Pilocarpine-Induced Status Epilepticus Is Associated with P-Glycoprotein Induction in Cardiomyocytes, Electrocardiographic Changes, and Sudden Death


Abstract
:1. Introduction
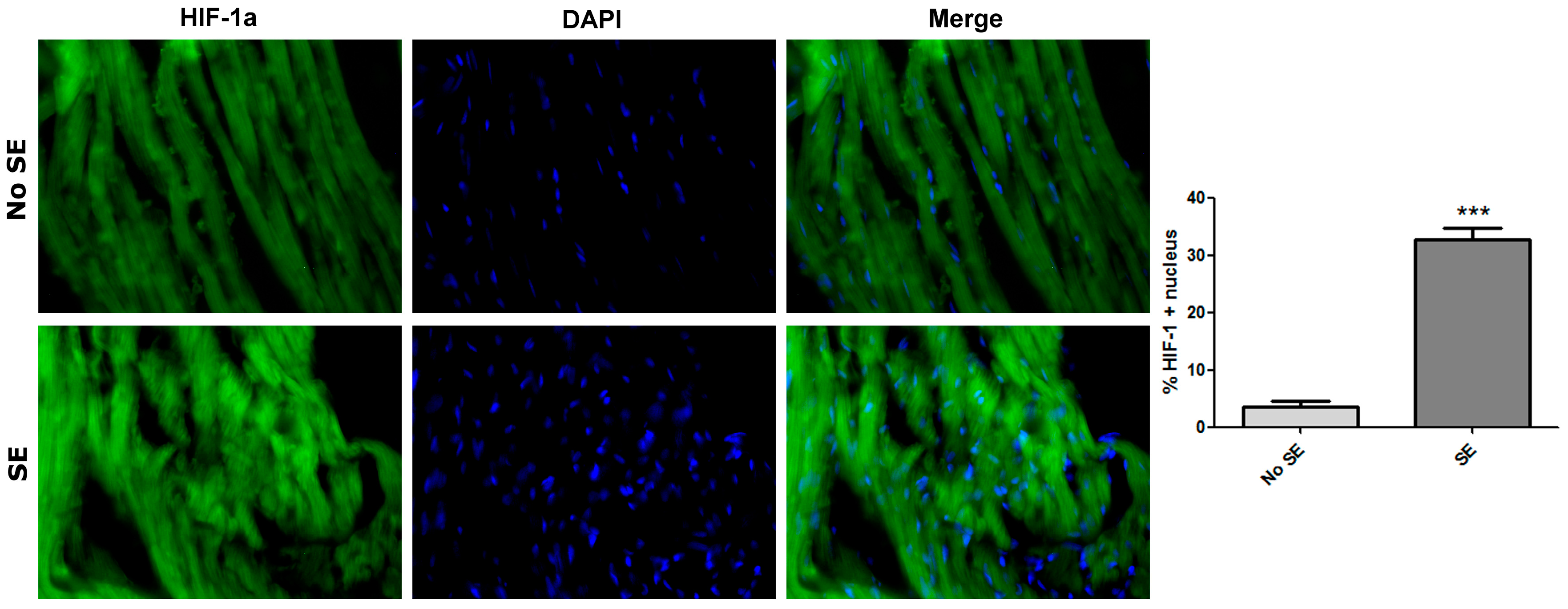
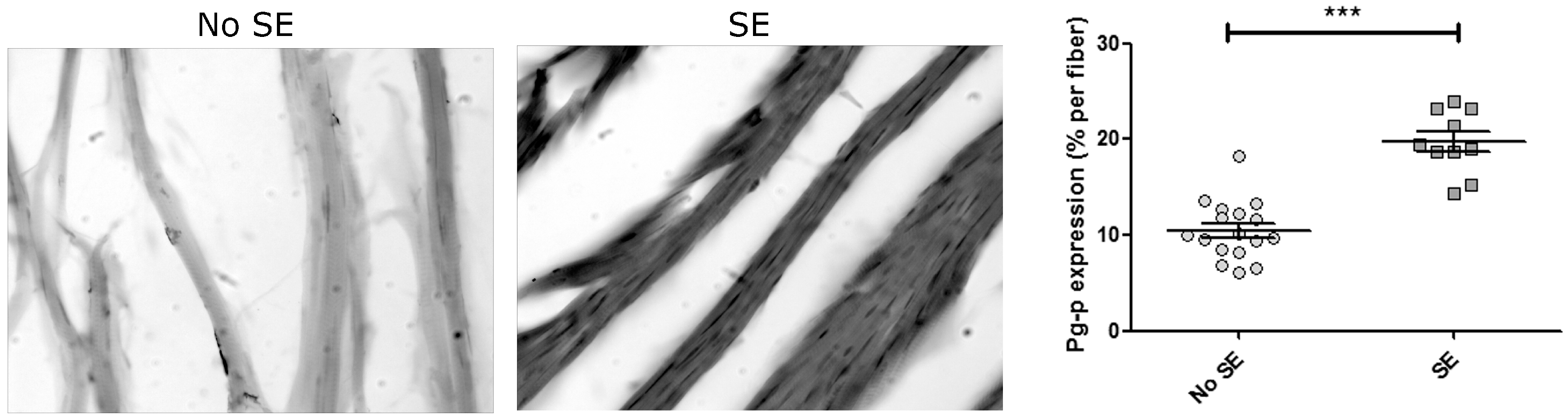
2. Results
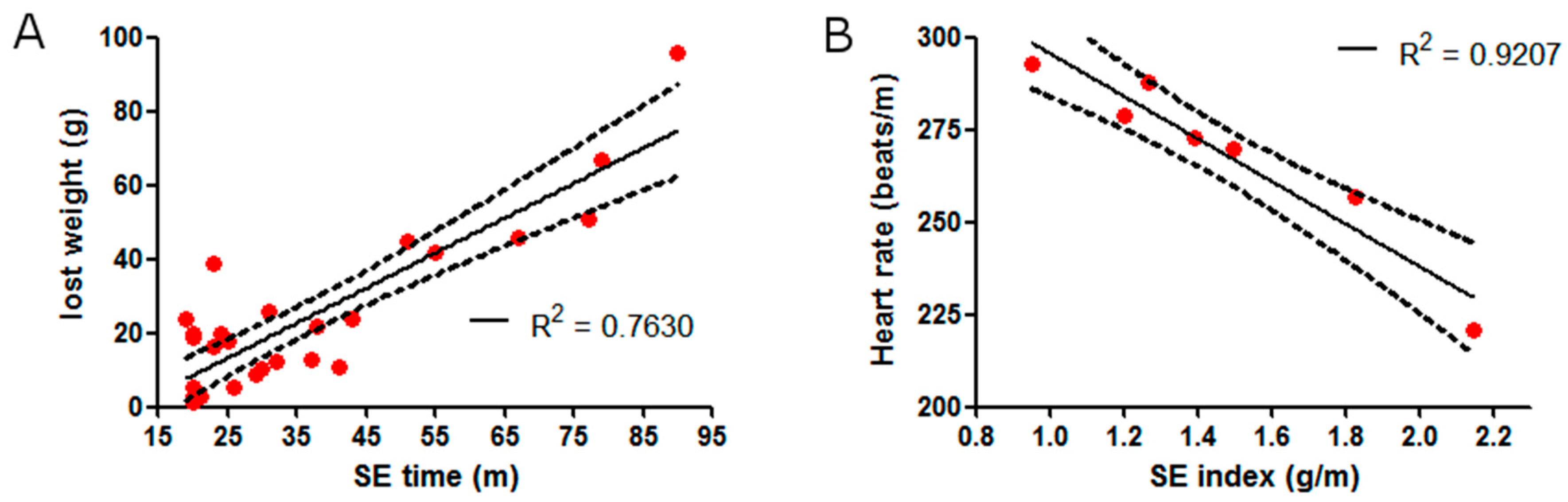
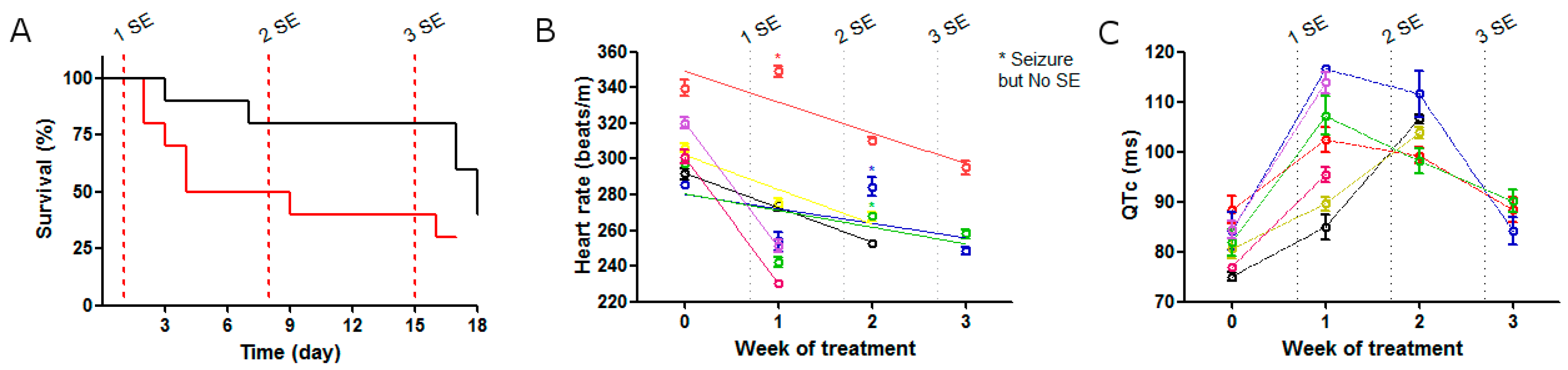
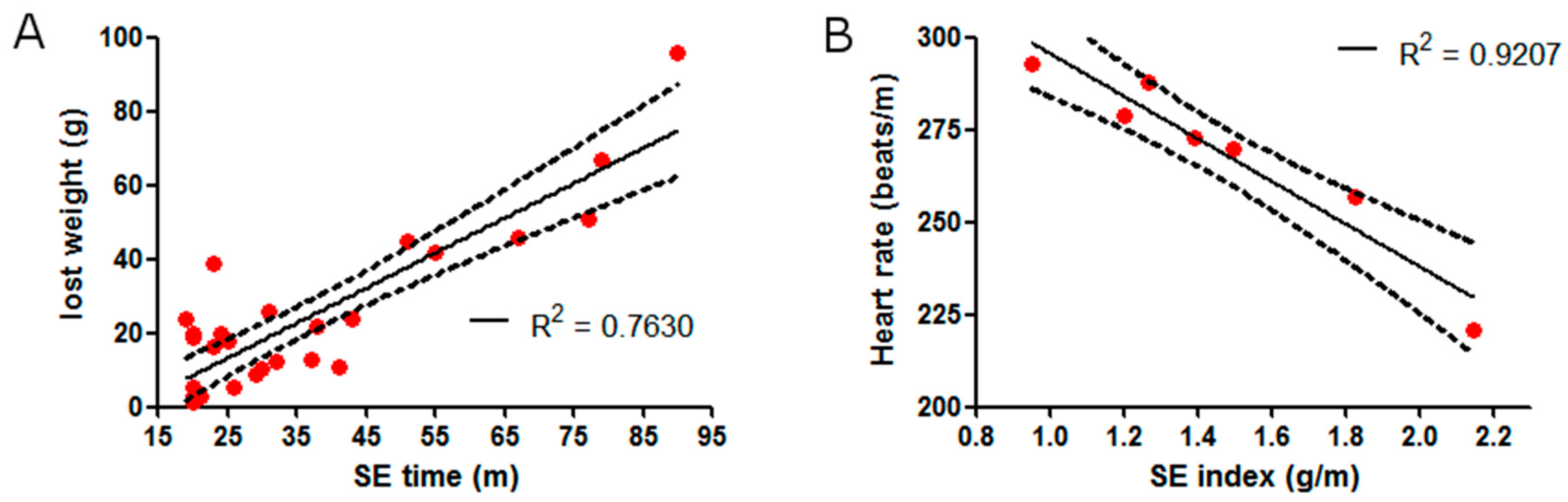
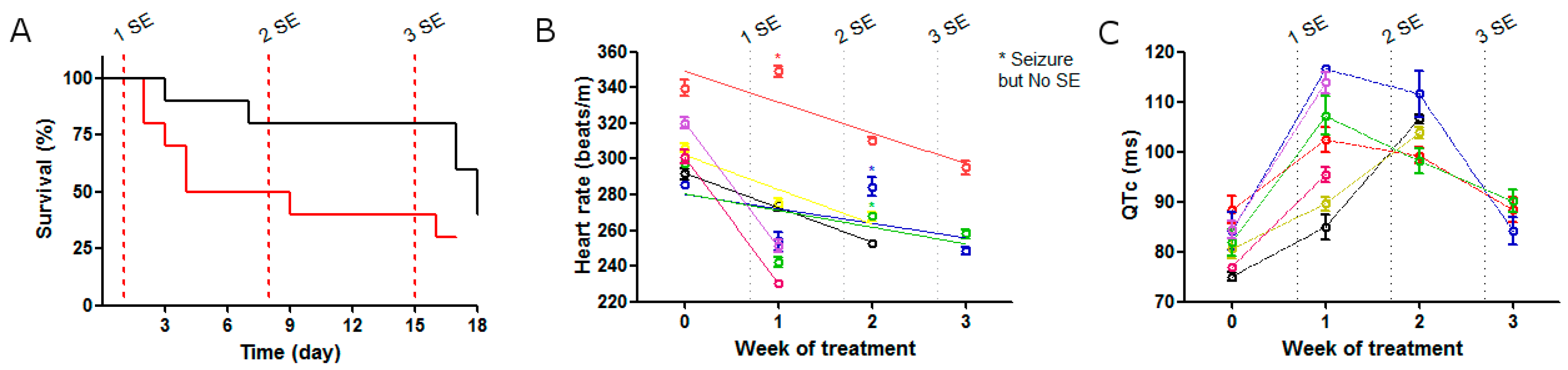
2.1. SE Strength Induces Heart Rate Variation
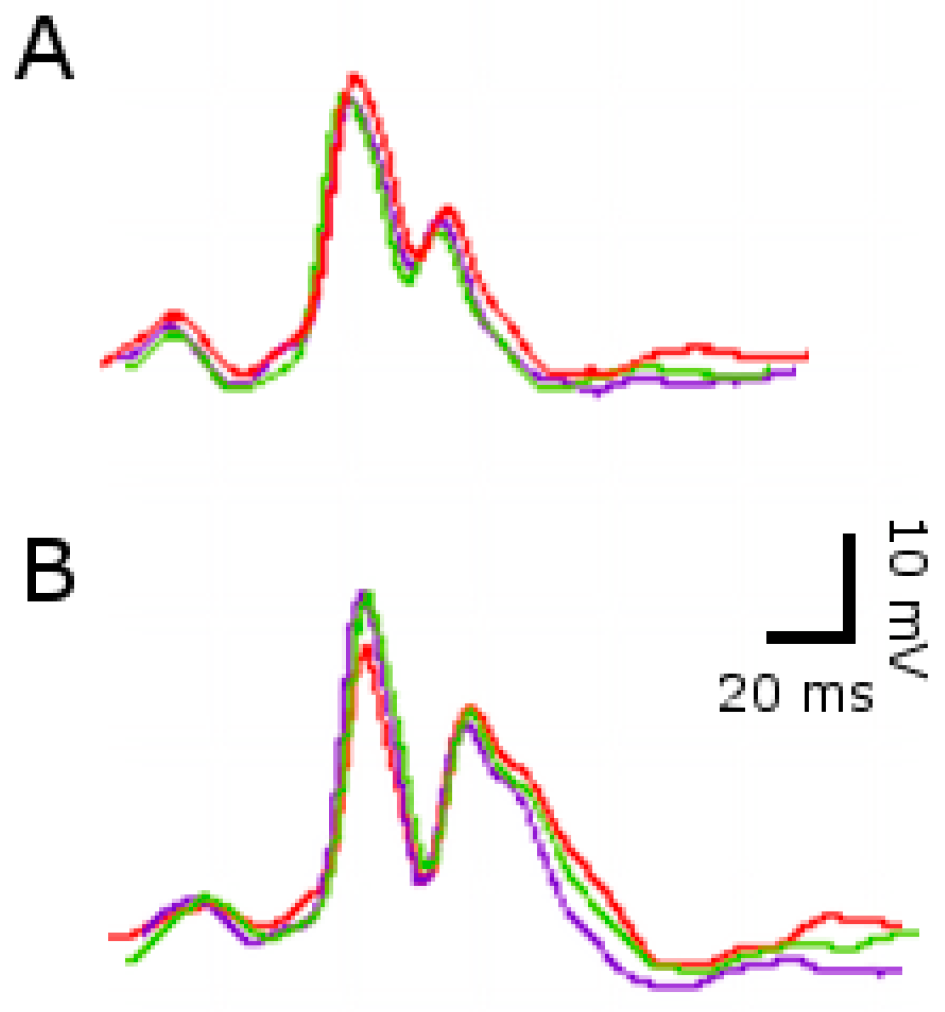
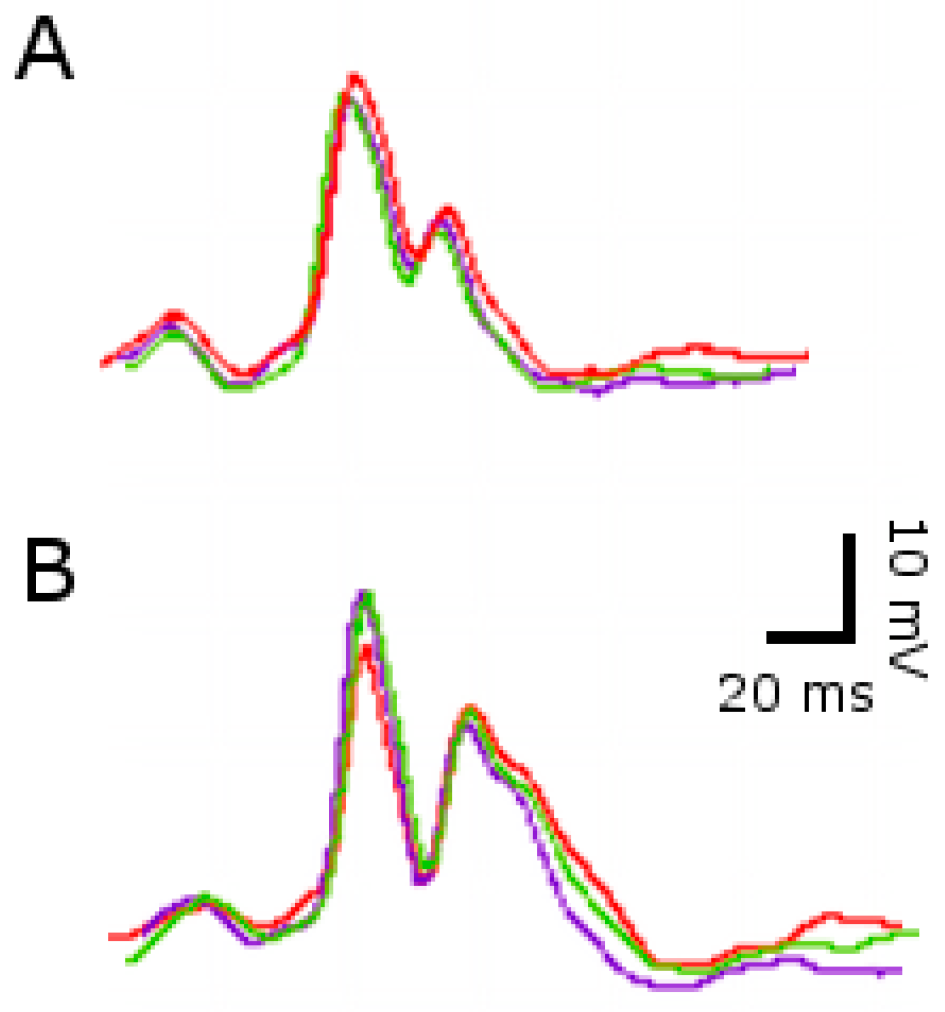
2.2. ECG Changes after Single SE
2.3. ECG Changes after Multiple SE
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Status Epilepticus Protocol
4.3. Experimental Scheme
4.4. Electrocardiographic and Heart Rate Evaluation
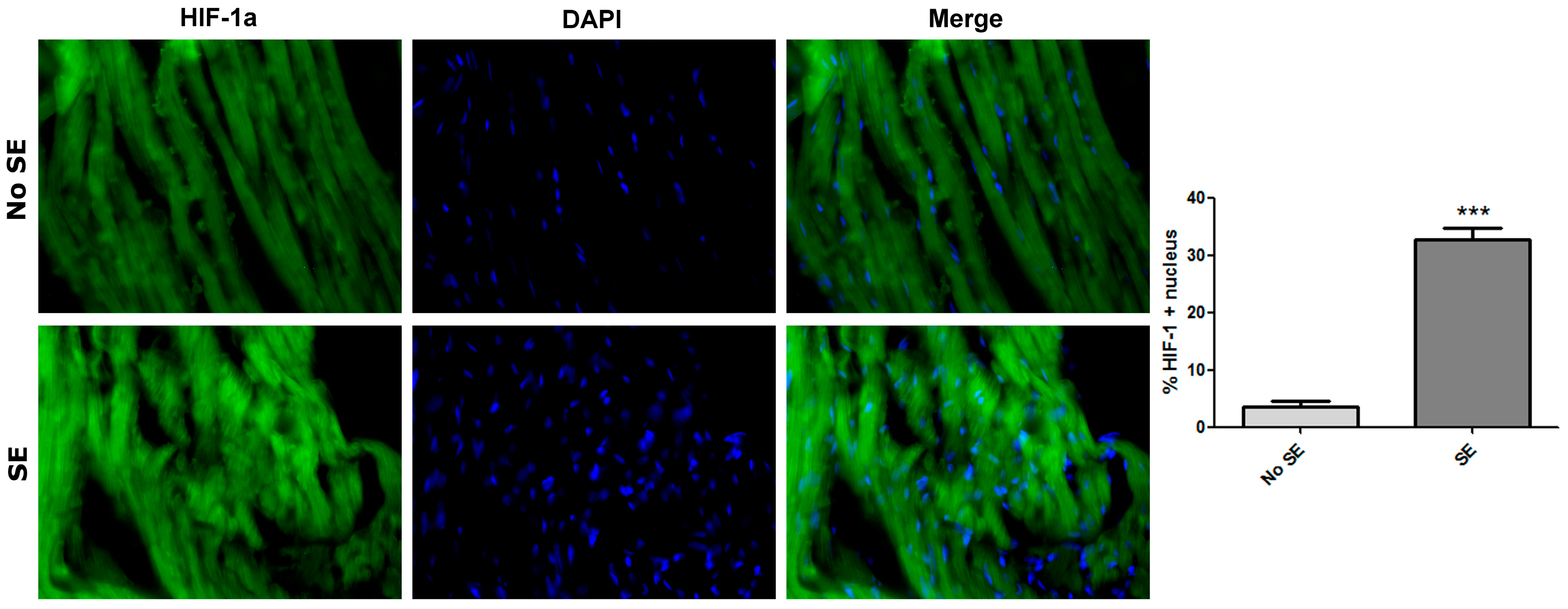
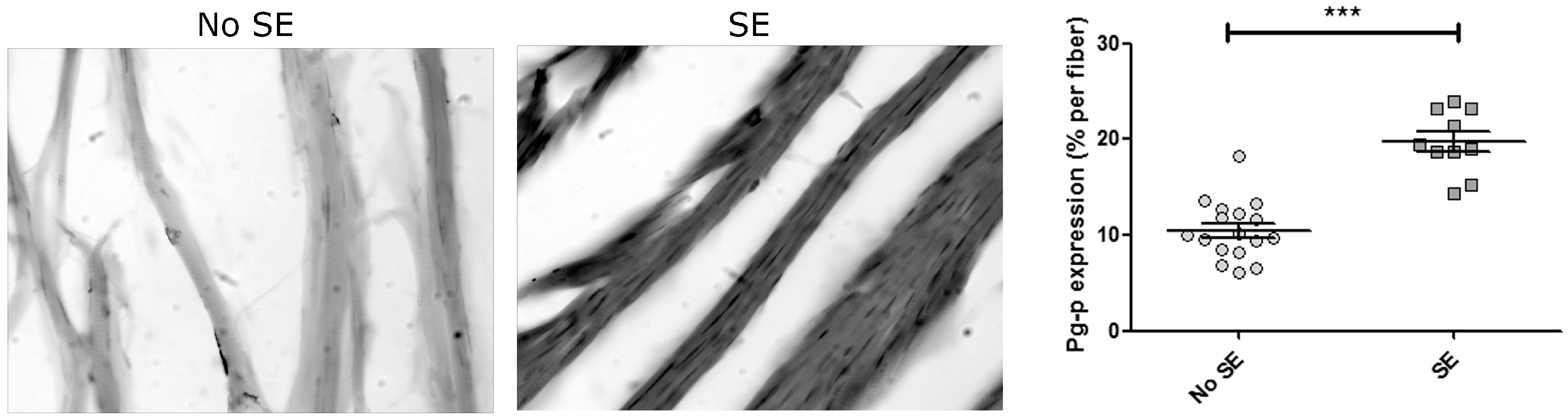
4.5. Immunohistochemistry
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Famularo, G. Status epilepticus and type 2 myocardial infarction. Am. J. Emerg. Med. 2016, 34, 1735.e3-4. [Google Scholar] [CrossRef] [PubMed]
- Hocker, S.; Prasad, A.; Rabinstein, A.A. Cardiac injury in refractory status epilepticus. Epilepsia 2013, 54, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Gautier, N.M.; Glasscock, E. Simultaneous Video-EEG-ECG Monitoring to Identify Neurocardiac Dysfunction in Mouse Models of Epilepsy. J. Vis. Exp. 2018, 131. [Google Scholar]
- Monté, C.P.J.A.; Arends, J.B.A.M.; Tan, I.Y.; Aldenkamp, A.P.; Limburg, M.; de Krom, M.C.T.F.M. Sudden unexpected death in epilepsy patients: Risk factors. Seizure 2007, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nashef, L. Sudden unexpected death in epilepsy: Terminology and definitions. Epilepsia 1997, 38, S6–S8. [Google Scholar] [CrossRef] [PubMed]
- Enrique, A.; Goicoechea, S.; Castaño, R.; Taborda, F.; Rocha, L.; Orozco, S.; Girardi, E.; Bruno Blanch, L. New model of pharmacoresistant seizures induced by 3-mercaptopropionic acid in mice. Epilepsy Res. 2017, 129, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, A.; Czornyj, L.; Lubieniecki, F.; Vazquez, S.; D’Giano, C.; Sevlever, G.; Lia Taratuto, A.; Brusco, A.; Elena, G. Multidrug-Resistance (MDR) Proteins Develops Refractory Epilepsy Phenotype: Clinical and Experimental Evidences. Curr. Drug Ther. 2006, 1, 291–309. [Google Scholar] [CrossRef]
- Shankar, R.; Donner, E.J.; McLean, B.; Nashef, L.; Tomson, T. Sudden unexpected death in epilepsy (SUDEP): What every neurologist should know. Epileptic Disord. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Auzmendi, J.A.; Orozco-Suárez, S.; Bañuelos-Cabrera, I.; González-Trujano, M.E.; Calixto González, E.; Rocha, L.; Lazarowski, A. P-glycoprotein contributes to cell membrane depolarization of hippocampus and neocortex in a model of repetitive seizures induced by pentylenetetrazole in rats. Curr. Pharm. Des. 2013, 19, 6732–6738. [Google Scholar] [CrossRef] [PubMed]
- Auzmendi, J.; Merelli, A.; Girardi, E.; Orozco-Suarez, S.; Rocha, L.; Lazarowski, A. Progressive heart P-glycoprotein (P-gp) overexpression after experimental repetitive seizures (ERS) associated with fatal status epilepticus (FSE). Is it related with SUDEP? Mol. Cell. Epilepsy 2014, 1. [Google Scholar] [CrossRef]
- Damasceno, D.D.; Savergnini, S.Q.; Gomes, E.R.M.; Guatimosim, S.; Ferreira, A.J.; Doretto, M.C.; Almeida, A.P. Cardiac dysfunction in rats prone to audiogenic epileptic seizures. Seizure 2013, 22, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.A.; Arida, R.M.; Cysneiros, R.M.; Terra, V.C.; Sonoda, E.Y.F.; de Albuquerque, M.; Cavalheiro, E.A. The brain-heart connection: Implications for understanding sudden unexpected death in epilepsy. Cardiol. J. 2009, 16, 394–399. [Google Scholar] [PubMed]
- Bealer, S.L.; Little, J.G. Seizures following hippocampal kindling induce QT interval prolongation and increased susceptibility to arrhythmias in rats. Epilepsy Res. 2013, 105, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Surges, R.; Adjei, P.; Kallis, C.; Erhuero, J.; Scott, C.A.; Bell, G.S.; Sander, J.W.; Walker, M.C. Pathologic cardiac repolarization in pharmacoresistant epilepsy and its potential role in sudden unexpected death in epilepsy: A case-control study. Epilepsia 2010, 51, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.A.; Sombati, S.; DeLorenzo, R.J. Glutamate injury-induced epileptogenesis in hippocampal neurons: An in vitro model of stroke-induced “epilepsy”. Stroke 2001, 32, 2344–2350. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, C.; Peng, J.; Patel, N.; Huang, Y.; Gao, X.; Aljarallah, S.; Eubanks, J.H.; McDonald, R.; Zhang, L. Early-Onset Convulsive Seizures Induced by Brain Hypoxia-Ischemia in Aging Mice: Effects of Anticonvulsive Treatments. PLoS ONE 2015, 10, e0144113. [Google Scholar] [CrossRef] [PubMed]
- López-Ramos, J.C.; Duran, J.; Gruart, A.; Guinovart, J.J.; Delgado-García, J.M. Role of brain glycogen in the response to hypoxia and in susceptibility to epilepsy. Front. Cell. Neurosci. 2015, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Juul, H.M.; Jensen, F.E. Models of hypoxia and ischemia-induced seizures. J. Neurosci. Methods 2016, 260, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Shetty, J. Neonatal seizures in hypoxic-ischaemic encephalopathy—Risks and benefits of anticonvulsant therapy. Dev. Med. Child Neurol. 2015, 57, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Tigaran, S.; Mølgaard, H.; McClelland, R.; Dam, M.; Jaffe, A.S. Evidence of cardiac ischemia during seizures in drug refractory epilepsy patients. Neurology 2003, 60, 492–495. [Google Scholar] [CrossRef] [PubMed]
- El Shorbagy, H.H.; Elsayed, M.A.; Kamal, N.M.; Azab, A.A.; Bassiouny, M.M.; Ghoneim, I.A. Heart-type fatty acid-binding protein as a predictor of cardiac ischemia in intractable seizures in children. J. Pediatr. Neurosci. 2016, 11, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Read, M.I.; McCann, D.M.; Millen, R.N.; Harrison, J.C.; Kerr, D.S.; Sammut, I.A. Progressive development of cardiomyopathy following altered autonomic activity in status epilepticus. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1554–H1564. [Google Scholar] [CrossRef] [PubMed]
- Merelli, A.; Caltana, L.; Girimonti, P.; Ramos, A.J.; Lazarowski, A.; Brusco, A. Recovery of motor spontaneous activity after intranasal delivery of human recombinant erythropoietin in a focal brain hypoxia model induced by CoCl2 in rats. Neurotox. Res. 2011, 20, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Aviles-Reyes, R.X.; Angelo, M.F.; Villarreal, A.; Rios, H.; Lazarowski, A.; Ramos, A.J. Intermittent hypoxia during sleep induces reactive gliosis and limited neuronal death in rats: Implications for sleep apnea. J. Neurochem. 2010, 112, 854–869. [Google Scholar] [CrossRef] [PubMed]
- De Lemos, M.L.; de la Torre, A.V.; Petrov, D.; Brox, S.; Folch, J.; Pallàs, M.; Lazarowski, A.; Beas-Zarate, C.; Auladell, C.; Camins, A. Evaluation of hypoxia inducible factor expression in inflammatory and neurodegenerative brain models. Int. J. Biochem. Cell Biol. 2013, 45, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Badowska-Kozakiewicz, AM.; Sobol, M.; Patera, J. Expression of multidrug resistance protein P-glycoprotein in correlation with markers of hypoxia (HIF-1α, EPO, EPO-R) in invasive breast cancer with metastasis to lymph nodes. Arch. Med. Sci. 2017, 13, 1303–1314. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.R.; Angelo, M.F.; Villarreal, A.; Lukin, J.; Ramos, A.J. Gabapentin administration reduces reactive gliosis and neurodegeneration after pilocarpine-induced status epilepticus. PLoS ONE 2013, 8, e78516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitsch, J.; Becker, A.J.; Schoch, S.; Müller, J.A.; de Curtis, M.; Gnatkovsky, V. Circadian clustering of spontaneous epileptic seizures emerges after pilocarpine-induced status epilepticus. Epilepsia 2017, 58, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.G.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- Ramos, A.J.; Lazarowski, A.; Villar, M.J.; Brusco, A. Transient expression of MDR-1/P-glycoprotein in a model of partial cortical devascularization. Cell. Mol. Neurobiol. 2004, 24, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Caltana, L.; Merelli, A.; Lazarowski, A.; Brusco, A. Neuronal and glial alterations due to focal cortical hypoxia induced by direct cobalt chloride (CoCl2) brain injection. Neurotox. Res. 2009, 15, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, A.; Caltana, L.; Merelli, A.; Rubio, M.D.; Ramos, A.J.; Brusco, A. Neuronal mdr-1 gene expression after experimental focal hypoxia: A new obstacle for neuroprotection? J. Neurol. Sci. 2007, 258, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, A.J.; García Rivello, H.J.; Vera Janavel, G.L.; Cuniberti, L.A.; Cabeza Meckert, P.M.; Yannarelli, G.G.; Mele, A.; Crottogini, A.J.; Laguens, R.P. Cardiomyocytes of chronically ischemic pig hearts express the MDR-1 gene-encoded P-glycoprotein. J. Histochem. Cytochem. 2005, 53, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Laguens, R.P.; Lazarowski, A.J.; Cuniberti, L.A.; Vera Janavel, G.L.; Cabeza Meckert, P.M.; Yannarelli, G.G.; del Valle, H.F.; Lascano, E.C.; Negroni, J.A.; Crottogini, A.J. Expression of the MDR-1 gene-encoded P-glycoprotein in cardiomyocytes of conscious sheep undergoing acute myocardial ischemia followed by reperfusion. J. Histochem. Cytochem. 2007, 55, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, S.; Nakae, I.; Tsutamoto, T.; Okamoto, N.; Horie, M. A novel clinical indicator using Tc-99m sestamibi for evaluating cardiac mitochondrial function in patients with cardiomyopathies. J. Nucl. Cardiol. 2007, 14, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.A.; Chiu, M.L.; Kronauge, J.F.; Kawamura, M.; Jones, A.G.; Holman, B.L.; Piwnica-Worms, D. Subcellular distribution and analysis of technetium-99m-MIBI in isolated perfused rat hearts. J. Nucl. Med. 1992, 33, 1516–1522. [Google Scholar] [PubMed]
- Piwnica-Worms, D.; Chiu, M.L.; Kronauge, J.F. Divergent kinetics of 201Tl and 99mTc-SESTAMIBI in cultured chick ventricular myocytes during ATP depletion. Circulation 1992, 85, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.L.; Kronauge, J.F.; Piwnica-Worms, D. Effect of mitochondrial and plasma membrane potentials on accumulation of hexakis (2-methoxyisobutylisonitrile) technetium(I) in cultured mouse fibroblasts. J. Nucl. Med. 1990, 31, 1646–1653. [Google Scholar] [PubMed]
- Hayashi, D.; Ohshima, S.; Isobe, S.; Cheng, X.W.; Unno, K.; Funahashi, H.; Shinoda, N.; Okumura, T.; Hirashiki, A.; Kato, K.; et al. Increased (99m)Tc-sestamibi washout reflects impaired myocardial contractile and relaxation reserve during dobutamine stress due to mitochondrial dysfunction in dilated cardiomyopathy patients. J. Am. Coll. Cardiol. 2013, 61, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Wadkins, R.M.; Roepe, P.D. Biophysical aspects of P-glycoprotein-mediated multidrug resistance. Int. Rev. Cytol. 1997, 171, 121–165. [Google Scholar] [PubMed]
- Roepe, P.D. What is the precise role of human MDR 1 protein in chemotherapeutic drug resistance? Curr. Pharm. Des. 2000, 6, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Auzmendi, J.; Salgueiro, J.; Canellas, C.; Zubillaga, M.; Men, P.; Alicia, R.; Merelli, A.; Buchholz, B.; Ricardo, G.; Ramos, A.J.; Lazarowski, A.L. Pilocarpine-induced Status Epilepticus (SE) induces functional and histological P-glycoprotein overexpression in cardiomyocytes, heart dysfunction and high ratio of sudden death in rats. In Proceeding of the Annual Meeting of American Epilepsy Society, Washington, DC, USA, 1–5 December 2017. [Google Scholar]
- Klabunde, R.E. Cardiac electrophysiology: Normal and ischemic ionic currents and the ECG. Adv. Physiol. Educ. 2017, 41, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Rudy, Y. Electrophysiologic effects of acute myocardial ischemia: A theoretical study of altered cell excitability and action potential duration. Cardiovasc. Res. 1997, 35, 256–272. [Google Scholar] [CrossRef]
- Nei, M. Cardiac effects of seizures. Epilepsy Curr. 2009, 9, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Massetani, R.; Strata, G.; Galli, R.; Gori, S.; Gneri, C.; Limbruno, U.; Di Santo, D.; Mariani, M.; Murri, L. Alteration of cardiac function in patients with temporal lobe epilepsy: Different roles of EEG-ECG monitoring and spectral analysis of RR variability. Epilepsia 1997, 38, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ansakorpi, H.; Korpelainen, J.T.; Huikuri, H. V; Tolonen, U.; Myllylä, V.V; Isojärvi, J.I.T. Heart rate dynamics in refractory and well controlled temporal lobe epilepsy. J. Neurol. Neurosurg. Psychiatry 2002, 72, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.; Kumlien, E.; Ericson, M.; Tomson, T. Preoperative heart rate variability in relation to surgery outcome in refractory epilepsy. Neurology 2005, 65, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Schuele, S.U.; Bermeo, A.C.; Alexopoulos, A. V; Locatelli, E.R.; Burgess, R.C.; Dinner, D.S.; Foldvary-Schaefer, N. Video-electrographic and clinical features in patients with ictal asystole. Neurology 2007, 69, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, B.K.; Britton, J.W.; Tigaran, S.; Cascino, G.D.; Burritt, M.F.; McConnell, J.P.; Ravkilde, J.; Molgaard, H.; Andreasen, F.; Dam, M.; et al. Cardiac troponin levels following monitored epileptic seizures. Neurology 2003, 60, 1690–1692. [Google Scholar] [CrossRef] [PubMed]
- Lotufo, P.A.; Valiengo, L.; Benseñor, I.M.; Brunoni, A.R. A systematic review and meta-analysis of heart rate variability in epilepsy and antiepileptic drugs. Epilepsia 2012, 53, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Z.; Huang, L.; Qu, W.; Hao, H.; Li, L. Heart-rate variability indices as predictors of the response to vagus nerve stimulation in patients with drug-resistant epilepsy. Epilepsia 2017, 58, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Donato, M.; Perez, V.; Deutsch, A.C.R.; Höcht, C.; Del Mauro, J.S.; Rodríguez, M.; Gelpi, R.J. Changes in the loading conditions induced by vagal stimulation modify the myocardial infarct size through sympathetic-parasympathetic interactions. Pflugers Arch. 2015, 467, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- DeMars, K.M.; Yang, C.; Hawkins, K.E.; McCrea, A.O.; Siwarski, D.M.; Candelario-Jalil, E. Spatiotemporal Changes in P-glycoprotein Levels in Brain and Peripheral Tissues Following Ischemic Stroke in Rats. J. Exp. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Patel, P.J.; Borovskiy, Y.; Killian, A.; Verdino, R.J.; Epstein, A.E.; Callans, D.J.; Marchlinski, F.E.; Deo, R. Optimal QT interval correction formula in sinus tachycardia for identifying cardiovascular and mortality risk: Findings from the Penn Atrial Fibrillation Free study. Heart Rhythm 2016, 13, 527–535. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECG | No SE (n = 3) | SE (n = 7) | ||||
|---|---|---|---|---|---|---|
| Heart rate (beats/m) | 321 | ± | 8.64 | 271 | ± | 9.03 * |
| RR interval (ms) | 187 | ± | 5.05 | 223 | ± | 8.32 * |
| PR interval (ms) | 50.5 | ± | 0.29 | 49 | ± | 1.22 |
| QRS amplitude (mV) | 27.3 | ± | 2.19 | 27.1 | ± | 1.03 |
| QT (ms) | 72 | ± | 0.50 | 85.3 | ± | 3.74 * |
| QTc (ms) | 71.9 | ± | 1.60 | 85.5 | ± | 3.68 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auzmendi, J.; Buchholz, B.; Salguero, J.; Cañellas, C.; Kelly, J.; Men, P.; Zubillaga, M.; Rossi, A.; Merelli, A.; Gelpi, R.J.; et al. Pilocarpine-Induced Status Epilepticus Is Associated with P-Glycoprotein Induction in Cardiomyocytes, Electrocardiographic Changes, and Sudden Death. Pharmaceuticals 2018, 11, 21. https://doi.org/10.3390/ph11010021
Auzmendi J, Buchholz B, Salguero J, Cañellas C, Kelly J, Men P, Zubillaga M, Rossi A, Merelli A, Gelpi RJ, et al. Pilocarpine-Induced Status Epilepticus Is Associated with P-Glycoprotein Induction in Cardiomyocytes, Electrocardiographic Changes, and Sudden Death. Pharmaceuticals. 2018; 11(1):21. https://doi.org/10.3390/ph11010021
Chicago/Turabian StyleAuzmendi, Jerónimo, Bruno Buchholz, Jimena Salguero, Carlos Cañellas, Jazmín Kelly, Paula Men, Marcela Zubillaga, Alicia Rossi, Amalia Merelli, Ricardo J. Gelpi, and et al. 2018. "Pilocarpine-Induced Status Epilepticus Is Associated with P-Glycoprotein Induction in Cardiomyocytes, Electrocardiographic Changes, and Sudden Death" Pharmaceuticals 11, no. 1: 21. https://doi.org/10.3390/ph11010021
APA StyleAuzmendi, J., Buchholz, B., Salguero, J., Cañellas, C., Kelly, J., Men, P., Zubillaga, M., Rossi, A., Merelli, A., Gelpi, R. J., Ramos, A. J., & Lazarowski, A. (2018). Pilocarpine-Induced Status Epilepticus Is Associated with P-Glycoprotein Induction in Cardiomyocytes, Electrocardiographic Changes, and Sudden Death. Pharmaceuticals, 11(1), 21. https://doi.org/10.3390/ph11010021