Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition


Abstract
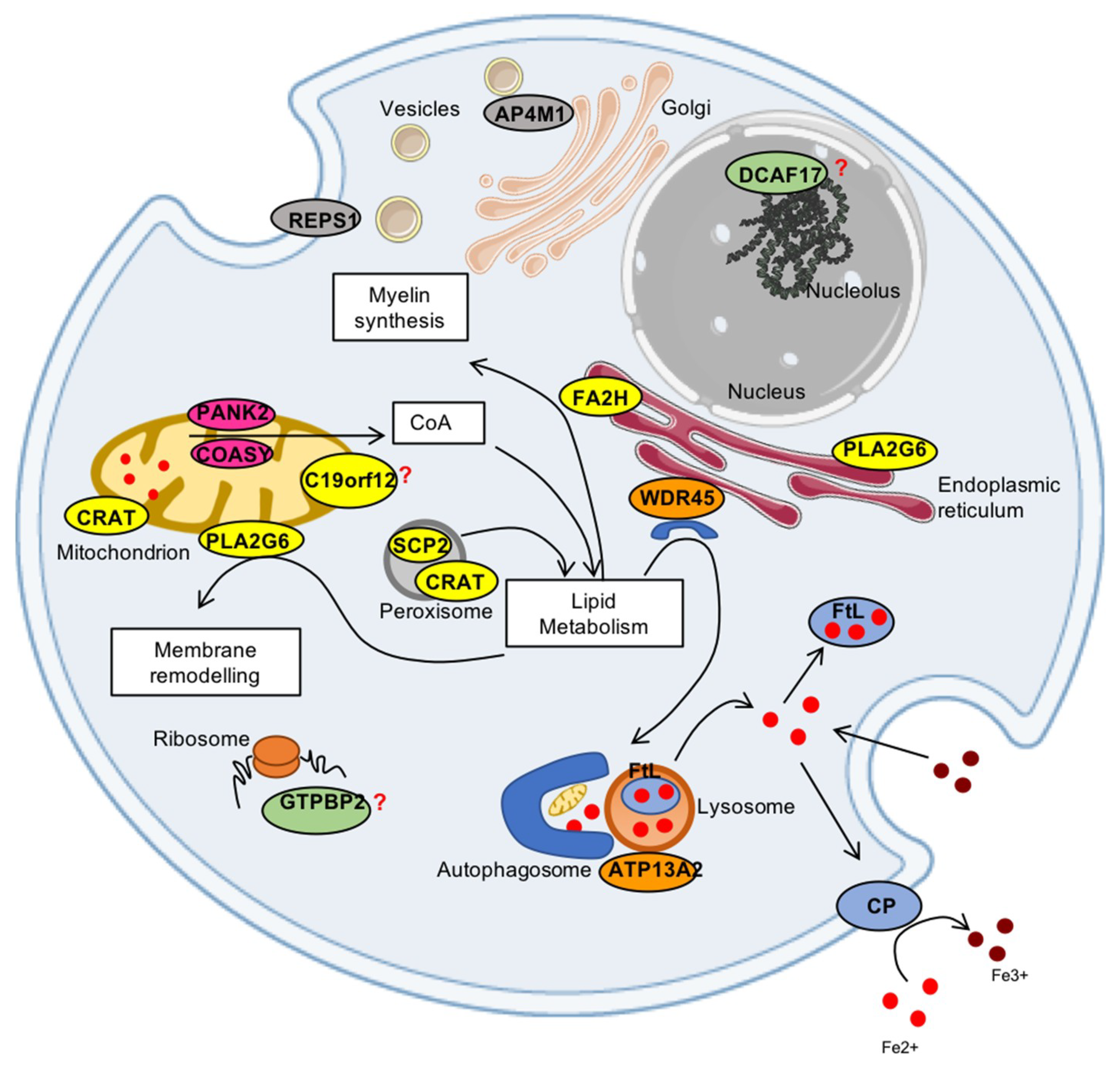
:1. Introduction
1.1. Iron Homeostasis in Diseases Caused by Mutations in Iron-Related Genes
1.1.1. CP (OMIM*117700)
1.1.2. FTL (OMIM*134790)
1.2. Iron Homeostasis in Inborn Errors of Coenzyme A Biosynthesis
PANK2 (OMIM*606157) and COASY (OMIM*609855)
1.3. Iron Homeostasis and Mutations in Lipid Metabolism Related Genes
1.3.1. PLA2G6 (OMIM*603604)
1.3.2. FA2H (OMIM*611026)
1.3.3. C19orf12 (OMIM*614297)
1.3.4. SCP2 (OMIM*184755) and CRAT (OMIM*600184)
1.4. Iron Homeostasis and Autophagosome/Lysosome Regulation
1.4.1. WDR45 (OMIM*300526)
1.4.2. ATP13A2 (OMIM*610513)
1.4.3. AP4M1 (OMIM*602296) and REPS1 (OMIM*614825)
1.5. Iron Homeostasis in Disease Genes with Unknown Functions
1.5.1. DCAF17 (OMIM*612515)
1.5.2. GTPBP2 (OMIM*607434)
2. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Gregory, A.; Hayflick, S. Neurodegeneration with Brain Iron Accumulation Disorders Overview; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2013. [Google Scholar]
- Arber, C.E.; Li, A.; Houlden, H.; Wray, S. Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathol. Appl. Neurobiol. 2016, 42, 220–241. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Nishimura, Y.; Mizoguchi, K.; Sakamoto, M.; Shimizu, T.; Honda, N. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology 1987, 37, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [Google Scholar] [PubMed]
- Miyajima, H. Aceruloplasminemia. Neuropathology 2015, 35, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.-P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef] [PubMed]
- Ogimoto, M.; Anzai, K.; Takenoshita, H.; Kogawa, K.; Akehi, Y.; Yoshida, R.; Nakano, M.; Yoshida, K.; Ono, J. Criteria for early identification of aceruloplasminemia. Intern. Med. 2011, 50, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Birchall, D.; Hayflick, S.J.; Gregory, A.; Schenk, J.F.; Zimmerman, E.A.; Shang, H.; Miyajima, H.; Chinnery, P.F.A. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology 2008, 70, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Tanaka, M.; Sotomatsu, A.; Hirai, S. Activated microglia cause superoxide-mediated release of iron from ferritin. Neurosci. Lett. 1995, 190, 21–24. [Google Scholar] [CrossRef]
- Morita, H.; Ikeda, S.; Yamamoto, K.; Morita, S.; Yoshida, K.; Nomoto, S.; Kato, M.; Yanagisawa, N. Hereditary ceruloplasmin deficiency with hemosiderosis: A clinicopathological study of a Japanese family. Ann. Neurol. 1995, 37, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cuyar, L.F.; Perry, G.; Miyajima, H.; Atwood, C.S.; Riveros-Angel, M.; Lyons, P.F.; Siedlak, S.L.; Smith, M.A.; Castellani, R.J. Redox active iron accumulation in aceruloplasminemia. Neuropathology 2008, 28, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Hineno, A.; Yoshida, K.; Ohara, S.; Morita, H.; Ikeda, S. Extensive brain pathology in a patient with aceruloplasminemia with a prolonged duration of illness. Hum. Pathol. 2012, 43, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Di Patti, M.C.B.; Maio, N.; Rizzo, G.; De Francesco, G.; Persichini, T.; Colasanti, M.; Polticelli, F.; Musci, G. Dominant mutants of ceruloplasmin impair the copper loading machinery in aceruloplasminemia. J. Biol. Chem. 2009, 284, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Hellman, N.E.; Kono, S.; Mancini, G.M.; Hoogeboom, A.J.; de Jong, G.J.; Gitlin, J.D. Mechanisms of copper incorporation into human ceruloplasmin. J. Biol. Chem. 2002, 277, 46632–46638. [Google Scholar] [CrossRef] [PubMed]
- Kono, S.; Suzuki, H.; Oda, T.; Shirakawa, K.; Takahashi, Y.; Kitagawa, M.; Miyajima, H. Cys-881 is essential for the trafficking and secretion of truncated mutant ceruloplasmin in aceruloplasminemia. J. Hepatol. 2007, 47, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yoshida, K.; Miyagoe, Y.; Ishikawa, A.; Hanaoka, K.; Nomoto, S.; Kaneko, K.; Ikeda, S.; Takeda, S. Quantitative evaluation of expression of iron-metabolism genes in ceruloplasmin-deficient mice. Biochim. Biophys. Acta 2002, 1588, 195–202. [Google Scholar] [CrossRef]
- Texel, S.J.; Camandola, S.; Ladenheim, B.; Rothman, S.M.; Mughal, M.R.; Unger, E.L.; Cadet, J.L.; Mattson, M.P. Ceruloplasmin deficiency results in an anxiety phenotype involving deficits in hippocampal iron, serotonin, and BDNF. J. Neurochem. 2012, 120, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Ropert, M.; le Lan, C.; Loreal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Kono, S.; Suzuki, H.; Oda, T.; Miyajima, H.; Takahashi, Y.; Shirakawa, K.; Ishikawa, K.; Kitagawa, M. Biochemical features of ceruloplasmin gene mutations linked to aceruloplasminemia. Neuromol. Med. 2006, 8, 361–374. [Google Scholar] [CrossRef]
- Hahn, P.; Qian, Y.; Dentchev, T.; Chen, L.; Beard, J.; Harris, Z.L.; Dunaief, J.L. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 13850–13855. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Vulpe, C.D.; Harris, L.Z.; David, S. Iron efflux from oligodendrocytes is differentially regulated in gray and white matter. J. Neurosci. 2011, 31, 13301–13311. [Google Scholar] [CrossRef] [PubMed]
- Hadziahmetovic, M.; Dentchev, T.; Song, Y.; Haddad, N.; He, X.; Hahn, P.; Pratico, D.; Wen, R.; Harris, Z.L.; Lambris, J.D.; et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest. Ophthalmol. Vis. Sci. 2008, 49, 2728–2736. [Google Scholar] [CrossRef] [PubMed]
- Hadziahmetovic, M.; Song, Y.; Wolkow, N.; Iacovelli, J.; Grieco, S.; Lee, J.; Lyubarsky, A.; Pratico, D.; Connelly, J.; Spino, M.; et al. The oral iron chelator deferiprone protects against iron overload-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci. 2011, 52, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Finkenstedt, A.; Wolf, E.; Höfner, E.; Gasser, B.I.; Bösch, S.; Bakry, R.; Creus, M.; Kremser, C.; Schocke, M.; Theurl, M.; et al. Hepatic but not brain iron is rapidly chelated by deferasirox in aceruloplasminemia due to a novel gene mutation. J. Hepatol. 2010, 53, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Do, R.F.; Roberti, M.; Borges Filho, H.M.; Goncalves, C.H.; Lima, F.L. Aceruloplasminemia: A rare disease—Diagnosis and treatment of two cases. Rev. Bras. Hematol. Hemoter. 2011, 33, 389–392. [Google Scholar]
- Zanardi, A.; Conti, A.; Cremonesi, M.; D’Adamo, P.; Gilberti, E.; Apostoli, P.; Cannistraci, C.V.; Piperno, A.; David, S.; Alessio, M. Ceruloplasmin replacement therapy ameliorates neurological symptoms in a preclinical model of aceruloplasminemia. EMBO Mol. Med. 2018, 10, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.R.J.; Fey, C.; Morris, C.M.; Bindoff, L.A.; Ince, P.G.; Chinnery, P.F.; Coulthard, A.; Jackson, M.J.; Jackson, A.P.; McHale, D.P.; et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 2001, 28, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Crompton, D.E.; Birchall, D.; Jackson, M.J.; Coulthard, A.; Lombès, A.; Quinn, N.; Wills, A.; Fletcher, N.; Mottershead, J.P.; et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain 2007, 130, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Davidzon, G.; Kurlan, R.M.; Tawil, R.; Bonilla, E.; Di Mauro, S.; Powers, J.M. Hereditary ferritinopathy: A novel mutation, its cellular pathology, and pathogenetic insights. J. Neuropathol. Exp. Neurol. 2005, 64, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Barbeito, A.G.; Muhoberac, B.B.; Vidal, R. Iron-mediated aggregation and a localized structural change characterize ferritin from a mutant light chain polypeptide that causes neurodegeneration. J. Biol. Chem. 2008, 283, 31679–31689. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Muhoberac, B.B.; Garringer, H.J.; Hurley, T.D.; Vidal, R. Unraveling of the E-helices and disruption of 4-fold pores are associated with iron mishandling in a mutant ferritin causing neurodegeneration. J. Biol. Chem. 2010, 285, 1950–1956. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, A.; Santambrogio, P.; Corsi, B.; Campanella, A.; Arosio, P.; Levi, S. Characterization of the l-ferritin variant 460InsA responsible of a hereditary ferritinopathy disorder. Neurobiol. Dis. 2006, 23, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, A.; Rovelli, E.; Frizzale, G.; Campanella, A.; Amendola, M.; Arosio, P.; Levi, S. Oxidative stress and cell death in cells expressing L-ferritin variants causing neuroferritinopathy. Neurobiol. Dis. 2010, 37, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Ghetti, B.; Takao, M.; Brefel-Courbon, C.; Uro-Coste, E.; Glazier, B.S.; Siani, V.; Benson, M.D.; Calvas, P.; Miravalle, L.; et al. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J. Neuropathol. Exp. Neurol. 2004, 63, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Vidal, R.; Englander, E.W. Accumulation of oxidative DNA damage in brain mitochondria in mouse model of hereditary ferritinopathy. Neurosci. Lett. 2010, 479, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Maccarinelli, F.; Pagani, A.; Cozzi, A.; Codazzi, F.; Di Giacomo, G.; Capoccia, S.; Rapino, S.; Finazzi, D.; Politi, L.S.; Cirulli, F.; et al. A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiol. Dis. 2015, 81, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Barbeito, A.G.; Garringer, H.J.; Baraibar, M.A.; Gao, X.; Arredondo, M.; Núñez, M.T.; Smith, M.A.; Ghetti, B.; Vidal, R. Abnormal iron metabolism and oxidative stress in mice expressing a mutant form of the ferritin light polypeptide gene. J. Neurochem. 2009, 109, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Miravalle, L.; Gao, X.; Barbeito, A.G.; Baraibar, M.A.; Hekmatyar, S.K.; Widel, M.; Bansal, N.; Delisle, M.B.; Ghetti, B.R. Expression of a mutant form of the ferritin light chain gene induces neurodegeneration and iron overload in transgenic mice. J. Neurosci. 2008, 28, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Barbeito, A.G.; Levade, T.; Delisle, M.B.; Ghetti, B.; Vidal, R. Abnormal iron metabolism in fibroblasts from a patient with the neurodegenerative disease hereditary ferritinopathy. Mol. Neurodegener. 2010, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Capoccia, S.; Maccarinelli, F.; Buffoli, B.; Rodella, L.F.; Cremona, O.; Arosio, P.; Cirulli, F. Behavioral characterization of mouse models of neuroferritinopathy. PLoS ONE 2015, 10, e0118990. [Google Scholar] [CrossRef] [PubMed]
- Garringer, H.J.; Irimia, J.M.; Li, W.; Goodwin, C.B.; Richine, B.; Acton, A.; Chan, R.J.; Peacock, M.; Muhoberac, B.B.; Ghetti, B.; et al. Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS ONE 2016, 11, e0161341. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Hida, A.; Ichikawa, Y.; Momose, Y.; Goto, J.; Igeta, Y.; Hashida, H.; Yoshida, K.; Ikeda, S.; Kanazawa, I.; et al. A novel ferritin light chain gene mutation in a Japanese family with neuroferritinopathy: Description of clinical features and implications for genotype-phenotype correlations. Mov. Disord. 2009, 24, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Dusek, P.; Hardy, J.; Westenberger, A.; Jankovic, J.; Bhatia, K.P. Genetics and Pathophysiology of Neurodegeneration with Brain Iron Accumulation (NBIA). Curr. Neuropharmacol. 2013, 11, 59–79. [Google Scholar] [PubMed]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Evers, C.; Seitz, A.; Assmann, B.; Opladen, T.; Karch, S.; Hinderhofer, K.; Granzow, M.; Paramasivam, N.; Eils, R.; Diessl, N.; et al. Diagnosis of CoPAN by whole exome sequencing: Waking up a sleeping tiger’s eye. Am. J. Med. Genet. A 2017. [Google Scholar] [CrossRef] [PubMed]
- Annesi, G.; Gagliardi, M.; Iannello, G.; Quattrone, A.; Iannello, G.; Quattrone, A. Mutational analysis of COASY in an Italian patient with NBIA. Parkinsonism Relat. Disord. 2016, 28, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, T.; Ferdinandusse, S.; Ruiter, J.P.N.; Alders, M.; Mathijssen, I.B.; Parboosingh, J.S.; Innes, A.M.; Meijers-Heijboer, H.; Poll-The, B.T.; Bernier, F.P.; et al. Biallelic loss of function variants in COASY cause prenatal onset pontocerebellar hypoplasia, microcephaly, and arthrogryposis. Eur. J. Hum. Genet. 2018, 26, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.A.; Hayflick, S.J. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): Review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int. Rev. Neurobiol. 2013, 110, 49–71. [Google Scholar] [PubMed]
- Zorzi, G.; Zibordi, F.; Chiapparini, L.; Bertini, E.; Russo, L.; Piga, A.; Longo, F.; Garavaglia, B.; Aquino, D.; Savoiardo, M.; et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: Results of a phase II pilot trial. Mov. Disord. 2011, 26, 1756–1759. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Hiken, M.; Gregory, A.; Malandrini, A.; Clark, D.; Hogarth, P.; Grafe, M.; Hayflick, S.J.; Woltjer, R.L. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011, 134, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-M.; Duncan, J.L.; Westaway, S.K.; Yang, H.; Nune, G.; Xu, E.Y.; Hayflick, S.J.; Gitschier, J. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum. Mol. Genet. 2005, 14, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; d’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate kinase-associated neurodegeneration: Altered mitochondria membrane potential and defective respiration in Pank2 knock-out mouse model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Leonardi, R.; Zhang, Y.-M.; Rehg, J.E.; Jackowski, S. Germline deletion of pantothenate kinases 1 and 2 reveals the key roles for CoA in postnatal metabolism. PLoS ONE 2012, 7, e40871. [Google Scholar] [CrossRef] [PubMed]
- Bosveld, F.; Rana, A.; van der Wouden, P.E.; Lemstra, W.; Ritsema, M.; Kampinga, H.H.; Sibon, O.C. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum. Mol. Genet. 2008, 17, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Z.; Kuo, Y.M.; Zhou, B. Dietary rescue of fumble—A Drosophila model for pantothenate-kinase-associated neurodegeneration. J. Inherit. Metab. Dis. 2005, 28, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial iron and energetic dysfunction distinguish fibroblasts and induced neurons from pantothenate kinase-associated neurodegeneration patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. iPSC-derived neuronal models of PANK2-associated neurodegeneration reveal mitochondrial dysfunction contributing to early disease. PLoS ONE 2017, 12, e0184104. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, G.; Cossu, G.; Balocco, M.; Marchese, R.; Murgia, D.; Melis, M.; Galanello, R.; Barella, S.; Matta, G.; Ruffinengo, U.; et al. A pilot trial of deferiprone for neurodegeneration with brain iron accumulation. Haematologica 2011, 96, 1708–1711. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Subramanian, C.; Yun, M.K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S.A. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef] [PubMed]
- Berti, C.C.; Dallabona, C.; Lazzaretti, M.; Dusi, S.; Tosi, E.; Tiranti, V.; Goffrini, P. Modeling human Coenzyme A synthase mutation in yeast reveals altered mitochondrial function, lipid content and iron metabolism. Microb. Cell (Graz, Austria) 2015, 2, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Khatri, D.; Zizioli, D.; Tiso, N.; Facchinello, N.; Vezzoli, S.; Gianoncelli, A.; Memo, M.; Monti, E.; Borsani, G.; Finazzi, D. Down-regulation of coasy, the gene associated with NBIA-VI, reduces Bmp signaling, perturbs dorso-ventral patterning and alters neuronal development in zebrafish. Sci. Rep. 2016, 6, 37660. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Wiersma, M.; Schüller, H.J.; Pode-Shakked, B.; Marek-Yagel, D.; Grigat, M.; Schwarzmayr, T.; Berutti, R.; Alhaddad, B.; Kanon, B.; et al. Mutations in PPCS, Encoding Phosphopantothenoylcysteine Synthetase, Cause Autosomal-Recessive Dilated Cardiomyopathy. Am. J. Hum. Genet. 2018, 102, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Schneider, S.A. Neurodegeneration with brain iron accumulation. Curr. Opin. Neurol. 2012, 25, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, X.; Zhao, C.; Choi, J.; Shi, J.; Song, K.; Turk, J.; Ma, Z.A. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology 2010, 151, 3038–3048. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Turk, J.; Mancuso, D.J.; Montier, L.; Wohltmann, M.; Wozniak, D.F.; Schmidt, R.E.; Gross, R.W.; Kotzbauer, P.T. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am. J. Pathol. 2008, 172, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Claypool, S.M.; Koehler, C.M. The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 2012, 37, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Beck, G.; Sugiura, Y.; Shinzawa, K.; Kato, S.; Setou, M.; Tsujimoto, Y.; Sakoda, S.; Sumi-Akamaru, H. Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 2011, 31, 11411–11420. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Biogenesis of iron-sulfur clusters in mammalian cells: New insights and relevance to human disease. Dis. Model. Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Paisán-Ruiz, C.; Boddaert, N.; Yoon, M.Y.; Hama, H.; Gregory, A.; Malandrini, A.; Woltjer, R.L.; Munnich, A.; Gobin, S.; et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann. Neurol. 2010, 68, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, M.; Yaghootfam, A.; Fewou, S.N.; Zoller, I.; Gieselmann, V. A mammalian fatty acid hydroxylase responsible for the formation of alpha-hydroxylated galactosylceramide in myelin. Biochem. J. 2005, 388, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Bhatia, K.P. Syndromes of neurodegeneration with brain iron accumulation. Semin. Pediatr. Neurol. 2012, 19, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Hayflick, S.J. Genetics of neurodegeneration with brain iron accumulation. Curr. Neurol. Neurosci. Rep. 2011, 11, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.A.; Kern, M.J.; Fullbright, G.; Bielawski, J.; Scherer, S.S.; Yum, S.W.; Li, J.J.; Cheng, H.; Han, X.; Venkata, J.K.; et al. Central nervous system dysfunction in a mouse model of FA2H deficiency. Glia 2011, 59, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Zoller, I.; Meixner, M.; Hartmann, D.; Büssow, H.; Meyer, R.; Gieselmann, V.; Eckhardt, M. Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration. J. Neurosci. 2008, 28, 9741–9754. [Google Scholar] [CrossRef] [PubMed]
- Mari, F.; Berti, B.; Romano, A.; Baldacci, J.; Rizzi, R.; Grazia Alessandrì, M.; Tessa, A.; Procopio, E.; Rubegni, A.; Lourenḉo, C.M.; et al. Clinical and neuroimaging features of autosomal recessive spastic paraplegia 35 (SPG35): Case reports, new mutations, and brief literature review. Neurogenetics 2018, 19, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M.; et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Panteghini, C.; Zorzi, G.; Venco, P.; Dusi, S.; Reale, C.; Brunetti, D.; Chiapparini, L.; Zibordi, F.; Siegel, B.; Garavaglia, B.; et al. C19orf12 and FA2H mutations are rare in Italian patients with neurodegeneration with brain iron accumulation. Semin. Pediatr. Neurol. 2012, 19, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Salih, M.A.; Mooney, C.; Alzahrani, J.; Elmalik, S.A.; Kabiraj, M.M.; Khan, A.O.; Paudel, R.; Houlden, H.; Azzedine, H.; et al. C19orf12 mutation leads to a pallido-pyramidal syndrome. Gene 2014, 537, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Landoure, G.; Zhu, P.P.; Lourenço, C.M.; Johnson, J.O.; Toro, C.; Bricceno, K.V.; Rinaldi, C.; Meilleur, K.G.; Sangaré, M.; Diallo, O.; et al. Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum. Mutat. 2013, 34, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Deschauer, M.; Gaul, C.; Behrmann, C.; Prokisch, H.; Zierz, S.; Haack, T.B. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J. Neurol. 2012, 259, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Holinski-Feder, E.; Neeve, V.C.; Pyle, A.; Griffin, H.; Ashok, D.; Foley, C.; Hudson, G.; Rautenstrauss, B.; Nürnberg, G.; et al. A new phenotype of brain iron accumulation with dystonia, optic atrophy, and peripheral neuropathy. Mov. Disord. 2012, 27, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Langwinska-Wosko, E.; Skowronska, M.; Kmiec, T.; Czlonkowska, A. Retinal and optic nerve abnormalities in neurodegeneration associated with mutations in C19orf12 (MPAN). J. Neurol. Sci. 2016, 370, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Kmiec, T.; Czlonkowska, A.; Kurkowska-Jastrzebska, I. Transcranial Sonography in Mitochondrial Membrane Protein-Associated Neurodegeneration. Clin. Neuroradiol. 2018, 28, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Venco, P.; Bonora, M.; Giorgi, C.; Papaleo, E.; Iuso, A.; Prokisch, H.; Pinton, P.; Tiranti, V. Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca(2)(+). Front. Genet. 2015, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Sibon, O.C.; Gorza, M.; Heim, K.; Organisti, C.; Meitinger, T.; Prokisch, H. Impairment of Drosophila orthologs of the human orphan protein C19orf12 induces bang sensitivity and neurodegeneration. PLoS ONE 2014, 9, e89439. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Kostopoulos, P.; Denis, S.; Rusch, H.; Overmars, H.; Dillmann, U.; Reith, W.; Haas, D.; Wanders, R.J.; Duran, M.; et al. Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am. J. Hum. Genet. 2006, 78, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Lewis-Smith, D.; Douroudis, K.; Duff, J.; Keogh, M.; Pyle, A.; Fletcher, N.; Chinnery, P.F. SCP2 mutations and neurodegeneration with brain iron accumulation. Neurology 2015, 85, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Seedorf, U.; Raabe, M.; Ellinghaus, P.; Kannenberg, F.; Fobker, M.; Engel, T.; Denis, S.; Wouters, F.; Wirtz, K.W.; Wanders, R.J.; et al. Defective peroxisomal catabolism of branched fatty acyl coenzyme A in mice lacking the sterol carrier protein-2/sterol carrier protein-x gene function. Genes Dev. 1998, 12, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Vreken, P.; Ferdinandusse, S.; Jansen, G.A.; Waterham, H.R.; van Roermund, C.W.; Van Grunsven, E.G. Peroxisomal fatty acid alpha- and beta-oxidation in humans: Enzymology, peroxisomal metabolite transporters Peroxisomal fatty acid alpha- and beta-oxidation in humans: Enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem. Soc. Trans. 2001, 29, 250–267. [Google Scholar] [CrossRef] [PubMed]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traoré, M.; Habarou, F.; Bole-Feysot, C.; Nitschké, P.; Ottolenghi, C.; et al. Impaired Transferrin Receptor Palmitoylation and Recycling in Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Hogarth, P.; Kruer, M.C.; Gregory, A.; Wieland, T.; Schwarzmayr, T.; Graf, E.; Sanford, L.; Meyer, E.; Kara, E.; et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012, 91, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Waddell, S.; Gaugel, A.; Frickey, T.; Lupas, A.; Nordheim, A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004, 23, 9314–9325. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Sun, L.; Miao, G.; Ji, C.; Zhao, H.; Sun, H.; Miao, L.; Yoshii, S.R.; Mizushima, N.; Wang, X.; Zhang, H. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015, 11, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, R.; Memo, M.; Garavaglia, B. Ferrous Iron Up-regulation in Fibroblasts of Patients with Beta Propeller Protein-Associated Neurodegeneration (BPAN). Front. Genet. 2017, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Paisán-Ruiz, C.; Quinn, N.P.; Lees, A.J.; Houlden, H.; Hardy, J.; Bhatia, K.P. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov. Disord. 2010, 25, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Bras, J.; Verloes, A.; Schneider, S.A.; Mole, S.E.; Guerreiro, R.J. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum. Mol. Genet. 2012, 21, 2646–2650. [Google Scholar] [CrossRef] [PubMed]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Münchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1843.e1–1843.e7. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, P.J.; Fleming, S.M.; Clippinger, A.K.; Lewis, J.; Tsunemi, T.; Giasson, B.; Dickson, D.W.; Mazzulli, J.R.; Bardgett, M.E.; Haik, K.L.; et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013, 22, 2067–2082. [Google Scholar] [CrossRef] [PubMed]
- Kett, L.R.; Stiller, B.; Bernath, M.M.; Tasset, I.; Blesa, J.; Jackson-Lewis, V.; Chan, R.B.; Zhou, B.; Di Paolo, G.; Przedborski, S.; et al. alpha-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J. Neurosci. 2015, 35, 5724–5742. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Rane, A.; Chinta, S.J.; Andersen, J.K. Regulation of ATP13A2 via PHD2-HIF1alpha Signaling Is Critical for Cellular Iron Homeostasis: Implications for Parkinson’s Disease. J. Neurosci. 2016, 36, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Roubertie, A.; Hieu, N.; Roux, C.J.; Leboucq, N.; Manes, G.; Charif, M.; Echenne, B.; Goizet, C.; Guissart, C.; Meyer, P.; et al. AP4 deficiency: A novel form of neurodegeneration with brain iron accumulation? Neurol. Genet. 2018, 4, e217. [Google Scholar] [CrossRef] [PubMed]
- Duerinckx, S.; Verhelst, H.; Perazzolo, C.; David, P.; Desmyter, L.; Pirson, I.; Abramowicz, M. Severe congenital microcephaly with AP4M1 mutation, a case report. BMC Med. Genet. 2017, 18, 48. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.M.H.; Schot, R.; Dumee, B.; Schellekens, K.; Swagemakers, S.; Bertoli-Avella, A.M.; Lequin, M.H.; Dudink, J.; Govaert, P.; van Zwol, A.L.; et al. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am. J. Hum. Genet. 2009, 85, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Al-Saif, A.; Al-Semari, A.; Bohlega, S.; Zlitni, S.; Alzahrani, F.; Bavi, P.; Kaya, N.; Colak, D.; Khalak, H.; et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am. J. Hum. Genet. 2008, 83, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Arias, E.E.; Chen, J.; Harper, J.W.; Walter, J.C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 2006, 23, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Mistry, B.V.; AAhmed, H.; Abdulla, R.; AAmer, H.; Prince, A.; MAlazami, A.; SAlkuraya, F.; Assiri, A. Deletion of DDB1- and CUL4-associated factor-17 (Dcaf17) gene causes spermatogenesis defects and male infertility in mice. Sci. Rep. 2018, 8, 9202. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, F.; Desai, S.; Diao, F.; Turkkahraman, D.; Wranitz, F.; Wood-Trageser, M.; Shin, Y.H.; Kotan, L.D.; Jiang, H.; Witchel, S.; et al. Novel inactivating mutations of the DCAF17 gene in American and Turkish families cause male infertility and female subfertility in the mouse model. Clin. Genet. 2018, 93, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Jaberi, E.; Rohani, M.; Shahidi, G.A.; Nafissi, S.; Arefian, E.; Soleimani, M.; Rasooli, P.; Ahmadieh, H.; Daftarian, N.; KaramiNejadRanjbar, M.; et al. Identification of mutation in GTPBP2 in patients of a family with neurodegeneration accompanied by iron deposition in the brain. Neurobiol. Aging 2016, 38, 216.e11–216.e18. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Garcia-Aznar, J.M.; Brandau, O.; Al-Hakami, F.; Yüksel, Z.; Marais, A.; Grüning, N.M.; Abbasi Moheb, L.; Paknia, O.; Alshaikh, N.; et al. Biallelic inactivating variants in the GTPBP2 gene cause a neurodevelopmental disorder with severe intellectual disability. Eur. J. Hum. Genet. 2018, 26, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.-C.; Kim, T.-D.; Lee, K.-H.; Kim, D.-Y.; Kim, S.; Lee, H.-R.; Kang, H.J.; Chung, S.J.; Senju, S.; Nishimura, Y.; et al. Modulation of exosome-mediated mRNA turnover by interaction of GTP-binding protein 1 (GTPBP1) with its target mRNAs. FASEB J. 2011, 25, 2757–2769. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Nagy, G.; Dotu, I.; Zhou, H.; Yang, X.-L.; Schimmel, P.; Senju, S.; Nishimura, Y.; Chuang, J.H.; Ackerman, S.L. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 2014, 345, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Zinoviev, A.; Goyal, A.; Jindal, S.; LaCava, J.; Komar, A.A.; Rodnina, M.V.; Hellen, C.U.T.; Pestova, T.V. Functions of unconventional mammalian translational GTPases GTPBP1 and GTPBP2. Genes Dev. 2018, 32, 1226–1241. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, C.; Forabosco, P.; Wiethoff, S.; Heidari, M.; Johnstone, D.M.; Botía, J.A.; Collingwood, J.F.; Hardy, J. Gene co-expression networks shed light into diseases of brain iron accumulation. Neurobiol. Dis. 2016, 87, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Tello, C.; Darling, A.; Lupo, V.; Perez-Duenas, B.; Espinos, C. On the complexity of clinical and molecular bases of neurodegeneration with brain iron accumulation. Clin. Genet. 2018, 93, 731–740. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Disease | Inheritance | Function | Protein localization | Brain Iron and Clinical Features |
|---|---|---|---|---|---|
| Genes related to iron homeostasis | |||||
| CP | Acerulo-plasminaemia | AR | Iron oxidation | Plasma membrane | Iron in the basal ganglia, liver, pancreas and myocardium. Movement disorders, dementia, diabetes mellitus, retinal degeneration, dysarthria, ataxia |
| FTL1 | Neuro-ferritinopathy (NF) | AD | Cellular iron storage | Cytoplasm | Iron deposition in basal ganglia, cerebellum, motor cortex; mild cerebral and cerebellar atrophy, cavitation of the putamen. Extrapyramidal movement disorders, dystonia, parkinsonisms, dysarthria |
| Genes related to Coenzyme A biosynthesis | |||||
| PANK2 | Pantothenate kinase-associated neurodegeneration (PKAN) | AR | Panthotenate phosphorylation; Coenzyme A synthesis | Mitochondria (inner membrane space) | Iron overload in GP; “eye of the tiger sign”. Dystonia, spasticity, cognitive decline, pigmentary retinopathy |
| COASY | COASY protein-associated neurodegeneration (CoPAN) | AR | 4’-PP adenyltran-sferase and dephospho-CoA kinase; Coenzyme A synthesis | Mitochondria (matrix), cytosol | Iron overload in GP. Oro-mandibular dystonia, dysarthria, spastic-dystonic paraparesis, obsessive-compulsive behavior |
| Genes related to lipid metabolism | |||||
| PLA2G6 | PLA2G6-associated neurodegeneration (PLAN) | AR | Hydrolysis of ester bonds at the sn-2 position of phospho-lipids; Membrane remodeling | Mitochondria, endoplasmic reticulum, cytosol | Iron overload in GP in <50% of cases. Infantile neuroaxonal dystrophy, hypotonia, gait disturbance and cerebellar atrophy. Dystonia, spasticity and parkinsonisms in adulthood |
| FA2H | Fatty acid hydroxylase-associated neurodegeneration | AR | Hydroxylation of fatty acids; Ceramide synthesis; Myelin formation | Endoplasmic reticulum | Iron overload in GP and SN. Profound ataxia, dystonia, dysarthria, spastic quadriplegia, axial hypotonia, optic atrophy |
| C19orf12 | Mitochondrial membrane protein-associated neurodegeneration (MPAN) | AR | Unknown; Lipid metabolism? Membrane remodeling? | Mitochondrial membranes, endoplasmic reticulum, MAM | Iron overload in GP and SN; abundant Lewy bodies. Global developmental delay, dystonia, parkinsonism, psychiatric symptoms, spastic paraparesis |
| SCP2 | Leukoencephalopathy with dystonia and motor neuropathy | AR | Thiolase activity; Breakdown of branched chain fatty acids | Peroxisomes | Iron deposition in thalamus; Dystonia and spasmodic torticollis, spinocerebellar ataxia, balance and gait impairment |
| CRAT | AR | Carnitine acetyltrasnferase, -oxidation | Mitochondria | Iron accumulation in GP and SN. Slowly progressive spinocerebellar degeneration. Cerebellar atrophy and posterior leukodystrophy | |
| Genes related to autophagy | |||||
| WDR45 | β-propeller-associated neurodegeneration (BPAN) | X-linked (de novo mutations) | Protein-protein interaction; Early autophagosome formation | Endoplasmic reticulum | Iron overload in GP and SN. Global developmental delay, neurological deterioration, dystonia, parkinsonism, cognitive decline, seizures |
| ATP13A2 | Kufor-Rakeb disease (KRS) | AR | Lysosomal cation pump; autophagosome formation | Lysosome, mitochondria | Often no iron overload. Early onset parkinsonism, pyramidal signs, altered eye movements, dementia |
| AP4M1 | AR | Vesicle formation | Endosome | Iron in globus pallidus reported in a single family. Early-onset developmental delay and deterioration of motor function, tetraparesis, intellectual disability | |
| REPS1 | AR | Endocytosis and vesicle transport | Cytoplasm, endosome | Iron accumulation in the globus pallidus and peduncles. Trunk hypotonia, progressive cerebellar ataxia, pyramidal syndrome. Cerebellar and cerebral atrophy. | |
| Genes with unknown function | |||||
| DCAF17 | Woodhouse-Sakati syndrome (WSS) | AR | Unknown | Nucleolus | Sometimes iron overload in GP and SN. Extrapyramidal symptoms, dystonia, cognitive impairment, hypogonadism, alopecia, diabetes mellitus |
| GTPBP2 | AR | Unknown; mRNA/ribosome stability? | Cytoplasm | Iron overload in GP and SN; cerebellar atrophy. Mental retardation, ataxia and dystonia | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. https://doi.org/10.3390/ph12010027
Levi S, Tiranti V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals. 2019; 12(1):27. https://doi.org/10.3390/ph12010027
Chicago/Turabian StyleLevi, Sonia, and Valeria Tiranti. 2019. "Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition" Pharmaceuticals 12, no. 1: 27. https://doi.org/10.3390/ph12010027
APA StyleLevi, S., & Tiranti, V. (2019). Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals, 12(1), 27. https://doi.org/10.3390/ph12010027