Procedures for the GMP-Compliant Production and Quality Control of [18F]PSMA-1007: A Next Generation Radiofluorinated Tracer for the Detection of Prostate Cancer


Abstract
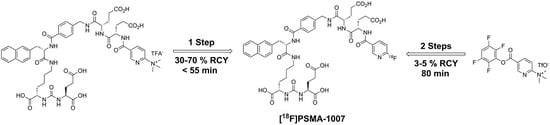
:
1. Introduction
2. Results
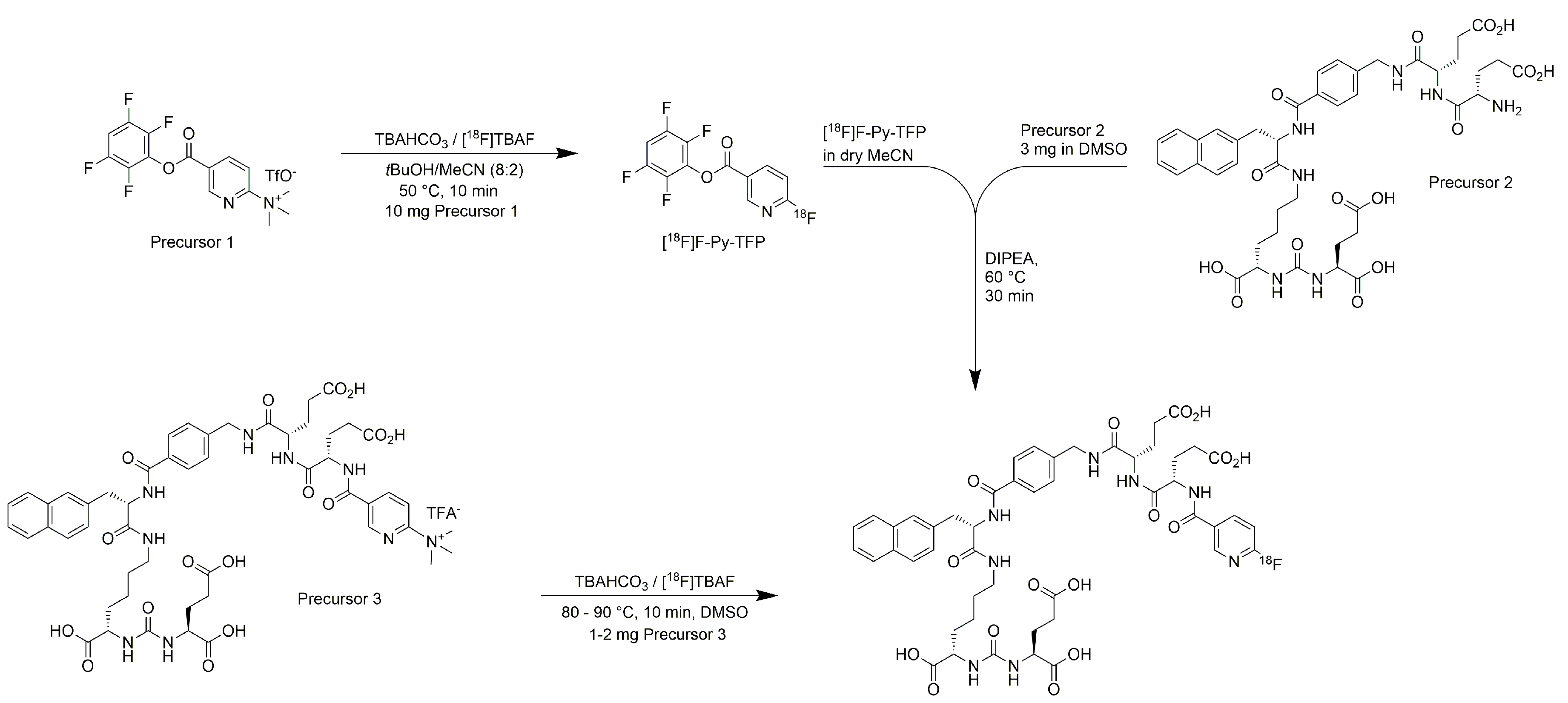
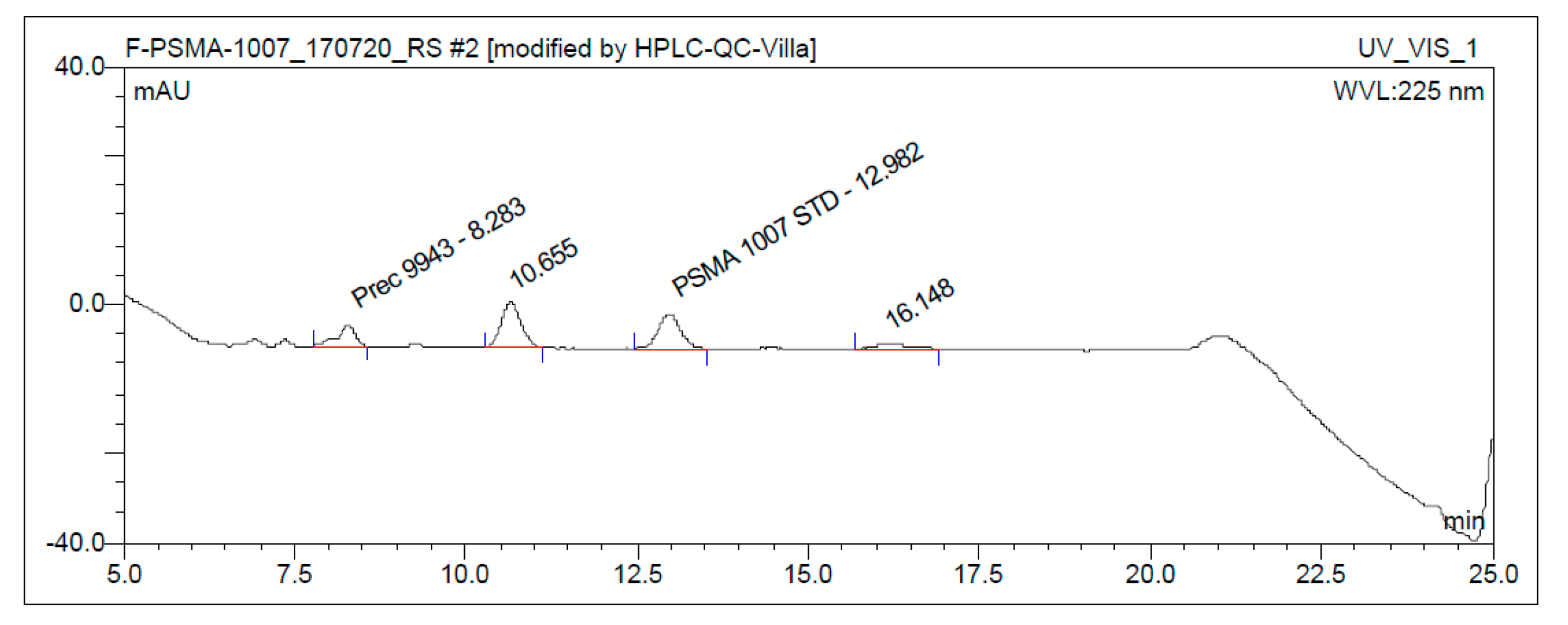
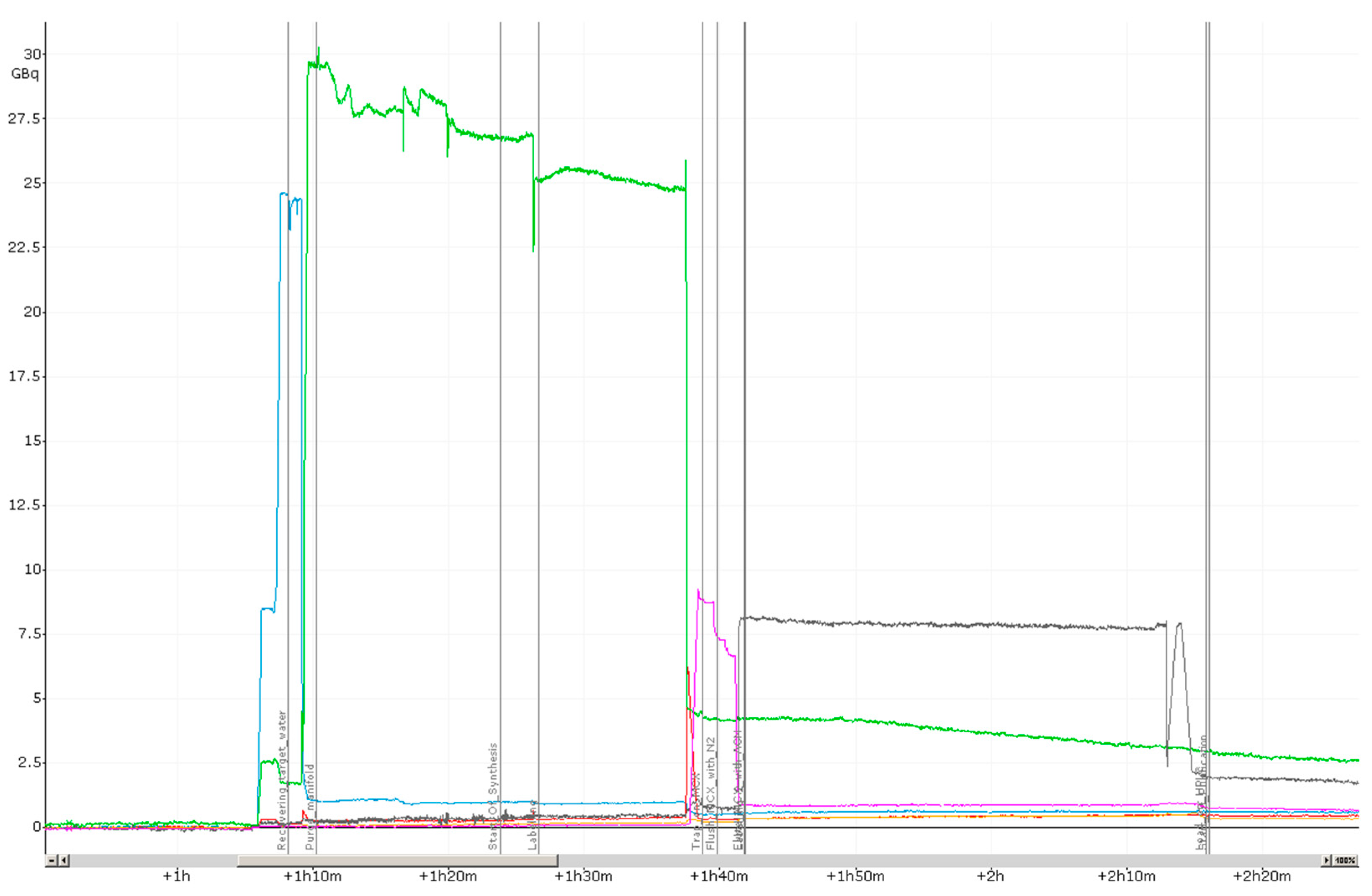
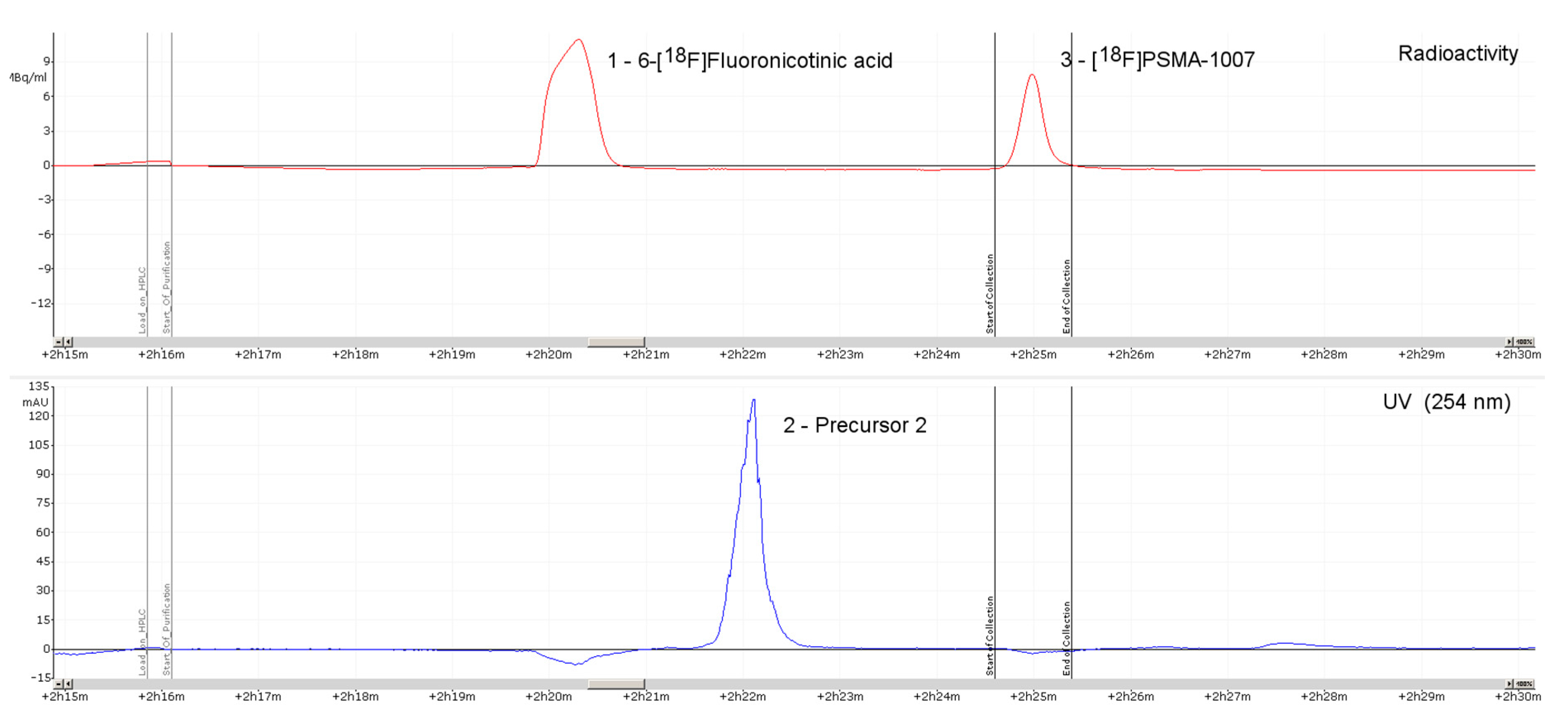
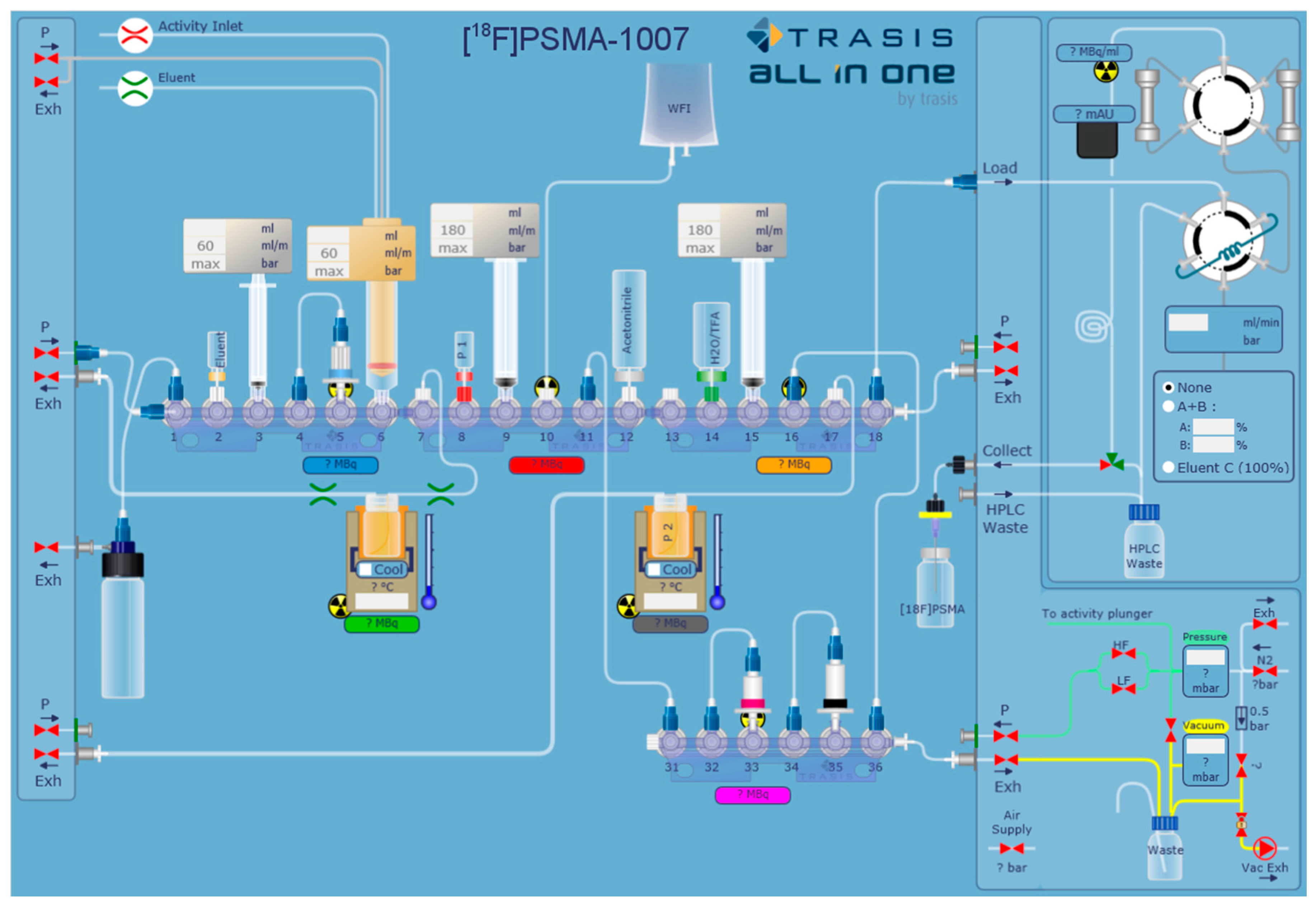
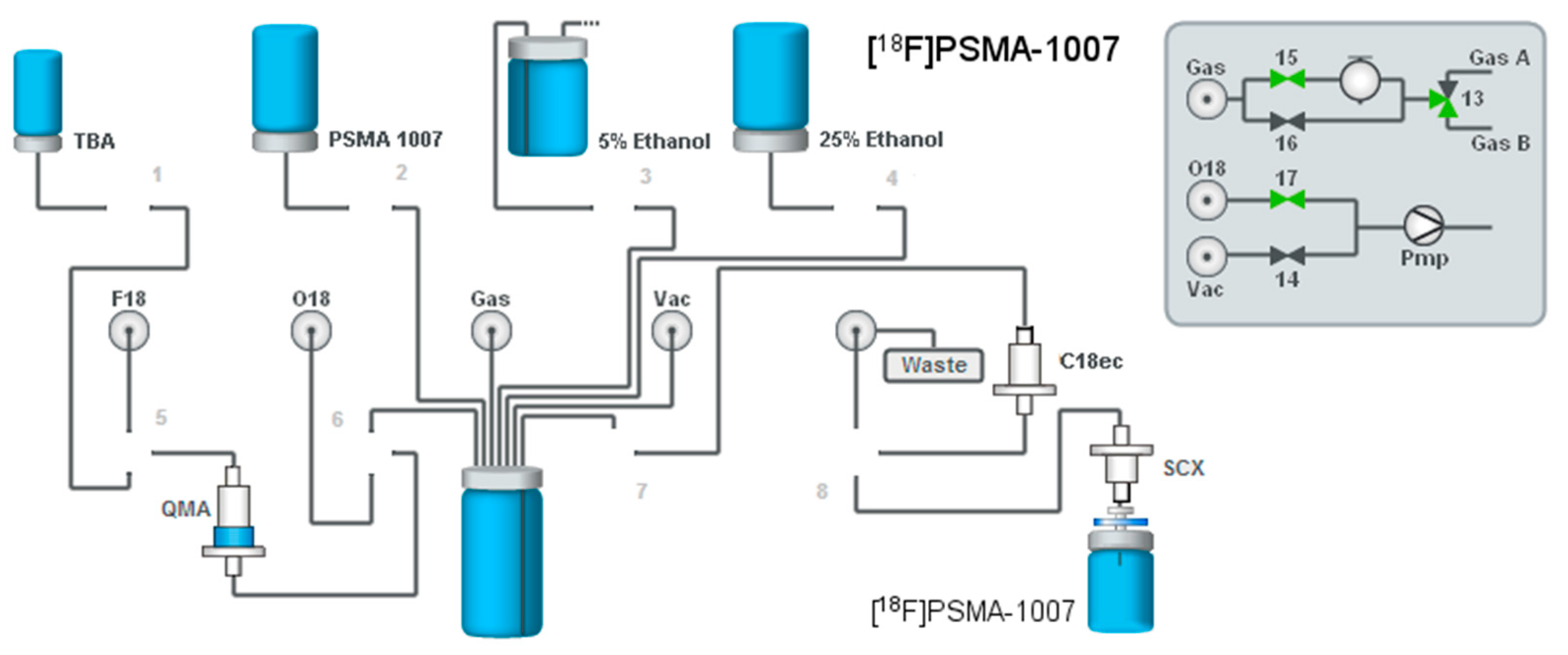
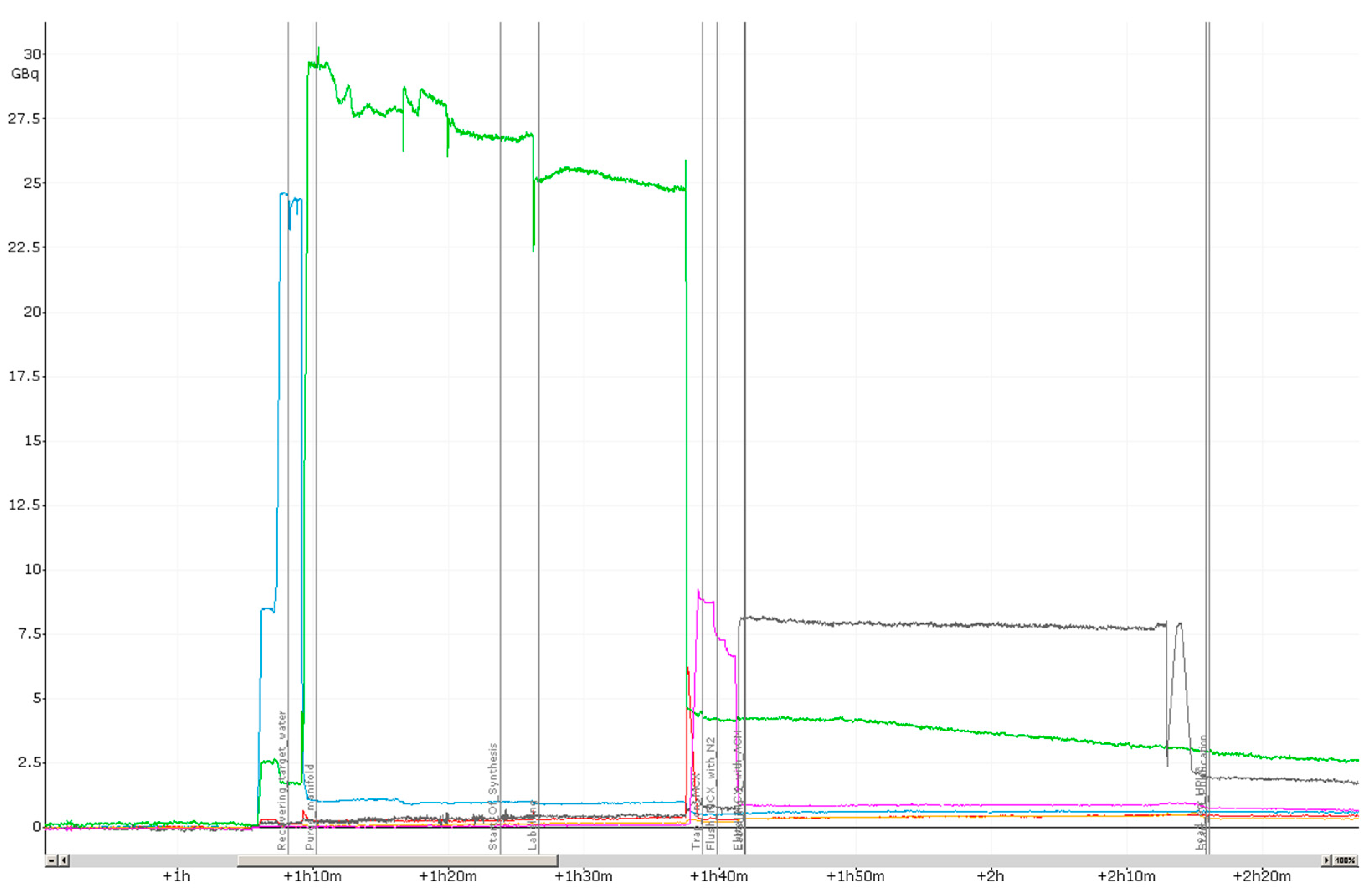
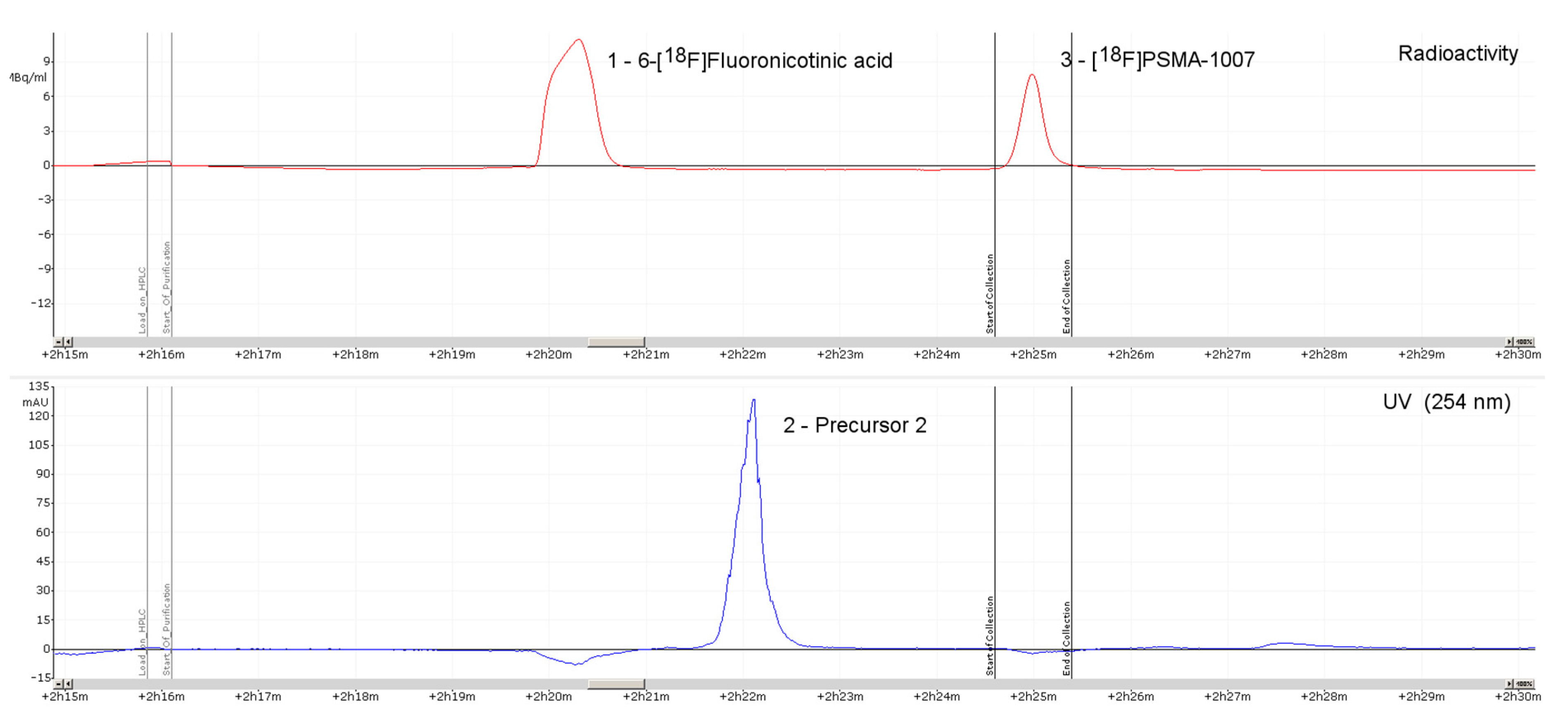
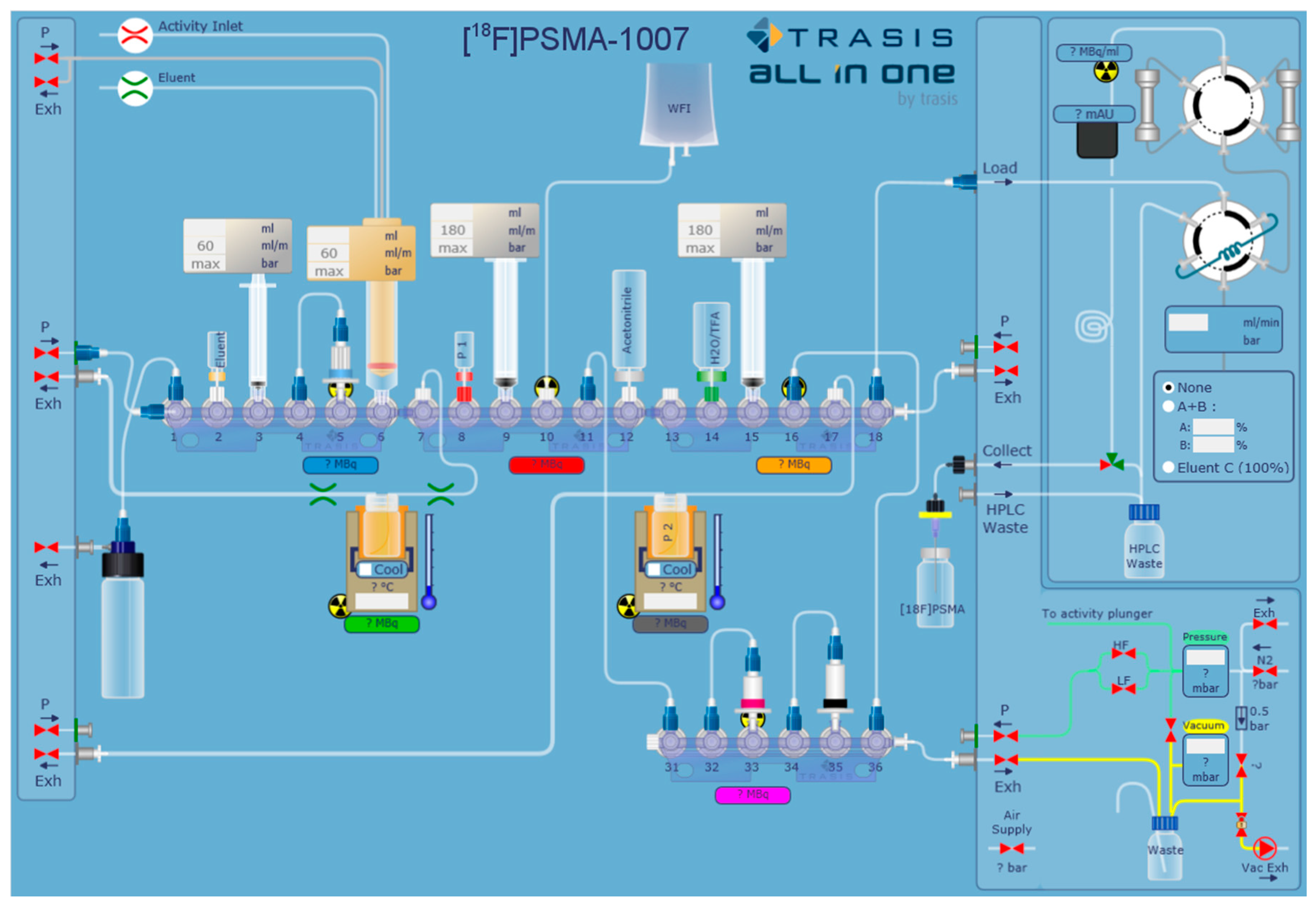
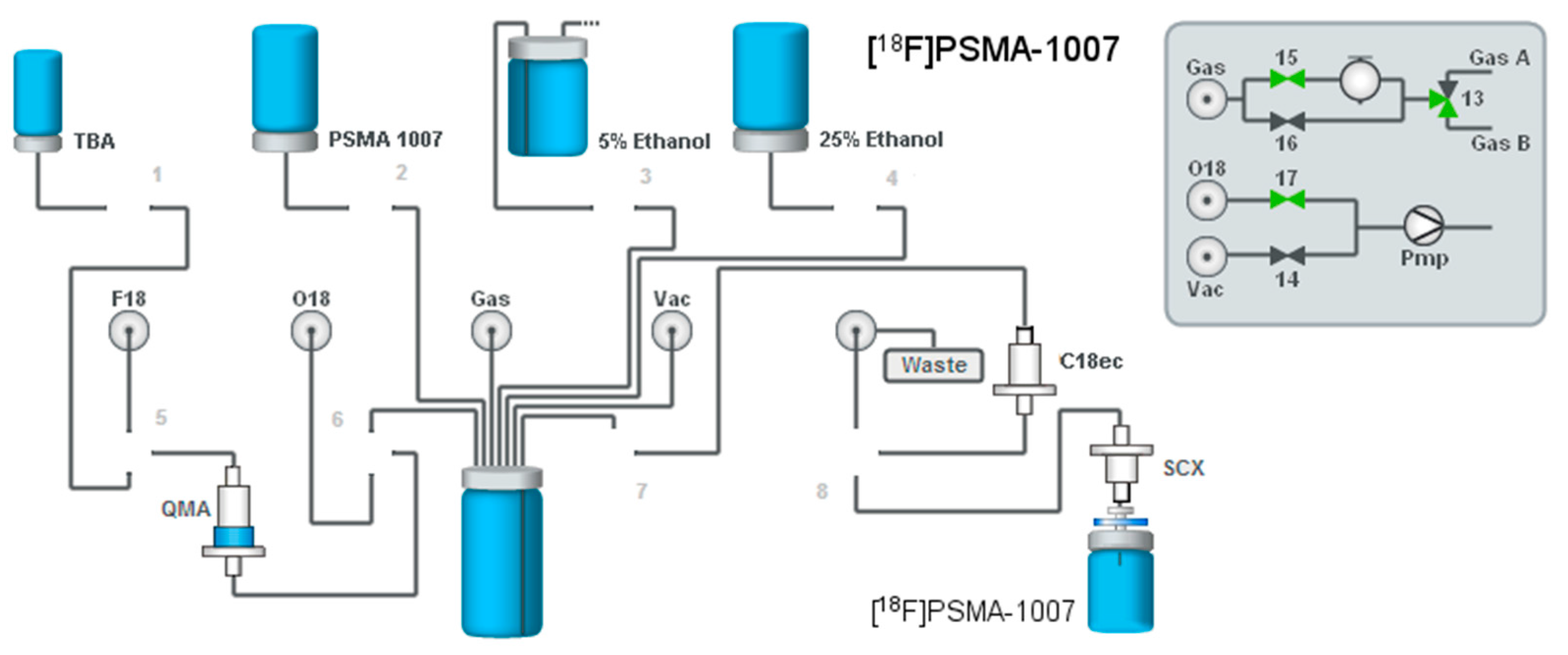
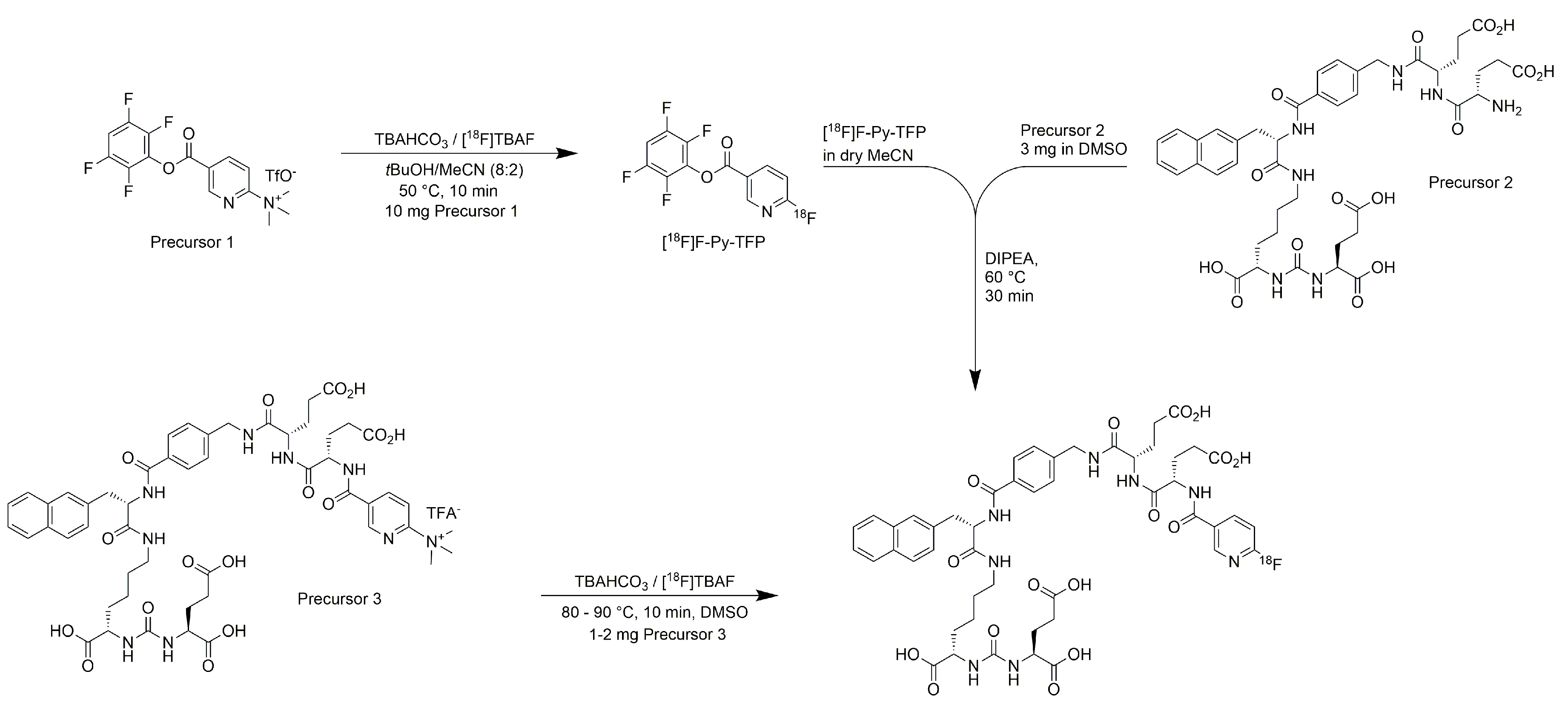
2.1. Production of [18F]PSMA-1007 via a Two-Step Procedure on a Trasis AllInOne Module
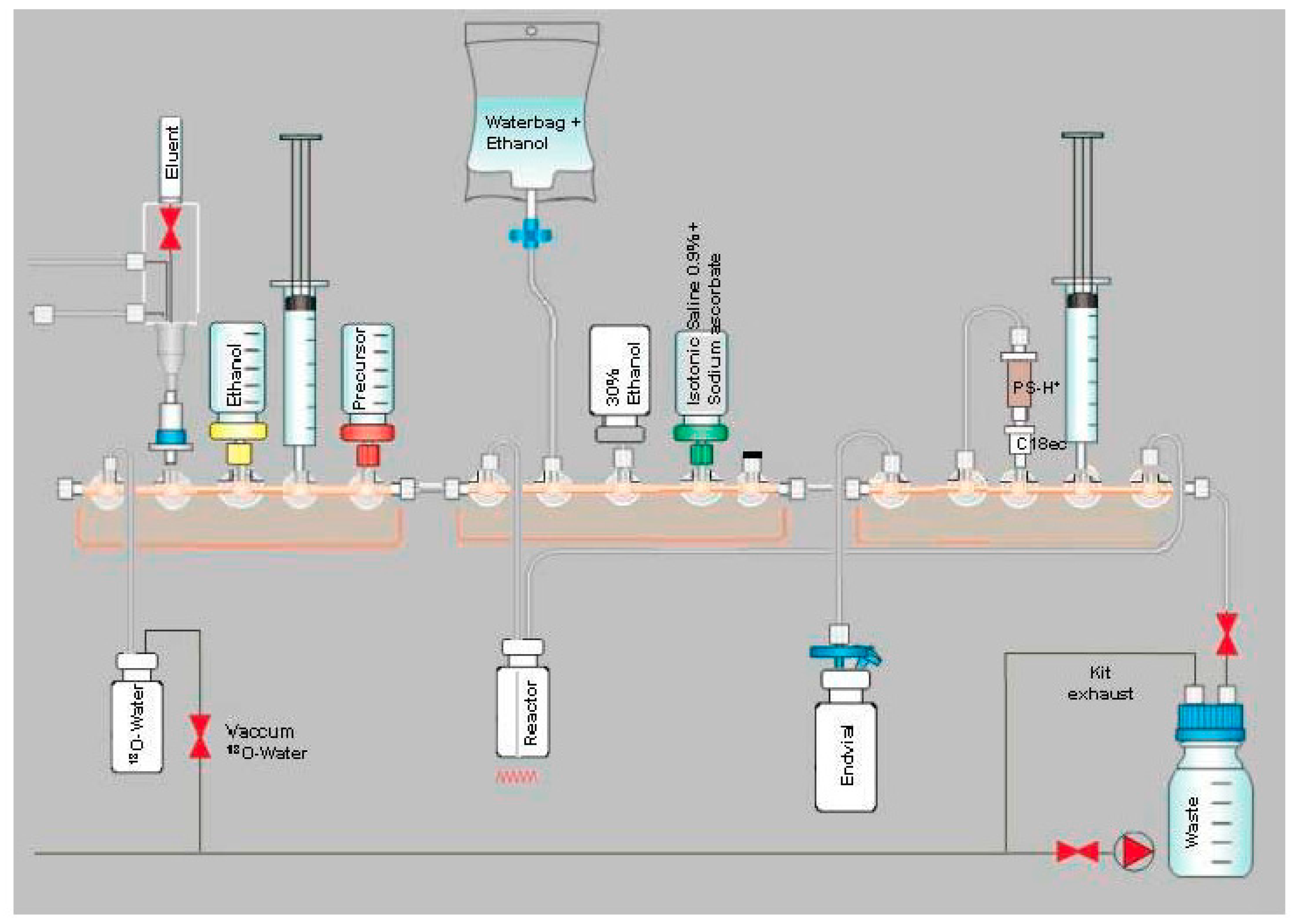
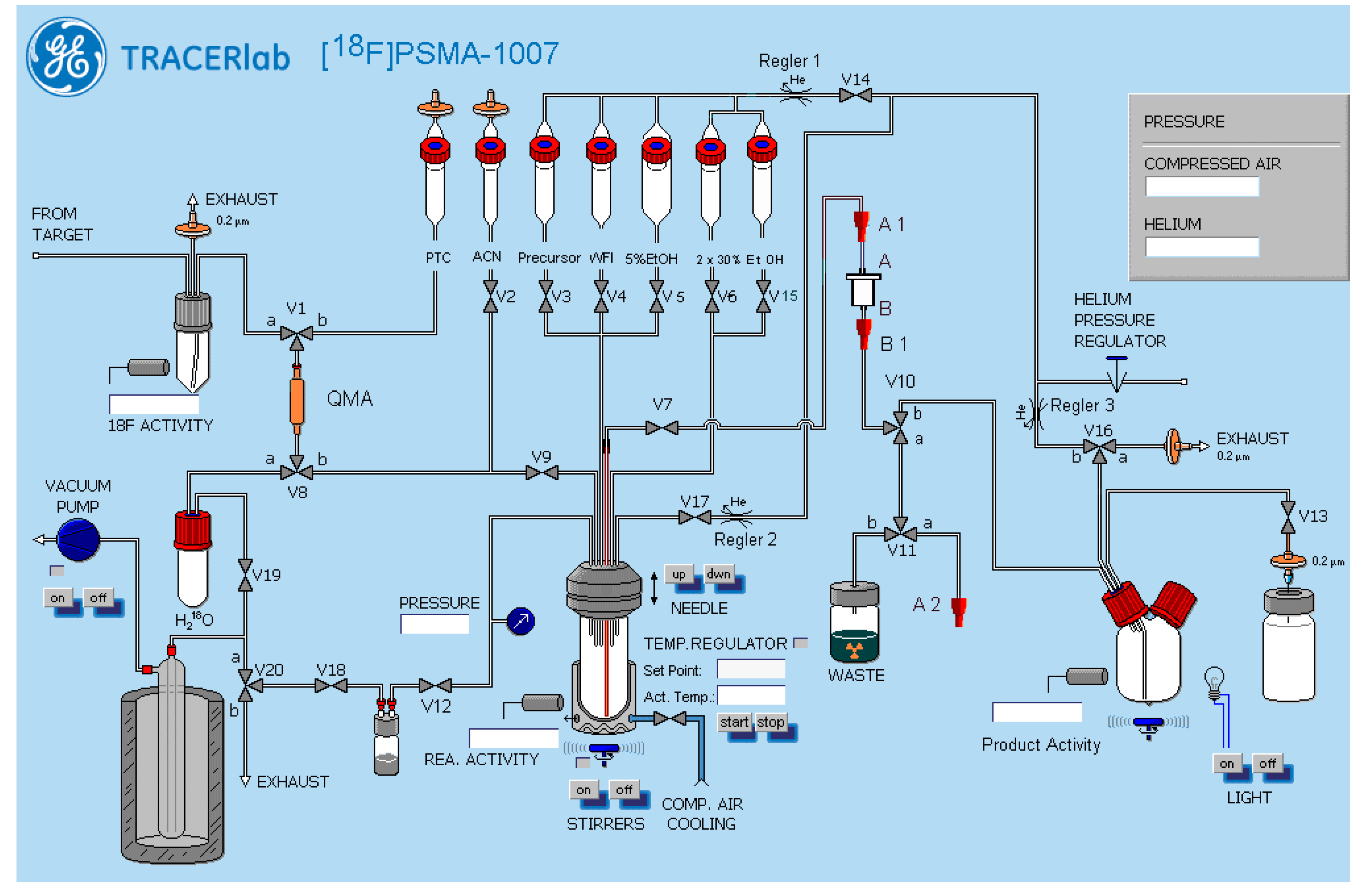
2.2. Production of [18F]PSMA-1007 via Direct Substitution on Tracerlab FX FN Module (SPE Purified)
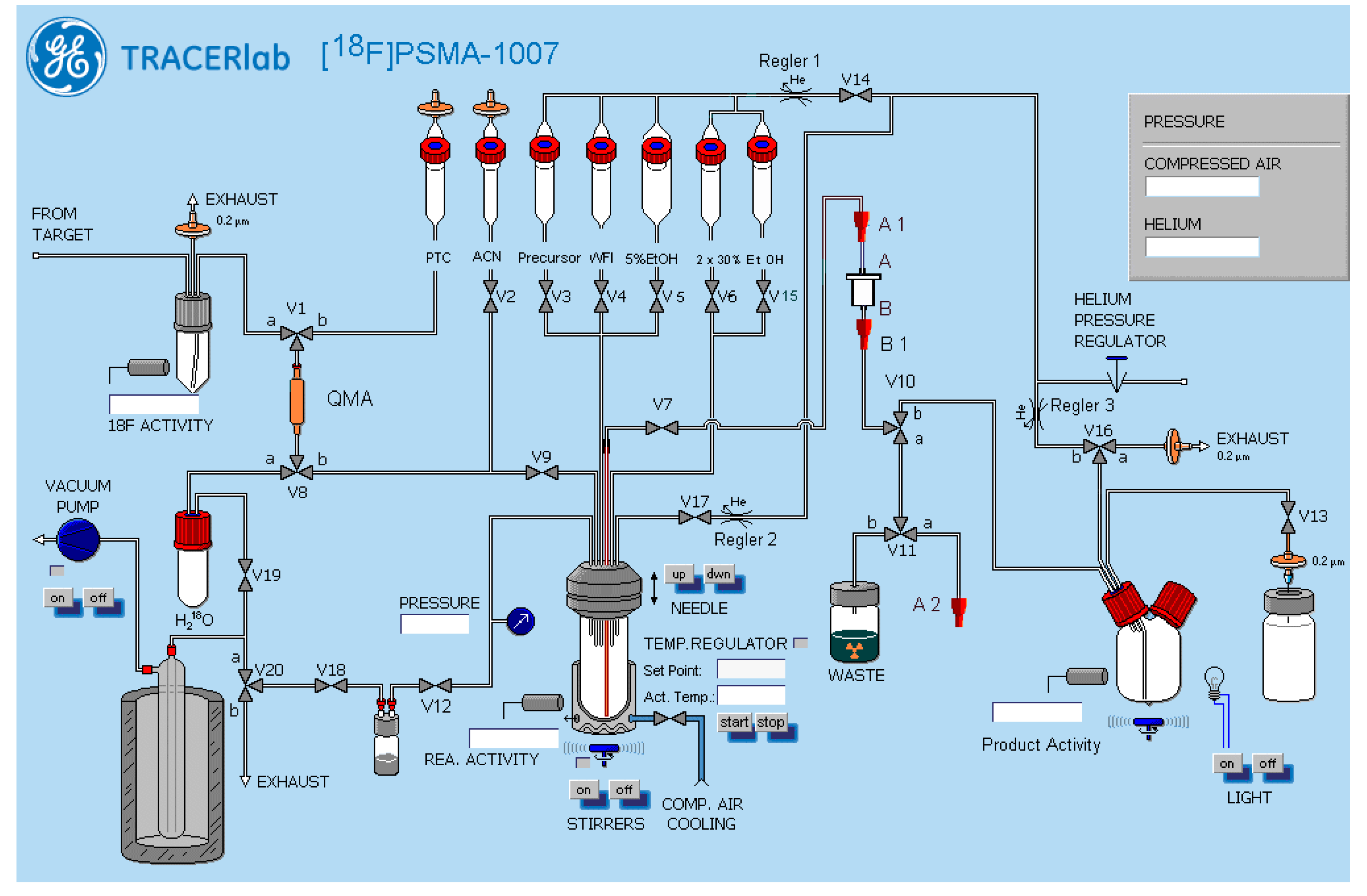
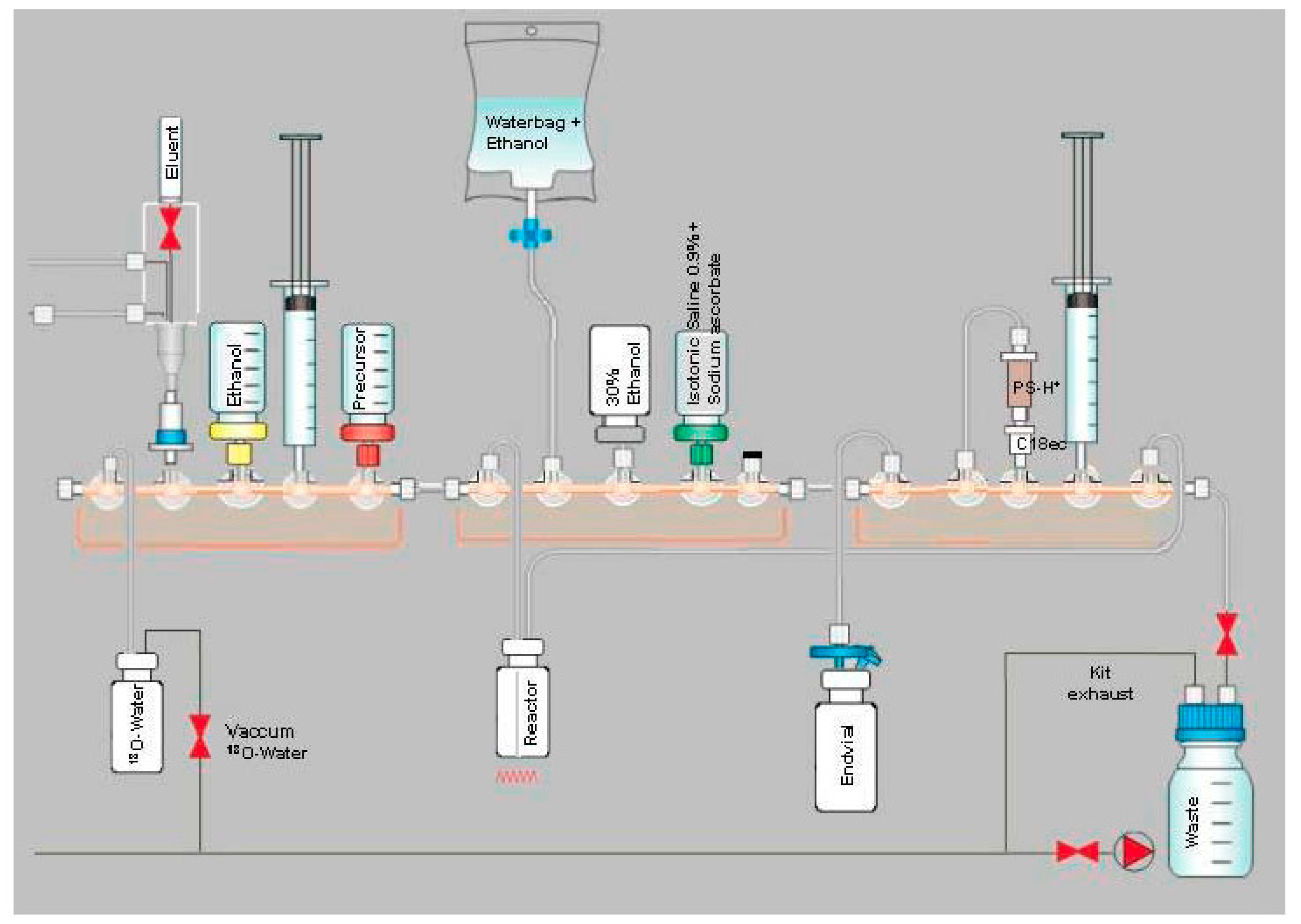
2.3. Production of [18F]PSMA-1007 via Direct Substitution on GE TRACERlab MX and NEPTIS Mosaic-RS (SPE Purified)
2.4. Production of [18F]PSMA-1007 via Direct Substitution on IBA SYNTHERA+ (SPE Purified)
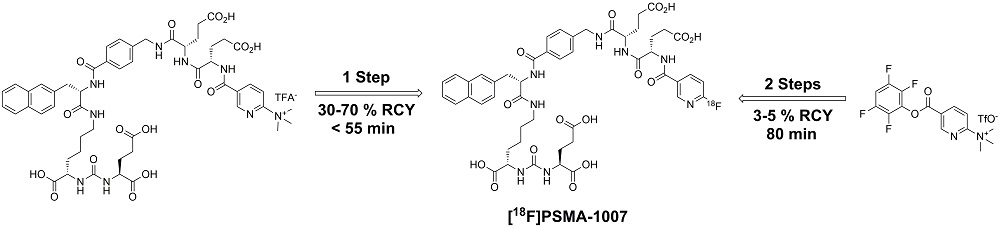
3. Discussion
3.1. Production of [18F]PSMA-1007 via a Two-Step Procedure on Trasis AllInOne Radiosynthesiser
3.2. Production of [18F]PSMA-1007 by Direct One-Step Synthesis on the GE Tracerlab FX FN Module, GE TRACERlab MX, NEPTIS Mosaic-RS and IBA SYNTHERA+
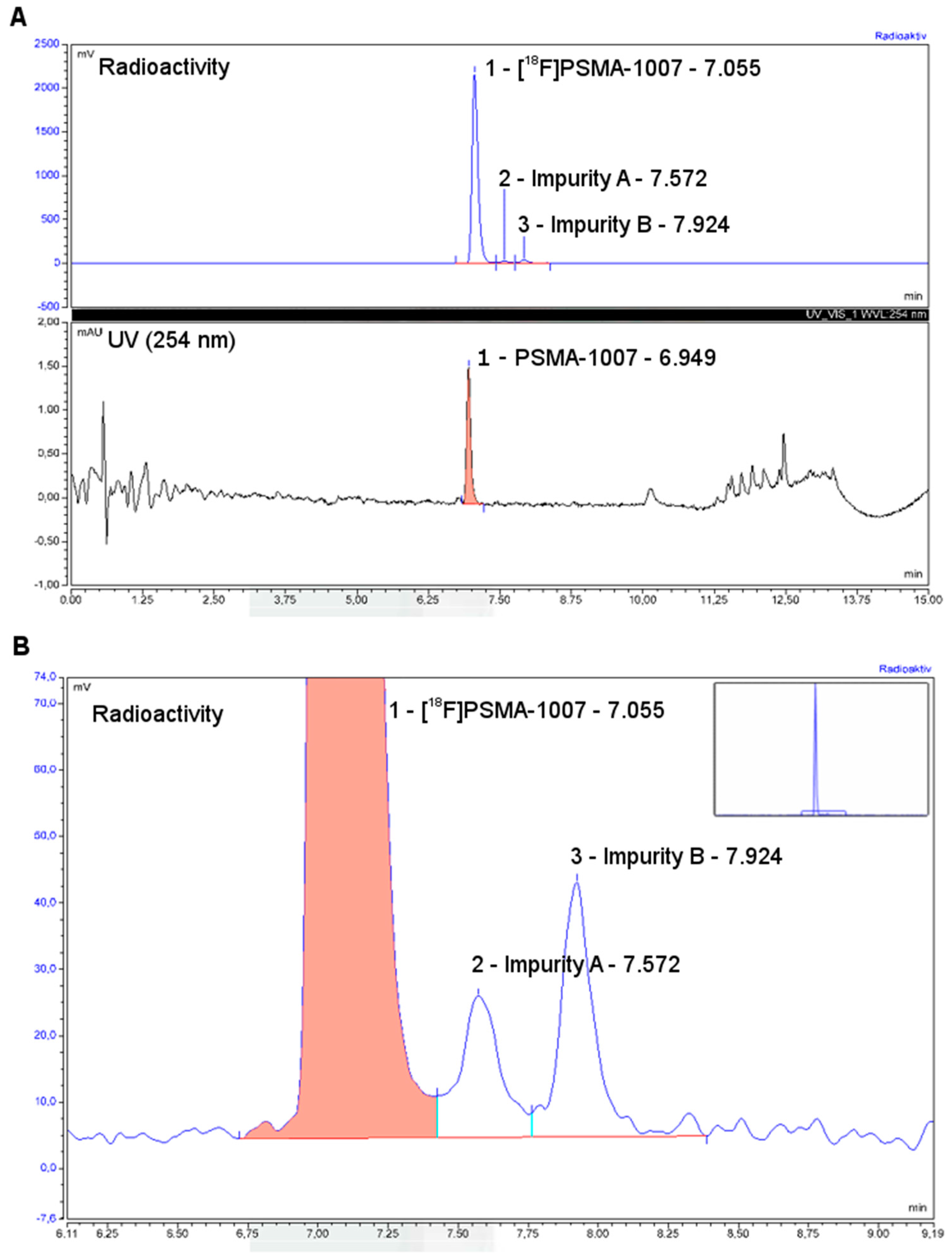
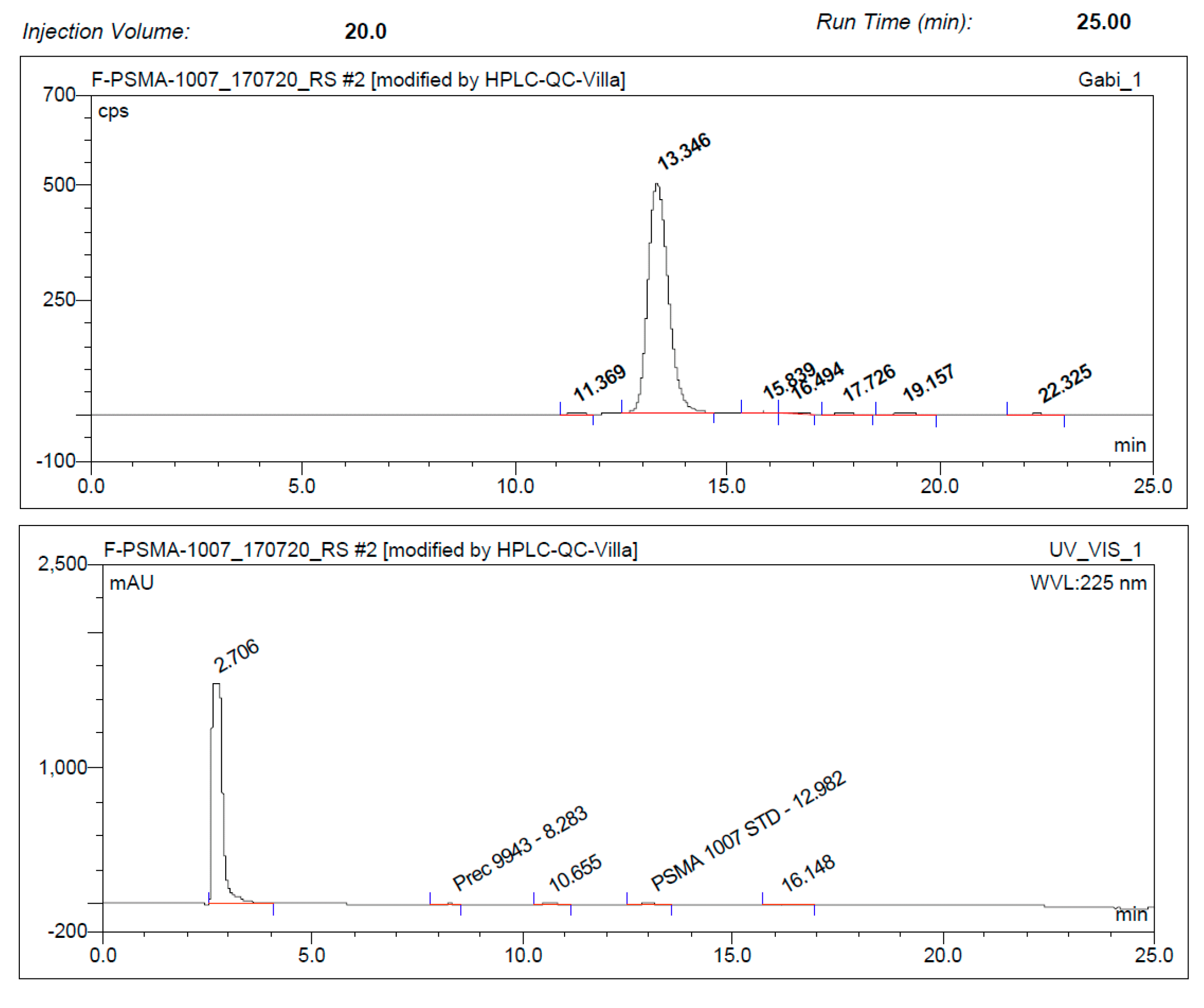
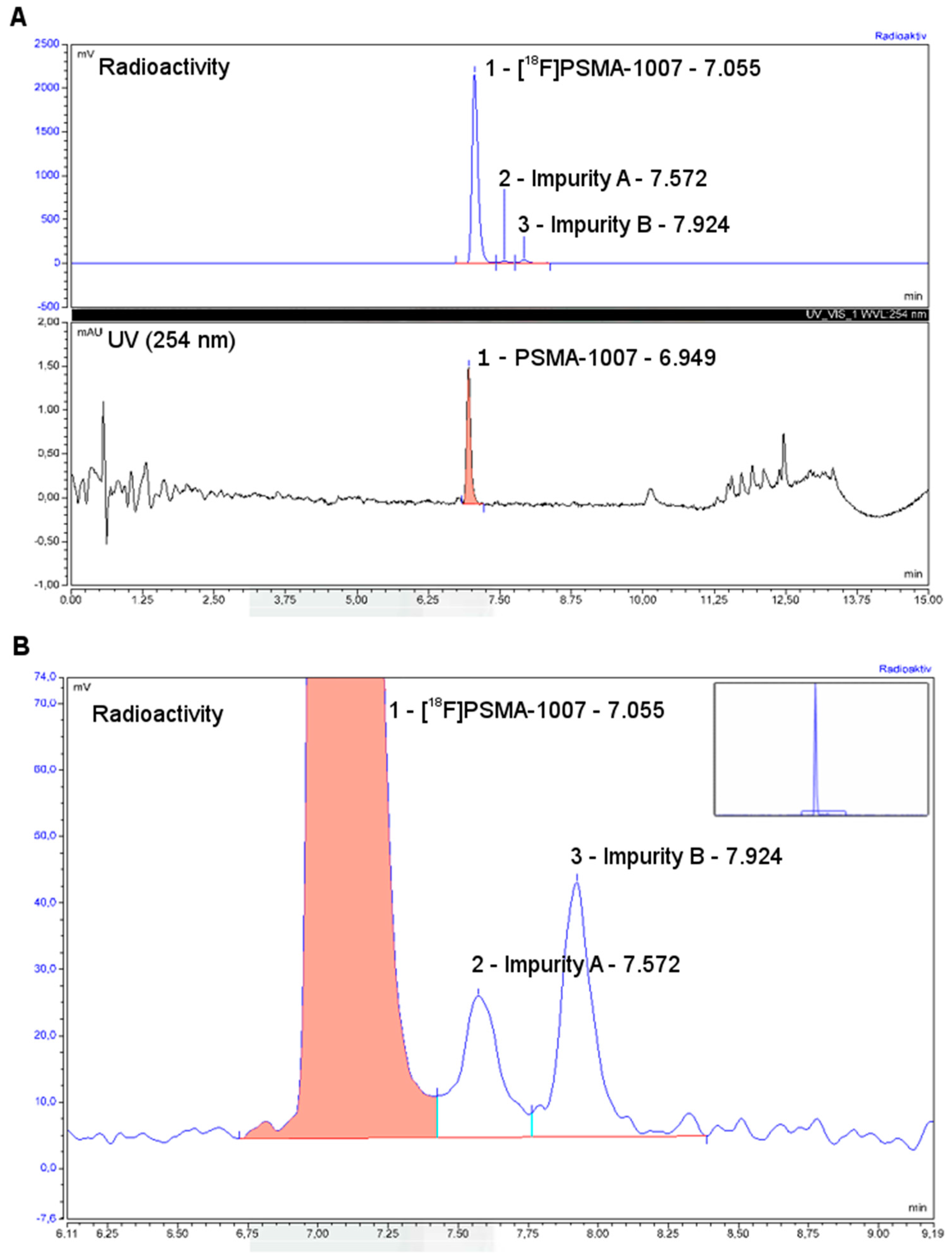
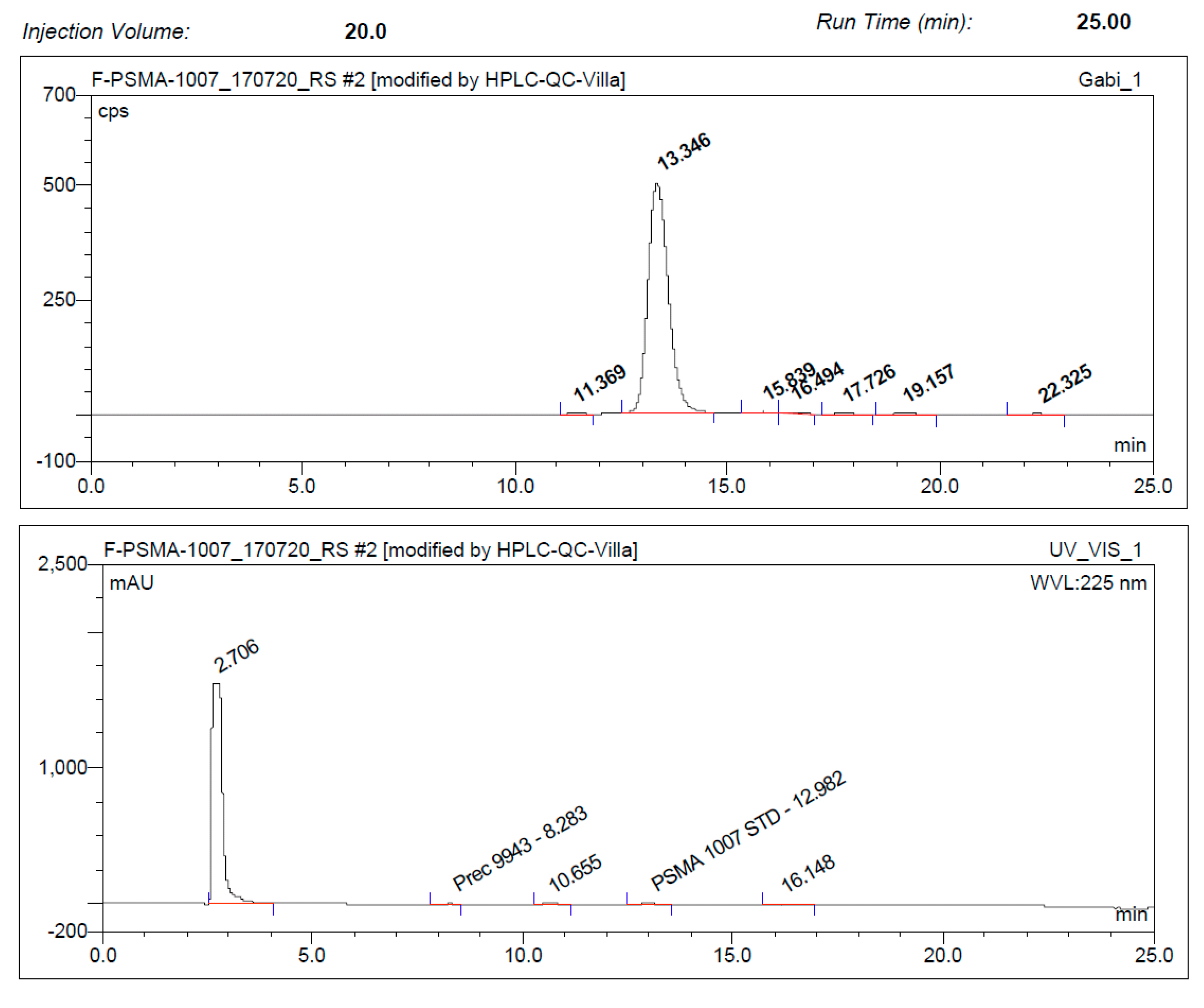
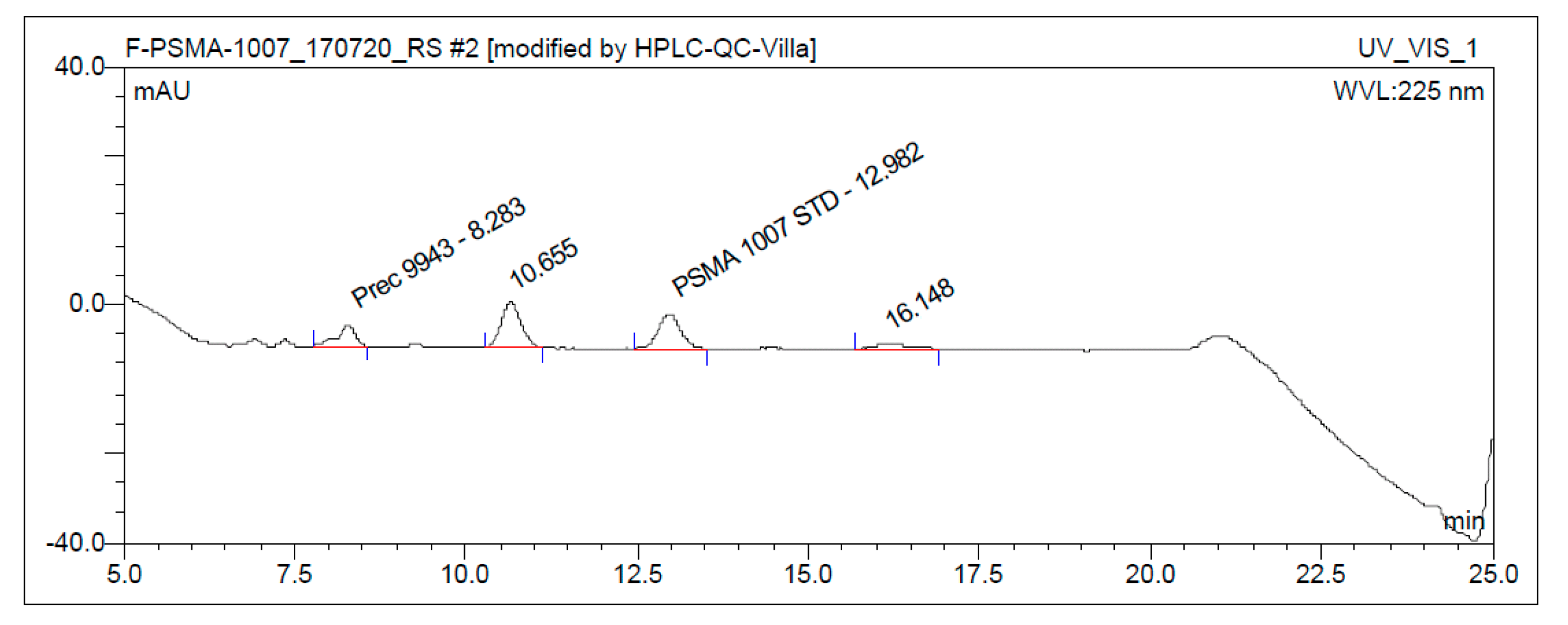
3.3. Quality Control
3.3.1. Acceptance Criteria
3.3.2. Discussion of Quality Control
4. Materials and Methods
4.1. Production of [18F]PSMA-1007 via a Two-Step Procedure on a Trasis AllInOne Module
4.1.1. Setup of the Radiosynthesiser
4.1.2. Reagents
4.1.3. Process Description
4.2. Production of [18F]PSMA-1007 via Direct Substitution on Modified Tracerlab FX FN Radiosynthesiser (Former Nuclear Interface FDG Radiosynthesiser) (SPE Purified)
4.2.1. Setup of the Radiosynthesiser
4.2.2. Reagents and Radionuclide Production
4.2.3. Process Description
4.3. Production of [18F]PSMA-1007 via Direct Substitution on GE TRACERlab MX and NEPTIS Mosaic-RS (SPE Purified)
4.3.1. Setup of the Radiosynthesiser
4.3.2. Reagents and Radionuclide Production
4.3.3. Process Description
4.4. Production of [18F]PSMA-1007 via Direct Substitution on IBA SYTHERA+
4.4.1. Setup of the Radiosynthesiser
4.4.2. Reagents and Radionuclide Production
4.4.3. Process Description
4.5. Quality Control
4.5.1. Standard Procedures
4.5.2. HPLC Analysis
4.5.3. Thin Layer Chromatography
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Afshar-Oromieh, A.; Haberkorn, U.; Eder, M.; Eisenhut, M.; Zechmann, C.M. [68Ga]Gallium-labelled PSMA ligand as superior PET tracer for the diagnosis of prostate cancer: Comparison with 18F-FECH. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Schwenck, J.; Rempp, H.; Reischl, G.; Kruck, S.; Stenzl, A.; Nikolaou, K.; Pfannenberg, C.; la Fougère, C. Comparison of 68Ga-labelled PSMA-11 and 11C-choline in the detection of prostate cancer metastases by PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Zechmann, C.M.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Holland-Letz, T.; Hadaschik, B.A.; Giesel, F.L.; Debus, J.; et al. Comparison of PET imaging with a 68Ga-labelled PSMA ligand and 18F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, L.; Briganti, A.; Fanti, S.; Joniau, S.; Reske, S.; Schiavina, R.; Stief, C.; Thalmann, G.N.; Picchio, M. New Clinical Indications for 18F/11C-choline, New Tracers for Positron Emission Tomography and a Promising Hybrid Device for Prostate Cancer Staging: A Systematic Review of the Literature. Eur. Urol. 2016, 70, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.M.; Gomes, C.; Faria, D.B.; Vieira, T.S.; Silva, F.A.; Vale, J.; Pimentel, F.L. 68Ga-prostate-specific Membrane Antigen Positron Emission Tomography/Computed Tomography for Prostate Cancer Imaging: A Narrative Literature Review. World J. Nucl. Med. 2017, 16, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Porres, D.; Heidenreich, A.; Heidegger, I.; Knuechel, R.; Steib, F.; Behrendt, F.F.; Verburg, F.A. Detection of recurrent prostate cancer lesions before salvage lymphadenectomy is more accurate with 68Ga-PSMA-HBED-CC than with 18F-Fluoroethylcholine PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Eder, M.; Neels, O.; Müller, M.; Bauder-Wüst, U.; Remde, Y.; Schäfer, M.; Hennrich, U.; Eisenhut, M.; Afshar-Oromieh, A.; Haberkorn, U.; et al. Novel Preclinical and Radiopharmaceutical Aspects of [68Ga]Ga-PSMA-HBED-CC: A New PET Tracer for Imaging of Prostate Cancer. Pharmaceuticals 2014, 7, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Hetzheim, H.; Kratochwil, C.; Benesova, M.; Eder, M.; Neels, O.C.; Eisenhut, M.; Kübler, W.; Holland-Letz, T.; Giesel, F.L.; et al. The Theranostic PSMA Ligand PSMA-617 in the Diagnosis of Prostate Cancer by PET/CT: Biodistribution in Humans, Radiation Dosimetry, and First Evaluation of Tumor Lesions. J. Nucl. Med. 2015, 56, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-Labeled PSMA I & T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Fendler, W.P.; Eiber, M.; Beheshti, M.; Bomanji, J.; Ceci, F.; Cho, S.; Giesel, F.; Haberkorn, U.; Hope, T.A.; Kopka, K.; et al. 68Ga-PSMA PET/CT: Joint EANM and SNMMI procedure guideline for prostate cancer imaging: Version 1.0. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Holland-Letz, T.; Giesel, F.L.; Kratochwil, C.; Mier, W.; Haufe, S.; Debus, N.; Eder, M.; Eisenhut, M.; Schäfer, M.; et al. Diagnostic performance of 68Ga-PSMA-11 (HBED-CC) PET/CT in patients with recurrent prostate cancer: Evaluation in 1007 patients. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Avtzi, E.; Giesel, F.L.; Holland-Letz, T.; Linhart, H.G.; Eder, M.; Eisenhut, M.; Boxler, S.; Hadaschik, B.A.; Kratochwil, C.; et al. The diagnostic value of PET/CT imaging with the 68Ga-labelled PSMA ligand HBED-CC in the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Eiber, M.; Maurer, T.; Souvatzoglou, M.; Beer, A.J.; Ruffani, A.; Haller, B.; Graner, F.P.; Kübler, H.; Haberhorn, U.; Eisenhut, M.; et al. Evaluation of Hybrid 68Ga-PSMA Ligand PET/CT in 248 Patients with Biochemical Recurrence After Radical Prostatectomy. J. Nucl. Med. 2015, 56, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.; Papa, N.; Christidis, D.; Wetherell, D.; Hofman, M.S.; Murphy, D.G.; Bolton, D.; Lawrentschuk, N. Sensitivity, Specificity, and Predictors of Positive 68Ga-Prostate-specific Membrane Antigen Positron Emission Tomography in Advanced Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. 2016, 70, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, J.; Schäfer, M.; Benešová, M.; Bauder-Wüst, U.; Leotta, K.; Eder, M.; Neels, O.C.; Haberkorn, U.; Kopka, K. Preclinical Evaluation of 18F-PSMA-1007, a New Prostate-Specific Membrane Antigen Ligand for Prostate Cancer Imaging. J. Nucl. Med. 2017, 58, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Mease, R.C.; Dusich, C.L.; Foss, C.A.; Ravert, H.T.; Dannals, R.F.; Seidel, J.; Prideaux, A.; Fox, J.J.; Sgouror, G.; Kozikowski, A.P.; et al. N-[N-[(S)-1,3-Dicarboxypropyl]Carbamoyl]-4-[18F]Fluorobenzyl-L-cysteine, [18F]DCFBC: A New Pmaging Probe for Prostate Cancer. Clin. Cancer Res. 2008, 14, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pullambhatla, M.; Foss, C.A.; Byun, Y.; Nimmagadda, S.; Senthamizhchelvan, S.; Sgouros, G.; Mease, R.C.; Pomper, M.G. 2-(3-{1-Carboxy-5-[(6-[18F]Fluoro-Pyridine-3-Carbonyl)-Amino]-Pentyl}-Ureido)-Pentanedioic Acid, [18F]DCFPyL, a PSMA-Based PET Imaging Agent for Prostate Cancer. Clin. Cancer Res. 2011, 17, 7645–7653. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Zlatopolskiy, B.; Machulla, H.J.; Reske, S.N.; Solbach, C. One pot radiofluorination of a new potential PSMA ligand [Al18F]NOTA-DUPA-Pep. J. Label. Compd. Radiopharm. 2012, 55, 320–325. [Google Scholar] [CrossRef]
- Graham, K.; Lesche, R.; Gromov, A.V.; Böhnke, N.; Schäfer, M.; Hassfeld, J. Radiofluorinated Derivatives of 2-(Phosphonomethyl)pentanedioic Acid as Inhibitor of Prostate Specific Membrane Antigen (PSMA) for the Imaging of Prostate Cancer. J. Med. Chem. 2012, 55, 9510–9520. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Cardinale, J.; Schäfer, M.; Neels, O.; Benešová, M.; Mier, W.; Haberkorn, U.; Kopka, K.; Kratochwil, C. 18F-Labelled PSMA-1007 shows similarity in structure, biodistribution and tumour uptake to the theragnostic compound PSMA-617. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1929–1930. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Hadaschik, B.; Cardinale, J.; Radtke, J.; Vinsensia, M.; Lehnert, W.; Kesch, C.; Tolstov, Y.; Singer, S.; Grabe, N.; et al. F-18 labelled PSMA-1007: Biodistribution, radiation dosimetry and histopathological validation of tumor lesions in prostate cancer patients. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Kesch, C.; Yun, M.; Cardinale, J.; Haberkorn, U.; Kopka, K.; Kratochwil, C.; Hadaschik, B.A. 18F-PSMA-1007 PET/CT Detects Micrometastases in a Patient With Biochemically Recurrent Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, e497–e499. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Kesch, C.; Cardinale, J.; Schlemmer, H.; Haberkorn, U.; Kopka, K. Giesel Simultaneous whole-body 18F-PSMA-1007-PET/MRI with integrated multi-parametric MRI of the prostatic fossa for comprehensive oncological staging of patients with prostate cancer. J. Nucl. Med. 2017, 58, 538. [Google Scholar]
- Martin, R.; Hesse, R.; Smits, R; Hübner, S.; Müller, M.; Hoepping, A. Präkursoren für die Radiofluorierung. German Patent Application No. DE 10 2016 122 273.9, 2016. submitted. [Google Scholar]
- The European Pharmacopoeia, 9.2; European Directorate for the Quality of the Medicines (EDQM): Strasbourg, France, 2017.
- Cardinale, J.; Schäfer, M.; Neels, O.C.; Remde, Y.; Haberkorn, U.; Giesel, F.L.; Kopka, K. F-18-PSMA-1007—Von der Präklinik zur ersten Humananwendung. Nuklearmedizin 2017, 56, A29. [Google Scholar]
- Olberg, D.E.; Arukwe, J.M.; Grace, D.; Hjelstuen, O.K.; Solbakken, M.; Kindberg, G.M.; Cuthbertson, A. One Step Radiosynthesis of 6-[18F]Fluoronicotinic Acid 2,3,5,6-Tetrafluorophenyl Ester ([18F]F-Py-TFP): A New Prosthetic Group for Efficient Labeling of Biomolecules with Fluorine-18. J. Med. Chem. 2010, 53, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Block, D.; Klatte, B.; Knöchel, A.; Beckmann, R.; Holm, U.N.C.A. [18F]-labelling of aliphatic compounds in high yields via aminopolyether—Supported nucleophilic substitution. J. Label. Compd. Radiopharm. 1986, 23, 467–477. [Google Scholar] [CrossRef]
- Kuntzsch, M.; Lamparter, D.; Brüggener, N.; Müller, M.; Kienzle, G.J.; Reischl, G. Development and Successful Validation of Simple and Fast TLC Spot Tests for Determination of Kryptofix® 2.2.2 and Tetrabutylammonium in 18F-Labeled Radiopharmaceuticals. Pharmaceuticals 2014, 7, 621–633. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Method | Acceptance Criteria |
|---|---|---|
| Appearance | Visual | Clear and colourless |
| Identity | HPLC | Rt ± 0.5 min of reference standard |
| Radiochemical purity | HPLC | ≥95% |
| TLC | ≥95% | |
| Radionuclidic purity | Half life | 110 ± 5 min |
| Gamma spectroscopy | 511 keV ≥ 99.9% (post-release) | |
| Chemical purity # | HPLC | PSMA-1007: ≤0.1 mg/Vmax |
| Any other impurity *: ≤0.1 mg/Vmax | ||
| Sum of all impurities *: ≤0.5 mg/Vmax | ||
| TLC | TBA: ≤2.6 mg/Vmax | |
| GC | Acetonitrile: ≤4.1 mg/Vmax | |
| DMSO: ≤50 mg/Vmax | ||
| EtOH: ≤10% V/V | ||
| Acetone: ≤50 mg/Vmax | ||
| pH | Potentiometric or strip indicator | 4.5–7.5 |
| Endotoxins | LAL test | ≤175 IU/Vmax |
| Filter integrity | Bubble point test | ≥3.5 bar (Cathivex-GV 0.22 µm) |
| Sterility | Post-release | Sterile (post-release) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardinale, J.; Martin, R.; Remde, Y.; Schäfer, M.; Hienzsch, A.; Hübner, S.; Zerges, A.-M.; Marx, H.; Hesse, R.; Weber, K.; et al. Procedures for the GMP-Compliant Production and Quality Control of [18F]PSMA-1007: A Next Generation Radiofluorinated Tracer for the Detection of Prostate Cancer. Pharmaceuticals 2017, 10, 77. https://doi.org/10.3390/ph10040077
Cardinale J, Martin R, Remde Y, Schäfer M, Hienzsch A, Hübner S, Zerges A-M, Marx H, Hesse R, Weber K, et al. Procedures for the GMP-Compliant Production and Quality Control of [18F]PSMA-1007: A Next Generation Radiofluorinated Tracer for the Detection of Prostate Cancer. Pharmaceuticals. 2017; 10(4):77. https://doi.org/10.3390/ph10040077
Chicago/Turabian StyleCardinale, Jens, René Martin, Yvonne Remde, Martin Schäfer, Antje Hienzsch, Sandra Hübner, Anna-Maria Zerges, Heike Marx, Ronny Hesse, Klaus Weber, and et al. 2017. "Procedures for the GMP-Compliant Production and Quality Control of [18F]PSMA-1007: A Next Generation Radiofluorinated Tracer for the Detection of Prostate Cancer" Pharmaceuticals 10, no. 4: 77. https://doi.org/10.3390/ph10040077
APA StyleCardinale, J., Martin, R., Remde, Y., Schäfer, M., Hienzsch, A., Hübner, S., Zerges, A.-M., Marx, H., Hesse, R., Weber, K., Smits, R., Hoepping, A., Müller, M., Neels, O. C., & Kopka, K. (2017). Procedures for the GMP-Compliant Production and Quality Control of [18F]PSMA-1007: A Next Generation Radiofluorinated Tracer for the Detection of Prostate Cancer. Pharmaceuticals, 10(4), 77. https://doi.org/10.3390/ph10040077