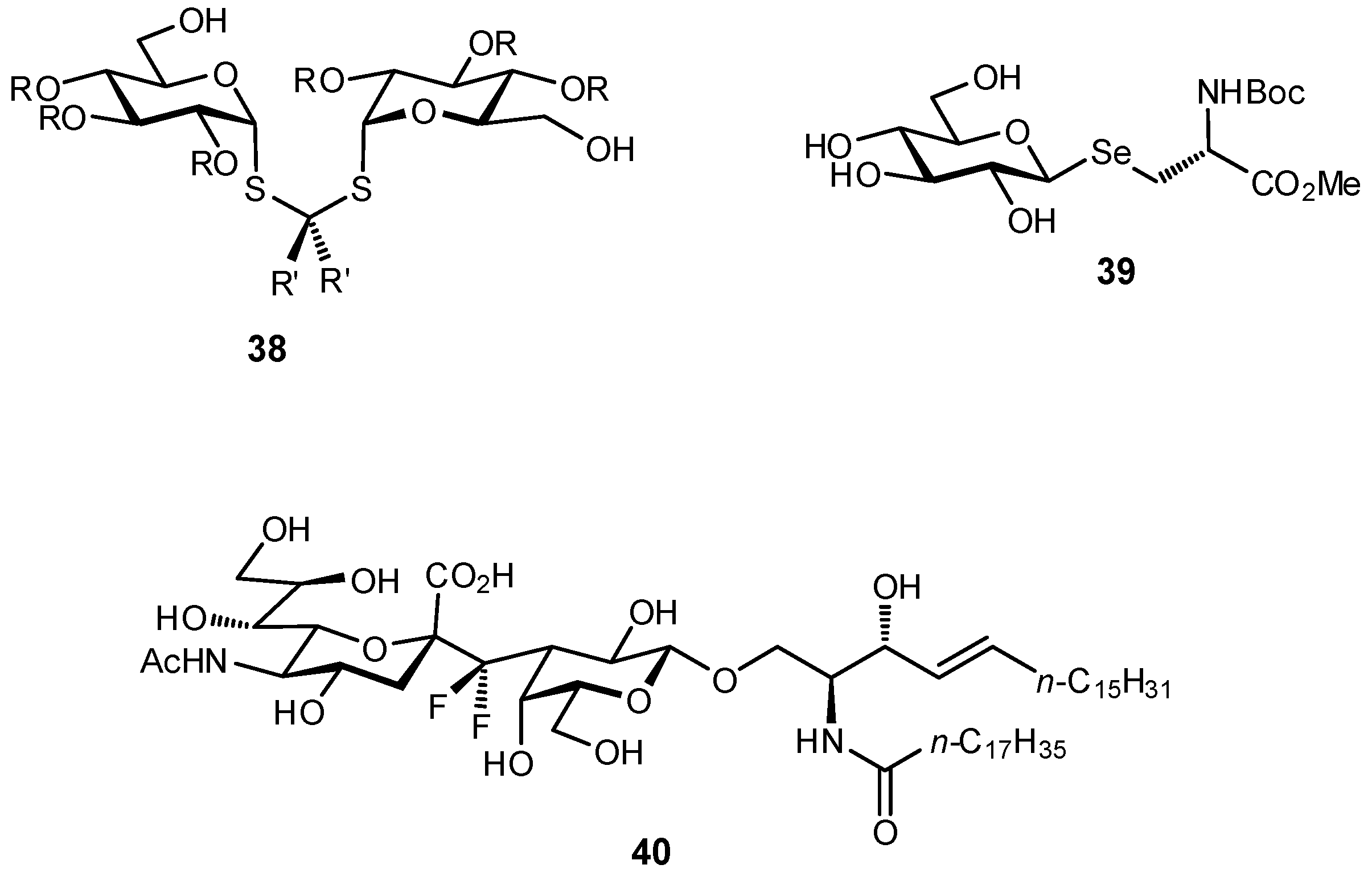
2.5. Multivalency
3. Glycomimetic Design—Strategies to Improve Pharmacokinetic Properties
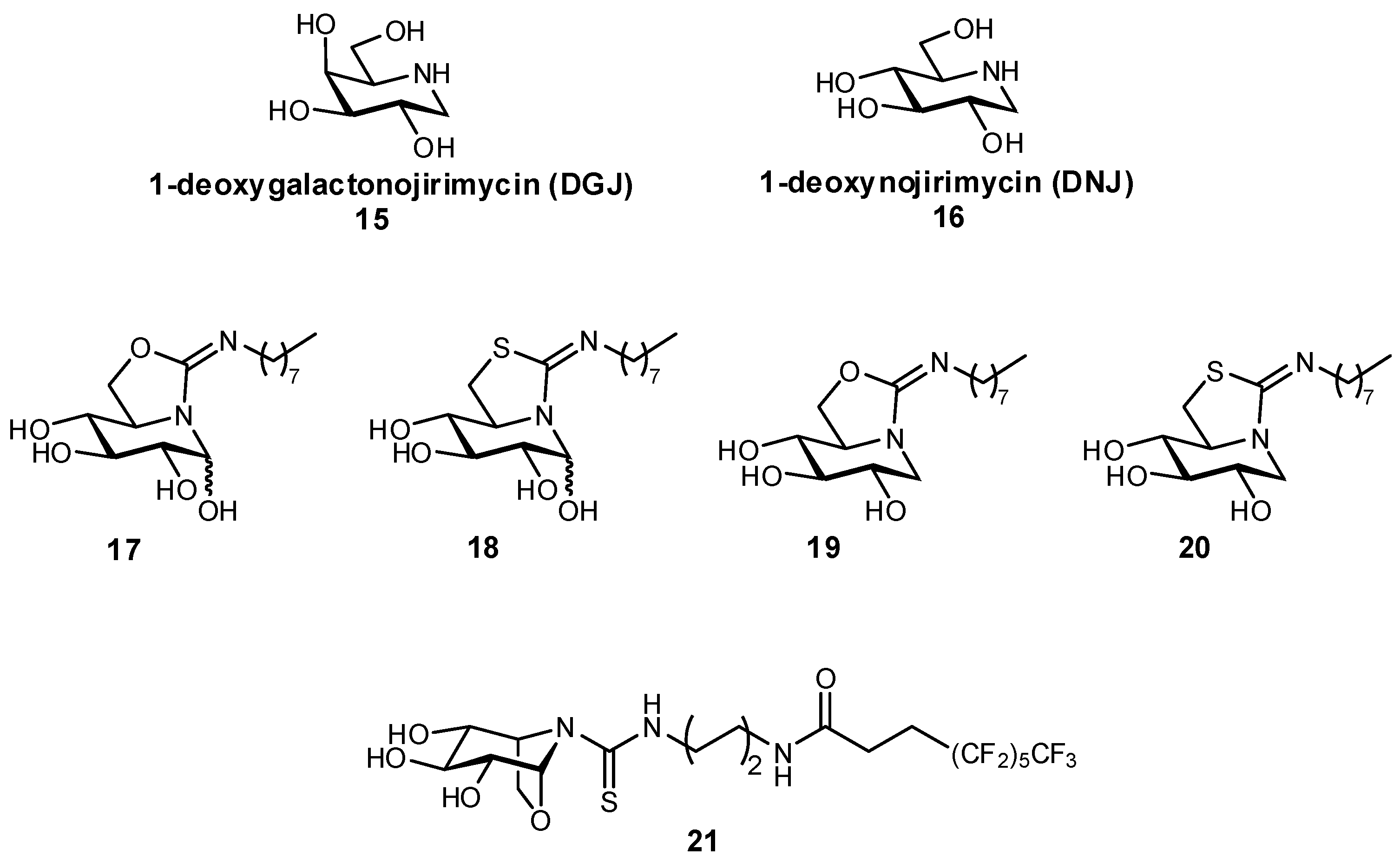
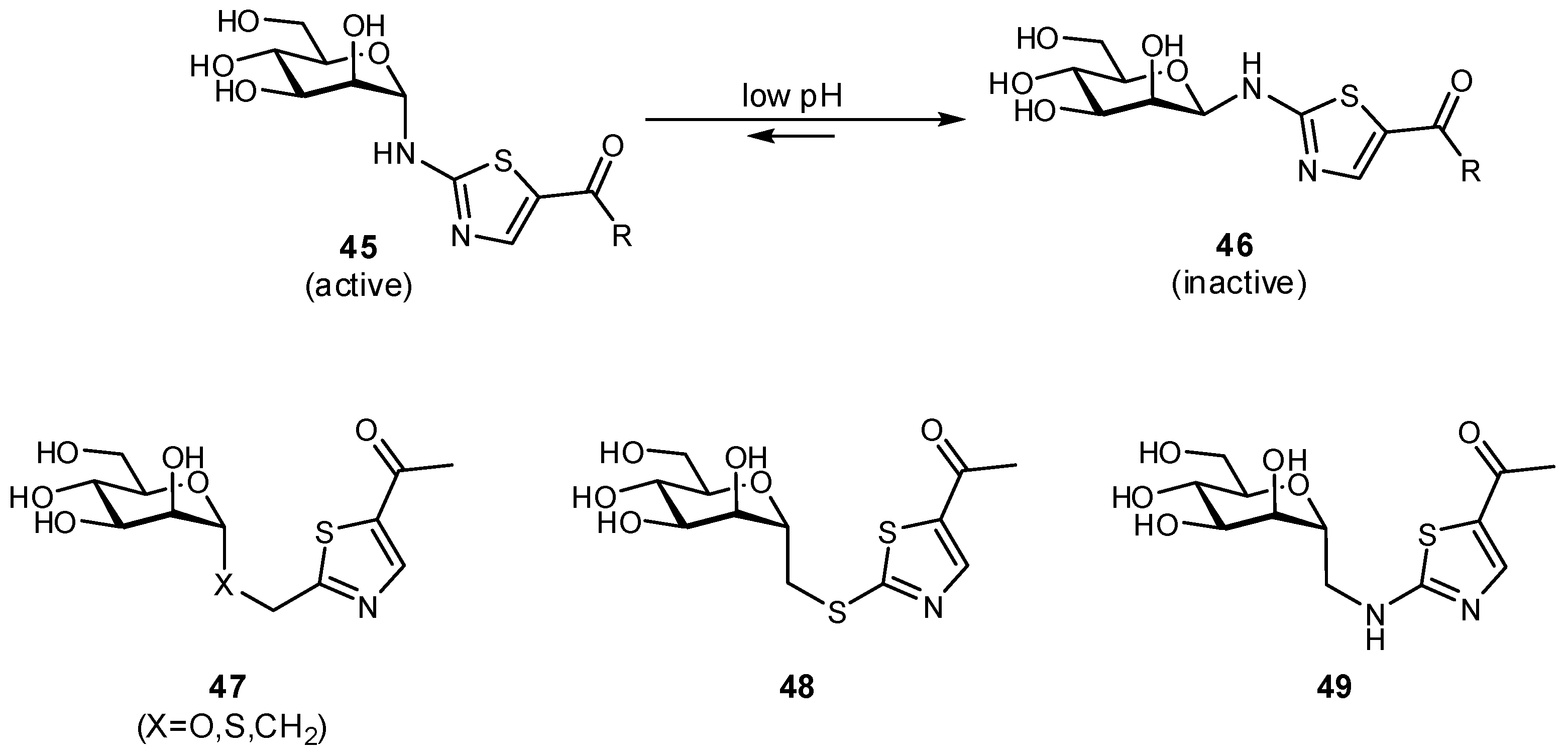
3.1. Preventing Glycosidic Hydrolysis
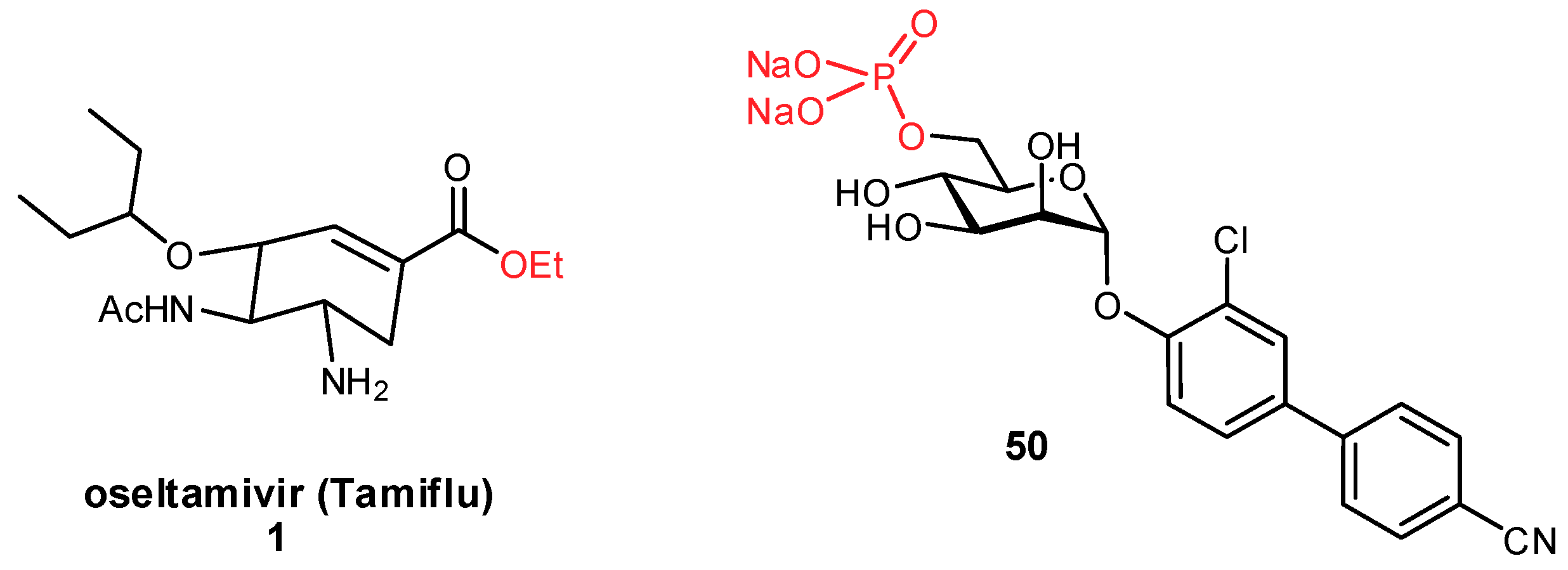
3.2. Improving Oral Bioavailability
3.3. Improving Residence Times and Plasma Half-lives
3.4. Other Considerations
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Nieuwdorp, M.; Meuwese, M.C.; Mooij, H.L.; Ince, C.; Broekhuizen, L.N.; Kastelein, J.J.P.; Stroes, E.S.G.; Vink, H. Measuring endothelial glycocalyx dimensions in humans: A potential novel tool to monitor vascular vulnerability. J. Appl. Physiol. 2008, 104, 845–852. [Google Scholar] [CrossRef]
- Van Teeffelen, J.W.; Brands, J.; Stroes, E.S.; Vink, H. Endothelial glycocalyx: Sweet shield of blood vessels. Trends Cardiovasc. Med. 2007, 17, 101–105. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. (Eds.) Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor: New York, NY, USA, 2017. [Google Scholar]
- Taylor, S.L.; McGuckin, M.A.; Wesselingh, S.; Rogers, G.B. Infection’s sweet tooth: How glycans mediate infection and disease susceptibility. Trends Microbiol. 2018, 26, 92–101. [Google Scholar] [CrossRef]
- Fuster, M.M.; Esko, J.D. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 2005, 5, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.H.; Bertozzi, C.R. Glycans in cancer and inflammation—Potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Lord, J.M.; Roberts, L.M.; Johannes, L. Glycosphingolipids as toxin receptors. Semin. Cell Dev. Biol. 2004, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, P.H.; Rademacher, C. (Eds.) Carbohydrates as Drugs; Springer: Berlin, Germany, 2014. [Google Scholar]
- Zhang, Y.; Wang, F. Carbohydrate drugs: Current status and development prospect. Drug Discov. Ther. 2015, 9, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef]
- Quiocho, F.A. Probing the atomic interactions between proteins and carbohydrates. Biochem. Soc. Trans. 1993, 21, 442–448. [Google Scholar] [CrossRef]
- Sears, P.; Wong, C.-H. Carbohydrate mimetics: A new strategy for tackling the problem of carbohydrate-mediated biological recognition. Angew. Chem. Int. Ed. 1999, 38, 2300–2324. [Google Scholar] [CrossRef]
- Schön, A.; Freire, E. Thermodynamics of intersubunit interactions in cholera toxin upon binding to the oligosaccharide portion of its cell surface receptor, ganglioside GM1. Biochemistry 1989, 28, 5019–5024. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Magnani, J.L.; Ernst, B. Glycomimetic drugs—A new source of therapeutic opportunities. Discov. Med. 2009, 8, 247–252. [Google Scholar]
- Sood, A.; Gerlits, O.O.; Ji, Y.; Bovin, N.V.; Coates, L.; Woods, R.J. Defining the specificity of carbohydrate-protein interactions by quantifying functional group contributions. J. Chem. Inf. Model. 2018, 58, 1889–1901. [Google Scholar] [CrossRef]
- Sager, C.P.; Eriş, D.; Smieško, M.; Hevey, R.; Ernst, B. What contributes to an effective mannose recognition domain? Beilstein J. Org. Chem. 2017, 13, 2584–2595. [Google Scholar] [CrossRef]
- Modenutti, C.; Gauto, D.; Radusky, L.; Blanco, J.; Turjanski, A.; Hajos, S.; Marti, M.A. Using crystallographic water properties for the analysis and prediction of lectin-carbohydrate complex structures. Glycobiology 2015, 25, 181–196. [Google Scholar] [CrossRef]
- Quiocho, F.A.; Wilson, D.K.; Vyas, N.K. Substrate specificity and affinity of a protein modulated by bound water molecules. Nature 1989, 340, 404–407. [Google Scholar] [CrossRef]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y.; et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef] [PubMed]
- McClellan, K.; Perry, C.M. Oseltamivir—A review of its use in influenza. Drugs 2001, 61, 263–283. [Google Scholar] [CrossRef]
- Von Itzstein, M.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Phan, T.V.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef]
- Cox, T.; Lachmann, R.; Hollak, C.; Aerts, J.; van Weely, S.; Hrebícek, M.; Platt, F.; Butters, T.; Dwek, R.; Moyses, C.; et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000, 355, 1481–1485. [Google Scholar] [CrossRef]
- Campbell, L.K.; Baker, D.E.; Campbell, R.K. Miglitol: Assessment of its role in the treatment of patients with diabetes mellitus. Ann. Pharmacother. 2000, 34, 1291–1301. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, Y.; Shen, Y. Voglibose (Basen, AO-128), one of the most important α-glucosidase inhibitors. Curr. Med. Chem. 2006, 13, 109–116. [Google Scholar] [CrossRef]
- Truscheit, E.; Frommer, W.; Junge, B.; Müller, L.; Schmidt, D.D.; Wingender, W. Chemistry and biochemistry of microbial α-glucosidase inhibitors. Angew. Chem. Int. Ed. Engl. 1981, 20, 744–761. [Google Scholar] [CrossRef]
- Sharon, N. Carbohydrates as future anti-adhesion drugs for infectious diseases. Biochim. Biophys. Acta 2006, 1760, 527–537. [Google Scholar] [CrossRef]
- Ofek, I.; Hasty, D.L.; Sharon, N. Anti-adhesion therapy of bacterial diseases: Prospects and problems. FEMS Immunol. Med. Microbiol. 2003, 38, 181–191. [Google Scholar] [CrossRef]
- Kalas, V.; Hibbing, M.E.; Maddirala, A.R.; Chugani, R.; Pinkner, J.S.; Mydock-McGrane, L.K.; Conover, M.S.; Janetka, J.W.; Hultgren, S.J. Structure-based discovery of glycomimetic FmlH ligands as inhibitors of bacterial adhesion during urinary tract infection. Proc. Natl. Acad. Sci. USA 2018, 115, E2819–E2828. [Google Scholar] [CrossRef]
- Hevey, R.; Ling, C.-C. Conjugation strategies used for the preparation of carbohydrate-conjugate vaccines. In Chemistry of Bioconjugates: Synthesis, Characterization, and Biomedical Applications; Narain, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Hevey, R.; Ling, C.-C. Recent advances in developing synthetic carbohydrate-based vaccines for cancer immunotherapies. Future Med. Chem. 2012, 4, 545–584. [Google Scholar] [CrossRef]
- Krug, L.M.; Ragupathi, G.; Ng, K.K.; Hood, C.; Jennings, H.J.; Guo, Z.; Kris, M.G.; Miller, V.; Pizzo, B.; Tyson, L.; et al. Vaccination of small cell lung cancer patients with polysialic acid or N-propionylated polysialic acid conjugated to keyhole limpet hemocyanin. Clin. Cancer Res. 2004, 10, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Auzanneau, F.-A. Synthesis of Lewis A trisaccharide analogues in which D-glucose and L-rhamnose replace D-galactose and L-fucose, respectively. Carbohydr. Res. 2006, 341, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Asnani, A.; Auzanneau, F.-A. Synthesis of a BSA-LeX glycoconjugate and recognition of LeX analogues by the anti-LeX monoclonal antibody SH1: The identification of a non-cross reactive analogue. Bioorg. Med. Chem. 2010, 18, 7174–7185. [Google Scholar] [CrossRef] [PubMed]
- Sahabuddin, S.; Chang, T.-C.; Lin, C.-C.; Jan, F.-D.; Hsiao, H.-Y.; Huang, K.-T.; Chen, J.-H.; Horng, J.-C.; Ho, J.A.; Lin, C.-C. Synthesis of N-modified sTn analogs and evaluation of their immunogenicities by microarray-based immunoassay. Tetrahedron 2010, 66, 7510–7519. [Google Scholar] [CrossRef]
- Cabani, S.; Gianni, P.; Mollica, V.; Lepori, L. Group contributions to the thermodynamic properties of non-ionic organic solutes in dilute aqueous solution. J. Solut. Chem. 1981, 10, 563–595. [Google Scholar] [CrossRef]
- Fitch, C.A.; Karp, D.A.; Lee, K.K.; Stites, W.E.; Lattman, E.E.; García-Moreno, B.E. Experimental pKa values of buried residues: Analysis with continuum methods and role of water penetration. Biophys. J. 2002, 82, 3289–3304. [Google Scholar] [CrossRef]
- Levitt, M.; Park, B.H. Water: Now you see it, now you don’t. Curr. Biol. 1993, 1, 223–226. [Google Scholar] [CrossRef]
- Pan, A.C.; Borhani, D.W.; Dror, R.O.; Shaw, D.E. Molecular determinants of drug-receptor binding kinetics. Drug Discov. Today 2013, 18, 667–673. [Google Scholar] [CrossRef]
- Schmidtke, P.; Luque, F.J.; Murray, J.B.; Barril, X. Shielded hydrogen bonds as structural determinants of binding kinetics: Application in drug design. J. Am. Chem. Soc. 2011, 133, 18903–18910. [Google Scholar] [CrossRef]
- Gao, J.; Qiao, S.; Whitesides, G.M. Increasing binding constants of ligands to carbonic anhydrase by using “greasy tails”. J. Med. Chem. 1995, 38, 2292–2301. [Google Scholar] [CrossRef]
- Biffinger, J.C.; Kim, H.W.; DiMagno, S.G. The polar hydrophobicity of fluorinated compounds. ChemBioChem 2004, 5, 622–627. [Google Scholar] [CrossRef]
- London, R.E.; Gabel, S.A. Fluorine-19 NMR studies of glucosyl fluoride transport in human erythrocytes. Biophys. J. 1995, 69, 1814–1818. [Google Scholar] [CrossRef]
- Oberbillig, T.; Mersch, C.; Wagner, S.; Hoffmann-Röder, A. Antibody recognition of fluorinated MUC1 glycopeptide antigens. Chem. Commun. 2012, 48, 1487–1489. [Google Scholar] [CrossRef]
- Sprinz, C.; Zanon, M.; Altmayer, S.; Watte, G.; Irion, K.; Marchiori, E.; Hochhegger, B. Effects of blood glucose level on 18F fluorodeoxyglucose (18F-FDG) uptake for PET/CT in normal organs: An analysis on 5623 patients. Sci. Rep. 2018, 8, 2126. [Google Scholar] [CrossRef]
- Maschauer, S.; Haubner, R.; Kuwert, T.; Prante, O. 18F-Glyco-RGD peptides for PET imaging of integrin expression: Efficient radiosynthesis by click chemistry and modulation of biodistribution by glycosylation. Mol. Pharm. 2014, 11, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Sadurní, A.; Gilmour, R. Stereocontrolled synthesis of 2-fluorinated C-glycosides. Eur. J. Org. Chem. 2018, 2018, 3684–3687. [Google Scholar] [CrossRef] [PubMed]
- Büll, C.; Boltje, T.J.; van Dinther, E.A.W.; Peters, T.; de Graaf, A.M.A.; Leusen, J.H.W.; Kreutz, M.; Figdor, C.G.; den Brok, M.H.; Adema, G.J. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano 2015, 9, 733–745. [Google Scholar] [CrossRef]
- Büll, C.; Boltje, T.J.; Wassink, M.; de Graaf, A.M.A.; van Delft, F.L.; den Brok, M.H.; Adema, G.J. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol. Cancer Ther. 2016, 12, 1935–1946. [Google Scholar] [CrossRef]
- Foxman, B. Epidemiology of urinary tract infections: Incidence, morbidity, and economic costs. Am. J. Med. 2002, 113, 5S–13S. [Google Scholar] [CrossRef]
- Mak, R.H.; Kuo, H.-J. Pathogenesis of urinary tract infection: An update. Curr. Opin. Pediatr. 2006, 18, 148–152. [Google Scholar] [CrossRef]
- Firon, N.; Ofek, I.; Sharon, N. Interaction of mannose-containing oligosaccharides with the fimbrial lectin of Escherichia coli. Biochem. Biophys. Res. Commun. 1982, 105, 1426–1432. [Google Scholar] [CrossRef]
- Wellens, A.; Garofalo, C.; Nguyen, H.; Van Gerven, N.; Slättegård, R.; Hernalsteens, J.-P.; Wyns, L.; Oscarson, S.; De Greve, H.; Hultgren, S.; et al. Intervening with urinary tract infections using anti-adhesives based on the crystal structure of the FimH-oligomannose-3 complex. PLoS ONE 2008, 3, e2040. [Google Scholar] [CrossRef]
- Han, Z.; Pinkner, J.S.; Ford, B.; Obermann, R.; Nolan, W.; Wildman, S.A.; Hobbs, D.; Ellenberger, T.; Cusumano, C.K.; Hultgren, S.J.; et al. Structure-based drug design and optimization of mannose bacterial FimH antagonists. J. Med. Chem. 2010, 53, 4779–4792. [Google Scholar] [CrossRef] [PubMed]
- Firon, N.; Ashkenazi, S.; Mirelman, D.; Ofek, I.; Sharon, N. Aromatic alpha-glycosides of mannose are powerful inhibitors of the adherence of type 1 fimbriated Escherichia coli to yeast and intestinal epithelial cells. Infect. Immun. 1987, 55, 472–476. [Google Scholar] [PubMed]
- Bouckaert, J.; Berglund, J.; Schembri, M.; De Genst, E.; Cools, L.; Wuhrer, M.; Hung, C.-S.; Pinkner, J.; Slättegård, R.; Zavialov, A.; et al. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 2005, 55, 441–455. [Google Scholar] [CrossRef]
- Sperling, O.; Fuchs, A.; Lindhorst, T.K. Evaluation of the carbohydrate recognition domain of the bacterial adhesin FimH: Design, synthesis and binding properties of mannoside ligands. Org. Biomol. Chem. 2006, 4, 3913–3922. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Abgottspon, D.; Wittwer, M.; Rabbani, S.; Herold, J.; Jiang, X.; Kleeb, S.; Lüthi, C.; Scharenberg, M.; Bezençon, J.; et al. FimH antagonists for the oral treatment of urinary tract infections: From design and synthesis to in vitro and in vivo evaluation. J. Med. Chem. 2010, 53, 8627–8641. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, M.; Schwardt, O.; Rabbani, S.; Ernst, B. Target selectivity of FimH antagonists. J. Med. Chem. 2012, 55, 9810–9816. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Abgottspon, D.; Kleeb, S.; Rabbani, S.; Scharenberg, M.; Wittwer, M.; Haug, M.; Schwardt, O.; Ernst, B. Antiadhesion therapy for urinary tract infections—A balanced PK/PD profile proved to be key for success. J. Med. Chem. 2012, 55, 4700–4713. [Google Scholar] [CrossRef]
- Mena-Barragán, T.; García-Moreno, M.I.; Sevšek, A.; Okazaki, T.; Nanba, E.; Higaki, K.; Martin, N.I.; Pieters, R.J.; García-Fernandez, J.M.; Ortiz Mellet, C. Probing the inhibitor versus chaperone properties of SP2-iminosugars towards human β-glucocerebrosidase: A picomolar chaperone for Gaucher disease. Molecules 2018, 23, 927. [Google Scholar] [CrossRef]
- Markham, A. Migalastat: First global approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; de la Mata, M.; Sánchez-Fernández, E.M.; Benito, J.M.; Díaz-Quintana, A.; Fustero, S.; Nanba, E.; Higaki, K.; Sánchez-Alcázar, J.A.; García Fernández, J.M.; et al. Fluorinated chaperone-β-cyclodextrin formulations for β-glucocerebrosidase activity enhancement in neuronopathic Gaucher disease. J. Med. Chem. 2017, 60, 1829–1842. [Google Scholar] [CrossRef]
- Prescher, H.; Frank, M.; Gütgemann, S.; Kuhfeldt, E.; Schweizer, A.; Nitschke, L.; Watzl, C.; Brossmer, R. Design, synthesis, and biological evaluation of small, high-affinity Siglec-7 ligands: Toward novel inhibitors of cancer immune evasion. J. Med. Chem. 2017, 60, 941–956. [Google Scholar] [CrossRef]
- Prescher, H.; Gütgemann, S.; Frank, M.; Kuhfeldt, E.; Watzl, C.; Brossmer, R. Synthesis and biological evaluation of 9-N-oxamyl sialosides as Siglec-7 ligands. Bioorg. Med. Chem. 2015, 23, 5915–5921. [Google Scholar] [CrossRef]
- Prescher, H.; Schweizer, A.; Kuhfeldt, E.; Nitschke, L.; Brossmer, R. New human CD22/Siglec-2 ligands with a triazole glycoside. ChemBioChem 2017, 18, 1216–1225. [Google Scholar] [CrossRef]
- Prescher, H.; Schweizer, A.; Kuhfeldt, E.; Nitschke, L.; Brossmer, R. Discovery of multifold modified sialosides as human CD22/Siglec-2 ligands with nanomolar activity on B-cells. ACS Chem. Biol. 2014, 9, 1444–1450. [Google Scholar] [CrossRef]
- Collins, B.E.; Blixt, O.; Han, S.; Duong, B.; Li, H.; Nathan, J.K.; Bovin, N.; Paulson, J.C. High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J. Immunol. 2006, 177, 2994–3003. [Google Scholar] [CrossRef]
- Sommer, R.; Wagner, S.; Rox, K.; Varrot, A.; Hauck, D.; Wamhoff, E.-C.; Schreiber, J.; Ryckmans, T.; Brunner, T.; Rademacher, C.; et al. Glycomimetic, orally bioavailable LecB inhibitors block biofilm formation of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2018, 140, 2537–2545. [Google Scholar] [CrossRef]
- Tomašić, T.; Hajšek, D.; Švajger, U.; Luzar, J.; Obermajer, N.; Petit-Haertlein, I.; Fieschi, F.; Anderluh, M. Monovalent mannose-based DC-SIGN antagonists: Targeting the hydrophobic groove of the receptor. Eur. J. Med. Chem. 2014, 75, 308–326. [Google Scholar] [CrossRef]
- Wagner, S.; Hauck, D.; Hoffmann, M.; Sommer, R.; Joachim, I.; Müller, R.; Imberty, A.; Varrot, A.; Titz, A. Covalent lectin inhibition and application in bacterial biofilm imaging. Angew. Chem. Int. Ed. 2017, 56, 16559–16564. [Google Scholar] [CrossRef]
- Chang, J.; Patton, J.T.; Sarkar, A.; Ernst, B.; Magnani, J.L.; Frenette, P.S. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 2010, 116, 1779–1786. [Google Scholar] [CrossRef]
- Telen, M.J.; Wun, T.; McCavit, T.L.; De Castro, L.M.; Krishnamurti, L.; Lanzkron, S.; Hsu, L.L.; Smith, W.R.; Rhee, S.; Magnani, J.L.; et al. Randomized phase 2 study of GMI-1070 in SCD: Reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 2015, 125, 2656–2664. [Google Scholar] [CrossRef]
- Gabius, H.-J.; Siebert, H.-C.; André, S.; Jiménez-Barbero, J.; Rüdiger, H. Chemical biology of the sugar code. ChemBioChem 2004, 5, 740–764. [Google Scholar] [CrossRef]
- Searle, M.S.; Williams, D.H. The cost of conformational order: Entropy changes in molecular associations. J. Am. Chem. Soc. 1992, 114, 10690–10697. [Google Scholar] [CrossRef]
- Perret, S.; Sabin, C.; Dumon, C.; Pokorná, M.; Gautier, C.; Galanina, O.; Ilia, S.; Bovin, N.; Nicaise, M.; Desmadril, M.; et al. Structural basis for the interaction between human milk oligosaccharides and the bacterial lectin PA-IIL of Pseudomonas aeruginosa. Biochem. J. 2005, 389, 325–332. [Google Scholar] [CrossRef]
- Marotte, K.; Sabin, C.; Préville, C.; Moumé-Pymbock, M.; Wimmerová, M.; Mitchell, E.P.; Imberty, A.; Roy, R. X-ray structures and thermodynamics of the interaction of PA-IIL from Pseudomonas aeruginosa with disaccharide derivatives. ChemMedChem 2007, 2, 1328–1338. [Google Scholar] [CrossRef]
- Imberty, A.; Chabre, Y.M.; Roy, R. Glycomimetics and glycodendrimers as high affinity microbial anti-adhesins. Chem. Eur. J. 2008, 14, 7490–7499. [Google Scholar] [CrossRef]
- Rinnbauer, M.; Ernst, B.; Wagner, B.; Magnani, J.; Benie, A.J.; Peters, T. Epitope mapping of sialyl LewisX bound to E-selectin using saturation transfer difference NMR experiments. Glycobiology 2003, 13, 435–443. [Google Scholar] [CrossRef]
- Somers, W.S.; Tang, J.; Shaw, G.D.; Camphausen, R.T. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLeX and PSGL-1. Cell 2000, 103, 467–479. [Google Scholar] [CrossRef]
- Kolb, H.C.; Ernst, B. Development of tools for the design of selectin antagonists. Chem. Eur. J. 1997, 3, 1571–1578. [Google Scholar] [CrossRef]
- Thoma, G.; Magnani, J.L.; Patton, J.T.; Ernst, B.; Jahnke, W. Preorganization of the bioactive conformation of sialyl LewisX analogues correlates with their affinity to E-selectin. Angew. Chem. 2001, 113, 1995–1999. [Google Scholar] [CrossRef]
- Sager, C.P.; Fiege, B.; Zihlmann, P.; Vannam, R.; Rabbani, S.; Jakob, R.P.; Preston, R.C.; Zalewski, A.; Maier, T.; Peczuh, M.W.; et al. The price of flexibility—A case study on septanoses as pyranose mimetics. Chem. Sci. 2018, 9, 646–654. [Google Scholar] [CrossRef]
- Moog, K.E.; Barz, M.; Bartneck, M.; Beceren-Braun, F.; Mohr, N.; Wu, Z.; Braun, L.; Dernedde, J.; Liehn, E.A.; Tacke, F.; et al. Polymeric selectin ligands mimicking complex carbohydrates: From selectin binders to modifiers of macrophage migration. Angew. Chem. Int. Ed. 2017, 56, 1416–1421. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, W.; Xu, X.; Sun, H.; Zhao, J.; Meng, X.; Wu, M.; Li, Z. Synthesis of fucosylated chondroitin sulfate glycoclusters: A robust route to new anticoagulant agents. Chem. Eur. J. 2018, 24, 1694–1700. [Google Scholar] [CrossRef]
- Bücher, K.S.; Konietzny, P.B.; Snyder, N.L.; Hartmann, L. Heteromultivalent glycooligomers as mimetics of blood group antigens. Chem. Eur. J. 2019, 25, 3301–3309. [Google Scholar] [CrossRef]
- Cecioni, S.; Imberty, A.; Vidal, S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 2015, 115, 525–561. [Google Scholar] [CrossRef]
- Bertolotti, B.; Sutkeviciute, I.; Ambrosini, M.; Ribeiro-Viana, R.; Rojo, J.; Fieschi, F.; Dvořáková, H.; Kašáková, M.; Parkan, K.; Hlaváčková, M.; et al. Polyvalent C-glycomimetics based on L-fucose or D-mannose as potent DC-SIGN antagonists. Org. Biomol. Chem. 2017, 15, 3995–4004. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; Ortega-Caballero, F.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernandez, J.M. The impact of heteromultivalency in lectin recognition and glycosidase inhibition: An integrated mechanistic study. Chem. Eur. J. 2017, 23, 6295–6304. [Google Scholar] [CrossRef]
- Ordanini, S.; Varga, N.; Porkolab, V.; Thépaut, M.; Belvisi, L.; Bertaglia, A.; Palmioli, A.; Berzi, A.; Trabattoni, D.; Clerici, M.; et al. Designing nanomolar antagonists of DC-SIGN-mediated HIV infection: Ligand presentation using molecular rods. Chem. Commun. 2015, 51, 3816–3819. [Google Scholar] [CrossRef]
- Boden, S.; Reise, F.; Kania, J.; Lindhorst, T.K.; Hartmann, L. Sequence-defined introduction of hydrophobic motifs and effects in lectin binding of precision glycomacromolecules. Macromol. Biosci. 2019, e1800425. [Google Scholar] [CrossRef]
- Berzi, A.; Ordanini, S.; Joosten, B.; Trabattoni, D.; Cambi, A.; Bernardi, A.; Clerici, M. Pseudo-mannosylated DC-SIGN ligands as immunomodulants. Sci. Rep. 2016, 6, 35373. [Google Scholar] [CrossRef]
- Pérez-Castells, J.; Hernández-Gay, J.J.; Denton, R.W.; Tony, K.A.; Mootoo, D.R.; Jiménez-Barbero, J. The conformational behaviour and P-selectin inhibition of fluorine-containing sialyl LeX glycomimetics. Org. Biomol. Chem. 2007, 5, 1087–1092. [Google Scholar] [CrossRef]
- Asensio, J.L.; Espinosa, J.F.; Dietrich, H.; Cañada, F.J.; Schmidt, R.R.; Martín-Lomas, M.; André, S.; Gabius, H.-J.; Jiménez-Barbero, J. Bovine heart galectin-1 selects a unique (syn) conformation of C-lactose, a flexible lactose analogue. J. Am. Chem. Soc. 1999, 121, 8995–9000. [Google Scholar] [CrossRef]
- Asensio, J.L.; Cañada, F.J.; Cheng, X.; Khan, N.; Mootoo, D.R.; Jiménez-Barbero, J. Conformational differences between O- and C-glycosides: The α-O-Man-(1➝1)-β-Gal/α-C-Man-(1➝1)-β-Gal case—A decisive demonstration of the importance of the exo-anomeric effect on the conformation of glycosides. Chem. Eur. J. 2000, 6, 1035–1041. [Google Scholar] [CrossRef]
- Espinosa, J.-F.; Bruix, M.; Jarreton, O.; Skrydstrup, T.; Beau, J.-M.; Jiménez-Barbero, J. Conformational differences between C- and O-glycosides: The α-C-mannobiose/α-O-mannobiose case. Chem. Eur. J. 1999, 5, 442–448. [Google Scholar] [CrossRef]
- O’Hagan, D.; Rzepa, H.S. Some influences of fluorine in bioorganic chemistry. Chem. Commun. 1997, 0, 645–652. [Google Scholar] [CrossRef]
- Berber, H.; Brigaud, T.; Lefebvre, O.; Plantier-Royon, R.; Portella, C. Reactions of difluoroenoxysilanes with glycosyl donors: Synthesis of difluoro-C-glycosides and difluoro-C-disaccharides. Chem. Eur. J. 2001, 7, 903–909. [Google Scholar] [CrossRef]
- Moreno, B.; Quehen, C.; Rose-Hélène, M.; Leclerc, E.; Quirion, J.-C. Addition of difluoromethyl radicals to glycals: A new route to alpha-CF2-D-glycosides. Org. Lett. 2007, 9, 2477–2480. [Google Scholar] [CrossRef]
- Hirai, G.; Watanabe, T.; Yamaguchi, K.; Miyagi, T.; Sodeoka, M. Stereocontrolled and convergent entry to CF2-sialosides: Synthesis of CF2-linked ganglioside GM4. J. Am. Chem. Soc. 2007, 129, 15420–15421. [Google Scholar] [CrossRef]
- Tony, K.A.; Denton, R.W.; Dilhas, A.; Jiménez-Barbero, J.; Mootoo, D.R. Synthesis of β-C-galacto-pyranosides with fluorine on the pseudoanomeric substituent. Org. Lett. 2007, 9, 1441–1444. [Google Scholar] [CrossRef]
- Dondoni, A.; Catozzi, N.; Marra, A. Concise and practical synthesis of C-glycosyl ketones from sugar benzothiazoles and their transformation into chiral tertiary alcohols. J. Org. Chem. 2005, 70, 9257–9268. [Google Scholar] [CrossRef]
- Poulain, F.; Serre, A.-L.; Lalot, J.; Leclerc, E.; Quirion, J.-C. Synthesis of α-CF2-mannosides and their conversion to fluorinated pseudoglycopeptides. J. Org. Chem. 2008, 73, 2435–2438. [Google Scholar] [CrossRef]
- Johnson, C.R.; Johns, B.A. Suzuki cross-coupling of carbohydrates: Synthesis of β-arylmethyl-C-glycosides and aryl-scaffolded trisaccharide mimics. Synlett 1997, 12, 1406–1408. [Google Scholar] [CrossRef]
- Dondoni, A.; Catozzi, N.; Marra, A. Stereoselective synthesis of α- and β-L-C-fucosyl aldehydes and their utility in the assembly of C-fucosides of biological relevance. J. Org. Chem. 2004, 69, 5023–5036. [Google Scholar] [CrossRef]
- Dondoni, A.; Scherrmann, M.-C. Thiazole-based synthesis of formyl C-glycosides. J. Org. Chem. 1994, 59, 6404–6412. [Google Scholar] [CrossRef]
- Redjdal, W.; Ibrahim, N.; Benmerad, B.; Alami, M.; Messaoudi, S. Convergent synthesis of N,S-bis glycosylquinolin-2-ones via a Pd-G3-XantPhos precatalyst catalysis. Molecules 2018, 23, 519. [Google Scholar] [CrossRef]
- Céspedes Dávila, M.F.; Schneider, J.P.; Godard, A.; Hazelard, D.; Compain, P. One-pot, highly stereoselective synthesis of dithioacetal-α,α-diglycosides. Molecules 2018, 23, 914. [Google Scholar] [CrossRef]
- Illyés, T.-Z.; Balla, S.; Bényei, A.; Kumar, A.A.; Timári, I.; Kövér, K.E.; Szilágyi, L. Exploring the syntheses of novel glycomimetics. Carbohydrate derivatives with Se-S- or Se-Se-glycosidic linkages. ChemistrySelect 2016, 1, 2383–2388. [Google Scholar] [CrossRef]
- Zhu, F.; O’Neill, S.; Rodriguez, J.; Walczak, M.A. Stereoretentive reactions at the anomeric position: Synthesis of selenoglycosides. Angew. Chem. Int. Ed. 2018, 57, 7091–7095. [Google Scholar] [CrossRef]
- Marcaurelle, L.A.; Bertozzi, C.R. New directions in the synthesis of glycopeptide mimetics. Chem. Eur. J. 1999, 5, 1384–1390. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.; Cusumano, Z.; Han, Z.; Binkley, J.; Kostakioti, M.; Hannan, T.; Pinkner, J.S.; Klein, R.; Kalas, V.; Crowley, J.; et al. Antivirulence C-mannosides as antibiotic-sparing, oral therapeutics for urinary tract infections. J. Med. Chem. 2016, 59, 9390–9408. [Google Scholar] [CrossRef]
- Brument, S.; Sivignon, A.; Dumych, T.I.; Moreau, N.; Roos, G.; Guérardel, Y.; Chalopin, T.; Deniaud, D.; Bilyy, R.O.; Darfeuille-Michaud, A.; et al. Thiazolylaminomannosides as potent antiadhesives of type 1 piliated Escherichia coli isolated from Crohn’s disease patients. J. Med. Chem. 2013, 56, 5395–5406. [Google Scholar] [CrossRef]
- Chalopin, T.; Alvarez Dorta, D.; Sivignon, A.; Caudan, M.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org. Biomol. Chem. 2016, 14, 3913–3925. [Google Scholar] [CrossRef]
- Sivignon, A.; Bouckaert, J.; Bernard, J.; Gouin, S.G.; Barnich, N. The potential of FimH as a novel therapeutic target for the treatment of Crohn’s disease. Expert. Opin. Ther. Targets 2017, 21, 837–847. [Google Scholar] [CrossRef]
- Alvarez Dorta, D.; Chalopin, T.; Sivignon, A.; de Ruyck, J.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Physiochemical tuning of potent Escherichia coli anti-adhesives by microencapsulation and methylene homologation. ChemMedChem 2017, 12, 986–998. [Google Scholar] [CrossRef]
- Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, M.E.; Huang, W.; Wang, H.; Ganapathy, V.; Leibach, F.H. Valacyclovir: A substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem. Biophys. Res. Commun. 1998, 246, 470–475. [Google Scholar] [CrossRef]
- Inui, K.; Terada, T.; Masuda, S.; Saito, H. Physiological and pharmacological implications of peptide transporters, PEPT1 and PEPT2. Nephrol. Dial. Transpl. 2000, 15, 11–13. [Google Scholar] [CrossRef]
- Nielsen, D.S.; Lohman, R.-J.; Hoang, H.N.; Hill, T.A.; Jones, A.; Lucke, A.J.; Fairlie, D.P. Flexibility versus rigidity for orally bioavailable cyclic hexapeptides. ChemBioChem 2015, 16, 2289–2293. [Google Scholar] [CrossRef]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; de Melo, T.R.F.; dos Santos, J.L.; Chung, M.C. The prodrug approach: A successful tool for improving drug solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef]
- He, G.; Massarella, J.; Ward, P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin. Pharmacokinet. 1999, 37, 471–484. [Google Scholar] [CrossRef]
- Kleeb, S.; Jiang, X.; Frei, P.; Sigl, A.; Bezençon, J.; Bamberger, K.; Schwardt, O.; Ernst, B. FimH antagonists: Phosphate prodrugs improve oral bioavailability. J. Med. Chem. 2016, 59, 3163–3182. [Google Scholar] [CrossRef]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef]
- Kleeb, S.; Pang, L.; Mayer, K.; Eris, D.; Sigl, A.; Preston, R.C.; Zihlmann, P.; Sharpe, T.; Jakob, R.P.; Abgottspon, D.; et al. FimH antagonists: Bioisosteres to improve the in vitro and in vivo PK/PD profile. J. Med. Chem. 2015, 58, 2221–2239. [Google Scholar] [CrossRef]
- Mizuno, N.; Niwa, T.; Yotsumoto, Y.; Sugiyama, Y. Impact of drug transporter studies on drug discovery and development. Pharmacol. Rev. 2003, 55, 425–461. [Google Scholar] [CrossRef]
- Holodniy, M.; Penzak, S.R.; Straight, T.M.; Davey, R.T.; Lee, K.K.; Goetz, M.B.; Raisch, D.W.; Cunningham, F.; Lin, E.T.; Olivo, N.; et al. Pharmacokinetics and tolerability of oseltamivir combined with probenecid. Antimicrob. Agents Chemother. 2008, 52, 3013–3021. [Google Scholar] [CrossRef]
- Chan, A.K.C.; Paredes, N.; Thong, B.; Chindemi, P.; Paes, B.; Berry, L.R.; Monagle, P. Binding of heparin to plasma proteins and endothelial surfaces is inhibited by covalent linkage to antithrombin. Thromb. Haemost. 2004, 91, 1009–1118. [Google Scholar] [CrossRef]
- Young, E.; Prins, M.; Levine, M.N.; Hirst, J. Heparin binding to plasma proteins, an important mechanism for heparin resistance. Thromb. Haemost. 1992, 67, 639–643. [Google Scholar] [CrossRef]
- Hazelard, D.; Compain, P. Square sugars: Challenges and synthetic strategies. Org. Biomol. Chem. 2017, 15, 3806–3827. [Google Scholar] [CrossRef]
- Eggink, L.L.; Spyroulias, G.A.; Jones, N.G.; Hanson, C.V.; Hoober, J.K. A peptide mimetic of 5-acetylneuraminic acid-galactose binds with high avidity to siglecs and NKG2D. PLoS ONE 2015, 10, e0130532. [Google Scholar] [CrossRef]
- Garber, K.C.A.; Wangkanont, K.; Carlson, E.E.; Kiessling, L.L. A general glycomimetic strategy yields non-carbohydrate inhibitors of DC-SIGN. Chem. Commun. 2010, 46, 6747–6749. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Wilkinson, F.L.; Jones, A.M.; Wilkinson, J.A.; Romero, M.; Duarte, J.; Alexander, M.Y. A novel role for small molecule glycomimetics in the protection against lipid-induced endothelial dysfunction: Involvement of Akt/eNOS and Nrf2/ARE signaling. Biochim. Biophys. Acta 2017, 1861, 3311–3322. [Google Scholar] [CrossRef]
- Dayde, B.; Pierra, C.; Gosselin, G.; Surleraux, D.; Ilagouma, A.T.; Laborde, C.; Volle, J.-N.; Virieux, D.; Pirat, J.-L. Synthesis of unnatural phosphonosugar analogues. Eur. J. Org. Chem. 2014, 2014, 1333–1337. [Google Scholar] [CrossRef]
- Eggink, L.L.; Roby, K.F.; Cote, R.; Hoober, J.K. An innovative immunotherapeutic strategy for ovarian cancer: CLEC10A and glycomimetic peptides. J. Immunother. Cancer 2018, 6, 28. [Google Scholar] [CrossRef]
- Theis, T.; Johal, A.S.; Kabat, M.; Basak, S.; Schachner, M. Enhanced neuronal survival and neurite outgrowth triggered by novel small organic compounds mimicking the LewisX glycan. Mol. Neurobiol. 2018, 55, 8203–8215. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, A.W.W.; Hasan, A.; Weston, R.; Edwards, N.; Jones, A.M.; Boulton, A.J.M.; Bowling, F.L.; Rashid, S.T.; Wilkinson, F.L.; Alexander, M.Y. Diabetic endothelial colony forming cells have the potential for restoration with glycomimetics. Sci. Rep. 2019, 9, 2309. [Google Scholar] [CrossRef]
- Kieber-Emmons, T.; Luo, P.; Qiu, J.; Agadjanyan, M.; Carey, L.; Hutchins, W.; Westerink, M.A.; Steplewski, Z. Peptide mimicry of adenocarcinoma-associated carbohydrate antigens. Hybridoma 1997, 16, 3–10. [Google Scholar] [CrossRef]
- Westerink, M.A.J.; Giardina, P.C.; Apicella, M.A.; Kieber-Emmons, T. Peptide mimicry of the meningococcal group C capsular polysaccharide. Proc. Natl. Acad. Sci. USA 1995, 92, 4021–4025. [Google Scholar] [CrossRef]
- Basak, S.; Birebent, B.; Purev, E.; Somasundaram, R.; Maruyama, H.; Zaloudik, J.; Swoboda, R.; Strittmatter, W.; Li, W.; Luckenbach, A.; et al. Induction of cellular immunity by anti-idiotypic antibodies mimicking GD2 ganglioside. Cancer Immunol. Immunother. 2003, 52, 145–154. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
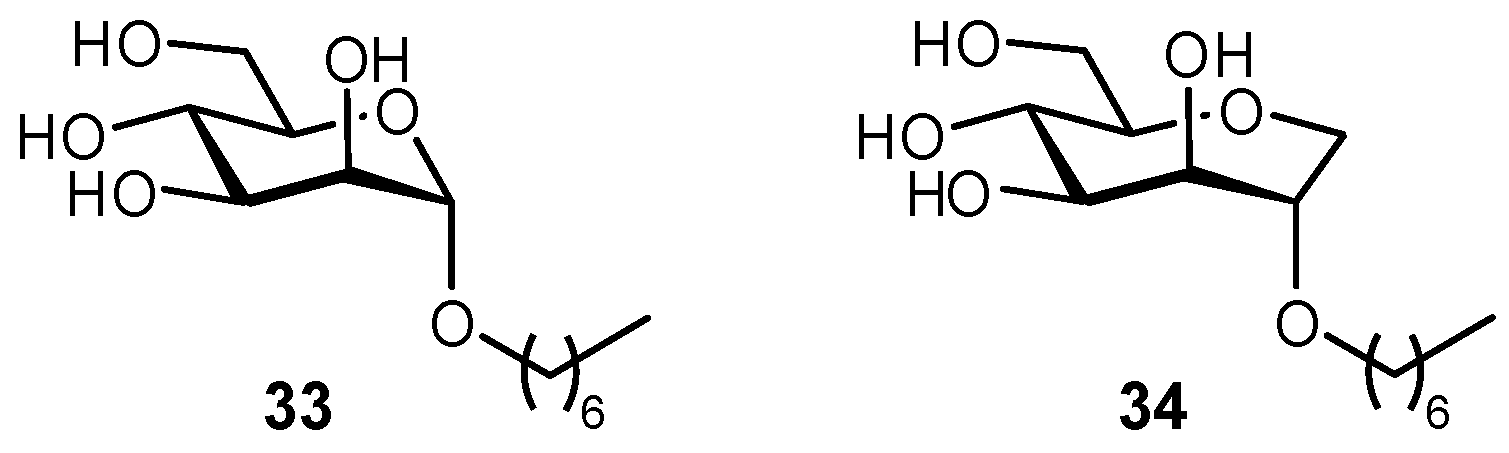{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original Group | Potential Replacements |
|---|---|
 |  |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hevey, R. Strategies for the Development of Glycomimetic Drug Candidates. Pharmaceuticals 2019, 12, 55. https://doi.org/10.3390/ph12020055
Hevey R. Strategies for the Development of Glycomimetic Drug Candidates. Pharmaceuticals. 2019; 12(2):55. https://doi.org/10.3390/ph12020055
Chicago/Turabian StyleHevey, Rachel. 2019. "Strategies for the Development of Glycomimetic Drug Candidates" Pharmaceuticals 12, no. 2: 55. https://doi.org/10.3390/ph12020055
APA StyleHevey, R. (2019). Strategies for the Development of Glycomimetic Drug Candidates. Pharmaceuticals, 12(2), 55. https://doi.org/10.3390/ph12020055