Heparin Mimetics: Their Therapeutic Potential

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
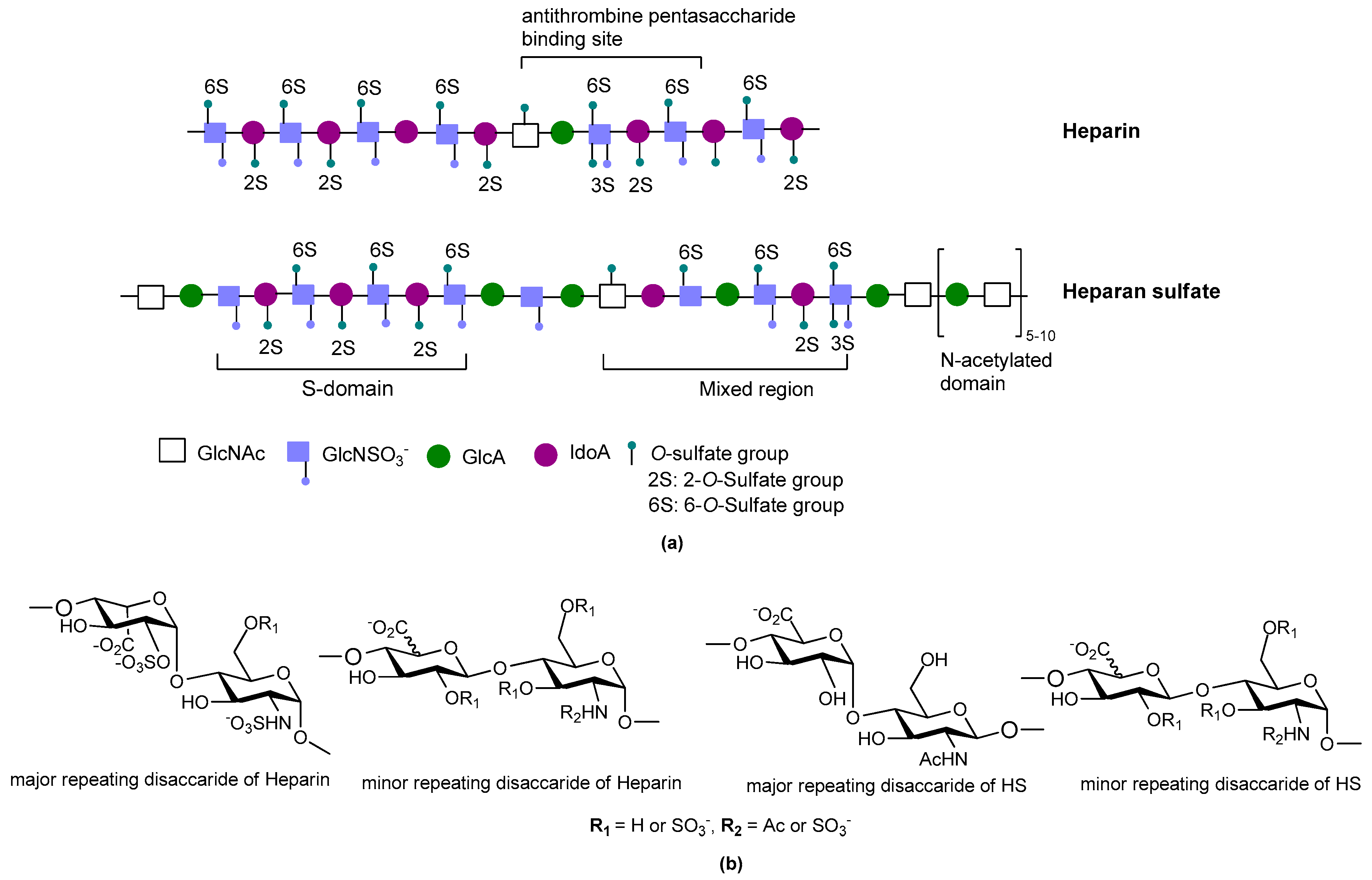
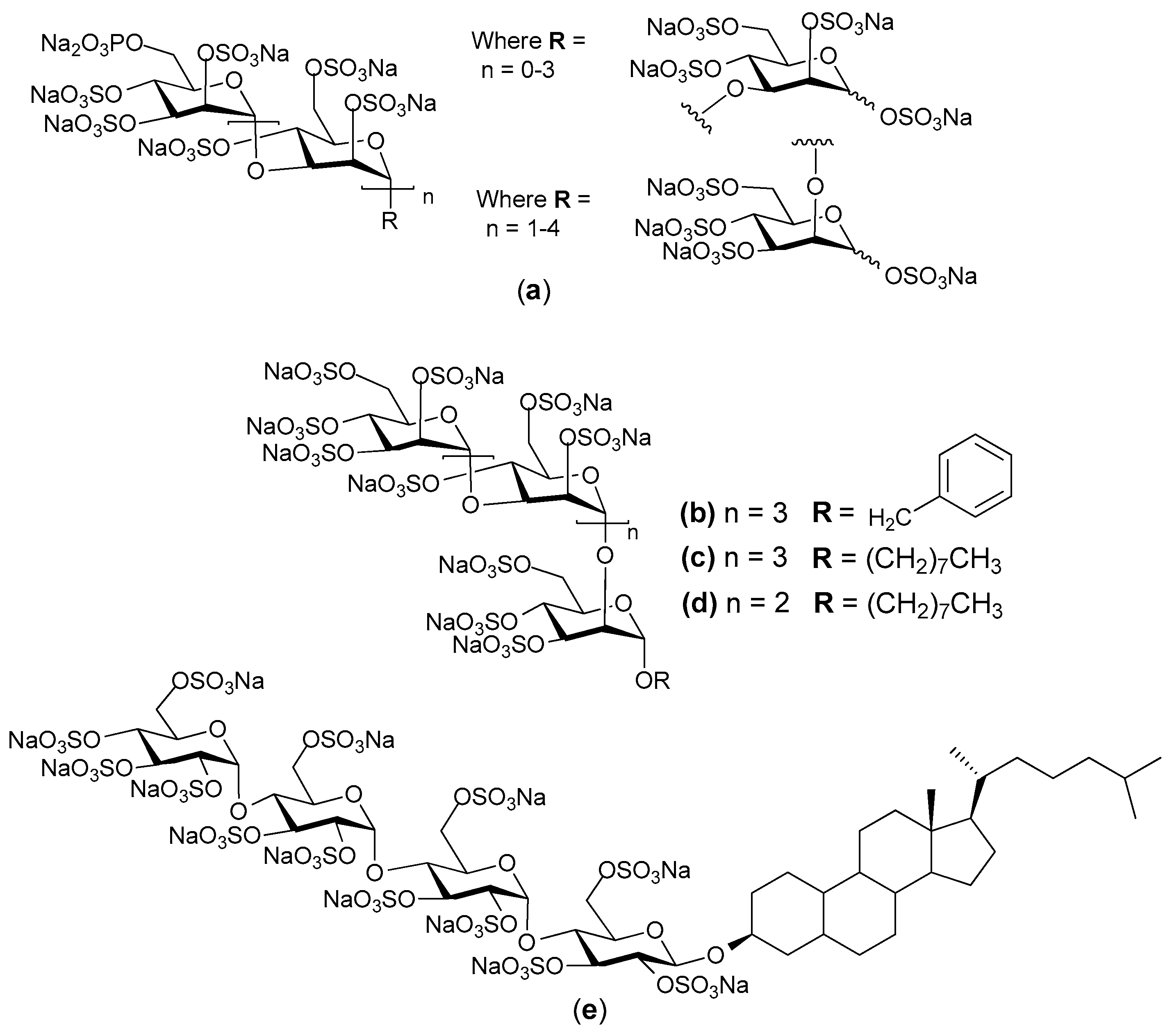
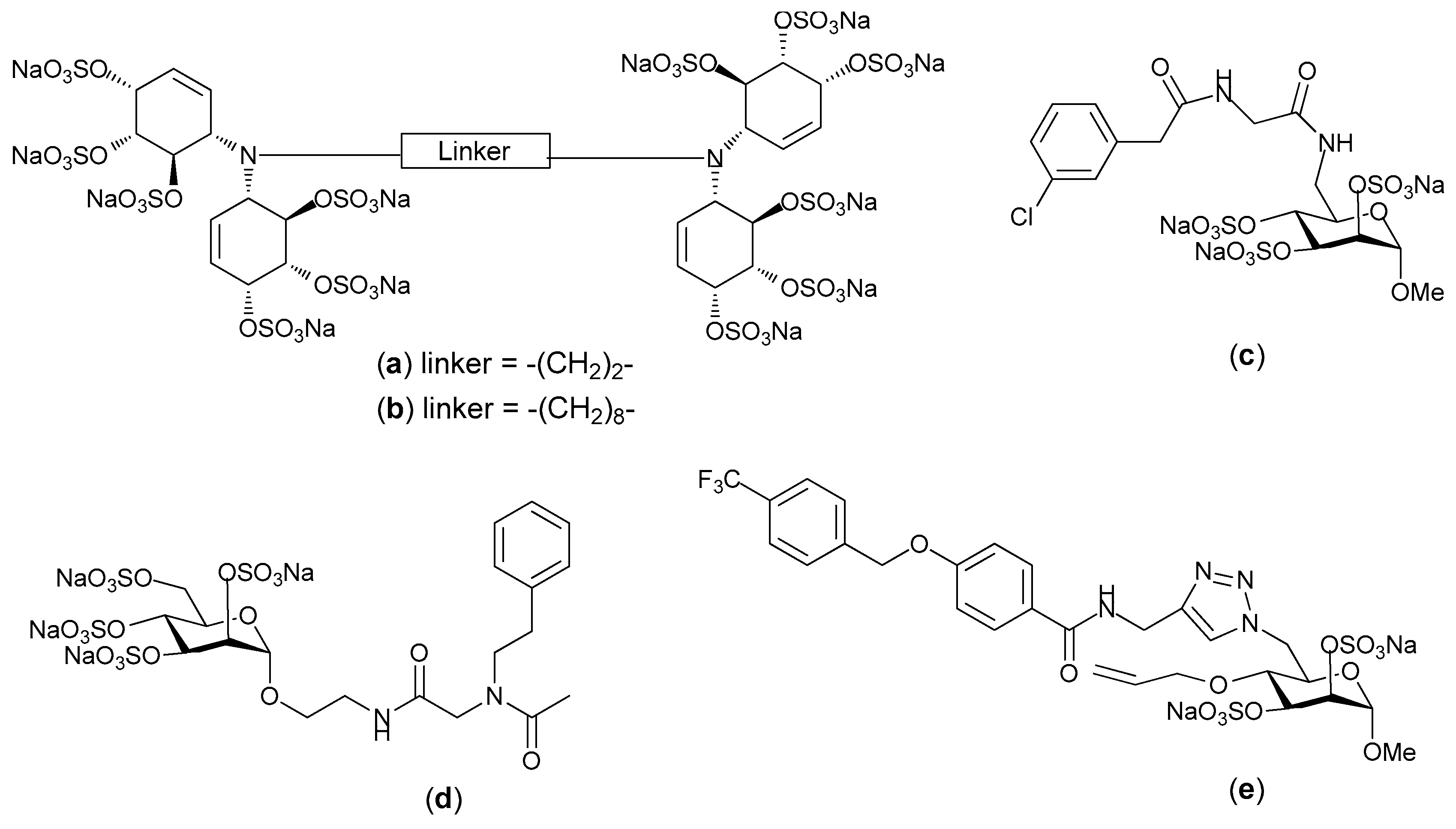
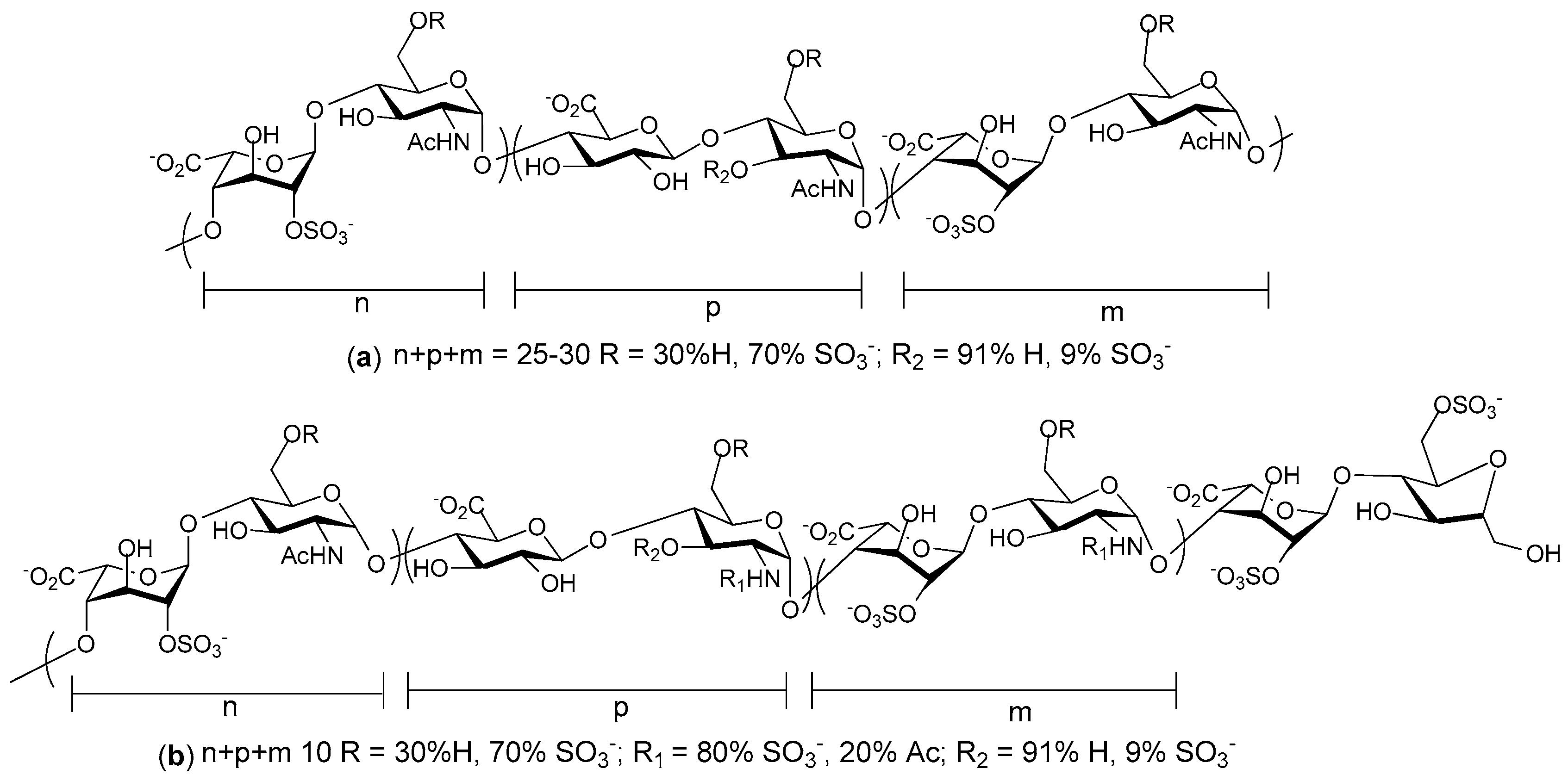
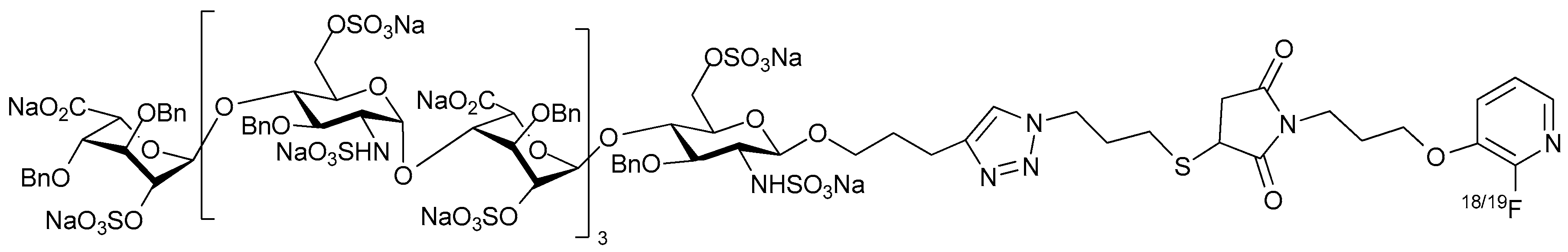
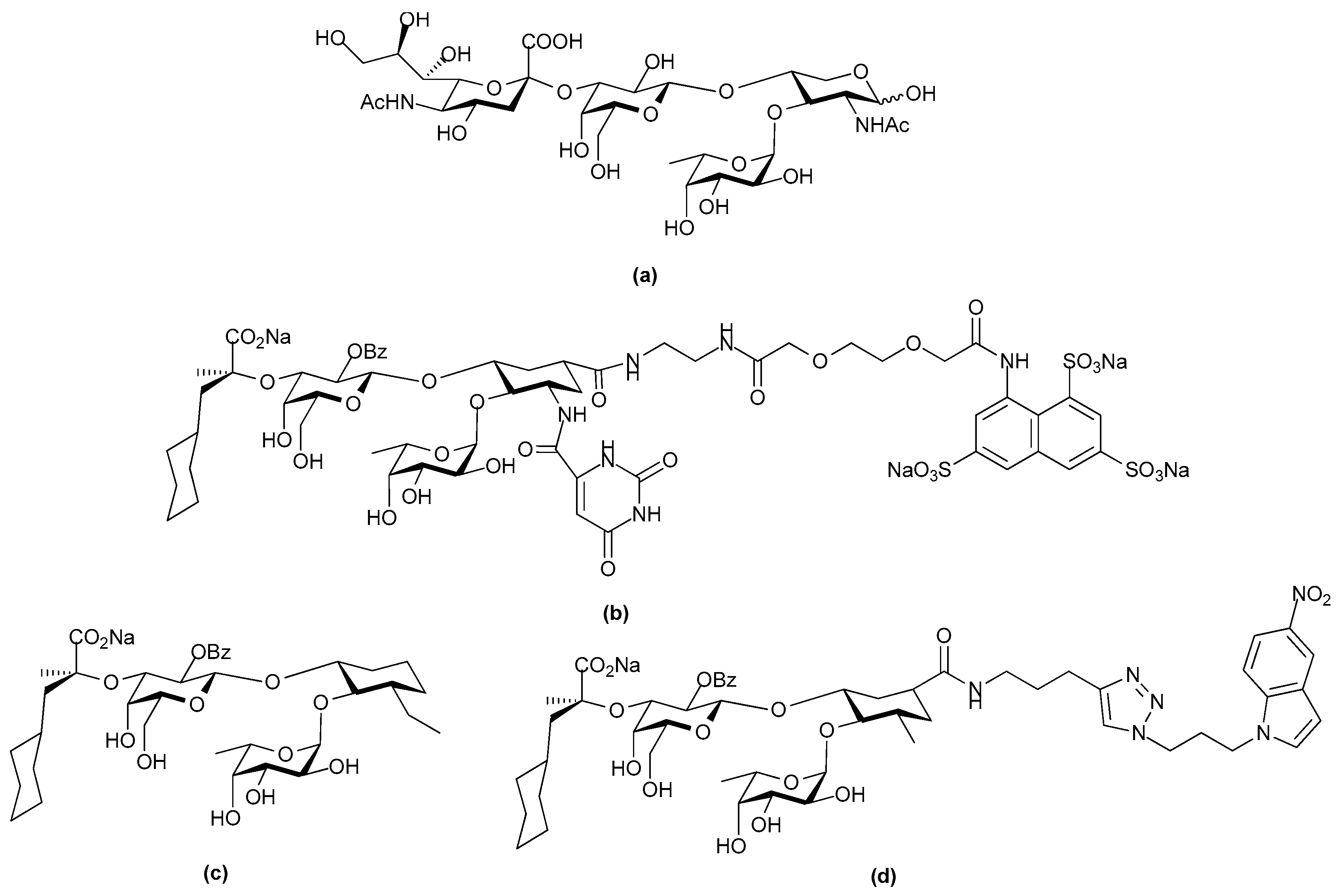
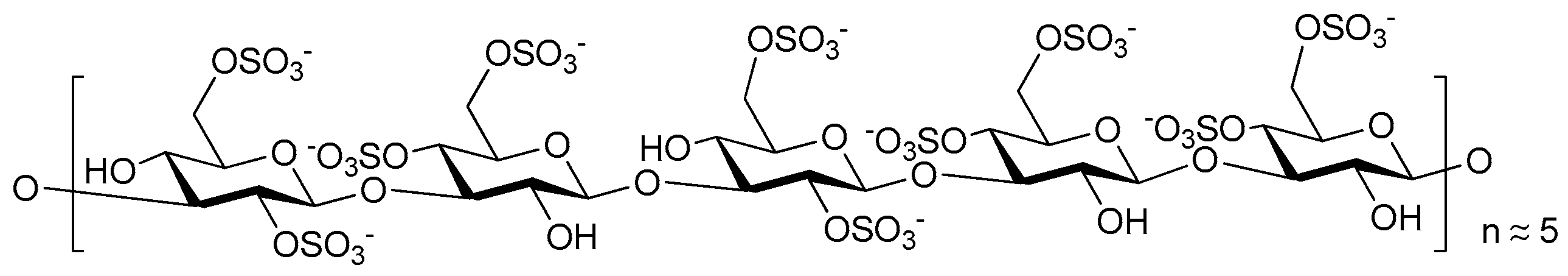
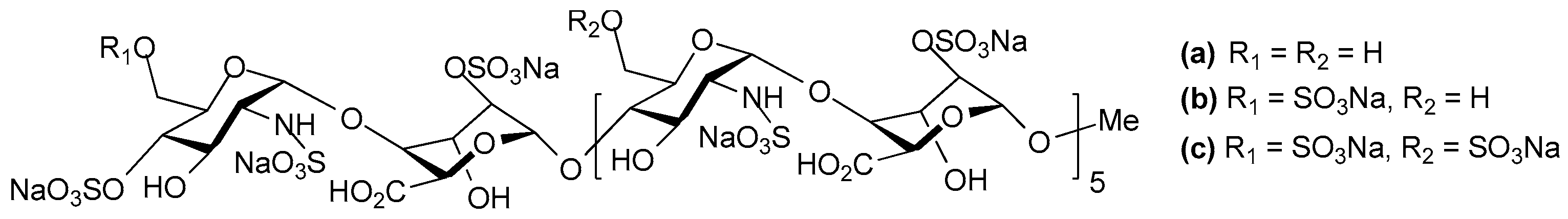
2. Structure and Diversity of Heparin Mimetics
- Size/molecular weight: The first step in any synthetic strategy for a heparin mimetic directed towards a specific target is to determine the size of the structure that is most likely to have the required biological activity. This decision requires knowledge of the shape and size of the heparin binding site on the protein target. Not all heparin binding sites resemble the small pockets on proteins surfaces that are traditionally targeted by drugs; for example, they may be a face on a protein surface. If the latter is true, the size of the heparin-like structure that is required for binding, and for modulating the protein’s function, will be larger than a disaccharide or a tetrasaccharide. It can also be that larger structures are required to provide the correct orientation of the entity that actively engages with the protein. Hence, smaller analogues may not always produce the anticipated increase in selectivity and potency.
- Heterogeneity: Heterogeneous mixtures are more likely to display a broad range of biological activities than structurally similar homogeneous products, but the technical challenges associated with their reproducible synthesis and the characterization of heterogeneous mixtures are far greater. This means proof of reproducible synthesis is required once a heterogeneous mimetic enters clinical development. Accordingly, current trends are towards the synthesis of structurally homogeneous heparin mimetics.
- Pattern and extent of sulfation: It is well known that the extent of sulfation influences the strength with which heparin or HS fragments bind proteins. This was concluded from studies where HS fragments were eluted off protein affinity columns with varying salt concentrations; the highly sulfated structures eluting at higher salt concentrations [22]. These studies also showed that when HS fragments bound some proteins, like Fibroblast growth factor-1 (FGF-1), a higher degree of sulfation did not necessarily translate into the fragments displaying higher affinity binding. Thus, not only the number of sulfates but also the positions of the sulfates were important [22]. We have also shown that the pattern and extent of sulfation has a marked effect on the location on a protein where heparin fragments prefer to bind and that not all fragments that bind affect the protein’s activity in the same way [15]. Given these findings with heparin fragments, it is probable that heparin mimetics will similarly vary in their activities in accordance with the patterns of sulfation. Techniques to control the degree of sulfation include; the choice of starting material, selective sulfation and limiting reaction conditions. Similarly, careful selection of different carbohydrate starting materials can result in different patterns of sulfation in a mimetic.
- Linkage patterns: The influence of anomers, or of linkage patterns on the biological activity of a polysaccharide can also be explored by careful selection of the starting material. Both of these aspects of GAG structure contribute profoundly to their solution structures, and in all probability also to the structures GAG fragments adopt when bound to proteins. The torsion angle values are altered by glycosidic linkages and the anomeric configuration of the linkage, and even small differences in these angles can contribute to differences of the structure in solution. This is illustrated by the more bent solution structure of HS compared to that of heparin [23], although here sulfation and monosaccharide differences also contribute.
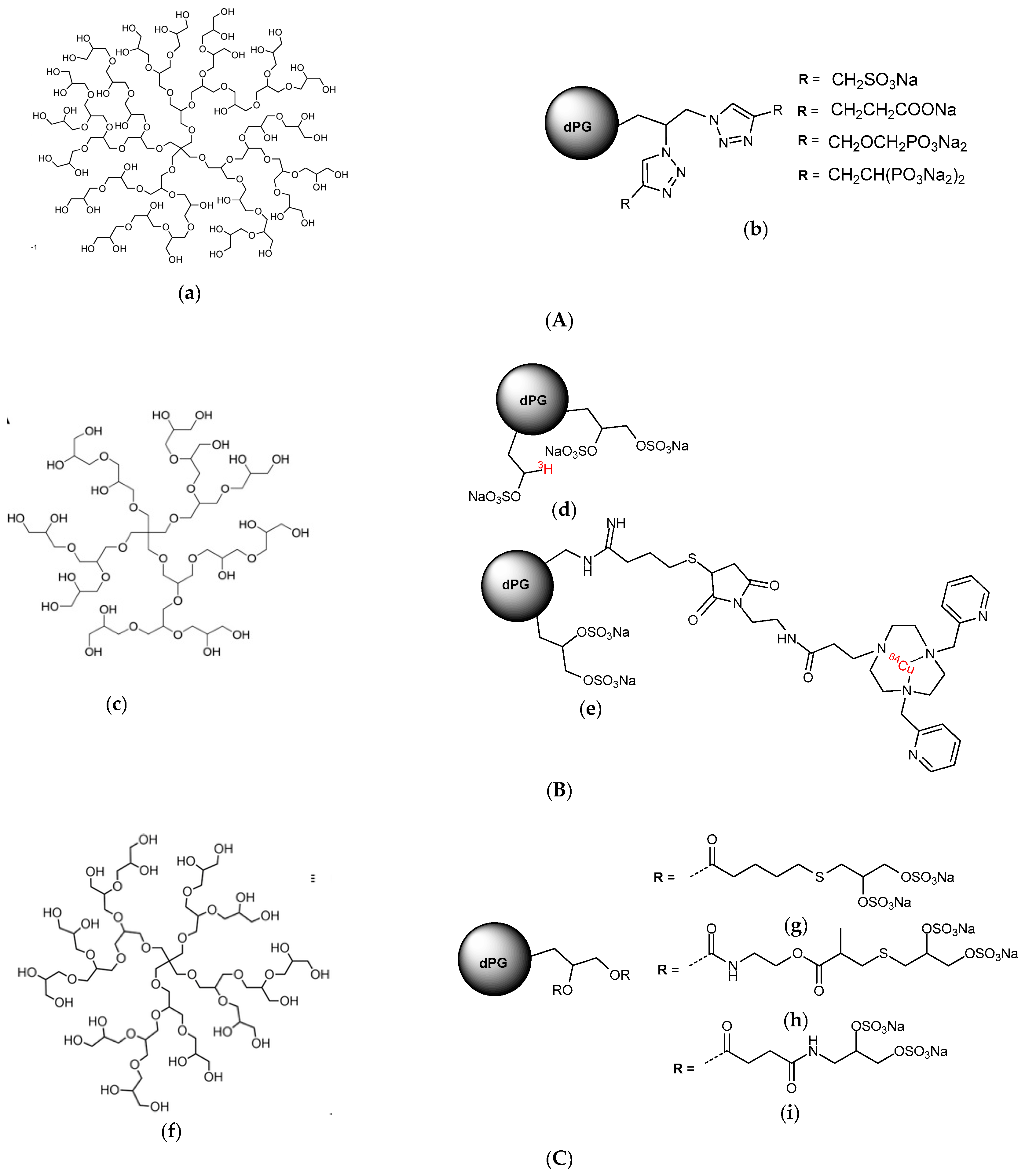
- Flexibility of the backbone: Polysaccharide chains are relatively inflexible due to the limited rotations allowed about a glycosidic linkage. Thus, more flexible heparin mimetics are synthesized by chemical modifications such as glycol splitting. Furthermore, synthetic non-carbohydrate chemical linkers of varying degrees of flexibility can be employed to link short carbohydrate chains, resulting in more flexible heparin mimetics. This approach was used to produce HS-mimics that bound interferon-γ. Two highly sulfated octasaccharide HS fragments linked by a spacer of 10 polyethylene glycol repeats were found to efficiently bind interferon-γ [24]. It was argued that when linked, the sulfated regions acted in a concerted manner and formed a functional unit, whereas when unlinked the octasaccharides did not bind efficiently.
3. Heparin Mimetics as Anticoagulants
4. Heparin Mimetics in Cancer
5. Heparin Mimetics as Anti-Inflammatories
6. Heparin Mimetics: Potential Toxicities
7. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Parish, C.R. The role of heparan sulfate in inflammation. Nat. Rev. Immunol. 2006, 6, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; San Antonio, J.D. Heparan sulfate proteoglycans: Heavy hitters in the angiogenesis arena. J. Clin. Investig. 2001, 108, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Nido, J.; Wandosell, F.; Avila, J. Glycosaminoglycans and β-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 2002, 23, 1323–1332. [Google Scholar] [CrossRef]
- Rosenberg, R.D.; Shworak, N.W.; Liu, J.; Schwartz, J.J.; Zhang, L. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J. Clin. Investig. 1997, 99, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Yip, G.W.; Smollich, M.; Goette, M. Therapeutic value of glycosaminoglycans in cancer. Mol. Cancer Ther. 2006, 5, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Rostand, K.S.; Esko, J.D. Microbial adherence to and invasion through proteoglycans. Infect. Immun. 1997, 65, 1–8. [Google Scholar] [PubMed]
- Kolset, S.O.; Tveit, H. Serglycin—Structure and biology. Cell. Mol. Life Sci. 2008, 65, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J. 2003 Claude S. Hudson award address in carbohydrate chemistry. Heparin: Structure and activity. J. Med. Chem. 2003, 46, 2551–2564. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.; Keeling, D. The story of the discovery of heparin and warfarin. Br. J. Haematol. 2008, 141, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page Clive, P. Pharmacology of heparin and related drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Cohen, M. Enoxaparin: A pharmacologic and clinical review. Expert Opin. Pharmacother. 2011, 12, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L. Heparin and heparan sulfate: Structure and function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef] [PubMed]
- El Masri, R.; Seffouh, A.; Lortat-Jacob, H.; Vives, R.R. The “in and out” of glucosamine 6-O-sulfation: The 6th sense of heparan sulfate. Glycoconj. J. 2017, 34, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Kett, W.C.; Coombe, D.R. A structural analysis of heparin-like glycosaminoglycans using MALDI-TOF mass spectrometry. Spectroscopy 2004, 18, 185–201. [Google Scholar] [CrossRef]
- Singh, A.; Kett, W.C.; Severin, I.C.; Agyekum, I.; Duan, J.; Amster, I.J.; Proudfoot, A.E.I.; Coombe, D.R.; Woods, R.J. The interaction of heparin tetrasaccharides with chemokine CCL5 is modulated by sulfation pattern and pH. J. Biol. Chem. 2015, 290, 15421–15436. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, H. Advances in selective chemical syntheses of complex oligosaccharides. Angew. Chem. Int. Ed. Engl. 1982, 21, 155–173. [Google Scholar] [CrossRef]
- Dulaney, S.B.; Huang, X. Strategies in synthesis of heparin/heparan sulfate oligosaccharides: 2000–present. Adv. Carbohydr. Chem. Biochem. 2012, 67, 95–136. [Google Scholar] [PubMed]
- Hu, Y.-P.; Zhong, Y.-Q.; Chen, Z.-G.; Chen, C.-Y.; Shi, Z.; Zulueta, M.M.L.; Ku, C.-C.; Lee, P.-Y.; Wang, C.-C.; Hung, S.-C. Divergent synthesis of 48 heparan sulfate-based disaccharides and probing the specific sugar-fibroblast growth factor-1 interaction. J. Am. Chem. Soc. 2012, 134, 20722–20727. [Google Scholar] [CrossRef] [PubMed]
- Arungundram, S.; Al-Mafraji, K.; Asong, J.; Leach, F.E., III; Amster, I.J.; Venot, A.; Turnbull, J.E.; Boons, G.-J. Modular synthesis of heparan sulfate oligosaccharides for structure activity relationship studies. J. Am. Chem. Soc. 2009, 131, 17394–17405. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.; Ferro, V. Synthetic approaches to L-iduronic acid and L-idose: Key building blocks for the preparation of glycosaminoglycan oligosaccharides. Adv. Carbohydr. Chem. Biochem. 2015, 72, 21–61. [Google Scholar] [PubMed]
- Coombe, D.; Kett, W.C. Heparin mimetics. Handb. Exp. Pharmacol. 2012, 207, 361–383. [Google Scholar]
- Kreuger, J.; Salmivirta, M.; Sturiale, L.; Gimenez-Gallego, G.; Lindahl, U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J. Biol. Chem. 2001, 276, 30744–30752. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Fung Ka, W.; Rodriguez, E.; Patel, R.; Gor, J.; Mulloy, B.; Perkins Stephen, J. The solution structure of heparan sulfate differs from that of heparin: Implications for function. J. Biol. Chem. 2013, 288, 27737–27751. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Bonnaffe, D.; Lubineau, A.; Lortat-Jacob, H. Heparan sulfate mimicry: A synthetic glycoconjugate that recognizes the heparin binding domain of interferon-γ inhibits the cytokine activity. J. Biol. Chem. 2005, 280, 37558–37564. [Google Scholar] [CrossRef] [PubMed]
- Blossom, D.B.; Kallen, A.J.; Patel, P.R.; Elward, A.; Robinson, L.; Gao, G.; Langer, R.; Perkins, K.M.; Jaeger, J.L.; Kurkjian, K.M.; et al. Outbreak of adverse reactions associated with contaminated heparin. N. Engl. J. Med. 2008, 359, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Chess, E.K.; Bairstow, S.; Donovan, S.; Havel, K.; Hu, P.; Johnson, R.J.; Lee, S.; McKee, J.; Miller, R.; Moore, E.; et al. Case study: Contamination of heparin with oversulfated chondroitin sulfate. Handb. Exp. Pharmacol. 2012, 207, 99–125. [Google Scholar]
- Hirsh, J.; Warkentin, T.E.; Raschke, R.; Granger, C.; Ohman, E.M.; Dalen, J.E. Heparin and low molecular weight heparin: Mechanisms of action, pharmacokinetics, dosing considerations, monitoring, efficacy, and safety. Chest 1998, 114, 489S–510S. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Levine, M.N.; Hirsh, J.; Horsewood, P.; Roberts, R.S.; Gent, M.; Kelton, J.G. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N. Engl. J. Med. 1995, 332, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
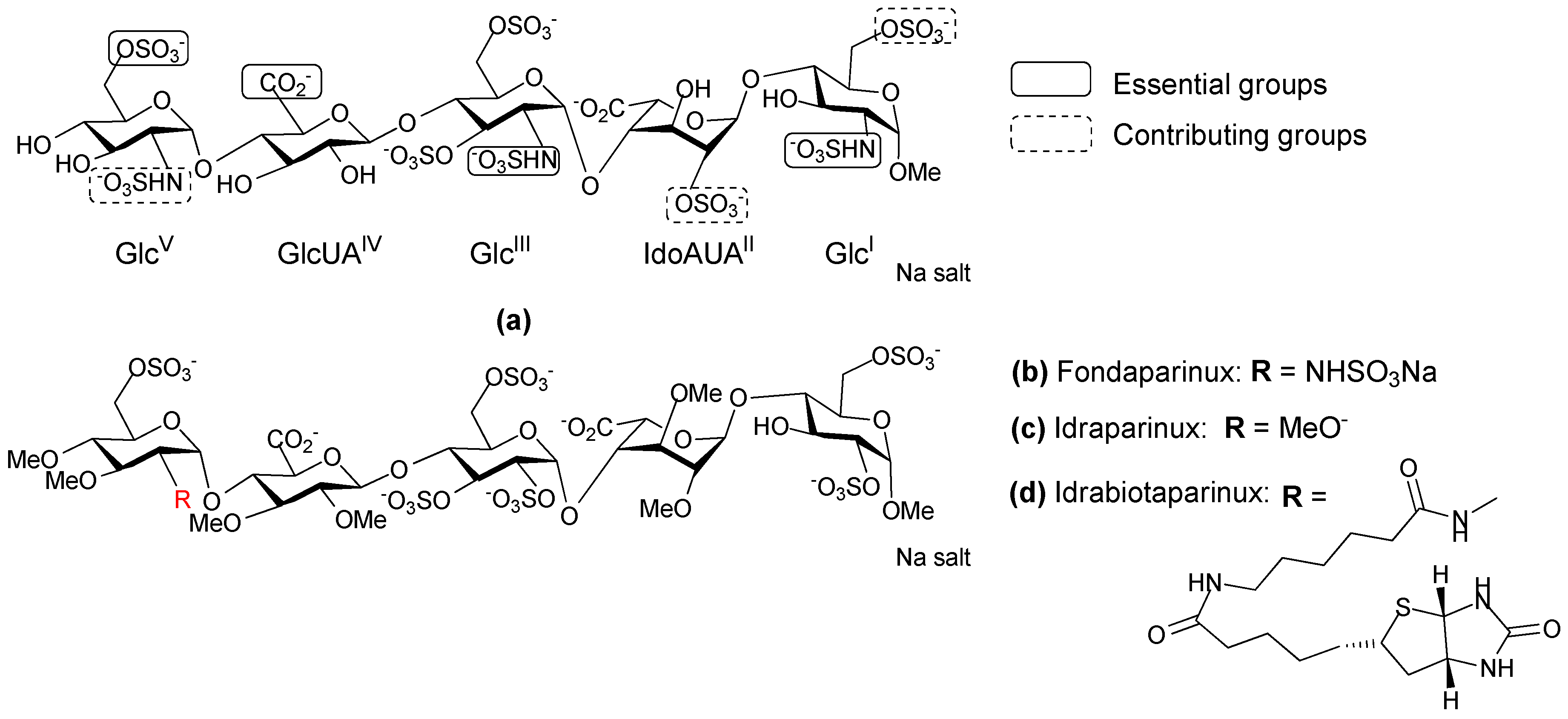
- Petitou, M.; van Boeckel Constant, A.A. A synthetic antithrombin III binding pentasaccharide is now a Drug! What comes next? Angew. Chem. Int. Ed. Engl. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Klement, P.; Rak, J. Emerging anticoagulants: Mechanism of action and future potential. Vnitr. Lek. 2006, 52, 119–122. [Google Scholar] [PubMed]
- Bauer, K.A. Selective inhibition of coagulation factors: Advances in antithrombotic therapy. Semin. Thromb. Hemostasis 2002, 28, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.M.; Petitou, M.; Lormeau, J.C.; Cariou, R.; Necciari, J.; Magnani, H.N.; Zandberg, P.; van Amsterdam, R.G.M.; van Boeckel, C.A.A.; Meuleman, D.G. SR 90107 A/Org 31540, A novel anti-factor Xa antithrombotic agent. Cardiovasc. Drug Rev. 1997, 15, 1–26. [Google Scholar] [CrossRef]
- Van Boeckel Constant, A.A.; Petitou, M. The unique antithrombin III binding domain of heparin: A lead to new synthetic antithrombotics. Angew. Chem. Int. Ed. Engl. 1993, 32, 1671–1818. [Google Scholar] [CrossRef]
- Herbert, J.M.; Herault, J.P.; Bernat, A.; Van Amsterdam, R.G.M.; Lormeau, J.C.; Petitou, M.; Van Boeckel, C.; Hoffmann, P.; Meuleman, D.G. Biochemical and pharmacological properties of SANORG 34006, a potent and long-acting synthetic pentasaccharide. Blood 1998, 91, 4197–4205. [Google Scholar] [PubMed]
- Westerduin, P.; van Boeckel, C.A.A.; Basten, J.E.M.; Broekhoven, M.A.; Lucas, H.; Rood, A.; van der Heijden, H.; van Amsterdam, R.G.M.; van Dinther, T.G.; et al. Feasible synthesis and biological properties of Six ‘non-glycosamino’ glycan analogs of the antithrombin III binding heparin pentasaccharide. Bioorg. Med. Chem. 1994, 2, 1267–1280. [Google Scholar] [CrossRef]
- Prandoni, P.; Tormene, D.; Perlati, M.; Brandolin, B.; Spiezia, L. Idraparinux: Review of its clinical efficacy and safety for prevention and treatment of thromboembolic disorders. Expert Opin. Investig. Drugs 2008, 17, 773–777. [Google Scholar] [CrossRef] [PubMed]
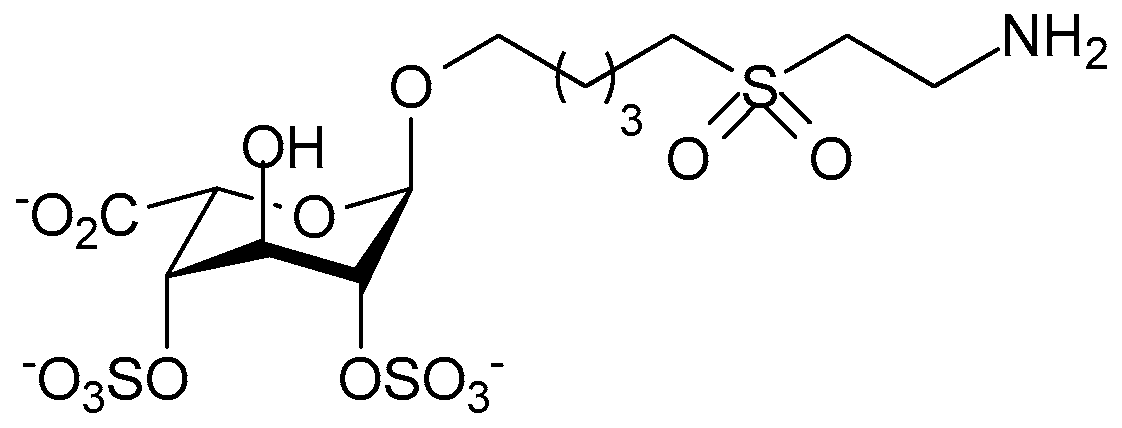
- Paty, I.; Trellu, M.; Destors, J.M.; Cortez, P.; Boelle, E.; Sanderink, G. Reversibility of the anti-FXa activity of idrabiotaparinux (biotinylated idraparinux) by intravenous avidin infusion. J. Thromb. Haemost. 2010, 8, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Savi, P.; Herault, J.P.; Duchaussoy, P.; Millet, L.; Schaeffer, P.; Petitou, M.; Bono, F.; Herbert, J.M. Reversible biotinylated oligosaccharides: A new approach for a better management of anticoagulant therapy. J. Thromb. Haemost. 2008, 6, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, X.; Pavithra, S.; Li, D. Idraparinux or idrabiotaparinux for long-term venous thromboembolism treatment: A systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2013, 8, e78972. [Google Scholar] [CrossRef] [PubMed]
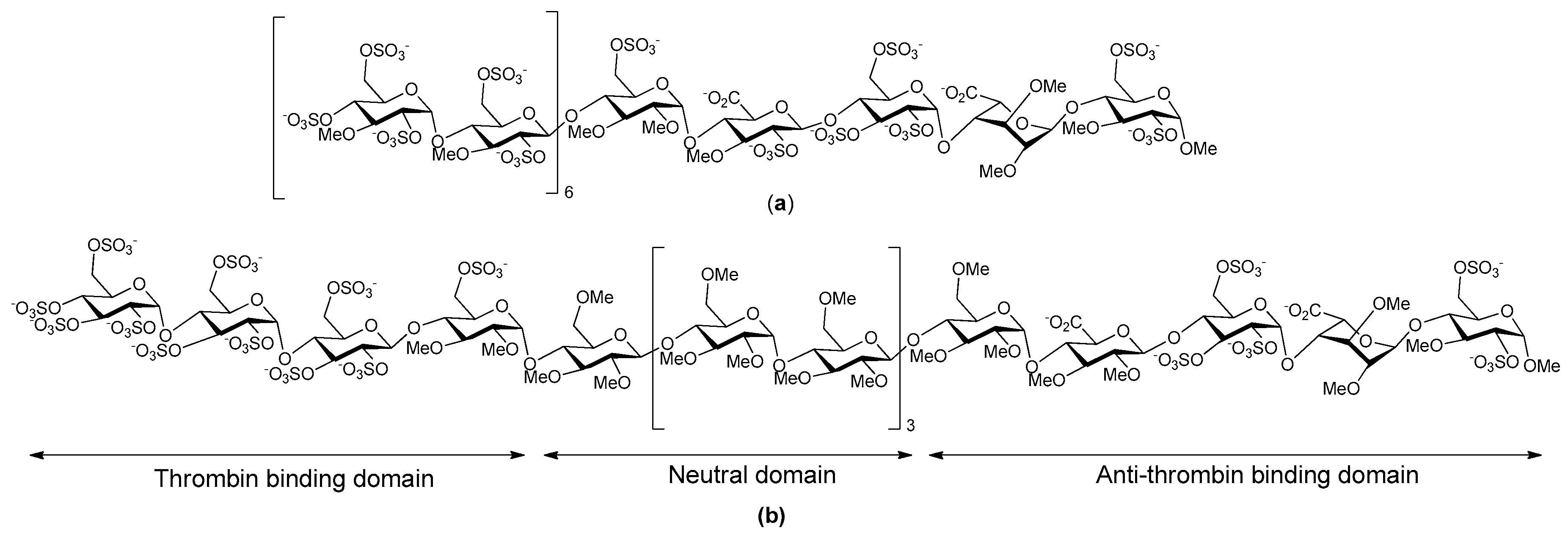
- Petitou, M.; Herault, J.-P.; Bernat, A.; Driguez, P.-A.; Duchaussoy, P.; Lormeau, J.-C.; Herbert, J.-M. Synthesis of thrombin-inhibiting heparin mimetics without side effects. Nature 1999, 398, 417–422. [Google Scholar] [PubMed]
- Casu, B. Structure and biological activity of heparin. Adv. Carbohydr. Chem. Biochem. 1985, 43, 51–134. [Google Scholar] [PubMed]
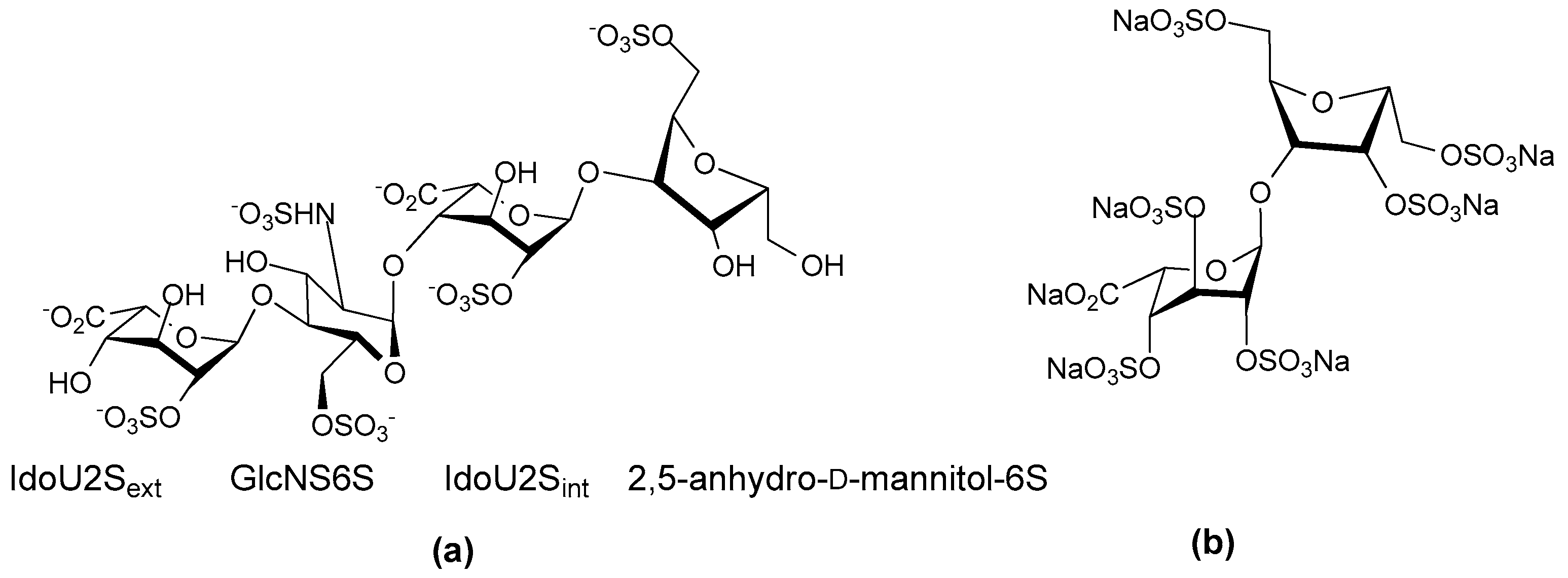
- Herbert, J.M.; Herault, J.P.; Bernat, A.; Savi, P.; Schaeffer, P.; Driguez, P.A.; Duchaussoy, P.; Petitou, M. SR123781A, A synthetic heparin mimetic. Thromb. Haemost. 2001, 85, 852–860. [Google Scholar] [PubMed]
- Bal dit Sollier, C.; Kang, C.; Berge, N.; Herault, J.P.; Bonneau, M.; Herbert, J.M.; Drouet, L. Activity of a synthetic hexadecasaccharide (SanOrg123781A) in a pig model of arterial thrombosis. J. Thromb. Haemost. 2004, 2, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Hoppensteadt, D.; Cunanan, J.; Geniaux, E.; Lorenz, M.; Viskov, C.; Ythier-Moury, P.; Brin, J.-F.; Jeske, W. AVE5026: A novel, extractive heparinoid with enriched anti-Xa activity and enhanced antithrombotic activity. Blood 2007, 110, 1881. [Google Scholar]
- Viskov, C.; Just, M.; Laux, V.; Mourier, P.; Lorenz, M. Description of the chemical and pharmacological characteristics of a new hemisynthetic ultra-low-molecular-weight heparin, AVE5026. J. Thromb. Haemost. 2009, 7, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Lassen, M.R.; Dahl, O.E.; Mismetti, P.; Destree, D.; Turpie, A.G.G. AVE5026, A new hemisynthetic ultra-low-molecular-weight heparin for the prevention of venous thromboembolism in patients after total knee replacement surgery—TREK: A dose-ranging study. J. Thromb. Haemost. 2009, 7, 566–572. [Google Scholar] [CrossRef] [PubMed]
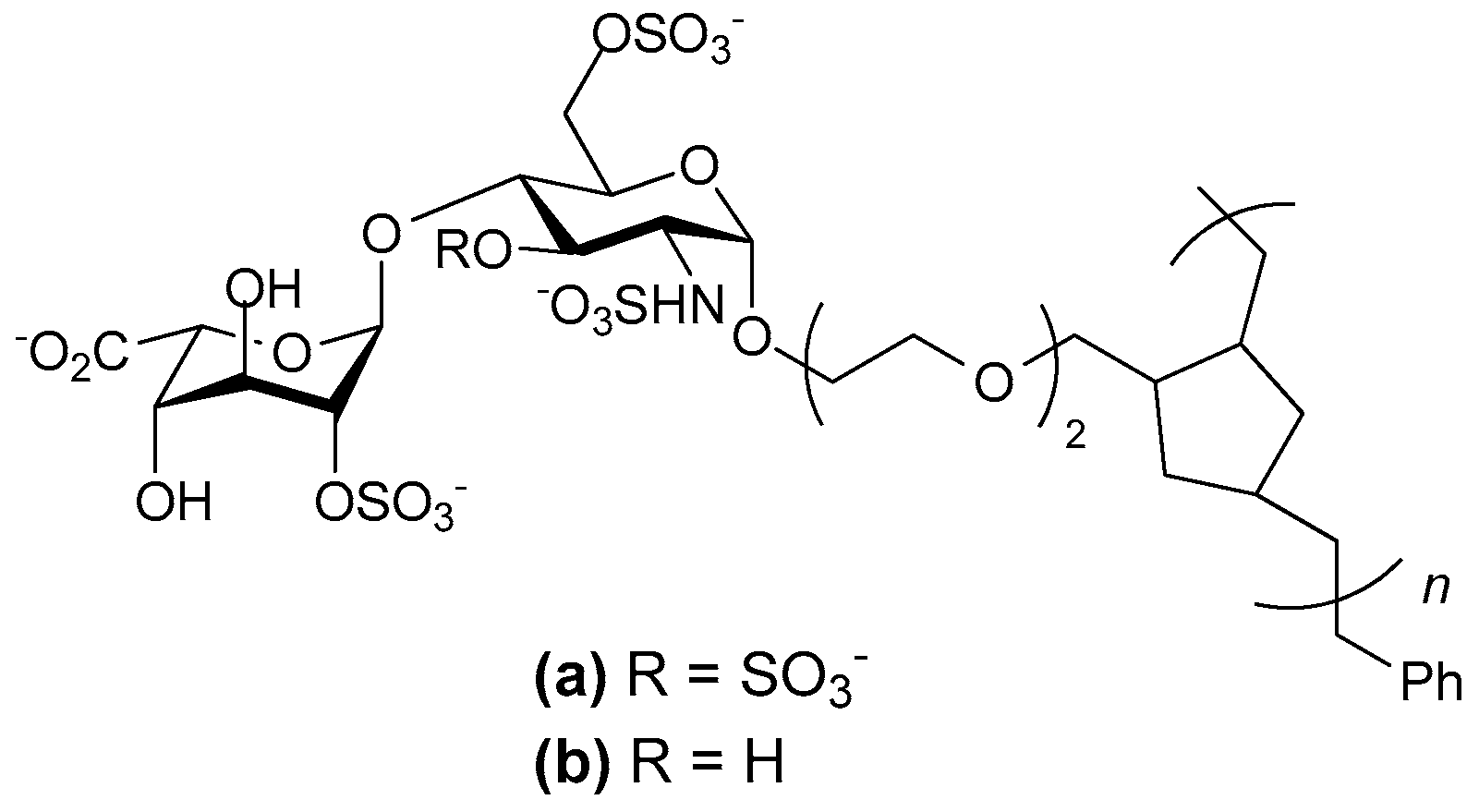
- Oh Young, I.; Sheng Gloria, J.; Chang, S.-K.; Hsieh-Wilson Linda, C. Tailored glycopolymers as anticoagulant heparin mimetics. Angew. Chem. Int. Ed. Engl. 2013, 52, 11796–11799. [Google Scholar]
- Brandt, S.; Krauel, K.; Jaax, M.; Renne, T.; Helm Christiane, A.; Hammerschmidt, S.; Delcea, M.; Greinacher, A. Polyphosphates form antigenic complexes with platelet factor 4 (PF4) and enhance PF4-binding to bacteria. Thromb. Haemost. 2015, 114, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Jaax, M.E.; Krauel, K.; Marschall, T.; Brandt, S.; Gansler, J.; Fuerll, B.; Appel, B.; Fischer, S.; Block, S.; Helm, C.A.; et al. Complex formation with nucleic acids and aptamers alters the antigenic properties of platelet factor 4. Blood 2013, 122, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Ornstein, D.L.; Mamourian, A.C. Low-molecular-weight heparin and cancer. Semin. Thromb. Hemost. 2000, 26, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Vlodavsky, I.; Sanderson, R.D. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol. Haemost. Thromb. 2008, 36, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, S.W.; Kim, S.K.; Lee, M.; Chang, H.W.; Moon, H.T.; Byun, Y.; Kim, S.Y. Antiangiogenic activity of orally absorbable heparin derivative in different types of cancer cells. Pharm. Res. 2009, 26, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Linhardt, R.; Francis, J.L.; Amirkhosravi, A. Anti-metastatic effect of a non-anticoagulant low-molecular-weight heparin versus the standard low-molecular-weight heparin, Enoxaparin. Thromb. Haemost. 2006, 96, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; Levine, M.N.; Kadziola, Z.; Lemoine, N.R.; Low, V.; Patel, H.K.; Rustin, G.; Thomas, M.; Quigley, M.; Williamson, R.C.N. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: The Fragmin Advanced Malignancy Outcome Study (FAMOUS). J. Clin. Oncol. 2004, 22, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Kuderer, N.M.; Ortel, T.L.; Francis, C.W. Impact of venous thromboembolism and anticoagulation on cancer and cancer survival. J. Clin. Oncol. 2009, 27, 4902–4911. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Ilan, N.; Naggi, A.; Casu, B. Heparanase: Structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr. Pharm. Des. 2007, 13, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.-Q.; Liu, H.; Navarro, E.; Kussie, P.; Zhu, Z. Development of heparanase inhibitors for anti-cancer therapy. Curr. Med. Chem. 2006, 13, 2101–2111. [Google Scholar] [PubMed]
- Kilarski, W.W.; Bikfalvi, A. Recent developments in tumor angiogenesis. Curr. Pharm. Biotechnol. 2007, 8, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Presta, M. Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr. Pharm. Des. 2007, 13, 2025–2044. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Fehrmann, F.; Bieker, R.; Berdel, W.E.; Mesters, R.M. Vascular endothelial growth factor and its receptor as drug targets in hematological malignancies. Curr. Drug Targets 2007, 8, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Ferro, V.; Fewings, K.; Palermo, M.C.; Li, C. Large-scale preparation of the oligosaccharide phosphate fraction of Pichia holstii NRRL Y-2448 phosphomannan for use in the manufacture of PI-88. Carbohydr. Res. 2001, 332, 183–189. [Google Scholar] [CrossRef]
- Ferro, V.; Li, C.; Fewings, K.; Palermo, M.C.; Linhardt, R.J.; Toida, T. Determination of the composition of the oligosaccharide phosphate fraction of Pichia (Hansenula) holstii NRRL Y-2448 phosphomannan by capillary electrophoresis and HPLC. Carbohydr. Res. 2002, 337, 139–146. [Google Scholar] [CrossRef]
- Khachigian, L.M.; Parish, C.R. Phosphomannopentaose sulfate (PI-88): Heparan sulfate mimetic with clinical potential in multiple vascular pathologies. Cardiovasc. Drug Rev. 2004, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Handley, P.N.; Carroll, A.; Ferro, V. New structural insights into the oligosaccharide phosphate fraction of Pichia (Hansenula) holstii NRRL Y2448 phosphomannan. Carbohydr. Res. 2017, 446–447, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Kudchadkar, R.; Gonzalez, R.; Lewis, K.D. PI-88: A novel inhibitor of angiogenesis. Expert Opin. Investig. Drugs 2008, 17, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.-Y.; Wang, Z.; Hu, J.; Liu, W.-F.; Shen, Z.-Z.; Zhang, X.; Yu, L.; Fan, J.; Zhou, J. PI-88 inhibits postoperative recurrence of hepatocellular carcinoma via disrupting the surge of heparanase after liver resection. Tumor Biol. 2016, 37, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-J.; Lee, P.-H.; Lin, D.-Y.; Wu, C.-C.; Jeng, L.-B.; Lin, P.-W.; Mok, K.-T.; Lee, W.-C.; Yeh, H.-Z.; Ho, M.-C.; et al. Heparanase inhibitor PI-88 as adjuvant therapy for hepatocellular carcinoma after curative resection: A randomized phase II trial for safety and optimal dosage. J. Hepatol. 2009, 50, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.D.; Robinson, W.A.; Millward, M.J.; Powell, A.; Price, T.J.; Thomson, D.B.; Walpole, E.T.; Haydon, A.M.; Creese, B.R.; Roberts, K.L.; et al. A phase II study of the heparanase inhibitor PI-88 in patients with advanced melanoma. Investig. New Drugs 2008, 26, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Karoli, T.; Liu, L.; Fairweather, J.K.; Hammond, E.; Li, C.P.; Cochran, S.; Bergefall, K.; Trybala, E.; Addison, R.S.; Ferro, V. Synthesis, biological activity, and preliminary pharmacokinetic evaluation of analogs of a phosphosulfomannan angiogenesis inhibitor (PI-88). J. Med. Chem. 2005, 48, 8229–8236. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, K.D.; Karoli, T.; Liu, L.; Dredge, K.; Copeman, E.; Li, C.P.; Davis, K.; Hammond, E.; Bytheway, I.; Kostewicz, E.; et al. Synthesis and biological evaluation of polysulfated oligosaccharide glycosides as inhibitors of angiogenesis and tumor growth. J. Med. Chem. 2010, 53, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Ferro, V.; Dredge, K.; Liu, L.; Hammond, E.; Bytheway, I.; Li, C.; Johnstone, K.; Karoli, T.; Davis, K.; Copeman, E.; et al. PI-88 and novel heparan sulfate mimetics inhibit angiogenesis. Semin. Thromb. Hemost. 2007, 33, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Hammond, E.; Davis, K.; Li, C.P.; Liu, L.; Johnstone, K.; Handley, P.; Wimmer, N.; Gonda, T.J.; Gautam, A.; et al. The PG500 series: Novel heparan sulfate mimetics as potent angiogenesis and heparanase inhibitors for cancer therapy. Investig. New Drugs 2010, 28, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Ferro, V.; Liu, L.; Johnstone, K.D.; Wimmer, N.; Karoli, T.; Handley, P.; Rowley, J.; Dredge, K.; Li, C.P.; Hammond, E.; et al. Discovery of PG545: A highly potent and simultaneous inhibitor of angiogenesis, tumor growth, and metastasis. J. Med. Chem. 2012, 55, 3804–3813. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Hammond, E.; Handley, P.; Gonda, T.J.; Smith, M.T.; Vincent, C.; Brandt, R.; Ferro, V.; Bytheway, I. PG545, A dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br. J. Cancer 2011, 104, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.; Handley, P.; Dredge, K.; Bytheway, I. Mechanisms of heparanase inhibition by the heparan sulfate mimetic PG545 and three structural analogues. FEBS Open Bio 2013, 3, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Brennan Todd, V.; Lin, L.; Brandstadter Joshua, D.; Rendell Victoria, R.; Dredge, K.; Huang, X.; Yang, Y. Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J. Clin. Investig. 2016, 126, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.-B.; Yun, M.; Kim, E.-O.; Kim, J.; Kim, B.; Jung Ji, H.; Kim, S.-H.; Kim, E.-O.; Wang, E.; Mukhopadhyay, D.; et al. The heparan sulfate mimetic PG545 interferes with Wnt/β-catenin signaling and significantly suppresses pancreatic tumorigenesis alone and in combination with gemcitabine. Oncotarget 2015, 6, 4992–5004. [Google Scholar] [CrossRef] [PubMed]
- Rudd, T.R.; Uniewicz, K.A.; Ori, A.; Guimond, S.E.; Skidmore, M.A.; Gaudesi, D.; Xu, R.; Turnbull, J.E.; Guerrini, M.; Torri, G.; et al. Comparable stabilization, structural changes and activities can be induced in FGF by a variety of HS and non-GAG analogues: Implications for sequence-activity relationships. Org. Biomol. Chem. 2010, 8, 5390–5397. [Google Scholar] [CrossRef] [PubMed]
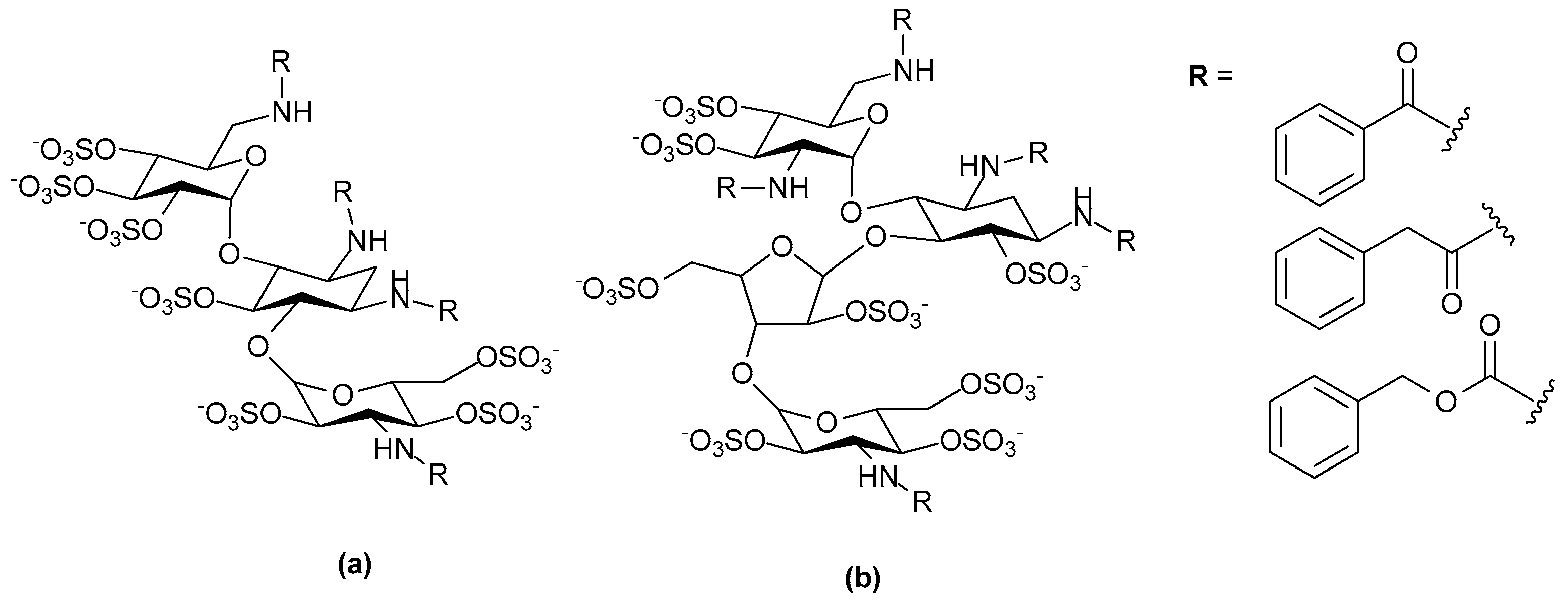
- Freeman, C.; Liu, L.; Banwell, M.G.; Brown, K.J.; Bezos, A.; Ferro, V.; Parish, C.R. Use of sulfated linked cyclitols as heparan sulfate mimetics to probe the heparin/heparan sulfate binding specificity of proteins. J. Biol. Chem. 2005, 280, 8842–8849. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, C.; Cochran, S.; Feder, D.; Guddat, L.W.; Ferro, V. A focused sulfated glycoconjugate ugi library for probing heparan sulfate-binding angiogenic growth factors. Bioorg. Med. Chem. Lett. 2012, 22, 6190–6194. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fernandez, C.; Kerns, R.J. Different protein-binding selectivities for N-acyl heparin derivatives having N-phenylacetyl and heterocycle analogs of N-phenylacetyl substituted in place of N-sulfo groups. Bioorg. Med. Chem. 2007, 17, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, C.; Cochran, S.; Jimmink, S.; Ferro, V. Synthesis of a heparan sulfate mimetic library targeting FGF and VEGF via click chemistry on a monosaccharide template. ChemMedChem 2012, 7, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Nancy-Portebois, V.; Cabannes, E.; Petitou, M.; Serin, G.; Mirjolet, J.-F. Antitumor Activity of EP80061, a Small-Glyco Drug in Preclinical Studies. In Proceedings of the 101st Annual Meeting of the American Association for Cancer Research, Washington, DC, USA, 17–21 April 2010; Cancer Research: Washington, DC, USA, 2010; p. 5459. [Google Scholar]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST0001, A chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Favini, E.; Dal Bo, L.; Tortoreto, M.; Zunino, F.; Zaffaroni, N.; Lanzi, C.; De Maglie, M.; De Maglie, M.; Dagrada, G.; et al. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget 2016, 7, 47848–47863. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Roy, S.; Cochran, E.; Zouaoui, R.; Chu, C.L.; Duffner, J.; Zhao, G.; Smith, S.; Galcheva-Gargova, Z.; Karlgren, J.; et al. M402, A novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS ONE 2011, 6, e21106. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Elli, S.; Naggi, A.; Guerrini, M.; Torri, G.; Petitou, M. Investigating glycol-split-heparin-derived inhibitors of heparanase: A study of synthetic trisaccharides. Molecules 2016, 21, 1602. [Google Scholar] [CrossRef] [PubMed]
- Cabannes, E.; Caravano, A.; Lewandowski, D.; Motte, V.; Nancy-Portebois, V.; Petitou, M.; Pierdet, P. Oligosaccharide compounds for use in mobilising stem cells. Patent No. WO2010029185 A1, 18 March 2010. [Google Scholar]
- Kuhnast, B.; El Hadri, A.; Boisgard, R.; Hinnen, F.; Richard, S.; Caravano, A.; Nancy-Portebois, V.; Petitou, M.; Tavitian, B.; Dolle, F. Synthesis, radiolabeling with fluorine-18 and preliminary in vivo evaluation of a heparan sulfate mimetic as potent angiogenesis and heparanase inhibitor for cancer applications. Org. Biomol. Chem. 2016, 14, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Capila, I.; Linhardt Robert, J. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Yan, Y.; Ji, Y.; Su, N.; Mei, X.; Wang, Y.; Du, S.; Zhu, W.; Zhang, C.; Lu, Y.; Xing, X.-H. Non-anticoagulant effects of low molecular weight heparins in inflammatory disorders: A review. Carbohydr. Polym. 2017, 160, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Chande, N.; MacDonald John, K.; Wang Josh, J.; McDonald John, W.D. Unfractionated or low molecular weight heparin for induction of remission in ulcerative colitis: A Cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm. Bowel. Dis. 2011, 17, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Reimann, S.; Groeger, D.; Kuehne, C.; Riese, S.B.; Dernedde, J.; Haag, R. Shell cleavable dendritic polyglycerol sulfates show high anti-inflammatory properties by inhibiting L-selectin binding and complement activation. Adv. Healthcare Mater. 2015, 4, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Severin India, C.; Soares, A.; Hantson, J.; Teixeira, M.; Sachs, D.; Valognes, D.; Scheer, A.; Schwarz Matthias, K.; Wells Timothy, N.C.; Proudfoot Amanda, E.I.; et al. Glycosaminoglycan analogs as a novel anti-inflammatory strategy. Front. Immunol. 2012, 3, 293. [Google Scholar] [CrossRef] [PubMed]
- Zaferani, A.; Talsma, D.; Richter, M.K.S.; Daha, M.R.; Navis, G.J.; Seelen, M.A.; van den Born, J. Heparin/heparan sulphate interactions with complement-A possible target for reduction of renal function loss? Nephrol. Dial. Transplant. 2014, 29, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, A.; Gaffney, P. Rheumatoid arthritis and heparin. Br. J. Rheumatol. 1996, 35, 808–809. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, P.R.; Doyle, C.T.; Gaffney, A.; Hogan, J.; Hayes, D.P.; Annis, P. Paradoxical response to heparin in 10 patients with ulcerative colitis. Am. J. Gastroenterol. 1995, 90, 220–223. [Google Scholar] [PubMed]
- Evans, R.C.; Shim Wong, V.; Morris, A.I.; Rhodes, J.M. Treatment of corticosteroid-resistant ulcerative colitis with heparin-A report of 16 cases. Aliment. Pharmacol. Ther. 1997, 11, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Mastruzzo, C.; Armato, F.; Tomaselli, V.; Magri, S.; Pistorio, P.; LaMicela, M.; D’Amico, L.; Crimi, N. Intranasal heparin reduces eosinophil recruitment after nasal allergen challenge in patients with allergic rhinitis. J. Allergy Clin. Immunol. 2001, 108, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Bendstrup, K.E.; Jensen, J.I. Inhaled heparin is effective in exacerbations of asthma. Respir. Med. 2000, 94, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Gonzalez, B.J.; Danta, I. Prevention of a exercise-induced bronchoconstriction by inhaled low-molecular-weight heparin. Am. J. Respir. Crit. Care. Med. 1999, 160, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Simanek, E.E.; McGarvey, G.J.; Jablonowski, J.A.; Wong, C.-H. Selectin-carbohydrate interactions: From natural ligands to designed mimics. Chem. Rev. 1998, 98, 833–862. [Google Scholar] [CrossRef] [PubMed]
- Nimrichter, L.; Burdick, M.M.; Aoki, K.; Laroy, W.; Fierro, M.A.; Hudson, S.A.; Von Seggern, C.E.; Cotter, R.J.; Bochner, B.S.; Tiemeyer, M.; et al. E-selectin receptors on human leukocytes. Blood 2008, 112, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Patton, J.T.; Sarkar, A.; Ernst, B.; Magnani, J.L.; Frenette, P.S. GMI-1070, A novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 2010, 116, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Magnani, J.L.; Patton, J.T.; Sarkar, A.K.; Svarovsky, S.A.; Ernst, B. Heterobifunctional Pan-Selectin Inhibitors. Patnent No. WO2007028050 A1, 8 March 2007. [Google Scholar]
- Wun, T.; Styles, L.; DeCastro, L.; Telen, M.J.; Kuypers, F.; Cheung, A.; Kramer, W.; Flanner, H.; Rhee, S.; Magnani, J.L.; et al. Phase 1 study of the E-selectin inhibitor GMI 1070 in patients with sickle cell anemia. PLoS ONE 2014, 9, e101301. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J.; Wun, T.; McCavit, T.L.; De Castro, L.M.; Krishnamurti, L.; Lanzkron, S.; Hsu, L.L.; Smith, W.R.; Rhee, S.; Magnani, J.L.; et al. Randomized phase 2 study of GMI-1070 in SCD: Reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 2015, 125, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Schwizer, D.; Patton, J.T.; Cutting, B.; Smiesko, M.; Wagner, B.; Kato, A.; Weckerle, C.; Binder, F.P.C.; Rabbani, S.; Schwardt, O.; et al. Pre-organization of the core structure of E-selectin antagonists. Chem. Eur. J. 2012, 18, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Somers, W.S.; Tang, J.; Shaw, G.D.; Camphausen, R.T. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLex and PSGL-1. Cell 2000, 103, 467–479. [Google Scholar] [CrossRef]
- Egger, J.; Weckerle, C.; Cutting, B.; Schwardt, O.; Rabbani, S.; Lemme, K.; Ernst, B. Nano-molar E-selectin antagonists with prolonged half-lives by a fragment-based approach. J. Am. Chem. Soc. 2013, 135, 9820–9828. [Google Scholar] [CrossRef] [PubMed]
- Kansas, G.S. Selectins and their ligands: Current concepts and controversies. Blood 1996, 88, 3259–3287. [Google Scholar] [PubMed]
- Fritzsche, J.; Alban, S.; Ludwig, R.J.; Rubant, S.; Boehncke, W.-H.; Schumacher, G.; Bendas, G. The influence of various structural parameters of semisynthetic sulfated polysaccharides on the P-selectin inhibitory capacity. Biochem. Pharmacol. 2006, 72, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Simonis, D.; Fritzsche, J.; Alban, S.; Bendas, G. Kinetic analysis of heparin and glucan sulfates binding to P-selectin and its impact on the general understanding of selectin inhibition. Biochemistry 2007, 46, 6156–6164. [Google Scholar] [CrossRef] [PubMed]
- Alban, S.; Ludwig, R.J.; Bendas, G.; Schoen, M.P.; Oostingh, G.J.; Radeke, H.H.; Fritzsche, J.; Pfeilschifter, J.; Kaufmann, R.; Boehncke, W.-H. PS3, A semisynthetic β-1,3-glucan sulfate, diminishes contact hypersensitivity responses through inhibition of L- and P-selectin functions. J. Investig. Dermatol. 2009, 129, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Weinhart, M.; Groger, D.; Enders, S.; Dernedde, J.; Haag, R. Synthesis of dendritic polyglycerol anions and their efficiency toward L-selectin inhibition. Biomacromolecules 2011, 12, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Groeger, D.; Bergmann, R.; Pietzsch, J.; Steinbach, J.; Graham, B.; Spiccia, L.; Berthon, F.; Czarny, B.; Devel, L.; et al. Synthesis and biodistribution studies of 3H- and 64Cu-labeled dendritic polyglycerol and dendritic polyglycerol sulfate. Bioconjugate Chem. 2015, 26, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.-K.; Lahrsen, E.; Alban, S. Regulation of complement and contact system activation via C1 inhibitor potentiation and factor XIIa activity modulation by sulfated glycans - structure-activity relationships. PLoS ONE 2016, 11, e0165493. [Google Scholar] [CrossRef] [PubMed]
- Beinrohr, L.; Harmat, V.; Dobó, J.; Löerincz, Z.; Gál, P.; Závodszky, P. C1 inhibitor serpin domain structure reveals the likely mechanism of heparin potentiation and conformational disease. J. Biol. Chem. 2007, 282, 21100–21109. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.J.; Oh, Y.I.; Chang, S.-K.; Hsieh-Wilson, L.C. Tunable heparan sulfate mimetics for modulating chemokine activity. J. Am. Chem. Soc. 2013, 135, 10898–10901. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Hansen, S.U.; Miller, G.J.; Cole, C.L.; Rushton, G.; Avizienyte, E.; Gardiner, J.M. Synthetic heparan sulfate dodecasaccharides reveal single sulfation site inter-converts CXCL8 and CXCL12 chemokine biology. Chem. Commun. 2015, 51, 13846–13849. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, M.; Bao, X.; Matsumura, F.; Götze, S.; Kandasamy, J.; Kononov, A.; Broide, D.H.; Nakayama, J.; Seeberger, P.H.; Fukuda, M. Synthetic di-sulfated iduronic acid attenuates asthmatic response by blocking T-cell recruitment to inflammatory sites. PNAS 2014, 111, 8173–8178. [Google Scholar] [CrossRef] [PubMed]
- De Paz, J.L.; Moseman, E.A.; Noti, C.; Polito, L.; Von Andrian, U.H.; Seeberger, P.H. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem. Biol. 2007, 2, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Smith, G.; Vlahov, I.; Abraham, W.M. Inhibition of allergic airway responses by heparin derived oligosaccharides: Identification of a ietrasaccharide sequence. Respir. Res. 2012, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Smith, G.; Abraham, W.M. Heparin-derived supersulfated disaccharide inhibits allergic airway responses in sheep. Pulm. Pharmacol. Ther. 2014, 28, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Shastri, M.D.; Peterson, G.M.; Stewart, N.; Sohal, S.S.; Patel, R.P. Non-anticoagulant derivatives of heparin for the management of asthma: Distant dream or close reality? Expert Opin. Investig. Drugs 2014, 23, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Craciun, I.; Fenner, A.M.; Kerns, R.J. N-arylacyl O-sulfonated aminoglycosides as novel inhibitors of human neutrophil elastase, cathepsin G and proteinase 3. Glycobiology 2016, 26, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Fryer, A.; Huang, Y.-C.; Rao, G.; Jacoby, D.; Mancilla, E.; Whorton, R.; Piantadosi, C.A.; Kennedy, T.; Hoidal, J. Selective O-desulfation produces nonanticoagulant heparin that retains pharmacological activity in the lung. J. Pharmacol. Exp. Ther. 1997, 282, 208–219. [Google Scholar] [PubMed]
- Jaseja, M.; Rej, R.N.; Sauriol, F.; Perlin, A.S. Novel regio- and stereoselective modifications of heparin in alkaline solution. Nuclear magnetic resonance spectroscopic evidence. Can. J. Chem. 1989, 67, 1449–1456. [Google Scholar] [CrossRef]
- Kennedy, T.P. Methods of Treating Acute Excerbations of Chronic Obstructive Pulmonary Disease. U.S. Patent No. 2009/0054374 A1, 26 February 2009. [Google Scholar]
- Larramendy-Gozalo, C.; Barret, A.; Daudigeos, E.; Mathieu, E.; Antonangeli, L.; Riffet, C.; Petit, E.; Papy-Garcia, D.; Barritault, D.; Brown, P.; et al. Comparison of CR36, a new heparan mimetic, and pentosan polysulfate in the treatment of prion diseases. J. Gen. Virol. 2007, 88, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Diamantini, G.; Fedeli, G.; Mantovani, M.; Oreste, P.; Pescador, R.; Porta, R.; Prino, G.; Torri, G.; Zoppetti, G. Retention of antilipemic activity by periodate-oxidized non-anticoagulant heparins. Arzneim.-Forsch. 1986, 36, 637–642. [Google Scholar]
- Casu, B.; Guerrini, M.; Naggi, A.; Perez, M.; Torri, G.; Ribatti, D.; Carminati, P.; Giannini, G.; Penco, S.; Pisano, C.; et al. Short Heparin Sequences Spaced by Glycol-Split Uronate Residues Are Antagonists of Fibroblast Growth Factor 2 and Angiogenesis Inhibitors. Biochemistry 2002, 41, 10519–10528. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Naggi, A. Old and new applications of non-anticoagulant heparin. Int. J. Cardiol. 2016, 212, S14–S21. [Google Scholar] [CrossRef]
- Pomin, V.H. Anticoagulant motifs of marine sulfated glycans. Glycoconj. J. 2014, 31, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Ciancia, M.; Quintana, I.; Cerezo, A.S. Overview of anticoagulant activity of sulfated polysaccharides from seaweeds in relation to their structures, focusing on those of green seaweeds. Curr. Med. Chem. 2010, 17, 2503–2529. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Q.; Abid, M.; Gupta, N.; Tyagi, T.; Ashraf, M.Z.; Jairajpuri, M.A. Polysulfated trehalose as a novel anticoagulant agent with dual mode of action. BioMed Res. Int. 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gracher, A.H.P.; Cipriani, T.R.; Carbonero, E.R.; Gorin, P.A.J.; Iacomini, M. Antithrombin and heparin cofactor II-mediated inactivation of α-thrombin by a synthetic, sulfated mannogalactan. Thromb. Res. 2010, 126, e180–e187. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, D.; Olson, S.T.; Gasiunas, N.; Gallagher, J.; Baglin, T.P.; Huntington, J.A. The Heparin Binding Properties of Heparin Cofactor II Suggest an Antithrombin-like Activation Mechanism. J. Biol. Chem. 2004, 279, 50267–50273. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E. Tissue Factor Pathway Inhibitor: Multiple Anticoagulant Activities for a Single Protein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Schmaier, A.H. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost, 2016, 14, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Long, A.T.; Kenne, E.; Jung, R.; Fuchs, T.A.; Renne, T. Contact system revisited: An interface between inflammation, coagulation, and innate immunity. J. Thromb. Haemost. 2016, 14, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qian, Y.; Zhou, X.; Lu, H.; Ramacciotti, E.; Zhang, L. Chemically Oversulfated Glycosaminoglycans Are Potent Modulators of Contact System Activation and Different Cell Signaling Pathways. J. Biol. Chem. 2010, 285, 22966–22975. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.K.; Viswanathan, K.; Ganguly, T.; Elankumaran, S.; Smith, S.; Pelzer, K.; Lansing, J.C.; Sriranganathan, N.; Zhao, G.; Galcheva-Gargova, Z.; et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N. Engl. J. Med. 2008, 358, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Ramacciotti, E.; Clark, M.; Sadeghi, N.; Hoppensteadt, D.; Thethi, I.; Gomes, M.; Fareed, J. Contaminants in heparin: Review of the literature, molecular profiling, and clinical implications. Clin. Appl. Thromb./Hemost. 2011, 17, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Corbier, A.; Le Berre, N.; Rampe, D.; Meng, H.; Lorenz, M.; Vicat, P.; Potdevin, S.; Doubovetzky, M. Oversulfated Chondroitin Sulfate and OSCS-Contaminated Heparin Cause Dose- and Route-Dependent Hemodynamic Effects in the Rat. Toxicol. Sci. 2011, 121, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Suwan, J.; Martin, J.G.; Zhang, F.; Zhang, Z.; Hoppensteadt, D.; Clark, M.; Fareed, J.; Linhardt, R.J. Oversulfated chondroitin sulfate interaction with heparin-binding proteins: New insights into adverse reactions from contaminated heparins. Biochem. Pharmacol. 2009, 78, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-H.; Rajabi, M.; Chen, T.; Karnaukhova, E.; Kozlowski, S. Oversulfated chondroitin sulfate inhibits the complement classical pathway by potentiating C1 inhibitor. PLoS ONE 2012, 7, e47296. [Google Scholar] [CrossRef] [PubMed]
- Arepally, G.M. Heparin-induced thrombocytopenia. Blood 2017, 129, 2864–2872. [Google Scholar] [CrossRef] [PubMed]
- Tardy-Poncet, B.; Tardy, B.; Grelac, F.; Reynaud, J.; Mismetti, P.; Bertrand, J.C.; Guyotat, D. Pentosan polysulfate-induced thrombocytopenia and thrombosis. Am. J. Hematol. 1994, 45, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Leroux, D.; Canepa, S.; Viskov, C.; Mourier, P.; Herman, F.; Rollin, J.; Gruel, Y.; Pouplard, C. Binding of heparin-dependent antibodies to PF4 modified by enoxaparin oligosaccharides: Evaluation by surface plasmon resonance and serotonin release assay. J. Thromb. Haemost. 2012, 10, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cai, C.; Chandarajoti, K.; Hsieh, P.-H.; Li, L.; Pham, T.Q.; Sparkenbaugh, E.M.; Sheng, J.; Key, N.S.; Pawlinski, R.; et al. Homogeneous low-molecular-weight heparins with reversible anticoagulant activity. Nat. Chem. Biol. 2014, 10, 248–250. [Google Scholar] [CrossRef] [PubMed]















© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, S.; Coombe, D.R. Heparin Mimetics: Their Therapeutic Potential. Pharmaceuticals 2017, 10, 78. https://doi.org/10.3390/ph10040078
Mohamed S, Coombe DR. Heparin Mimetics: Their Therapeutic Potential. Pharmaceuticals. 2017; 10(4):78. https://doi.org/10.3390/ph10040078
Chicago/Turabian StyleMohamed, Shifaza, and Deirdre R. Coombe. 2017. "Heparin Mimetics: Their Therapeutic Potential" Pharmaceuticals 10, no. 4: 78. https://doi.org/10.3390/ph10040078
APA StyleMohamed, S., & Coombe, D. R. (2017). Heparin Mimetics: Their Therapeutic Potential. Pharmaceuticals, 10(4), 78. https://doi.org/10.3390/ph10040078