Brain Iron Homeostasis: A Focus on Microglial Iron

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Brain Iron
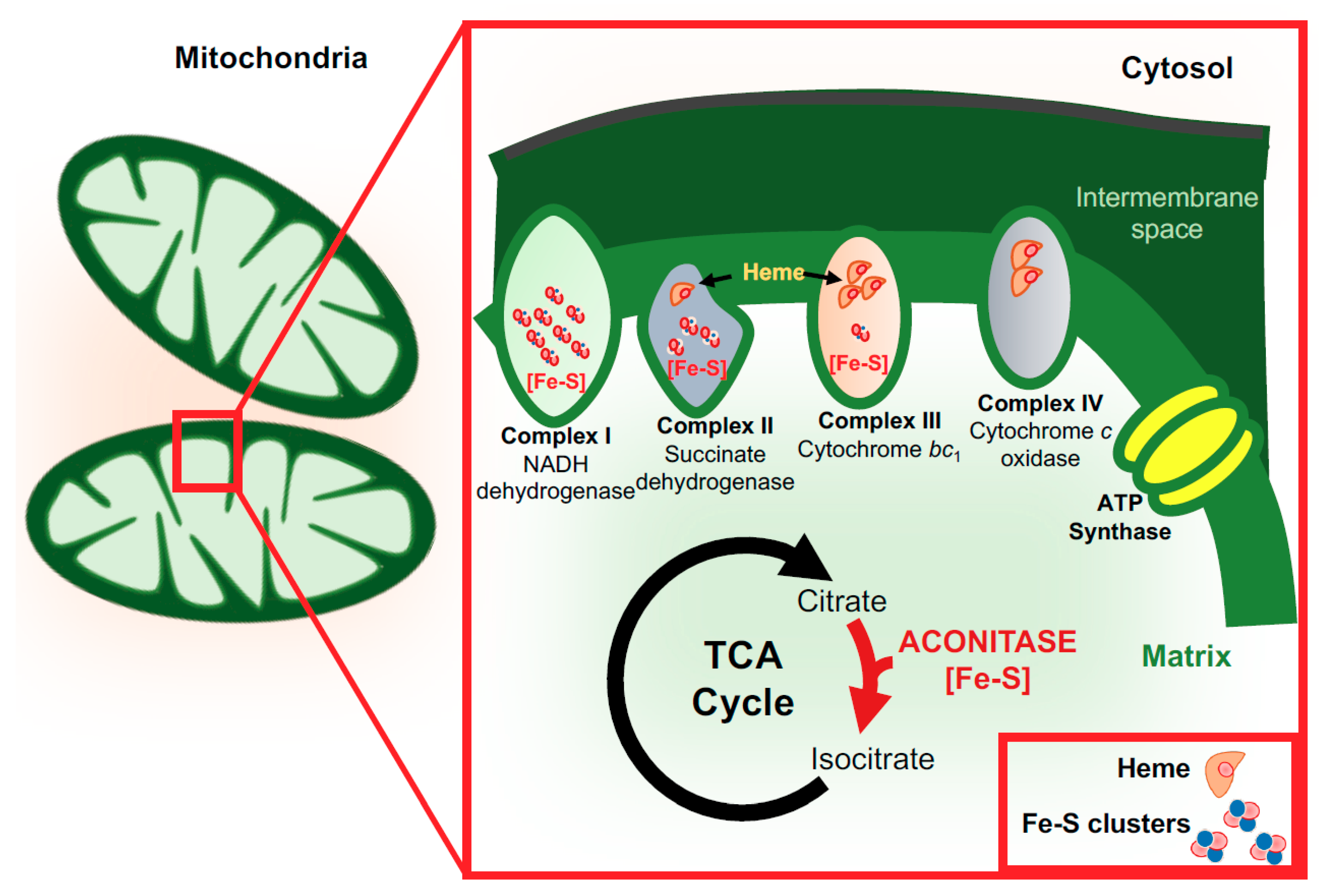
3. Functions of Iron in the Brain
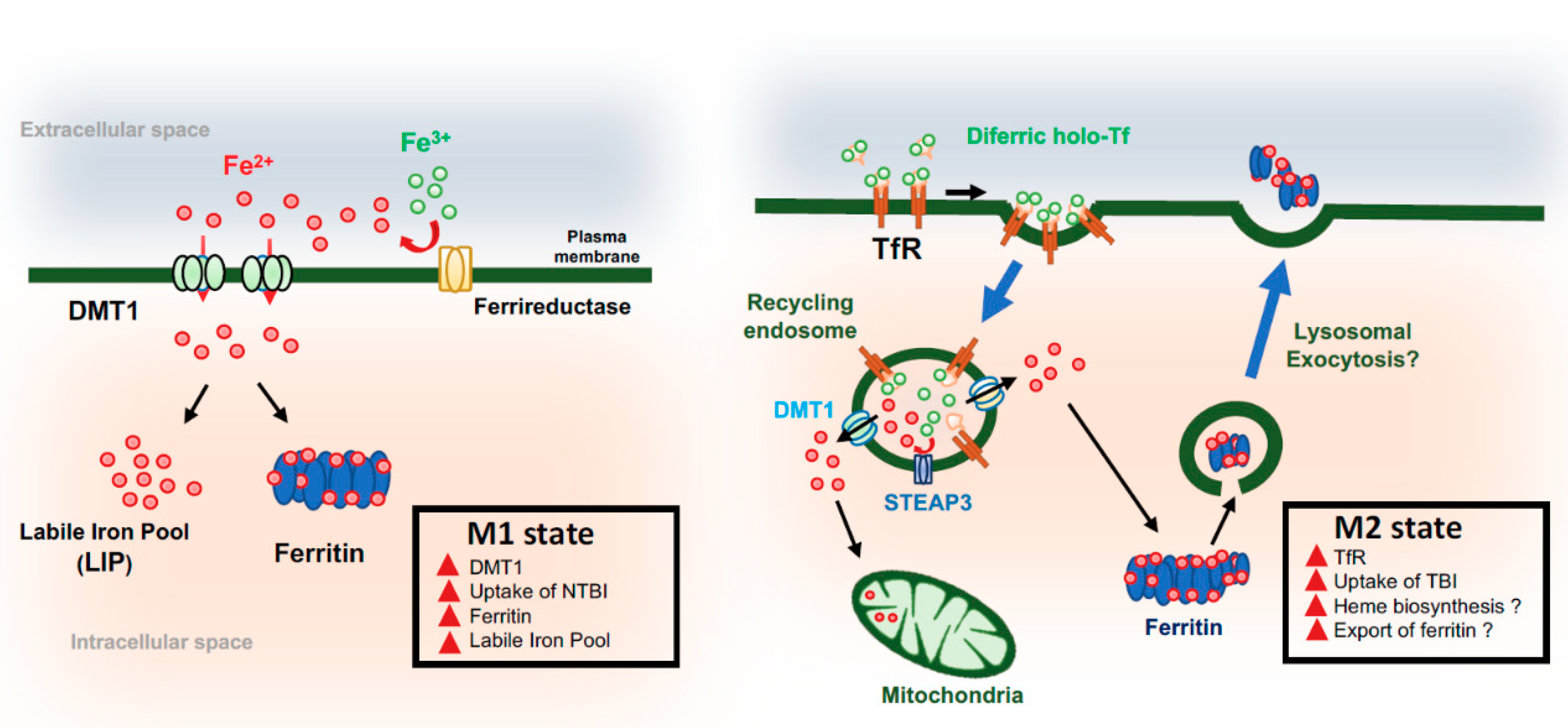
4. Microglia and Iron
5. Microglia Activation and Alzheimer’s Disease
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Alzheimer’s disease | AD |
| familial Alzheimer’s disease | FAD |
| sporadic Alzheimer’s disease | SAD |
| neurodegeneration with brain iron accumulation | NBIA |
| Parkinson’s disease | PD |
| Huntington’s disease | HD |
| amyotrophic lateral sclerosis | ALS |
| multiple sclerosis | MS |
| amyloid beta | Aβ |
| transferrin | Tf |
| Tf-bound iron | TBI |
| transferrin receptor | TfR |
| non-transferrin-bound iron | NTBI |
| divalent metal transporter-1 | DMT1 |
| labile iron pool | LIP |
| central nervous system | CNS |
| Blood–brain barrier | BBB |
| brain microvascular endothelial cell | BMVEC |
| transient receptor potential mucolipin-1 | TRPML1 |
| ferritin | Ftn |
| reactive oxygen species | ROS |
| gamma-aminobutyric acid | GABA |
| inducible nitric oxidase | iNOS |
| N-methyl-D-aspartate | NMDA |
| adenosine triphosphate | ATP |
| 3-hydroxy-3-methyl-glutaryl-coenzyme A | HMG-CoA |
| tricarboxylic acid | TCA |
| interferon gamma | IFN-γ |
| toll-like receptor | TLR |
| tumor necrosis factor-alpha | TNF-α |
| interleukin-6 | IL-6 |
| interleukin 1-beta | IL-1β |
| interleukin-12 | IL-12 |
| interleukin-10 | IL-10 |
| C-C motif chemokine ligand-2 | CCL2 |
| transforming growth factor-beta | TGF-β |
| insulin-like growth factor-1 | IGF-1 |
| fibroblast growth factor | FGF |
| nerve growth factor | NGF |
| brain-derived growth factor | BDNF |
| toll-like receptor 2 | TLR2 |
| toll-like receptor 4 | TLR4 |
| toll-like receptor 6 | TLR6 |
| cluster of differentiation 33 | CD33 |
| cluster of differentiation 36 | CD36 |
| triggering receptor expressed on myeloid cells 2 | TREM2 |
| apolipoprotein E | apoE |
| apolipoprotein E4 | apoE4 |
| apolipoprotein E2 | apoE2 |
References
- Beard, J.L.; Connor, J.R. Iron status and neural functioning. Annu. Rev. Nutr. 2003, 23, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Carrasco, M.A.; Muñoz, P.; Núñez, M.T. A Role for Reactive Oxygen/Nitrogen Species and Iron on Neuronal Synaptic Plasticity. Antioxid. Redox Signal. 2007, 9, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy Metabolism of the Brain, Including the Cooperation between Astrocytes and Neurons, Especially in the Context of Glycogen Metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Madsen, E.; Gitlin, J.D. Copper and Iron Disorders of the Brain. Annu. Rev. Neurosci. 2007, 30, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Zhang, X.; Slagle-Webb, B.; Seaman, W.E.; Connor, J.R. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem. 2008, 107, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Salvador, G.A. Iron in neuronal function and dysfunction. Biofactors 2010, 36, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, R.; Spinelli, F.; Contino, M.; Colabufo, N.A. The Pivotal Role of Copper in Neurodegeneration: A New Strategy for the Therapy of Neurodegenerative Disorders. Mol. Pharm. 2018, 15, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Bowman, A.B.; Kwakye, G.F.; Herrero Hernandez, E.; Aschner, M. Role of manganese in neurodegenerative diseases. J. Trace Elem. Med. Biol. 2011, 25, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 2013, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Muhoberac, B.B.; Vidal, R. Abnormal iron homeostasis and neurodegeneration. Front. Aging Neurosci. 2013, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Andersen, M.V.; Christoffersen, P.R.; Jensen, M.D.; Lichota, J.; Moos, T. Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol. Dis. 2015, 81, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Reichmann, H. Role of Iron in Neurodegenerative Diseases. J. Neural Transm. 2016, 123, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [PubMed]
- Lloyd, A.F.; Davies, C.L.; Miron, V.E. Microglia: Origins, homeostasis, and roles in myelin repair. Curr. Opin. Neurobiol. 2017, 47, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Tay, T.L.; Wolf, Y.; Jung, S. Microglia: Unique and common features with other tissue macrophages. Acta Neuropathol. 2014, 128, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, P.; Thion, M.S.; Garel, S. Neuronal and microglial regulators of cortical wiring: Usual and novel guideposts. Front. Neurosci. 2015, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The role of microglia in the healthy brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Miyamoto, A.; Nabekura, J. Microglia: Actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013, 36, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Rathnasamy, G.; Ling, E.-A.; Kaur, C. Consequences of iron accumulation in microglia and its implications in neuropathological conditions. CNS Neurol. Dis. Drug Targets 2013, 12, 785–798. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Sosa, J.C.; Gardeck, A.M.; Baez, A.S.; Lee, C.H.; Wessling-Resnick, M. Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 2018, 293, 7853–7863. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Review Balancing Acts: Molecular Control of Mammalian Iron Metabolism sequences of systemic iron overload result from chronic iron accumulation in tissues. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- Moos, T.; Nielsen, T.R.; Skjørringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Wessling-Resnick, M. Iron imports. III. Transfer of iron from the mucosa into circulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1–G6. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Kosman, D.J. Iron transport across the blood-brain barrier: Development, neurovascular regulation and cerebral amyloid angiopathy. Cell. Mol. Life Sci. 2015, 72, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Cooperman, S. Brain Iron Metabolism. Semin. Pediatr. Neurol. 2006, 13, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Qian, Z.M. Brain iron metabolism: Neurobiology and neurochemistry. Prog. Neurobiol. 2007, 83, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Burdo, J.R.; Menzies, S.L.; Simpson, I.A.; Garrick, L.M.; Garrick, M.D.; Dolan, K.G.; Haile, D.J.; Beard, J.L.; Connor, J.R. Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J. Neurosci. Res. 2001, 66, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Siddappa, A.J.M.; Rao, R.B.; Wobken, J.D.; Leibold, E.A.; Connor, J.R.; Georgieff, M.K. Developmental changes in the expression of iron regulatory proteins and iron transport proteins in the perinatal rat brain. J. Neurosci. Res. 2002, 68, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Enerson, B.E.; Drewes, L.R. The Rat Blood—Brain Barrier Transcriptome. J. Cereb. Blood Flow Metab. 2006, 26, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Skjoerringe, T.; Gosk, S.; Morgan, E.H. Brain capillary endothelial cells mediate iron transport into the brain by segregating iron from transferrin without the involvement of divalent metal transporter 1. J. Neurochem. 2006, 98, 1946–1958. [Google Scholar] [CrossRef] [PubMed]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [PubMed]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Grishchuk, Y.; Pena, K.A.; Coblentz, J.; King, V.E.; Humphrey, D.M.; Wang, S.L.; Kiselyov, K.I.; Slaugenhaupt, S.A. Impaired myelination and reduced brain ferric iron in the mouse model of mucolipidosis IV. Dis. Models Mech. 2015, 8, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Ponnuru, P.; Klinger, M.E.; Myers, R.L.; Devraj, K.; Coe, C.L.; Lubach, G.R.; Carruthers, A.; Connor, J.R. A novel model for brain iron uptake: Introducing the concept of regulation. J. Cereb. Blood Flow Metab. 2015, 35, 48–57. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Kosman, D.J. Ferroportin and exocytoplasmic ferroxidase activity are required for brain microvascular endothelial cell iron efflux. J. Biol. Chem. 2013, 288, 17932–17940. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Kosman, D.J. Glial cell ceruloplasmin and hepcidin differentially regulate iron efflux from brain microvascular endothelial cells. PLoS ONE 2014, 9, e89003. [Google Scholar] [CrossRef] [PubMed]
- Duck, K.A.; Connor, J.R. Iron uptake and transport across physiological barriers. Biometals 2016, 29, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M. Ferrous ions detected in cerebrospinal fluid by using bleomycin and DNA damage. Clin. Sci. 1992, 82, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.; Bucchini, D.; Martin, M.E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Menzies, S.; Muckenthaler, M.; Torti, F.M.; Wood, T.; Torti, S.V.; Hentze, M.W.; Beard, J.; Connor, J. Mouse brains deficient in H-ferritin have normal iron concentration but a protein profile of iron deficiency and increased evidence of oxidative stress. J. Neurosci. Res. 2003, 71, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Garringer, H.J.; Goodwin, C.B.; Richine, B.; Acton, A.; VanDuyn, N.; Muhoberac, B.B.; Irimia-Dominguez, J.; Chan, R.J.; Peacock, M.; et al. Systemic and Cerebral Iron Homeostasis in Ferritin Knock-Out Mice. PLoS ONE 2015, 10, e0117435. [Google Scholar] [CrossRef] [PubMed]
- Chiou, B.; Neal, E.H.; Bowman, A.B.; Lippmann, E.S.; Simpson, I.A.; Connor, J.R. Endothelial cells are critical regulators of iron transport in a model of the human blood-brain barrier. J. Cereb. Blood Flow Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Zhang, X.; Connor, J.R. H-ferritin is the major source of iron for oligodendrocytes. Glia 2011, 59, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebron, J.A.; Bjorkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Kawabata, H.; Masuda, T.; Uchiyama, T.; Mizumoto, C.; Ohmori, K.; Koeffler, H.P.; Kadowaki, N.; Takaori-Kondo, A. H-Ferritin Is Preferentially Incorporated by Human Erythroid Cells through Transferrin Receptor 1 in a Threshold-Dependent Manner. PLoS ONE 2015, 10, e0139915. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The role of mitochondria in cellular iron–sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [PubMed]
- Alle, H.; Roth, A.; Geiger, J.R.P. Energy-efficient action potentials in hippocampal mossy fibers. Science 2009, 325, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and myelination: The role of iron. Glia 2009, 57, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.S.; Steinkirchner, T.M.; Rao, G.A.; Larkin, E.C. Effect of prenatal iron deficiency on myelination in rat pups. Am. J. Pathol. 1986, 125, 620–624. [Google Scholar] [PubMed]
- Ortiz, E.; Pasquini, J.M.; Thompson, K.; Felt, B.; Butkus, G.; Beard, J.; Connor, J.R. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J. Neurosci. Res. 2004, 77, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, R.; Ben-Shachar, D.; Youdim, M.B.H. Nutritional iron and dopamine binding sites in the rat brain. Pharmacol. Biochem. Behav. 1982, 17, 43–47. [Google Scholar] [CrossRef]
- Adhami, V.M.; Husain, R.; Husain, R.; Seth, P.K. Influence of Iron Deficiency and Lead Treatment on Behavior and Cerebellar and Hippocampal Polyamine Levels in Neonatal Rats. Neurochem. Res. 1996, 21, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Kwik-Uribe, C.L.; Gietzen, D.; German, J.B.; Golub, M.S.; Keen, C.L. Chronic Marginal Iron Intakes during Early Development in Mice Result in Persistent Changes in Dopamine Metabolism and Myelin Composition. J. Nutr. 2000, 130, 2821–2830. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wessling-Resnick, M. Iron and mechanisms of emotional behavior. J. Nutr. Biochem. 2014, 25, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.M.; Ruskin, B.; Lovenberg, W. Tryptophan Hydroxylase The role of oxygen, iron, and sulfhydryl groups as determinants of stability and catalytic activity. J. Biol. Chem. 1980, 255, 4137–4143. [Google Scholar] [PubMed]
- Glinka, Y.; Gassen, M.; Youdim, M.B.H. Iron and Neurotransmitter Function in the Brain. In Metals and Oxidative Damage in Neurological Disorders; Connor, J.R., Ed.; Springer: Boston, MA, USA, 1997; pp. 1–22. [Google Scholar]
- Bianco, L.E.; Wiesinger, J.; Earley, C.J.; Jones, B.C.; Beard, J.L. Iron deficiency alters dopamine uptake and response to L-DOPA injection in Sprague-Dawley rats. J. Neurochem. 2008, 106, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, S.; Youdim, M.B. Brain iron: A lesson from animal models. Am. J. Clin. Nutr. 1989, 50, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, S. Neurochemical basis of behavioral effects of brain iron deficiency in animals. In Brain, Behavior and Iron in the Infant Diet; Dobbing, J., Ed.; Springer: London, UK, 1990; pp. 63–76. [Google Scholar]
- Erikson, K.M.; Jones, B.C.; Hess, E.J.; Zhang, Q.; Beard, J.L. Iron deficiency decreases dopamine D 1 and D 2 receptors in rat brain. Pharmacol. Biochem. Behav. 2001, 69, 409–418. [Google Scholar] [CrossRef]
- Chen, Q.; Connor, J.R.; Beard, J.L. Brain Iron, Transferrin and Ferritin Concentrations Are Altered in Developing Iron-Deficient Rats. J. Nutr. 1995, 125, 1529–1535. [Google Scholar] [PubMed]
- Waldmeier, P.C.; Buchle, A.M.; Steulet, A.F. Inhibition of catechol-O-methyltransferase (COMT) as well as tyrosine and tryptophan hydroxylase by the orally active iron chelator, 1,2-dimethyl-3-hydroxypyridin-4-one (L1, CP20), in rat brain in vivo. Biochem. Pharmacol. 1993, 45, 2417–2424. [Google Scholar] [CrossRef]
- Taneja, V.; Mishra, K.; Agarwal, K.N. Effect of early iron deficiency in rat on the gamma-aminobutyric acid shunt in brain. J. Neurochem. 1986, 46, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Li, D. Effects of iron deficiency on iron distribution and gamma-aminobutyric acid (GABA) metabolism in young rat brain tissues. Hokkaido J. Med. Sci. 1998, 73, 215–225. [Google Scholar] [PubMed]
- Appel, S.H.; Zhao, W.; Beers, D.R.; Henkel, J.S. The Microglial-Motoneuron dialogue in ALS. Acta Myol. 2011, 30, 4. [Google Scholar] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Farber, N.B.; Kim, S.H.; Dikranian, K.; Jiang, X.P.; Heinkel, C. Receptor mechanisms and circuitry underlying NMDA antagonist neurotoxicity. Mol. Psychiatry 2002, 7, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Aida, T.; Yanagisawa, M.; Katou, S.; Sakimura, K.; Mishina, M.; Tanaka, K. NMDA receptor subunits have different roles in NMDA-induced neurotoxicity in the retina. Mol. Brain 2013, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Franca-Costa, J.; Van Weyenbergh, J.; Boaventura, V.S.; Luz, N.F.; Malta-Santos, H.; Oliveira, M.C.; Santos de Campos, D.C.; Saldanha, A.C.; dos-Santos, W.L.; Bozza, P.T.; et al. Arginase I, polyamine, and prostaglandin E2 pathways suppress the inflammatory response and contribute to diffuse cutaneous leishmaniasis. J. Infect. Dis. 2015, 211, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Wes, P.D.; Holtman, I.R.; Boddeke, E.W.; Moller, T.; Eggen, B.J. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 2016, 64, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.; Borowsky, M.; Wang, L.-C.; Means, T.K.; Khoury, J.E. The Microglial Sensome Revealed by Direct RNA Sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Morimoto, E.T.A.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Wessling-Resnick, M. Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 2010, 30, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2018, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Ling, E.A. Increased expression of transferrin receptors and iron in amoeboid microglial cells in postnatal rats following an exposure to hypoxia. Neurosci. Lett. 1999, 262, 183–186. [Google Scholar] [CrossRef]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; Gonzalez-Billault, C.; Nunez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.; McIntosh, A.L.; Finucane, O.M.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.A.; Gun’ko, Y.K.; Lynch, M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Gaasch, J.A.; Lockman, P.R.; Geldenhuys, W.J.; Allen, D.D.; Van der Schyf, C.J. Brain Iron Toxicity: Differential Responses of Astrocytes, Neurons, and Endothelial Cells. Neurochem. Res. 2007, 32, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Wessling-Resnick, M. Crossing the Iron Gate: Why and How Transferrin Receptors Mediate Viral Entry. Annu. Rev. Nutr. 2018, 38, 431–458. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimers Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Good, P.F.; Werner, P.; Hsu, A.; Olanow, C.W.; Perl, D.P. Evidence for Neuronal Oxidative Damage in Alzheimer’s Disease. Am. J. Pathol. 1996, 149, 21–28. [Google Scholar] [PubMed]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 1995, 45, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Su, B.; Wang, X.; Smith, M.A.; Perry, G. Causes of oxidative stress in Alzheimer disease. Cell. Mol. Life Sci. 2007, 64, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Wang, X.; Perry, G.; Nunomura, A.; Tabaton, M.; Zhu, X.; Smith, M.A. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 2010, 59, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I. The metal theory of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S277–S281. [Google Scholar] [CrossRef] [PubMed]
- Zeineh, M.M.; Chen, Y.; Kitzler, H.H.; Hammond, R.; Vogel, H.; Rutt, B.K. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging 2015, 36, 2483–2500. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Bolaños, J.P.; Mastroberardino, P.G. Redox Status and Bioenergetics Liaison in Cancer and Neurodegeneration. Int. J. Cell Biol. 2012, 2012, 659645. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.G.; Pollack, A.N.; Cheng, K.C.; Sun, D.; Saido, T.; Haaf, M.P.; Yang, Q.X.; Connor, J.R.; Meadowcroft, M.D. Dietary lipophilic iron alters amyloidogenesis and microglial morphology in Alzheimer’s disease knock-in APP mice. Metallomics 2018, 10, 426–443. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Guo, Y.; Sun, M.; Li, B.; Zhang, Y.; Li, C. Iron is a potential key mediator of glutamate excitotoxicity in spinal cord motor neurons. Brain Res. 2009, 1257, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell. Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. Alzheimer Genetic Analysis Group, T.A.G.A. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Atagi, Y.; Liu, C.-C.; Painter, M.M.; Chen, X.-F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.-C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Holtzman, D.M. Apolipoprotein E levels and Alzheimer risk. Ann. Neurol. 2015, 77, 204–205. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Steffensen, K.R.; Verghese, P.B.; Kummer, M.P.; Gustafsson, J.-A.; Holtzman, D.M.; Heneka, M.T. Critical Role of Astroglial Apolipoprotein E and Liver X Receptor-α Expression for Microglial Aβ Phagocytosis. J. Neurosci. 2011, 31, 7049–7059. [Google Scholar] [CrossRef] [PubMed]
- Cammer, W. Oligodendrocyte-Associated Enzymes. In Oligodendroglia. Advances in Neurochemistry; William, N., Ed.; Springer: Boston, MA, USA, 1984; pp. 199–232. [Google Scholar]
- Xu, Q.; Li, Y.; Cyras, C.; Sanan, D.A.; Cordell, B. Isolation and characterization of apolipoproteins from murine microglia. Identification of a low density lipoprotein-like apolipoprotein J-rich but E-poor spherical particle. J. Biol. Chem. 2000, 275, 31770–31777. [Google Scholar] [CrossRef] [PubMed]
- Cudaback, E.; Li, X.; Montine, K.S.; Montine, T.J.; Keene, C.D. Apolipoprotein E isoform-dependent microglia migration. FASEB J. 2011, 25, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Karlo, J.C.; Landreth, G.E. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 2012, 32, 10117–10128. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Faux, N.G.; Bush, A.I. Alzheimer’s Disease Neuroimaging Initiative, A.s.D.N. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 2015, 6, 6760. [Google Scholar] [CrossRef] [PubMed]
- Ali-Rahmani, F.; Schengrund, C.-L.; Connor, J.R. HFE gene variants, iron, and lipids: A novel connection in Alzheimer’s disease. Front. Pharmacol. 2014, 5, 165. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, D.R.C.; Kruck, T.P.A.; Kalow, W.; Andrews, D.F.; Dalton, A.J.; Bell, M.Y.; Smith, W.L. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Palanimuthu, D.; Poon, R.; Sahni, S.; Anjum, R.; Hibbs, D.; Lin, H.Y.; Bernhardt, P.V.; Kalinowski, D.S.; Richardson, D.R. A novel class of thiosemicarbazones show multi-functional activity for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 139, 612–632. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nnah, I.C.; Wessling-Resnick, M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals 2018, 11, 129. https://doi.org/10.3390/ph11040129
Nnah IC, Wessling-Resnick M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals. 2018; 11(4):129. https://doi.org/10.3390/ph11040129
Chicago/Turabian StyleNnah, Israel C., and Marianne Wessling-Resnick. 2018. "Brain Iron Homeostasis: A Focus on Microglial Iron" Pharmaceuticals 11, no. 4: 129. https://doi.org/10.3390/ph11040129
APA StyleNnah, I. C., & Wessling-Resnick, M. (2018). Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals, 11(4), 129. https://doi.org/10.3390/ph11040129