Iron in Lung Pathology

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Iron Regulation
2.1. Systemic Iron Homeostasis
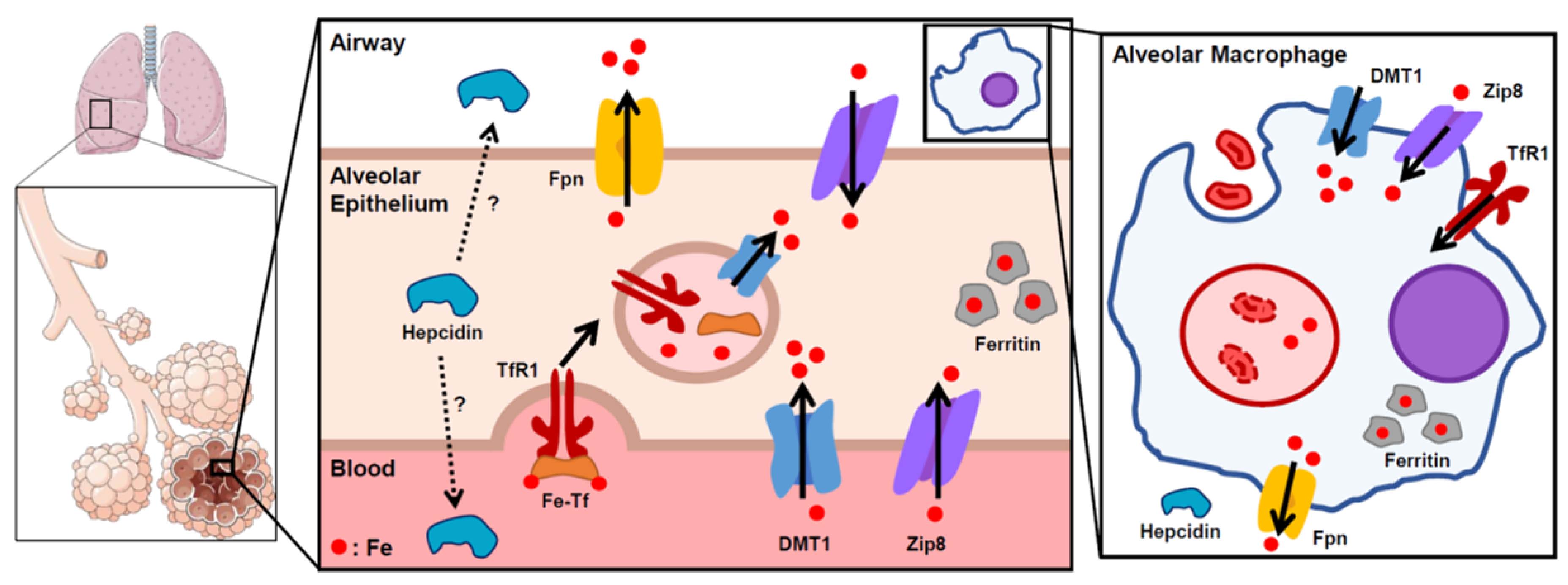
2.2. Lung Iron Regulation
3. Iron in Lung Pathology
3.1. Acute Lung Injury
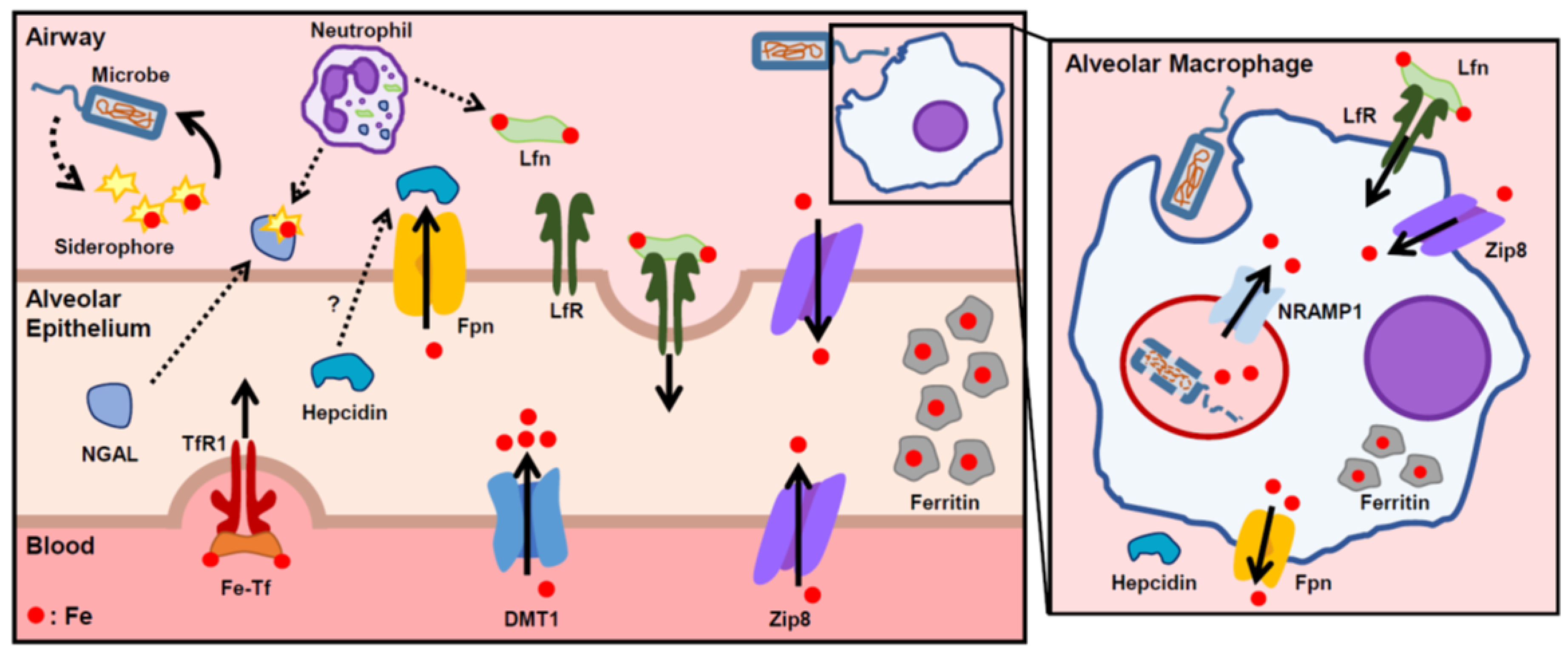
3.2. Lung Infections
3.3. Cystic Fibrosis
3.4. Chronic Obstructive Pulmonary Disease
4. Potential Therapeutics
Funding
Conflicts of Interest
References
- Ganz, T.; Nemeth, E. Regulation of iron acquisition and iron distribution in mammals. Biochim. Biophys. Acta 2006, 1763, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Ramm, G.A.; Ruddell, R.G. Hepatotoxicity of iron overload: Mechanisms of iron-induced hepatic fibrogenesis. Semin. Liver Dis. 2005, 25, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.Y.; Flynn, D.M.; Hoffbrand, A.V.; Politis, D. Infection with Yersinia enterocolitica in patients with iron overload. Br. Med. J. (Clin. Res. Ed.) 1986, 292, 97. [Google Scholar] [CrossRef]
- Parrow, N.L.; Fleming, R.E.; Minnick, M.F. Sequestration and scavenging of iron in infection. Infect. Immun. 2013, 81, 3503–3514. [Google Scholar] [CrossRef]
- Michels, K.R.; Zhang, Z.; Bettina, A.M.; Cagnina, R.E.; Stefanova, D.; Burdick, M.D.; Vaulont, S.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin-mediated iron sequestration protects against bacterial dissemination during pneumonia. JCI Insight 2017, 2, e92002. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Finberg, K.E. Unraveling mechanisms regulating systemic iron homeostasis. Hematol. Am. Soc. Hematol. Educ. Progr. 2011, 2011, 532–537. [Google Scholar] [CrossRef]
- Finch, C. Regulators of iron balance in humans. Blood 1994, 84, 1697–1702. [Google Scholar]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematol. Am. Soc. Hematol. Educ. Progr. 2011, 2011, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Jung, C.L.; Gabayan, V.; Deng, J.C.; Ganz, T.; Nemeth, E.; Bulut, Y. Hepcidin induction by pathogens and pathogen-derived molecules is strongly dependent on interleukin-6. Infect. Immun. 2014, 82, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef]
- Heilig, E.A.; Thompson, K.J.; Molina, R.M.; Ivanov, A.R.; Brain, J.D.; Wessling-Resnick, M. Manganese and iron transport across pulmonary epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L1247–L1259. [Google Scholar] [CrossRef]
- Ghio, A.J.; Wang, X.; Silbajoris, R.; Garrick, M.D.; Piantadosi, C.A.; Yang, F. DMT1 expression is increased in the lungs of hypotransferrinemic mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L938–L944. [Google Scholar] [CrossRef]
- Wang, X.; Ghio, A.J.; Yang, F.; Dolan, K.G.; Garrick, M.D.; Piantadosi, C.A. Iron uptake and Nramp2/DMT1/DCT1 in human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L987–L995. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, G.; D’Anna, M.C.; Roque, M.E. Iron homeostasis and its disruption in mouse lung in iron deficiency and overload. Exp. Physiol. 2015, 100, 1199–1216. [Google Scholar] [CrossRef]
- Ghio, A.J.; Piantadosi, C.A.; Wang, X.; Dailey, L.A.; Stonehuerner, J.D.; Madden, M.C.; Yang, F.; Dolan, K.G.; Garrick, M.D.; Garrick, L.M. Divalent metal transporter-1 decreases metal-related injury in the lung. Am. J. Physiol. Lung Cell Mol. Physiol 2005, 289, L460–L467. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Molina, R.M.; Donaghey, T.C.; Buckett, P.D.; Brain, J.D.; Wessling-Resnick, M. Influence of DMT1 and iron status on inflammatory responses in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L659–L665. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Stonehuerner, J.G.; Richards, J.H.; Nguyen, N.B.; Callaghan, K.D.; Haile, D.J.; Ghio, A.J. Deficiency in the divalent metal transporter 1 increases bleomycin-induced lung injury. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2010, 23, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Haile, D.J.; Wang, X.; Dailey, L.A.; Stonehuerner, J.G.; Ghiom, A.J. Apical location of ferroportin 1 in airway epithelia and its role in iron detoxification in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L14–L23. [Google Scholar] [CrossRef]
- Neves, J.; Leitz, D.; Kraut, S.; Brandenberger, C.; Agrawal, R.; Weissmann, N.; Muhlfeld, C.; Mall, M.A.; Altamura, S.; Muckenthaler, M.U. Disruption of the Hepcidin/Ferroportin Regulatory System Causes Pulmonary Iron Overload and Restrictive Lung Disease. EBioMedicine 2017, 20, 230–239. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Ma, Y.; Wu, X.; Jin, L.; Yu, F. Increased hepcidin expression in non-small cell lung cancer tissue and serum is associated with clinical stage. Thorac. Cancer 2014, 5, 14–24. [Google Scholar] [CrossRef]
- Nguyen, N.B.; Callaghan, K.D.; Ghio, A.J.; Haile, D.J.; Yang, F. Hepcidin expression and iron transport in alveolar macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L417–L425. [Google Scholar] [CrossRef]
- Frazier, M.D.; Mamo, L.B.; Ghio, A.J.; Turi, J.L. Hepcidin expression in human airway epithelial cells is regulated by interferon-gamma. Respir. Res. 2011, 12, 100. [Google Scholar] [CrossRef]
- Deschemin, J.C.; Mathieu, J.R.R.; Zumerle, S.; Peyssonnaux, C.; Vaulont, S. Pulmonary Iron Homeostasis in Hepcidin Knockout Mice. Front. Physiol. 2017, 8, 804. [Google Scholar] [CrossRef] [PubMed]
- Altamura, S.; Kessler, R.; Grone, H.J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.J.; Bao, S.; Galvez-Peralta, M.; Pyle, C.J.; Rudawsky, A.C.; Pavlovicz, R.E.; Killilea, D.W.; Li, C.; Nebert, D.W.; Wewers, M.D.; et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Rep. 2013, 3, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Pyle, C.J.; Akhter, S.; Bao, S.; Dodd, C.E.; Schlesinger, L.S.; Knoell, D.L. Zinc Modulates Endotoxin-Induced Human Macrophage Inflammation through ZIP8 Induction and C/EBPbeta Inhibition. PLoS ONE 2017, 12, e0169531. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef]
- Chabot, F.; Mitchell, J.A.; Gutteridge, J.M.; Evans, T.W. Reactive oxygen species in acute lung injury. Eur. Respir. J. 1998, 11, 745–757. [Google Scholar]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Ghio, A.J.; Carter, J.D.; Richards, J.H.; Richer, L.D.; Grissom, C.K.; Elstad, M.R. Iron and iron-related proteins in the lower respiratory tract of patients with acute respiratory distress syndrome. Crit. Care Med. 2003, 31, 395–400. [Google Scholar] [CrossRef]
- Connelly, K.G.; Moss, M.; Parsons, P.E.; Moore, E.E.; Moore, F.A.; Giclas, P.C.; Seligman, P.A.; Repine, J.E. Serum ferritin as a predictor of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1997, 155, 21–25. [Google Scholar] [CrossRef]
- Sharkey, R.A.; Donnelly, S.C.; Connelly, K.G.; Robertson, C.E.; Haslett, C.; Repine, J.E. Initial serum ferritin levels in patients with multiple trauma and the subsequent development of acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1999, 159, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Jean, G.; Charra, B.; Chazot, C.; Vanel, T.; Terrat, J.C.; Hurot, J.M.; Laurent, G. Risk factor analysis for long-term tunneled dialysis catheter-related bacteremias. Nephron 2002, 91, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Gangaidzo, I.T.; Moyo, V.M.; Mvundura, E.; Aggrey, G.; Murphree, N.L.; Khumalo, H.; Saungweme, T.; Kasvosve, I.; Gomo, Z.A.; Rouault, T.; et al. Association of pulmonary tuberculosis with increased dietary iron. J. Infect. Dis. 2001, 184, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.J.; Murray, A.B.; Murray, M.B.; Murray, C.J. The adverse effect of iron repletion on the course of certain infections. Br. Med. J. 1978, 2, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.D. Iron and susceptibility to infectious disease. Science 1974, 184, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Bao, G.H.; Barasch, J.; Xu, J.; Wang, W.; Hu, F.L.; Deng, S.X. Purification and Structural Characterization of “Simple Catechol”, the NGAL-Siderocalin Siderophore in Human Urine. RSC Adv. 2015, 5, 28527–28535. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Santoni-Rugiu, E.; Ralfkiaer, E.; Porse, B.T.; Moser, C.; Hoiby, N.; Borregaard, N.; Cowland, J.B. Lipocalin 2 is protective against E. coli pneumonia. Respir. Res. 2010, 11, 96. [Google Scholar] [CrossRef]
- Martineau, A.R.; Newton, S.M.; Wilkinson, K.A.; Kampmann, B.; Hall, B.M.; Nawroly, N.; Packe, G.E.; Davidson, R.N.; Griffiths, C.J.; Wilkinson, R.J. Neutrophil-mediated innate immune resistance to mycobacteria. J. Clin. Investig. 2007, 117, 1988–1994. [Google Scholar] [CrossRef]
- Baynes, R.; Bezwoda, W.; Bothwell, T.; Khan, Q.; Mansoor, N. The non-immune inflammatory response: Serial changes in plasma iron, iron-binding capacity, lactoferrin, ferritin and C-reactive protein. Scand. J. Clin. Lab. Investig. 1986, 46, 695–704. [Google Scholar] [CrossRef]
- Cellier, M.F.; Courville, P.; Campion, C. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect. Inst. Pasteur 2007, 9, 1662–1670. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Nramp1 and Other Transporters Involved in Metal Withholding during Infection. J. Biol. Chem. 2015, 290, 18984–18990. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and Host Defense against Infectious Diseases. PLoS Pathog. 2015, 11, e1004998. [Google Scholar] [CrossRef] [PubMed]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, D.; Raychev, A.; Deville, J.; Humphries, R.; Campeau, S.; Ruchala, P.; Nemeth, E.; Ganz, T.; Bulut, Y. Hepcidin Protects against Lethal Escherichia coli Sepsis in Mice Inoculated with Isolates from Septic Patients. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, D.; Raychev, A.; Arezes, J.; Ruchala, P.; Gabayan, V.; Skurnik, M.; Dillon, B.J.; Horwitz, M.A.; Ganz, T.; Bulut, Y.; et al. Endogenous hepcidin and its agonist mediate resistance to selected infections by clearing non-transferrin-bound iron. Blood 2017, 130, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.X.; Song, S.W.; Chen, Q.H.; Zeng, C.L.; Zheng, X.; Wang, J.L.; Fang, X.M. Silencing airway epithelial cell-derived hepcidin exacerbates sepsis induced acute lung injury. Crit. Care 2014, 18, 470. [Google Scholar] [CrossRef]
- Paradkar, P.N.; De Domenico, I.; Durchfort, N.; Zohn, I.; Kaplan, J.; Ward, D.M. Iron depletion limits intracellular bacterial growth in macrophages. Blood 2008, 112, 866–874. [Google Scholar] [CrossRef]
- Chlosta, S.; Fishman, D.S.; Harrington, L.; Johnson, E.E.; Knutson, M.D.; Wessling-Resnick, M.; Cherayil, B.J. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect. Immun. 2006, 74, 3065–3067. [Google Scholar] [CrossRef]
- Stites, S.W.; Plautz, M.W.; Bailey, K.; O’Brien-Ladner, A.R.; Wesselius, L.J. Increased concentrations of iron and isoferritins in the lower respiratory tract of patients with stable cystic fibrosis. Am. J. Respir. Crit. Care Med. 1999, 160, 796–801. [Google Scholar] [CrossRef]
- Reid, D.W.; Lam, Q.T.; Schneider, H.; Walters, E.H. Airway iron and iron-regulatory cytokines in cystic fibrosis. Eur. Respir. J. 2004, 24, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; Anderson, G.J.; Lamont, I.L. Role of lung iron in determining the bacterial and host struggle in cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L795–L802. [Google Scholar] [CrossRef] [PubMed]
- Mussalo-Rauhamaa, H.; Leppanen, A.; Salmela, S.S.; Pyysalo, H. Cigarettes as a source of some trace and heavy metals and pesticides in man. Arch. Environ. Health 1986, 41, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Mumby, S.; Adcock, I.M.; Choi, A.M.K.; Chung, K.F.; Quinlan, G.J. The “Iron”-y of Iron Overload and Iron Deficiency in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 196, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Hilborn, E.D.; Stonehuerner, J.G.; Dailey, L.A.; Carter, J.D.; Richards, J.H.; Crissman, K.M.; Foronjy, R.F.; Uyeminami, D.L.; Pinkerton, K.E. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am. J. Respir. Crit. Care Med. 2008, 178, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Mumby, S.; Saito, J.; Adcock, I.M.; Chung, K.F.; Quinlan, G.J. Decreased breath excretion of redox active iron in COPD: A protective failure? Eur. Respir. J. 2016, 47, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- DeMeo, D.L.; Mariani, T.; Bhattacharya, S.; Srisuma, S.; Lange, C.; Litonjua, A.; Bueno, R.; Pillai, S.G.; Lomas, D.A.; Sparrow, D.; et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am. J. Hum. Genet. 2009, 85, 493–502. [Google Scholar] [CrossRef]
- Kim, W.J.; Wood, A.M.; Barker, A.F.; Brantly, M.L.; Campbell, E.J.; Eden, E.; McElvaney, G.; Rennard, S.I.; Sandhaus, R.A.; Stocks, J.M.; et al. Association of IREB2 and CHRNA3 polymorphisms with airflow obstruction in severe alpha-1 antitrypsin deficiency. Respir Res. 2012, 13, 16. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabon, M.A.; Konrad, C.; Polverino, F.; Perez, E.; Mizumura, K.; et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef]
- Ghio, A.J.; Jaskot, R.H.; Hatch, G.E. Lung injury after silica instillation is associated with an accumulation of iron in rats. Am. J. Physiol. 1994, 267, L686–L692. [Google Scholar] [CrossRef]
- Kim, J.; Wessling-Resnick, M. The Role of Iron Metabolism in Lung Inflammation and Injury. J. Allergy 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Moreau-Marquis, S.; O’Toole, G.A.; Stanton, B.A. Tobramycin and FDA-approved iron chelators eliminate Pseudomonas aeruginosa biofilms on cystic fibrosis cells. Am. J. Respir. Cell Mol. Biol. 2009, 41, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Andrianaki, A.M.; Kyrmizi, I.; Thanopoulou, K.; Baldin, C.; Drakos, E.; Soliman, S.S.M.; Shetty, A.C.; McCracken, C.; Akoumianaki, T.; Stylianou, K.; et al. Iron restriction inside macrophages regulates pulmonary host defense against Rhizopus species. Nat. Commun. 2018, 9, 3333. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef] [PubMed]
- Wetli, H.A.; Buckett, P.D.; Wessling-Resnick, M. Small-molecule screening identifies the selanazal drug ebselen as a potent inhibitor of DMT1-mediated iron uptake. Chem. Biol. 2006, 13, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Investig. 2011, 121, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B.E.; Singh, P.K. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J. Clin. Investig. 2007, 117, 877–888. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, V.; Nemeth, E.; Kim, A. Iron in Lung Pathology. Pharmaceuticals 2019, 12, 30. https://doi.org/10.3390/ph12010030
Zhang V, Nemeth E, Kim A. Iron in Lung Pathology. Pharmaceuticals. 2019; 12(1):30. https://doi.org/10.3390/ph12010030
Chicago/Turabian StyleZhang, Vida, Elizabeta Nemeth, and Airie Kim. 2019. "Iron in Lung Pathology" Pharmaceuticals 12, no. 1: 30. https://doi.org/10.3390/ph12010030
APA StyleZhang, V., Nemeth, E., & Kim, A. (2019). Iron in Lung Pathology. Pharmaceuticals, 12(1), 30. https://doi.org/10.3390/ph12010030