Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Normal Iron Metabolism
1.1. The Ferroportin/Hepcidin Duo Controls Systemic Iron Metabolism
1.2. Regulation of Hepcidin Expression
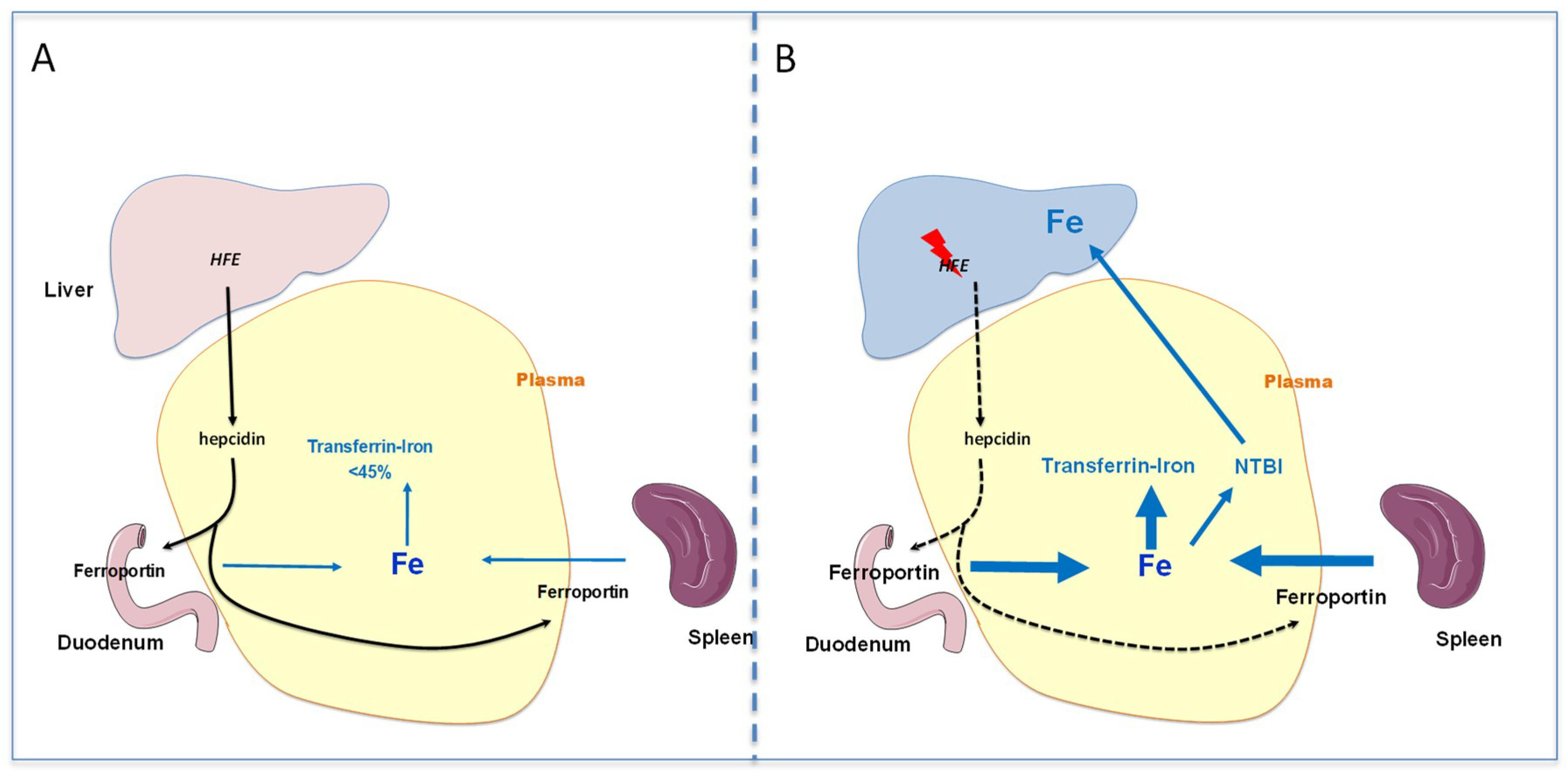
2. Pathophysiology of HFE Hemochromatosis
2.1. HFE Hemochromatosis
2.2. Pathophysiology of Iron Overload during HFE Hemochromatosis
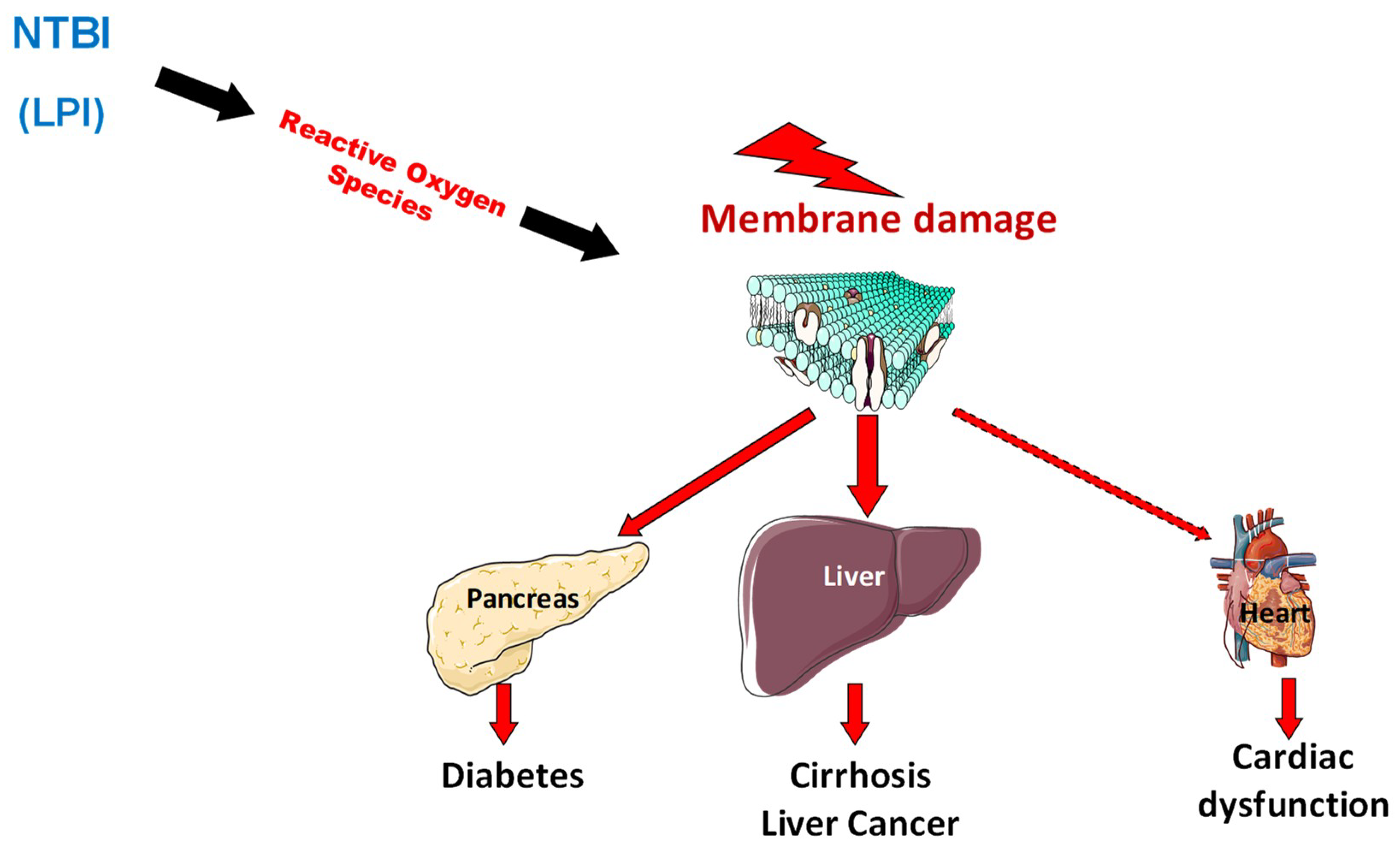
2.3. Pathophysiology of Organ Damage in Hemochromatosis
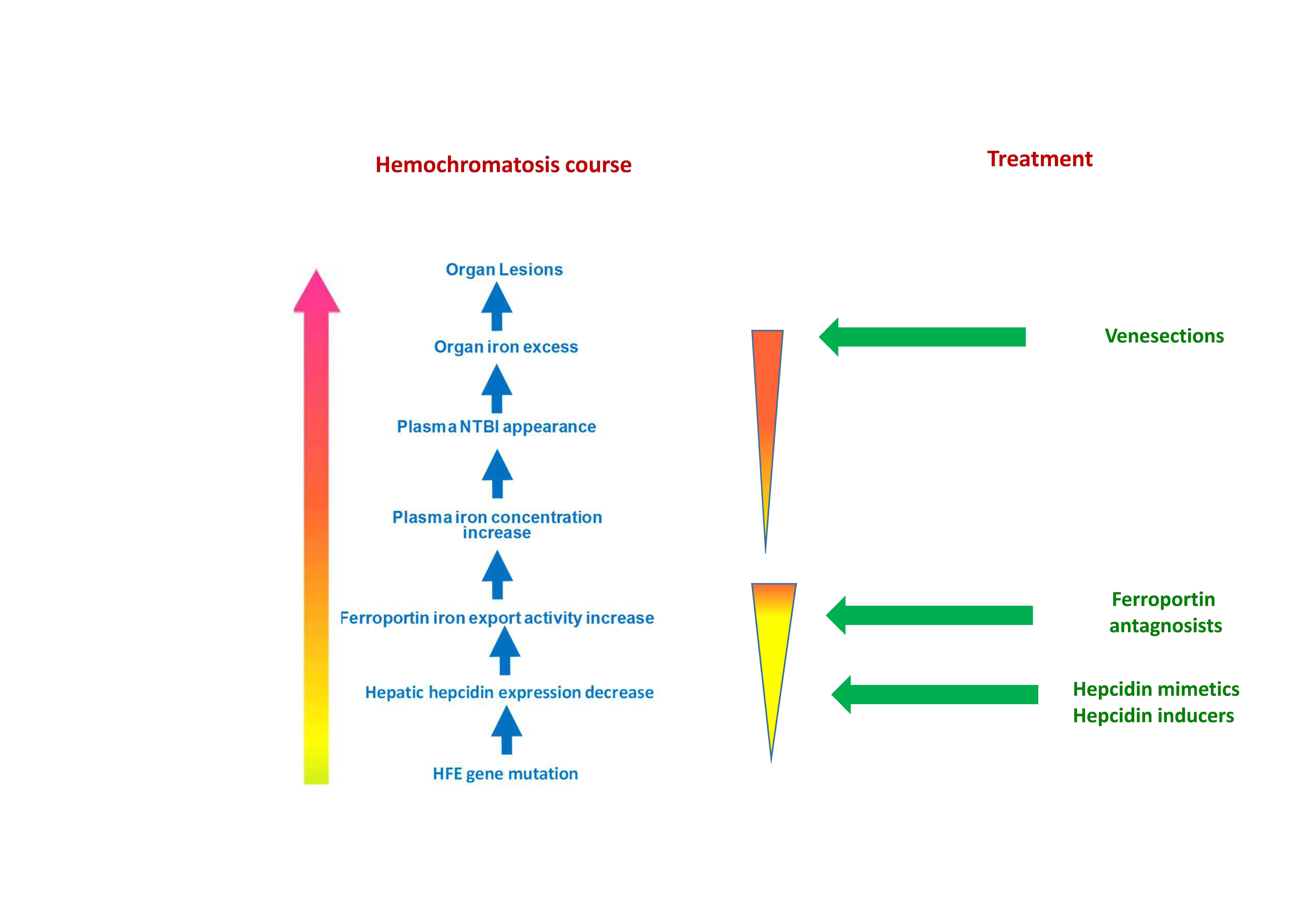
3. Iron Is Presently the Main Therapeutic Target in HFE-Related Hemochromatosis
3.1. Venesections Are the Mainstay Treatment for Iron Removal
3.2. Use of Chelation Therapy
3.3. Biochemical Follow-Up of Venesection Therapy
4. Iron Removal Is not the Unique Therapeutic Target in HFE-Related Hemochromatosis
4.1. Other Preventive Actions to Avoid Iron Overload Complications
4.2. The Development of a Pathophysiological Treatment Is a Major Goal
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, G.; Di Sabatino, A.; Pasini, A.; Ubezio, C.; Costanzo, F.; Grataroli, D.; Masotti, M.; Alvisi, C.; Corazza, G.R. Intestinal expression of genes implicated in iron absorption and their regulation by hepcidin. Clin. Nutr. Edinb. Scotl. 2017, 36, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, T.H.; Pirzio-Biroli, G.; Finch, C.A. Iron absorption. I. Factors influencing absorption. J. Lab. Clin. Med. 1958, 51, 24–36. [Google Scholar] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [PubMed]
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751. [Google Scholar] [PubMed]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Roetto, A.; Cali, A.; De Gobbi, M.; Garozzo, G.; Carella, M.; Majorano, N.; Totaro, A.; Gasparini, P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 2000, 25, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Cox, T.M. Co-localization of the mammalian hemochromatosis gene product (HFE) and a newly identified transferrin receptor (TFR2) in intestinal tissue and cells. J. Histochem. Cytochem. 2003, 51, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Neitz, S.; Magert, H.J.; Schulz, A.; Forssmann, W.G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loreal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Viatte, L.; Bennoun, M.; Beaumont, C.; Kahn, A.; Vaulont, S. Hepcidin, A New Iron Regulatory Peptide. Blood Cells Mol. Dis. 2002, 29, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.G.; Kulaksiz, H.; Herrmann, T.; Riedel, H.D.; Bents, K.; Veltkamp, C.; Stremmel, W. Expression of hepcidin in hereditary hemochromatosis: Evidence for a regulation in response to serum transferrin saturation and non-transferrin-bound iron. Blood 2003, 102, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Ramey, G.; Deschemin, J.C.; Vaulont, S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009, 94, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Tsuchihashi, Z.; Irrinki, A.; Lee, V.K.; Mapa, F.A.; Morikang, E.; Prass, C.E.; Starnes, S.M.; Wolff, R.K.; Parkkila, S.; et al. The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J. Biol. Chem. 1997, 272, 14025–14028. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Penny, D.M.; Irrinki, A.; Lee, V.K.; Lebron, J.A.; Watson, N.; Tsuchihashi, Z.; Sigal, E.; Bjorkman, P.J.; Schatzman, R.C. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc. Natl. Acad. Sci. USA 1998, 95, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates HFE-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.; Andrews, N.C. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem. 2006, 281, 28494–28498. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Fleming, M.D. Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor-2. Am. J. Hematol. 2012, 87, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 2009, 41, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B., Jr.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, G.; Mleczko-Sanecka, K.; Altamura, S.; Hentze, M.W.; Muckenthaler, M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 2009, 87, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Island, M.L.; Jouanolle, A.M.; Mosser, A.; Deugnier, Y.; David, V.; Brissot, P.; Loreal, O. A new mutation in the hepcidin promoter impairs its BMP response and contributes to a severe phenotype in HFE related hemochromatosis. Haematologica 2009, 94, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.-Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Rausa, M.; Pagani, A.; Nai, A.; Campanella, A.; Gilberti, M.E.; Apostoli, P.; Camaschella, C.; Silvestri, L. Bmp6 expression in murine liver non parenchymal cells: A mechanism to control their high iron exporter activity and protect hepatocytes from iron overload? PLoS ONE 2015, 10, e0122696. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.-S.; Olsavszky, V.; Ulbrich, F.; Sticht, C.; Demory, A.; Leibing, T.; Henzler, T.; Meyer, M.; Zierow, J.; Schneider, S.; et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2017, 129, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Meynard, D.; Monnier, A.; Darnaud, V.; Bouvet, R.; Wang, R.H.; Deng, C.; Vaulont, S.; Mosser, J.; Coppin, H.; et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 2008, 112, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Verga Falzacappa, M.V.; Casanovas, G.; Hentze, M.W.; Muckenthaler, M.U. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. 2008, 86, 531–540. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, F.; Hentze, M.W.; Muckenthaler, M.U. The hemochromatosis proteins HFE, TFR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Iron regulation of hepcidin through HFE and HJV: Common or distinct pathways? Hepatology 2015, 62, 1922–1923. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Loreal, O. Iron metabolism and related genetic diseases: A cleared land, keeping mysteries. J. Hepatol 2016, 64, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Hamdi-Roze, H.; Beaumont-Epinette, M.P.; Ben Ali, Z.; Le Lan, C.; Loustaud-Ratti, V.; Causse, X.; Loreal, O.; Deugnier, Y.; Brissot, P.; Jouanolle, A.M.; et al. Rare HFE variants are the most frequent cause of hemochromatosis in non-c282y homozygous patients with hemochromatosis. Am. J. Hematol. 2016, 91, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Primer 2018, 4, 18016. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.J.; Gurrin, L.C.; Constantine, C.C.; Osborne, N.J.; Delatycki, M.B.; Nicoll, A.J.; McLaren, C.E.; Bahlo, M.; Nisselle, A.E.; Vulpe, C.D.; et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N. Engl. J. Med. 2008, 358, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, B.A.C.; Laarakkers, C.M.M.; Klaver, S.M.; Jacobs, E.M.G.; van Tits, L.J.H.; Janssen, M.C.H.; Swinkels, D.W. Serum hepcidin levels are innately low in HFE-related haemochromatosis but differ between C282Y-homozygotes with elevated and normal ferritin levels. Br. J. Haematol. 2008, 142, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Kuhn, L.C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Cellular iron metabolism. Kidney Int. Suppl. 1999, 69, S2–S11. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Lok, C.N. The transferrin receptor: Role in health and disease. Int. J. Biochem. Cell Biol. 1999, 31, 1111–1137. [Google Scholar] [CrossRef]
- Arosio, P.; Carmona, F.; Gozzelino, R.; Maccarinelli, F.; Poli, M. The importance of eukaryotic ferritins in iron handling and cytoprotection. Biochem. J. 2015, 472, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Peto, T.E. Non-transferrin plasma iron. Br. J. Haematol 1987, 66, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Loreal, O.; Gosriwatana, I.; Guyader, D.; Porter, J.; Brissot, P.; Hider, R.C. Determination of non-transferrin-bound iron in genetic hemochromatosis using a new HPLC-based method. J. Hepatol. 2000, 32, 727–733. [Google Scholar] [CrossRef]
- Grootveld, M.; Bell, J.D.; Halliwell, B.; Aruoma, O.I.; Bomford, A.; Sadler, P.J. Non-transferrin-bound iron in plasma or serum from patients with idiopathic hemochromatosis. Characterization by high performance liquid chromatography and nuclear magnetic resonance spectroscopy. J. Biol. Chem. 1989, 264, 4417–4422. [Google Scholar] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Wright, T.L.; Ma, W.L.; Weisiger, R.A. Efficient clearance of non-transferrin-bound iron by rat liver. Implications for hepatic iron loading in iron overload states. J. Clin. Investig. 1985, 76, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Bolder, U.; Schteingart, C.D.; Arnaud, J.; Hofmann, A.F. Intestinal absorption and enterohepatic cycling of biliary iron originating from plasma non-transferrin-bound iron in rats. Hepatology 1997, 25, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Hubert, N.; Lescoat, G.; Sciot, R.; Moirand, R.; Jego, P.; Leroyer, P.; Brissot, P. Regulation of ferritin and transferrin receptor expression by iron in human hepatocyte cultures. J. Hepatol. 1993, 18, 301–312. [Google Scholar] [CrossRef]
- Sciot, R.; Verhoeven, G.; Van Eyken, P.; Cailleau, J.; Desmet, V.J. Transferrin receptor expression in rat liver: Immunohistochemical and biochemical analysis of the effect of age and iron storage. Hepatology 1990, 11, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Jenkitkasemwong, S.; Wang, C.Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003, 33, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Samuels, M.E.; Ludwig, E.H.; MacDonald, M.L.; Franchini, P.L.; Dube, M.P.; Andres, L.; MacFarlane, J.; Sakellaropoulos, N.; Politou, M.; et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 2004, 36, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Cunat, S.; Beaumont-Epinette, M.-P.; Kannengiesser, C.; Causse, X.; Sauvion, S.; Pouliquen, B.; Deugnier, Y.; David, V.; Loréal, O.; et al. Variable age of onset and clinical severity in transferrin receptor 2 related haemochromatosis: Novel observations. Br. J. Haematol. 2013, 162, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Sonnenberg, A.; Stremmel, W.; Trampisch, H.J.; Strohmeyer, G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N. Engl. J. Med. 1985, 313, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Purschel, A.; Stremmel, W.; Haussinger, D.; Strohmeyer, G. Long-term survival in patients with hereditary hemochromatosis [see comments]. Gastroenterology 1996, 110, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Loreal, O.; Deugnier, Y.; Moirand, R.; Lauvin, L.; Guyader, D.; Jouanolle, H.; Turlin, B.; Lescoat, G.; Brissot, P. Liver fibrosis in genetic hemochromatosis. Respective roles of iron and non-iron-related factors in 127 homozygous patients. J. Hepatol. 1992, 16, 122–127. [Google Scholar] [PubMed]
- Guyader, D.; Jacquelinet, C.; Moirand, R.; Turlin, B.; Mendler, M.H.; Chaperon, J.; David, V.; Brissot, P.; Adams, P.; Deugnier, Y. Noninvasive prediction of fibrosis in C282Y homozygous hemochromatosis. Gastroenterology 1998, 115, 929–936. [Google Scholar] [CrossRef]
- Deugnier, Y.M.; Loreal, O.; Turlin, B.; Guyader, D.; Jouanolle, H.; Moirand, R.; Jacquelinet, C.; Brissot, P. Liver pathology in genetic hemochromatosis: A review of 135 homozygous cases and their bioclinical correlations. Gastroenterology 1992, 102, 2050–2059. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Houglum, K.; Bedossa, P.; Chojkier, M. TGF-beta and collagen-alpha 1 (I) gene expression are increased in hepatic acinar zone 1 of rats with iron overload. Am. J. Physiol. 1994, 267, G908–G913. [Google Scholar] [CrossRef] [PubMed]
- Gualdi, R.; Casalgrandi, G.; Montosi, G.; Ventura, E.; Pietrangelo, A. Excess iron into hepatocytes is required for activation of collagen type I gene during experimental siderosis. Gastroenterology 1994, 107, 1118–1124. [Google Scholar] [CrossRef]
- Deugnier, Y.M.; Guyader, D.; Crantock, L.; Lopez, J.M.; Turlin, B.; Yaouanq, J.; Jouanolle, H.; Campion, J.P.; Launois, B.; Halliday, J.W.; et al. Primary liver cancer in genetic hemochromatosis: A clinical, pathological, and pathogenetic study of 54 cases. Gastroenterology 1993, 104, 228–234. [Google Scholar] [CrossRef]
- Guggenbuhl, P.; Brissot, P.; Loreal, O. Miscellaneous non-inflammatory musculoskeletal conditions. Haemochromatosis: The bone and the joint. Best Pract. Res. Clin. Rheumatol 2011, 25, 6649–6664. [Google Scholar] [CrossRef]
- Wardman, P.; Candeias, L.P. Fenton chemistry: An introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Britton, R.S. Hepatic injury in chronic iron overload. Role of lipid peroxidation. Chem. Biol. Interact. 1989, 70, 183–226. [Google Scholar] [CrossRef]
- Bacon, B.R.; Tavill, A.S.; Brittenham, G.M.; Park, C.H.; Recknagel, R.O. Hepatic lipid peroxidation in vivo in rats with chronic iron overload. J. Clin. Investig. 1983, 71, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; O’Neill, R.; Britton, R.S. Hepatic mitochondrial energy production in rats with chronic iron overload. Gastroenterology 1993, 105, 1134–1140. [Google Scholar] [CrossRef]
- Le Lan, C.; Loreal, O.; Cohen, T.; Ropert, M.; Glickstein, H.; Laine, F.; Pouchard, M.; Deugnier, Y.; Le Treut, A.; Breuer, W.; et al. Redox active plasma iron in C282Y/C282Y hemochromatosis. Blood 2005, 105, 4527–4531. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I.; Breuer, W.; Zanninelli, G.; Cianciulli, P. LPI-labile plasma iron in iron overload. Best Pr. Res. Clin. Haematol 2005, 18, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Milet, J.; Dehais, V.; Bourgain, C.; Jouanolle, A.M.; Mosser, A.; Perrin, M.; Morcet, J.; Brissot, P.; David, V.; Deugnier, Y.; et al. Common variants in the BMP2, BMP4, and HJV genes of the hepcidin regulation pathway modulate HFE hemochromatosis penetrance. Am. J. Hum. Genet. 2007, 81, 799–807. [Google Scholar] [CrossRef] [PubMed]
- De Tayrac, M.; Roth, M.-P.; Jouanolle, A.-M.; Coppin, H.; le Gac, G.; Piperno, A.; Férec, C.; Pelucchi, S.; Scotet, V.; Bardou-Jacquet, E.; et al. Genome-wide association study identifies TF as a significant modifier gene of iron metabolism in HFE hemochromatosis. J. Hepatol. 2015, 62, 664–672. [Google Scholar] [CrossRef] [PubMed]
- McLaren, C.E.; Emond, M.J.; Subramaniam, V.N.; Phatak, P.D.; Barton, J.C.; Adams, P.C.; Goh, J.B.; McDonald, C.J.; Powell, L.W.; Gurrin, L.C.; et al. Exome sequencing in HFE C282Y homozygous men with extreme phenotypes identifies a GNPAT variant associated with severe iron overload. Hepatology 2015, 62, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Tchernitchko, D.; Scotet, V.; Lefebvre, T.; L’Hostis, C.; Gourlaouen, I.; Merour, M.-C.; Rebah, K.; Peoc’h, K.; Assari, S.; Ferec, C.; et al. GNPAT polymorphism rs11558492 is not associated with increased severity in a large cohort of HFE p.Cys282Tyr homozygous patients. Hepatology 2017, 65, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Levstik, A.; Stuart, A.; Adams, P.C. GNPAT variant (D519G) is not associated with an elevated serum ferritin or iron removed by phlebotomy in patients referred for C282Y-linked hemochromatosis. Ann. Hepatol. 2016, 15, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Greni, F.; Valenti, L.; Mariani, R.; Pelloni, I.; Rametta, R.; Busti, F.; Ravasi, G.; Girelli, D.; Fargion, S.; Galimberti, S.; et al. GNPAT rs11558492 is not a Major Modifier of Iron Status: Study of Italian Hemochromatosis Patients and Blood Donors. Ann. Hepatol. 2017, 16, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, L.M.; Dixon, J.L.; Purdie, D.M.; Powell, L.W.; Crawford, D.H.G. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology 2002, 122, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Bhattacharya, R.; Lindor, K.D.; Chalasani, N.; Raaka, S.; Heathcote, E.J.; Miskovsky, E.; Shaffer, E.; Rulyak, S.J.; Kowdley, K.V. HFE C282Y mutations are associated with advanced hepatic fibrosis in Caucasians with nonalcoholic steatohepatitis. Hepatology 2007, 46, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Vegetti, A.; Saitta, C.; Ferrara, F.; Corradini, E.; Raffa, G.; Pietrangelo, A.; Raimondo, G. Hepatitis B virus DNA integration in tumour tissue of a non-cirrhotic HFE-haemochromatosis patient with hepatocellular carcinoma. J. Hepatol. 2013, 58, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Diwakaran, H.H.; Befeler, A.S.; Britton, R.S.; Brunt, E.M.; Bacon, B.R. Accelerated hepatic fibrosis in patients with combined hereditary hemochromatosis and chronic hepatitis C infection. J. Hepatol. 2002, 36, 687–691. [Google Scholar] [CrossRef]
- Stickel, F.; Buch, S.; Zoller, H.; Hultcrantz, R.; Gallati, S.; Österreicher, C.; Finkenstedt, A.; Stadlmayr, A.; Aigner, E.; Sahinbegovic, E.; et al. Evaluation of genome-wide loci of iron metabolism in hereditary hemochromatosis identifies PCSK7 as a host risk factor of liver cirrhosis. Hum. Mol. Genet. 2014, 23, 3883–3890. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Maggioni, P.; Piperno, A.; Rametta, R.; Pelucchi, S.; Mariani, R.; Dongiovanni, P.; Fracanzani, A.L.; Fargion, S. Patatin-like phospholipase domain containing-3 gene I148M polymorphism, steatosis, and liver damage in hereditary hemochromatosis. World J. Gastroenterol. 2012, 18, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.; Altes, A.; Brissot, P.; Butzeck, B.; Cabantchik, I.; Cançado, R.; Distante, S.; Evans, P.; Evans, R.; Ganz, T.; et al. Contributors and Hemochromatosis International Taskforce Therapeutic recommendations in HFE hemochromatosis for p.Cys282Tyr (C282Y/C282Y) homozygous genotype. Hepatol. Int. 2018, 12, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Falize, L.; Guillygomarc’h, A.; Perrin, M.; Laine, F.; Guyader, D.; Brissot, P.; Turlin, B.; Deugnier, Y. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: A study of 36 cases. Hepatology 2006, 44, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Phatak, P.; Brissot, P.; Wurster, M.; Adams, P.C.; Bonkovsky, H.L.; Gross, J.; Malfertheiner, P.; McLaren, G.D.; Niederau, C.; Piperno, A.; et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010, 52, 1671–1779. [Google Scholar] [CrossRef] [PubMed]
- Rombout-Sestrienkova, E.; Koek, G.H.; Neslo, R.; van Kraaij, M.; Menheere, P.P.; Masclee, A.; Swinkels, D.W. Course of iron parameters in HFE-hemochromatosis patients during initial treatment with erythrocytapheresis compared to phlebotomy. J. Clin. Apheresis 2016, 31, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E.; Schlumpf, K.S.; Wright, D.J.; Johnson, B.; Glynn, S.A.; Busch, M.P.; Olbina, G.; Westerman, M.; Nemeth, E.; Ganz, T. NHLBI Retrovirus Epidemiology Donor Study-II (REDS-II) Hepcidin level predicts hemoglobin concentration in individuals undergoing repeated phlebotomy. Haematologica 2013, 98, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Kaltwasser, J.P.; Werner, E.; Schalk, K.; Hansen, C.; Gottschalk, R.; Seidl, C. Clinical trial on the effect of regular tea drinking on iron accumulation in genetic haemochromatosis. Gut 1998, 43, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Ludwiczek, S.; Theurl, I.; Muckenthaler, M.U.; Jakab, M.; Mair, S.M.; Theurl, M.; Kiss, J.; Paulmichl, M.; Hentze, M.W.; Ritter, M.; et al. Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat. Med. 2007, 13, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Vanclooster, A.; van Deursen, C.; Jaspers, R.; Cassiman, D.; Koek, G. Proton Pump Inhibitors Decrease Phlebotomy Need in HFE Hemochromatosis: Double-Blind Randomized Placebo-Controlled Trial. Gastroenterology 2017, 153, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, H.R. Articular cartilage in the degenerative arthropathy of hemochromatosis. Arthritis Rheum. 1982, 25, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Heiland, G.R.; Aigner, E.; Dallos, T.; Sahinbegovic, E.; Krenn, V.; Thaler, C.; Weiss, G.; Distler, J.H.; Datz, C.; Schett, G.; et al. Synovial immunopathology in haemochromatosis arthropathy. Ann. Rheum. Dis. 2010, 69, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- Carroll, G.J.; Sharma, G.; Upadhyay, A.; Jazayeri, J.A. Ferritin concentrations in synovial fluid are higher in osteoarthritis patients with HFE gene mutations (C282Y or H63D). Scand. J. Rheumatol. 2010, 39, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Chung, J.W.; Lapointe, R.; Santos, M.M. The hemochromatosis protein HFE 20 years later: An emerging role in antigen presentation and in the immune system. Immun. Inflamm. Dis. 2017, 5, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Lainé, F.; Guggenbuhl, P.; Morcet, J.; Jézéquel, C.; Guyader, D.; Moirand, R.; Deugnier, Y. Worse Outcomes of Patients with HFE Hemochromatosis With Persistent Increases in Transferrin Saturation During Maintenance Therapy. Clin. Gastroenterol. Hepatol. 2017, 15, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Philip, J.; Lorho, R.; Ropert, M.; Latournerie, M.; Houssel-Debry, P.; Guyader, D.; Loreal, O.; Boudjema, K.; Brissot, P. Liver transplantation normalizes serum hepcidin level and cures iron metabolism alterations in HFE hemochromatosis. Hepatology 2014, 59, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Investig. 2011, 121, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Schmidt, P.J.; Meynard, D.; Garuti, C.; Montosi, G.; Chen, S.; Vukicevic, S.; Pietrangelo, A.; Lin, H.Y.; Babitt, J.L. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in HFE knockout mice. Gastroenterology 2010, 139, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.L.; Biswas, K.; Rottman, J.; Allen, J.R.; Long, J.; Miranda, L.P.; Winters, A.; Arvedson, T.L. Identification of Antibody and Small Molecule Antagonists of Ferroportin-Hepcidin Interaction. Front. Pharmacol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loréal, O.; Cavey, T.; Robin, F.; Kenawi, M.; Guggenbuhl, P.; Brissot, P. Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals 2018, 11, 131. https://doi.org/10.3390/ph11040131
Loréal O, Cavey T, Robin F, Kenawi M, Guggenbuhl P, Brissot P. Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals. 2018; 11(4):131. https://doi.org/10.3390/ph11040131
Chicago/Turabian StyleLoréal, Olivier, Thibault Cavey, François Robin, Moussa Kenawi, Pascal Guggenbuhl, and Pierre Brissot. 2018. "Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects" Pharmaceuticals 11, no. 4: 131. https://doi.org/10.3390/ph11040131
APA StyleLoréal, O., Cavey, T., Robin, F., Kenawi, M., Guggenbuhl, P., & Brissot, P. (2018). Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals, 11(4), 131. https://doi.org/10.3390/ph11040131